

In the present study, we demonstrate that ablation of Toll-like receptor 9 (TLR9) causes cardiac rupture after myocardial infarction. Although the TLR9 signaling pathway is not involved in the acute inflammatory response in infarct hearts, it promotes proliferation and differentiation of cardiac fibroblasts during postinfarct repair.
Keywords: Toll-like receptor 9, myocardial infarction, cardiac rupture, inflammation, cardiac fibroblast
Abstract
We have reported that the Toll-like receptor 9 (TLR9) signaling pathway plays an important role in the development of pressure overload-induced inflammatory responses and heart failure. However, its role in cardiac remodeling after myocardial infarction has not been elucidated. TLR9-deficient and control C57Bl/6 wild-type mice were subjected to left coronary artery ligation. The survival rate 14 days postoperation was significantly lower in TLR9-deficient mice than that in wild-type mice with evidence of cardiac rupture in all dead mice. Cardiac magnetic resonance imaging showed no difference in infarct size and left ventricular wall thickness and function between TLR9-deficient and wild-type mice. There were no differences in the number of infiltrating inflammatory cells and the levels of inflammatory cytokine mRNA in infarct hearts between TLR9-deficient and wild-type mice. The number of α-smooth muscle actin (αSMA)-positive myofibroblasts and αSMA/Ki67-double-positive proliferative myofibroblasts was increased in the infarct and border areas in infarct hearts compared with those in sham-operated hearts in wild-type mice, but not in TLR9-deficient mice. The class B CpG oligonucleotide increased the phosphorylation level of NF-κB and the number of αSMA-positive and αSMA/Ki67-double-positive cells and these increases were attenuated by BAY1-7082, an NF-κB inhibitor, in cardiac fibroblasts isolated from wild-type hearts. The CpG oligonucleotide showed no effect on NF-κB activation or the number of αSMA-positive and αSMA/Ki67-double-positive cells in cardiac fibroblasts from TLR9-deficient hearts. Although the TLR9 signaling pathway is not involved in the acute inflammatory response in infarct hearts, it ameliorates cardiac rupture possibly by promoting proliferation and differentiation of cardiac fibroblasts.
Listen to this article’s corresponding podcast @ http://ajpheart.podbean.com/e/tlr9-in-post-infarct-cardiac-rupture/.
NEW & NOTEWORTHY
In the present study, we demonstrate that ablation of Toll-like receptor 9 (TLR9) causes cardiac rupture after myocardial infarction. Although the TLR9 signaling pathway is not involved in the acute inflammatory response in infarct hearts, it promotes proliferation and differentiation of cardiac fibroblasts during postinfarct repair.
revascularization along with pharmacological therapies has improved survival in acute myocardial infarction (MI). However, the patients with MI still remain at substantial risk for ventricular arrhythmia and sudden cardiac death, unpredictable complications of acute MI. A recent study indicated that cardiac rupture and recurrent MI accounts for a higher proportion of sudden death in the early phase of acute MI (23).
MI results in an acute loss of a large number of cardiomyocytes. Cardiomyocyte death triggers a reparative response that leads to the activation of innate immune mechanisms initiating inflammation and ultimately the formation of a collagen-based scar. Macrophages phagocytose the necrotic myocardium and cardiac fibroblasts differentiate into myofibroblasts, proliferate, and migrate into the infarct area (30). This repair process is associated with left ventricular (LV) remodeling (5). Toll-like receptors (TLRs) play a crucial role in the induction of innate immunity and inflammatory responses (11). We have previously shown that the TLR9-mediated inflammatory response plays an important role in the genesis of pressure overload-induced heart failure (21). TLR9 is localized in endosomes and endoplasmic reticulum and recognizes unmethylated 2′-deoxyribo(cytidine-phosphate-guanosine) (CpG) DNA motifs, which are frequently present in bacteria, viruses, and mitochondria but are rare in mammalian nuclei (11, 21).
In the present study, we hypothesized that innate immune responses mediated through the TLR9 signaling pathway might contribute to the repair process after MI. We compared LV remodeling in infarcted hearts in TLR9-deficient (TLR9-KO) mice with that in C57Bl/6 wild-type (WT) mice. The results of the current study identify the protective effect of the TLR9-mediated signaling pathway against cardiac rupture during the early phase after acute MI. Surprisingly, inflammation was not involved in the protective effect of TLR9 in infarct hearts.
MATERIALS AND METHODS
Experimental animals.
All in vivo and in vitro experimental protocols were approved by King’s College London Ethical Review Process Committee and UK Home Office (Project License No. PPL70/7260) and were carried out in accordance with the Guidance on the Operation of the Animals (Scientific Procedures) Act, 1986 (UK Home Office). We used 8- to 10-wk-old male WT C57Bl/6 mice and TLR9-KO mice with a C57Bl/6 background (8). Mice were given food and water ad libitum.
A Vevo 2100 system with a 22- to 55-MHz linear transducer (Visual Sonics, Toronto, Canada) was used to perform echocardiography on conscious mice 1 day before MI operation. Noninvasive measurement of tail blood pressure was also performed on conscious mice using a BP Monitor for Rats and Mice (model MK-2000; Muromachi Kikai, Tokyo, Japan) as previously described (21).
Myocardial infarction.
WT and TLR9-KO mice were subjected to permanent ligation of the left coronary artery ~1 mm below the edge of the left atrial appendage using an 8-0 nylon suture to achieve MI as we previously described (22, 33). Mice were anesthetized with ketamine (75 mg/kg) and medetomidine (1 mg/kg) by intraperitoneal injection before the operation and the full depth of anesthesia was checked by the pedal reflex. The body temperature of mice was maintained constant at 37°C on a warming mat. Mice underwent endotracheal intubation and ventilation using an animal ventilator during the operation. A sham operation without coronary artery ligation was performed on both WT and TLR9-KO mice. Mice were closely monitored postoperatively and reviewed at the end of the day of surgery. Thereafter, mice were monitored daily during the animal study. At the end of the study, mice were euthanized by CO2 exposure followed by cervical dislocation.
Survival analysis.
A survival analysis was performed for the WT and TLR9-KO mice after MI and the sham-operated WT and TLR9-KO mice for the 2-wk study period. The mice showed no clinical cardiorespiratory symptoms until they underwent sudden death. Autopsies were performed on all dead mice to identify the cause of death. Cardiac rupture was indicated by blood coagulation in the chest cavity and a small slit in LV wall (13, 14).
Ischemic area at risk.
Evans blue dye (1%, 0.5 ml) was infused into the heart from the inferior vena cava just after the MI operation. The heart was cut cross-sectionally into six sections. The ischemic area at risk was defined as the ratio of the area lacking Evans blue dye to the whole LV area (32).
Real-time quantitative reverse transcription PCR.
Total RNA was isolated from LVs using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA). The mRNA expression levels were determined by real-time quantitative reverse transcription PCR (qRT-PCR) as previously described (18). Briefly, SuperScript II Reverse Transcriptase (Thermo Fisher Scientific) was used for reverse transcription. qRT-PCR reaction was performed using a Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) and the PCR primers were designed as follows: forward 5′-ACAACCACGGCCTTCCCTACTT-3′ and reverse 5′-CACGATTTCCCAGAGAACATGTG-3′ for interleukin 6 (Il6), forward 5′-TCCCAGGTTCTCTTCAAGGGA-3′ and reverse 5′-GGTGAGGAGCACGTAGTCGG-3′ for tumor necrosis factor-α (Tnfa), forward 5′-AAGAGCTTCAGGCAGGCAGTATCA-3′ and reverse 5′-TAATGGGAACGTCACACACCAGCA-3′ for interleukin-1β (Il1b), forward 5′-ACGCGGACTCTGTTGCTGCT-3′ and reverse 5′-GCGGGACCCCTTTGTCCACG-3′ for collagen type Iα2 (Col1a2), forward 5′-CCCGGGTGCTCCTGGACAGA-3′ and reverse 5′-CACCCTGAGGACCAGGCGGA-3′ for collagen type IIIα1 (Col3a1), forward 5′-GTAAGGCCTGTAGCTGTGCC-3′ and reverse 5′-GTCTCGTTGATTTCTGGGGA-3′ for tissue inhibitor of metalloproteinase 1 (Timp1), forward 5′-CACAGACTTCAGCGAATGGA-3′ and reverse 5′-CTTGGGAAGCTTGAGAGTGG-3′ for Timp2, forward 5′-CAGCCCTGTGATACTTGGGT-3′ and reverse 5′-CAAGCTTCCAGCCAAACTTC-3′ for Timp3, forward 5′-ACCTCCGGAAGGAGTACGTT-3′ and reverse 5′-TTATCTGGCAGCAACACAGC-3′ for Timp4, forward 5′-GATAACCTGGATGCCGTCGTG-3′ and reverse 5′-CTTCACGCTCTTGAGACTTTGGTTC-3′ for matrix metalloproteinase (Mmp) 2, forward 5′-GAAGGCAAACCCTGTGTGTT-3′ and reverse 5′-AGAGTACTGCTTGCCCAGGA-3′ for Mmp9, and forward 5′-ATGACAACTTTGTCAAGCTCATTT-3′ and reverse 5′-GGTCCACCACCCTGTTGCT-3′ for Gapdh. The PCR primers for interleukin-10 (Il10) were purchased from Thermo Fisher Scientific (Mm01288386_m1). For amplification of Il10, the TaqMan Gene Expression Master Mix (Thermo Fisher Scientific) was used. PCR standard curves were constructed using the corresponding complementary DNA and all data were normalized to Gapdh mRNA content and are expressed as fold increase over the control group.
Cardiac magnetic resonance imaging.
Three days after MI, late gadolinium enhanced cardiac magnetic resonance imaging (MRI) was performed on mice using a 7T horizontal MR scanner (Varian, Palo Alto, CA) under monitoring of electrocardiography and respiratory motion as previously described (24, 25). Anesthesia was maintained with 1.5% isoflurane and 98.5% oxygen. Systolic and diastolic frames were obtained, and then LV end-diastolic volume, LV end-systolic volume, and LV ejection fraction were analyzed. Infarct size was evaluated in end-diastolic phase using a semiautomated in-house-developed cardiac preclinical computer software program and expressed as percentage of infarct myocardial mass on total myocardial mass as described previously (25). The average LV wall thickness of 20 different points in an end-diastolic slice at 3 mm from apex was used for the evaluation of LV wall thickness in infarct area, and papillary muscles were excluded from the analysis.
Gelatin zymography.
The MMP2 and MMP9 activities were evaluated in the infarcted LV. The tissue was homogenized in 150 μl of an ice-cold lysis buffer containing Tris·HCl (pH 7.4, 50 mM), NaCl (150 mM), CaCl2 (10 mM), and 0.25% Triton X-100. After centrifugation, the supernatant was collected and a 20-μg amount of protein was used for assay. Gelatin zymography analysis was performed using a Gelatin Zymography kit (PMC-AK47-COS; Cosmo Bio, Tokyo, Japan) according to the manufacturer’s instructions. The results were evaluated by densitometric analysis of the lytic areas obtained from the gelatin electrophoresis using ImageJ software (ver. 1.50; National Insistutes of Health).
Primary cultured cardiac fibroblasts.
LVs were minced into small pieces ~1 mm3 by scissors and digested using collagenase type 2 (Worthington Biochemical, Lakewood, NJ) (17). The collected cells were suspended in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum and seeded on 6-cm tissue culture dishes (Primaria; BD Biosciences, San Jose, CA). The adherent cells were considered as cardiac fibroblasts, and the cells of the first passage were used for the following experiments. The cells were seeded on collagen I (A1048301; Thermo Fisher Scientific)-precoated cover glasses in 24-well plates for 24 h and then subjected to starvation for 12 h using DMEM without FCS. The cells were incubated with ODN1826 (0.1 μg/ml; class B CpG oligonucleotide; tlrl-1826; Invivo Gen, San Diego, CA), ODN2138 (0.1 μg/ml; ODN1826 control; tlrl-1826c; Invivo Gen), or lipopolysaccharide (LPS; 0.1 ng/ml; tlrl-eblps; Invivo Gen) with or without BAY1-7082, an NF-κB inhibitor (tlrl-b82; Invivo Gen), for 24 or 48 h.
Western blot analysis.
Collected cultured cardiac fibroblasts were lysed in homogenization buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% Triton X) with protease inhibitors cocktail (P8340; Sigma-Aldrich, St. Louis, MO) and phosphatase inhibitor cocktail (no. 5870; Cell Signaling Technology, Danvers, MA). Specific antibodies were used for Western blot analysis: α-tubulin (mouse monoclonal, no. 3873; Cell Signaling Technology), NF-κB p65 (rabbit monoclonal, no. 8242; Cell Signaling Technology), and phospho-NF-κB p65 (rabbit monoclonal, no. 3033; Cell Signaling Technology). Incubation with secondary antibodies was followed by developing with an infrared imaging system, ODYSSEY CLx (LI-COR Biosciences, Lincoln, NE). Image Studio software (LI-COR Biosciences) was used for quantitative analysis of each protein expression level.
Histological and cytological analyses.
LV samples were embedded in the OCT compound (Cryomatrix; Thermo Fisher Scientific) and then frozen in liquid nitrogen immediately. The samples were sectioned into 5-μm thickness using a cryostat (Microm, HM560; Thermo Fisher Scientific) and fixed with acetone. The sections were used for hematoxylin-eosin, Masson’s trichrome (Masson’s Trichrome Stain Kit; Polysciences, Warrington, PA), immunohistochemical, and immunofluorescence staining. Cultured cardiac fibroblasts were fixed with 2% paraformaldehyde. The antibodies used were anti-CD45 (MAB114; R&D Systems, Minneapolis, MN), anti-CD68 (MCA1957GA; AbD Serotec, Raleigh, NC), anti-Ly6G/6C (550291; BD Biosciences), anti-CD3 (ab16669; Abcam, Cambridge, MA), anti-fibroblast-specific protein 1 (ab27957; Abcam), anti-α-smooth muscle actin (αSMA) (ab21027; Abcam), and anti-Ki67 (ab15580; Abcam). For immunohistochemical staining, avidin-peroxidase (VECTASTAIN Elite ABC Kit; PK-6100; Vector Laboratories, Burlingame, CA) and DAB peroxidase substrate kit (SK-4100; Vector Laboratories) were used, followed by the counterstaining with hematoxylin as described previously (21). For immunofluorescence staining, secondary antibodies such as Alexa Fluor 568 (A-11057; Life Technologies, Carlsbad, CA) and Alexa Fluor 488 (A-21441; Life Technologies), and DAPI (ProLong Gold Antifade Reagent with DAPI; P36935; Life Technologies) were used. Quantitative analyses of inflammatory cells and myofibroblasts infiltrating into infarct area were examined by counting the number of immunopositive cells in five different areas (magnification, ×200) per section and expressed as the number per millimeters squared (10). All staining of cardiac fibroblasts was performed in duplicate and the number of immunopositive cells was quantitatively analyzed by counting the number of cells in different five areas (magnification: ×200) in each sample.
Statistical analysis.
Results are shown as means ± SE. Paired data were evaluated by Student's t-test, and one-way ANOVA with the Bonferroni's post hoc test was used for multiple comparisons. The Kaplan-Meier method with the log-rank test was used for survival analysis. P < 0.05 was considered statistically significant.
RESULTS
Ablation of Tlr9 increased mortality due to cardiac rupture after MI.
Baseline physiological characteristics such as body weight, systolic blood pressure, and heart rate and echocardiographic parameters were not different between TLR9-KO and control WT mice (Table 1). TLR9-KO and WT mice were subjected to the left coronary artery ligation. Fourteen days after operation, 36.4% of TLR-KO (n = 11) survived, while 88.9% of WT (n = 9) survived (P < 0.05) (Fig. 1A). The mice died suddenly without showing any signs of clinical cardiorespiratory symptoms. Autopsy revealed that all of dead mice in both groups showed signs of cardiac rupture such as blood coagulation in the chest cavity and a small slit in LV wall (Fig. 1B). There was no difference in physiological and echocardiographic parameters at the baseline state before operation between TLR9-KO mice with rupture and without rupture (Table 2).
Table 1.
Baseline characteristics of TLR9-KO mice
| WT Sham (n = 6) | WT MI (n = 9) | TLR9-KO Sham (n = 7) | TLR9-KO MI (n = 11) | |
|---|---|---|---|---|
| Body weight, g | 24.2 ± 0.6 | 24.3 ± 0.4 | 24.6 ± 0.5 | 24.8 ± 0.3 |
| Systolic blood pressure, mmHg | 106 ± 4 | 114 ± 3 | 110 ± 4 | 114 ± 4 |
| Heart rate, beats/min | 726 ± 6 | 724 ± 7 | 701 ± 11 | 700 ± 9 |
| IVSd, mm | 0.79 ± 0.05 | 0.72 ± 0.01 | 0.74 ± 0.02 | 0.75 ± 0.01 |
| LVIDd, mm | 2.89 ± 0.07 | 3.02 ± 0.06 | 3.14 ± 0.12 | 3.13 ± 0.07 |
| LVIDs, mm | 1.54 ± 0.10 | 1.48 ± 0.05 | 1.51 ± 0.09 | 1.60 ± 0.07 |
| LVPWd, mm | 0.76 ± 0.05 | 0.72 ± 0.02 | 0.72 ± 0.03 | 0.73 ± 0.01 |
| LVFS, % | 50.2 ± 1.1 | 51.1 ± 0.9 | 52.1 ± 1.5 | 49.1 ± 1.4 |
Data are expressed as means ± SE. WT, wild type; TLR9-KO, Toll-like receptor 9 knockout; MI, myocardial infarction; IVSd, end-diastolic interventricular septum thickness; LVIDd, end-diastolic left ventricular internal dimension; LVIDs, end-systolic left ventricular internal dimension; LVPWd, end-diastolic left ventricular posterior wall thickness; LVFS, left ventricular fractional shortening. There are no significant differences in all parameters among all groups.
Fig. 1.

A: Kaplan-Meier survival curve after myocardial infarction (MI) in wild-type (WT; n = 9) and Toll-like receptor 9 knockout (TLR9-KO; n = 11) mice analyzed by log rank test. *P < 0.05 vs MI mice in WT group. B: representative image of TLR9-KO hearts with postinfarct cardiac rupture (left), and the scale bar indicates 1 mm. Black arrow indicates the slit of rupture. Black suture used for coronary ligation remains on the heart. Hematoxylin-eosin stain of the ruptured heart (right), and the scale bar = 100 μm. Black arrow indicates rupture site, and “T” indicates thrombus in the left ventricular cavity. C: the ischemic areas at risk evaluated by Evans blue stain in WT (n = 5) and TLR9-KO (n = 6) hearts after left coronary artery ligation were analyzed by Student’s t-test. Data are expressed as means ± SE.
Table 2.
Baseline characteristics of TLR9-KO mice with or without postinfarct rupture
| Nonrupture (n = 4) | Rupture (n = 7) | |
|---|---|---|
| Body weight, g | 25.0 ± 0.6 | 24.7 ± 0.4 |
| Systolic blood pressure, mmHg | 106 ± 4 | 114 ± 3 |
| Heart rate, beats/min | 684 ± 20 | 709 ± 7 |
| IVSd, mm | 0.74 ± 0.02 | 0.75 ± 0.02 |
| LVIDd, mm | 2.98 ± 0.11 | 3.22 ± 0.07 |
| LVIDs, mm | 1.48 ± 0.14 | 1.66 ± 0.07 |
| LVPWd, mm | 0.73 ± 0.02 | 0.73 ± 0.02 |
| LVFS, % | 50.5 ± 3.2 | 48.4 ± 1.3 |
WT, wild type; TLR9-KO, Toll-like receptor 9 knockout; IVSd, end-diastolic interventricular septum thickness; LVIDd, end-diastolic left ventricular internal dimension; LVIDs, end-systolic left ventricular internal dimension; LVPWd, end-diastolic left ventricular posterior wall thickness; LVFS, left ventricular fractional shortening. Data are expressed as means ± SE. There are no significant differences in all parameters between the groups by t-test.
The mortality in WT mice after MI was similar to that in our previous report (33). No sham-operated WT (n = 6) and TLR9-KO (n = 7) mice died (data not shown). Since cardiac rupture occurred on or after postoperative day 4, we analyzed the cardiac phenotypes 1 and 3 days after MI in this study.
Ischemic area at risk, LV morphologies, and LV systolic function were not different between WT and TLR9-KO mice.
Evans blue staining performed just after the MI operation revealed that the ischemic area at risk was not different between WT and TLR9-KO hearts after MI (Fig. 1C). Cardiac MRI performed 3 days after MI revealed that LV geometry such as LV end-diastolic volume and LV end-systolic volume and cardiac function indicated by LV ejection fraction were not different between WT and TLR9-KO mice (Fig. 2, A and B). Furthermore, infarct size and LV wall thickness in the infarct area were similar in both groups (Fig. 2B).
Fig. 2.

Left ventricular (LV) geometry, LV systolic function, infarct size, and LV wall thickness in the infarct areas of WT (n = 4) and TLR9-KO (KO) (n = 4) mice evaluated by cardiac magnetic resonance imaging (MRI) 3 days after myocardial infarction. A: typical cross-sectional images of late gadolinium enhanced cardiac MRI from the apex to the base in end-diastolic phase. Blue area indicates myocardial infarction. Pink area indicates intra-LV cavity. B: quantitative analysis of LV end-diastolic volume, LV end-systolic volume, LV ejection fraction (LVEF), infarct size, and LV wall thickness of the infarct regions. Data were analyzed by Student’s t-test and are expressed as means ± SE.
Noncardiomyocytes infiltrated in the infarct area.
Cardiac inflammation, damage to extracellular matrix proteins, and blunted fibrotic healing constitute the central mechanism of the genesis of cardiac rupture (6). Hematoxylin-eosin and Masson’s trichrome staining indicated that there was no obvious cell infiltration and cardiac fibrosis in sham-operated WT and TLR9-KO mice (Fig. 3, A and B). Cell infiltration in infarct and border areas was observed 1 and 3 days after MI in WT and TLR9-KO mice (Fig. 3A). Cardiac fibrosis in infarct area was negligible in WT and TLR9-KO hearts 1 and 3 days after MI (Fig. 3B). A heart of WT mouse 4 wk after transverse aortic constriction served as a positive control for Masson’s trichrome stain (Fig. 3B).
Fig. 3.

Hematoxylin-eosin and Masson’s trichrome staining in WT and TLR9-KO (KO) hearts 1 day and 3 days after myocardial infarction (MI). A: typical hematoxylin-eosin stain images. B: typical Masson’s trichrome stain images. A heart of WT mouse 4 wk after transverse aortic constriction served as a positive control for Masson’s trichrome stain. S, sham-operated mice; B, border area; I, infarct area; R, remote area; Day 1, 1 day after MI; Day 3, 3 days after MI; P.C., positive control. Scale bars = 100 μm.
The infiltration of inflammatory cells into the infarct area was not different between WT and TLR9-KO mice.
To characterize the infiltrating cells in infarct hearts, we performed immunohistochemical analysis on heart sections. CD45-positive leukocytes increased in the border areas of MI in WT and TLR9-KO hearts 1 day after operation compared with the corresponding sham-operated hearts (P < 0.05), and there were further increases 3 days after MI (P < 0.05) (Fig. 4, A and B). In infarct area, CD45-positive leukocytes increased 3 days after MI in WT and TLR9-KO hearts compared with the corresponding sham-operated hearts (P < 0.05) (Fig. 4, A and B). However, there was no significant difference in the number of infiltrated CD45-positive leukocytes between WT and TLR9-KO mice in each area 1 or 3 days after operation. Leukocytes showed no increase in the remote noninfarct areas both in WT and TLR9-KO mice. The changes in the number of CD68-positive macrophages showed a similar pattern to that of the leukocytes (Fig. 4, A and B). The numbers of CD45-positive and CD68-positive cells indicated that majority of infiltrating leukocytes into the infarct and border areas consisted of CD68-positive macrophages. The number of Ly6G/6C-positive neutrophils increased only in the border areas 1 day and 3 days after MI compared with the corresponding sham-operated hearts (P < 0.05) (Fig. 4, A and B) and showed no difference between WT and TLR9-KO hearts. CD3-positive T cells did not significantly increase after MI in WT or TLR9-KO mice (Fig. 4, A and B). These results suggest that the infiltration of the inflammatory cells into the infarct myocardium in the acute phase after MI was not affected by Tlr9 ablation.
Fig. 4.

Inflammatory cell infiltration in WT and TLR9-KO (KO) hearts after myocardial infarction (MI). A: typical immunohistochemical stain images using anti-CD45 antibody for leukocytes, anti-CD68 for macrophages, anti-Ly6G/6C for neutrophils, and anti-CD3 for T cells. Black arrows indicate CD3-positive cells. S, sham-operated mice; B, border area; I, infarct area; R, remote area; Day 1, 1 day after MI; Day 3, 3 days after MI. Scale bar = 100 μm. B: Quantitative analysis of each infiltrating inflammatory cell-type (n = 4 in each group). Data were analyzed by one-way ANOVA, and are expressed as means ± SE *P < 0.05 vs corresponding sham-operated mice. †P < 0.05 vs border area of the same genotype on Day 1. ‡P < 0.05 vs infarct area of KO hearts on Day 1.
Evaluation of inflammatory cytokine and fibrosis-related gene mRNA levels after MI in TLR9-KO and WT mice.
To further examine inflammatory and fibrotic responses in infarct hearts, we evaluated the levels of inflammatory cytokines, collagens, MMPs, and TIMPs mRNAs. The mRNA levels of proinflammatory cytokines such as Il6, Tnfa, and Il1b and those of the anti-inflammatory cytokine Il10 were elevated after MI compared with corresponding sham-operated mice 1 and/or 3 days after MI, but there were no differences between WT and TLR9-KO hearts (Fig. 5, A–D). These results were in agreement with the results of the immunohistochemical staining in Fig. 4. Col1a2 and Col3a1 mRNA levels were upregulated 3 days after MI compared with the corresponding sham-operated mice, but there were no differences between WT and TLR9-KO hearts (Fig. 5, E and F). This suggests that although collagen transcription was activated 3 days after MI, the deposition of mature collagen was below the detection level by Masson’s trichrome staining (Fig. 3B). MMPs are involved in the healing and remodeling process of LV after MI (4). Activated MMPs can be inhibited by interaction with naturally occurring, specific inhibitors, TIMPs. We examined mRNA expression levels of MMP2, MMP9, and four TIMPs. The MMP2 mRNA level on day 1 after MI in WT hearts was downregulated compared with the corresponding sham-operated hearts. The MMP9 mRNA level on days 1 and 3 after MI in WT hearts was upregulated than those corresponding sham-operated hearts. However, there were no significant differences in MMP2 and MMP9 mRNA levels between WT and TLR9-KO MI hearts. Timp1 mRNA level was elevated on days 1 and 3 after MI in WT hearts and on day 3 in TLR9-KO hearts after MI compared with corresponding controls (Fig. 5I) but showed no difference between WT and TLR9-KO hearts after MI. The Timp2 mRNA level in WT hearts was upregulated on day 3 in comparison with day 1 after MI, while that in TLR9-KO hearts showed no changes after MI (Fig. 5J). As a result, the level of Timp2 mRNA in WT hearts was higher than that in TLR9-KO hearts 3 days after MI (Fig. 5J). Timp3 and Timp4 mRNA levels were not changed after MI and showed no difference between WT and their corresponding TLR9-KO hearts. (Fig. 5, K and L).
Fig. 5.

mRNA expression levels of interleukin-6 (Il6; A), tumor necrosis factor-α (Tnfa; B), interleukin-1β (Il1b; C), interleukin-10 (Il10; D), collagen type Iα2 (Col1a2; E), collagen type IIIα1 (Col3a1; F), matrix metalloproteinase (Mmp) 2 (G), Mmp9 (H), tissue inhibitor of metalloproteinase (Timp) 1 (I), Timp2 (J), Timp3 (K), and Timp4, (L) corrected by Gapdh levels 1 and 3 days after myocardial infarction (MI) in WT and TLR9-KO (KO) hearts (n = 4–7 mice for each group). Day 1: 1 day after MI; Day 3: 3 days after MI. M: the typical image (left) and the result of densitometoric analysis (right, n = 3 mice for each group) of gelatin zymography using heart homogenates on Day 3. Data were analyzed by one-way ANOVA and are expressed as means ± SE *P < 0.05 vs corresponding sham-operated hearts. †P < 0.05 vs MI mice on Day 1 in the same genotype. ‡P < 0.05 vs MI mice in WT group on Day 3.
To evaluate MMP2 and -9 activities, we performed gelatin zymography assay using heart homogenates. MMP2 and -9 were activated in infarct hearts of TLR9-KO and WT mice 3 days after MI (P < 0.05) (Fig. 5M). While MMP9 activity was not different between WT and TLR9-KO hearts after MI, MMP2 activity in TLR9-KO infarct hearts was higher than that in WT infarct hearts (P < 0.05) (Fig. 5M).
Proliferative myofibroblasts increased in WT hearts but not in TLR9-KO hearts after MI.
Myofibroblasts play a central role in fibrotic healing (30). The number of myofbroblasts in infarct TLR9-KO hearts was determined to assess whether the TLR9 signaling has an effect on the proliferation and/or differentiation of myofibroblasts. αSMA and Ki67 are the markers for myofibroblast differentiation and cell proliferation, respectively. The number of αSMA-positive myofibroblasts significantly increased both in border and infarct areas in WT hearts 3 days after MI compared with corresponding sham-operated hearts (P < 0.05) and the same area in KO hearts on 3 days (P < 0.05) but not in infarct TLR9-KO hearts (Fig. 6, A and B). In addition, the number of αSMA/Ki67-double-positive proliferative myofibroblasts increased in border and infarct areas in WT hearts 3 days after MI compared with corresponding sham (P < 0.05), the same area in WT on 1 day (P < 0.05), and the same area in KO on 3 days (P < 0.05) but not in TLR9-KO hearts (Fig. 6, A and B).
Fig. 6.
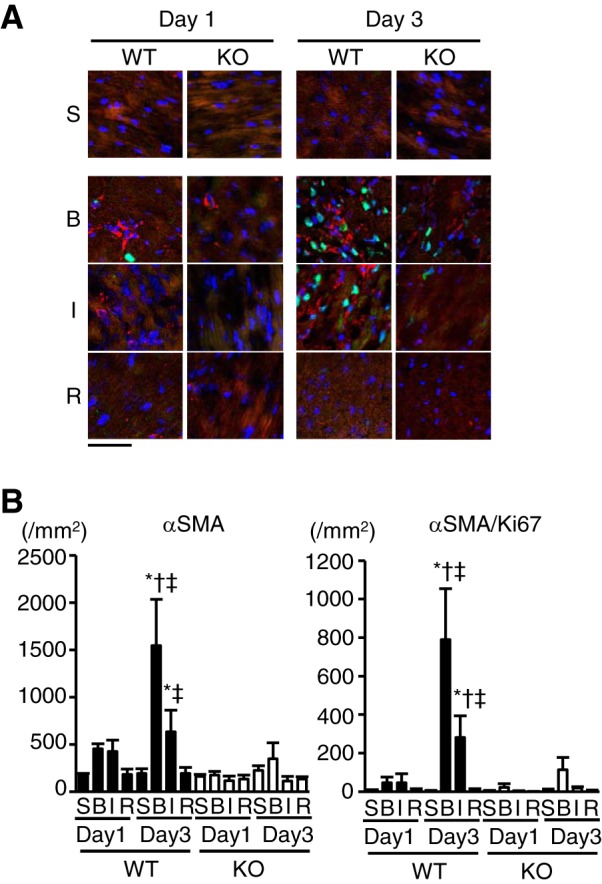
Prevalence of α-smooth muscle actin (αSMA, red)-positive myofibroblasts exhibiting Ki67 (green) immunoreactivity in TLR9-KO (KO) hearts after myocardial infarction (MI) compared with WT hearts. A: typical images of immunofluorescence staining with anti-αSMA and anti-Ki67 antibodies. DAPI-positive nuclei are expressed in blue. The areas of infarct, border and noninfarct remote were separately examined in MI mouse hearts. S, sham-operated mice; B, border area; I, infarct area; R, remote area; Day 1, 1 day after MI; Day 3, 3 days after MI. Scale bar = 50 μm. B: the quantitatively analyzed number of αSMA-positive and αSMA/Ki67-double-positive cells (n = 3–4 for each group). Data were analyzed by one-way ANOVA, and are expressed as means ± SE *P < 0.05 vs corresponding sham. †P < 0.05 vs the same area in WT on Day 1. ‡P < 0.05 vs the same area in KO on Day 3.
TLR9 stimulation promoted proliferation and differentiation of cardiac fibroblasts through the activation of NF-κB pathway.
To investigate the direct relationship between TLR9 signaling and proliferation and differentiation of cardiac fibroblasts, we isolated cardiac fibroblasts from mouse hearts and examined their response to TLR9 stimulation. Almost all the cells were immunopositive for fibroblast-specific protein 1 and had fibroblast-like morphology (Fig. 7A). Synthetic CpG-oligonucleotide (ODN) has been shown to activate TLR9 (8). NF-κB transcription factors, which play important roles in various physiological and pathological conditions, are also known to be activated by CpG-ODN through TLR9 (27, 34). Stimulation with ODN1826, a class B CpG oligonucleotide, induced upregulation of phospho-NF-κB p65 expression over α-tubulin expression in WT cardiac fibroblasts (P < 0.05 vs. control), which was attenuated by 1.0 or 2.0 μM BAY11-7082, an NF-κB inhibitor (P < 0.05) (Fig. 7, B and C). ODN1826 also increased phospho-NF-κB p65 expression over total NF-κB p65 expression (P < 0.05) in WT cardiac fibroblasts, which was attenuated by 0.5, 1.0, or 2.0 μM BAY11-7082 (P < 0.05) (Fig. 7, B and C). ODN1826 did not alter phospho-NF-κB p65 expression in TLR9-KO cardiac fibroblasts (Fig. 7, B and C). ODN control did not affect the NF-κB pathway both in WT or TLR9-KO cardiac fibroblasts (Fig. 7, B and C). From these results, we chose 1.0 μM of BAY11-7082, which is the minimum concentration to obtain significant inhibition of NF-κB activation for the following immunocytochemical analysis. ODN1826 increased the number of αSMA-positive cells (P < 0.05), Ki67-positive cells (P < 0.05), and αSMA/Ki67-double-positive cardiac proliferative myofibroblasts (P < 0.05) compared with control, and these increases were attenuated by the treatment with BAY11-7082 in WT cardiac fibroblasts (P < 0.05) (Fig. 7, D and E). On the other hand, ODN1826 did not increase the number of αSMA-positive or Ki67-positive cells in TLR9-KO cardiac fibroblasts (Fig. 7, D and E). The ODN control did not change the number of αSMA-positive or Ki67-positive cells in WT and TLR9-KO cardiac fibroblasts (Fig. 7, D and E). These results suggest that the TLR9-mediated signaling pathway promotes the differentiation and proliferation of cardiac fibroblasts through the activation of NF-κB pathway.
Fig. 7.

NF-κB activation and prevalence of αSMA and/or Ki67-positive cells in WT and TLR9-KO (KO) cardiac fibroblasts after TLR9 stimulation. A: immunocytochemical stain with anti-fibroblast-specific protein 1 in primary cultured cardiac fibroblasts from WT and KO hearts. Scale bar = 200 μm. B: representative Western blotting images. α-Tubulin (α-tub), NF-κB p65, and phospho-NF-κB p65 expressions were evaluated at 24 h after incubation with ODN1826, class B CpG oligonucleotide (ODN: 0.1 μg/ml), ODN control (ODN-C: 0.1 μg/ml), and BAY11-7082 (BAY0.5: 0.5 μM; BAY1.0: 1.0 μM; BAY2.0: 2.0 μM). Control (Ctl) was incubated with medium without ODN or BAY11-7082. C: quantitative analyses of each protein expression level (n = 3 in each group). D: typical images of immunocytochemical stain. αSMA (red) and Ki67 (green) expressions were evaluated 48 h after incubation with ODN-C (0.1 μg/ml), ODN (0.1 μg/ml) and BAY11-7082 (1.0 μM). DAPI-positive nuclei are expressed in blue. Yellow arrows indicate αSMA and Ki67 double-positive cells. Scale bar = 100 μm. E: quantitative analyses of the percentage of αSMA-positive, Ki67-positive, and αSMA/Ki67-double-positive cells per DAPI-positive cells (n = 3 in each group). Data were analyzed by one-way ANOVA and are expressed as means ± SE *P < 0.05 vs all other groups. †P < 0.05 vs other groups except for BAY0.5.
LPS activated NF-κB and promoted proliferation and differentiation of TLR9-KO cardiac fibroblasts.
To examine the effect of other NF-κB activators than TLR9 signaling pathway on differentiation and proliferation in TLR9-KO cardiac fibroblasts, we stimulated TLR9-KO cardiac fibroblasts with LPS, a TLR4 agonist (2, 28, 31). LPS increased phospho-NF-κB p65 expression over α-tubulin expression (P < 0.05) or phospho-NF-κB p65 expression over total NF-κB p65 expression (P < 0.05) both in WT and TLR9-KO cardiac fibroblasts (Fig. 8, A and B). This increase was attenuated by BAY11-7082 treatment both in WT and TLR9-KO cardiac fibroblasts (P < 0.05) (Fig. 8, A and B). Furthermore, LPS increased the number of αSMA-positive cells (P < 0.05), Ki67-positive cells (P < 0.05), and αSMA/Ki67-double-positive cardiac proliferative myofibroblasts (Fig. 8, C and D). These increases were attenuated by BAY11-7082 treatment (P < 0.05) (Fig. 8, C and D). These results suggest that another NF-κB activator than the TLR9 agonist can differentiate and proliferate TLR9-KO cardiac fibroblasts.
Fig. 8.

NF-κB activation and prevalence of α-smooth muscle actin (αSMA) and/or Ki67-positive cells in WT and TLR9-KO (KO) cardiac fibroblasts stimulated with lipopolysaccharides (LPS). A: representative Western blotting images. α-Tubulin (α-tub), NF-κB p65, and phospho-NF-κB p65 expressions were evaluated at 24 h after incubation with LPS (0.1 ng/ml) and BAY11-7082 (BAY0.5: 0.5 μM, BAY1.0: 1.0 μM, BAY2.0: 2.0 μM, BAY10: 10 μM). Control (Ctl) was incubated with medium without LPS or BAY11-7082. B: quantitative analyses of each protein expression level (n = 3 in each group). C: typical images of immunocytochemical stain. αSMA (red) and Ki67 (green) expressions were evaluated 48 h after incubation with LPS (0.1 ng/ml) and BAY11-7082 (2.0 μM). DAPI-positive nuclei are expressed in blue. Yellow arrows indicate αSMA and Ki67 double-positive cells. Scale bar = 100 μm. D: quantitative analyses of the percentage of αSMA-positive, Ki67-positive, and αSMA/Ki67-double-positive cells per DAPI-positive cells (n = 3 in each group). Data were analyzed by one-way ANOVA and are expressed as means ± SE. *P < 0.05 vs Ctl. †P < 0.05 vs LPS. ‡P < 0.05 vs LPS + BAY0.5.
DISCUSSION
Extensive cardiomyocyte necrosis and LV remodeling occur following coronary artery occlusion. Cardiac repair following MI is dependent on the activation of inflammatory pathways (5). The main objective of the present study was to elucidate the role of TLR9-mediated signaling pathway in the postinfarct LV remodeling process after MI. Our current study demonstrates that TLR9 prevents cardiac rupture in the early phase after MI. This is the first study to report that TLR9 signaling pathway is involved in the genesis of cardiac rupture after MI. Unexpectedly, the TLR9-mediated signaling pathway is not involved in the inflammatory response in the early phase after MI but appears to protect hearts from cardiac rupture by facilitating differentiation of cardiac fibroblasts to myofibroblasts and their proliferation in infarct hearts. In this study, we did not examine cardiac remodeling at the chronic stage, because there will be survivorship bias due to high rupture rate in TLR9-KO mice.
Although the incidence of postinfarct cardiac rupture has been thought to be relatively low, autopsy-proven rupture out of all autopsied patients who died of acute MI is not rare (6). In the VALLIANT trial, 24% of post-MI deaths were due to cardiac rupture as confirmed by autopsy, indicating the lack of effective therapeutics for rupture prevention (6). Thus it is necessary to elucidate the molecular mechanism underlying cardiac rupture after MI to develop strategies for its prevention. A study using the mouse models revealed risk factors for post-MI cardiac rupture such as gender, aging, infarct size, and hemodynamic stress (6). In the clinical setting, most of acute MI patients receive reperfusion therapy. However, we used permanent coronary ligation model to induce MI, since the ischemia-reperfusion model of a specific gene modified mice might result in the different MI size compared with control mice. The cardiac MRI analyses showed that the infarct size, LV systolic function, and LV wall thickness in the infarcted area were not different between WT and TLR9-KO mice. In addition, the ischemic areas at risk were not different between the two groups. Systolic blood pressure was similar in WT and TLR9-KO mice. There was no difference in systolic blood pressure and echocardiographic parameters before myocardial infarction between TLR9-KO mice with cardiac rupture and without cardiac rupture. Therefore, hemodynamic and LV macro-morphological changes are not considered to contribute to the higher incidence of cardiac rupture in TLR9-KO mice. It is unclear which factors would cause the difference between surviving TLR9-KO mice and the dead ones.
The histological data showed the accumulation of leukocytes, including neutrophils and macrophages in infarct hearts after MI. Surprisingly, the accumulation of inflammatory cells into the infarcted myocardium in the acute phase after MI was not affected by Tlr9 ablation. Although the mRNA levels of proinflammatory cytokines such as Il6, Tnfa, and Il1b and the anti-inflammatory cytokine, Il10 were elevated after MI, there were no differences between WT and TLR9-KO hearts. These results of our study suggest that TLR9-independent signaling pathways are dominant, inducing cardiac inflammation in the early phase after MI.
Cardiac fibroblasts comprise one-quarter to two-thirds of mammalian cardiac cells (1). They play a pivotal role in cardiac repair after MI and affect various aspects of cardiac repair responses from deposition of extracellular matrix proteins to scar maturation. Although collagen mRNA levels were already elevated, our histological analysis showed only a negligible level of cardiac fibrosis and no difference between WT and TLR9-KO hearts 3 days after MI. This is in agreement with previous reports that collagen deposition was not obvious by 3 days after MI (12, 26).
During the repair stage after MI, cardiac fibroblasts are activated and undergo differentiation to myofibroblasts with drastic phenotypic and functional changes such as highly proliferative and migratory activity, acquisition of myofibroblast phenotype, and augmented matrix-synthetic capacity (29). It is widely recognized that myofibroblasts are critically involved in the reparative response following MI and are implicated in the pathogenesis of cardiac remodeling (3, 29). In the present study, we found an increased number of myofibroblasts in the infarcted myocardium of WT but not of TLR9-KO. Our in vitro results using isolated cardiac fibroblasts suggest that TLR9 activation directly induces differentiation of cardiac fibroblasts to myofibroblasts and their proliferation through the NF-κB activation. In agreement with our results, TLR9 activation has been shown to stimulate invasion in glioblastoma, astrocytoma and breast cancer epithelial cells (16) and cause differentiation of pulmonary fibroblasts into a myofibroblast phenotype (15). In contrast, Ohm et al. (20) showed that TLR9 stimulation by CpG oligonucleotide attenuated proliferation of cardiac fibroblasts isolated from mouse hearts using a bromodeoxyuridine incorporation assay. In addition, CpG-induced TLR9 activation did not lead to αSMA mRNA induction. Although we do not know the exact reason for this discrepancy, phenotypes of fibroblasts may be altered during cell isolation. However, our in vivo results clearly showed that Tlr9 ablation resulted in the attenuation of proliferation and differentiation of cardiac fibroblasts in infarct hearts. Our in vitro study also showed that NF-κB was activated by TLR9 stimulation in cardiac fibroblasts, and this NF-κB activation can promote the differentiation and proliferation of cardiac fibroblasts.
There are many signals that are able to activate NF-κB signaling in addition to TLR9 pathway. TLR9-KO cardiac fibroblasts as well as WT fibroblast were able to react to other NF-κB activator than TLR9 agonist and differentiate/proliferate through the NF-κB signaling pathway in this study. This appears to be incompatible with the fact that cardiac fibroblast activation/differentiation and proliferation were completely blunted in TLR9 KO hearts. The contribution of each signaling pathway leading to NF-κB activation to cardiac fibroblast differentiation/proliferation may vary with time after myocardial infarction. In the acute phase after MI, TLR9-dependent NF-κB signaling would be important for cardiac fibroblast differentiation and proliferation and prevention of cardiac rupture.
To elucidate the molecular mechanism of how TLR9-induced NF-κB activation promotes the differentiation/proliferation of cardiac fibroblasts, we investigated the involvement of c-myc, which was reported to be upregulated by TLR9 agonist through NF-κB activation and contributed to the differentiation/proliferation in fibroblasts (19, 34). ODN1826 (0.1 μg/ml) stimulation failed to upregulate the c-myc expression compared with control and ODN control groups (data not shown). NF-κB may directly regulate the expression of the genes related to the differentiation/proliferation of fibroblasts or indirectly regulate them mediated though the induction of regulatory proteins. The mechanism of how NF-κB regulates the differentiation and proliferation still needs to be investigated in the future. Thus the protective role of the TLR9-mediated signaling pathway in postinfarct cardiac rupture is related to its function to stimulate differentiation and proliferation of cardiac fibroblasts.
The extracellular matrix is degraded by MMPs, and enhanced MMP activity is known to reduce cardiac stiffness and increase the incidence of cardiac rupture (7, 9, 14). In this study, we found that MMP2 and -9 activities were upregulated in infarct TLR9-KO and WT hearts. The activity of MMP2, but not MMP9, in infarct TLR9-KO hearts was higher than that in infarct WT hearts. The increased MMP2 activity observed in TLR9-KO hearts may contribute to cardiac rupture after MI. TIMPs can bind to pro-MMPs and inhibit their activation. The mRNA level of Timp2, which can inhibit MMP2 activity, was lower in TLR9-KO infarct hearts than that in WT hearts 3 days after MI. Cardiac fibroblasts and myofibroblasts are rich sources of TIMP2 in myocardium (12). Decreased n umbers of myofibroblasts may result in lower Timp2 mRNA expression level and higher activity of MMP2 in TLR9-KO infarct hearts.
The results presented here are in contrast to our previous study regarding the role of TLR9 in pressure overload-induced inflammation and heart failure, in which mitochondrial DNA that escapes from autophagy-medicated degradation activates TLR9 and induces inflammation (21). Thus the role of TLR9 signaling pathway is disease dependent, and each stress-activated intracellular signaling mechanism to induce inflammation has a distinct pathophysiological role during cardiac remodeling. Our previous report suggested long-term beneficial effects of targeted deletion of TLR9 on cardiac function in pressure-overloaded hearts (21). However, the low survival rate in this MI model did not allow for obtaining long-term data.
In conclusion, the inflammatory responses such as proinflammatory cytokine production and inflammatory cell infiltration are not mediated by TLR9 in the early phase after MI. The TLR9 signaling pathway plays a crucial role in the genesis of cardiac rupture in the early phase after MI, probably by facilitating differentiation of cardiac fibroblasts into myofibroblasts and their proliferation through the activation of NF-κB pathway. As the main effector cells of the postinfarction reparative response, the TLR9-mediated signaling pathway in cardiac fibroblasts may be a therapeutic target for the prevention of cardiac rupture in the acute phase after MI, which is fatal and has no specific therapy.
GRANTS
This work was supported by the British Heart Foundation (CH/11/3/29051, RG/11/12/29052, RE/13/2/30182) and Japan Society for the Promotion of Science (JSPS) KAKENHI (15H04822).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.O. conception and design of research; S.O.,Y.O., M.T. performed the experiments and analyzed the data; A.P. performed cardiac MRI; S.O., Y.O., M.T, O.Y., A.S., K.N. and K.O. interpreted results of the experiments; S.O. and Y.O. prepared the figures; S.O., Y.O. and K.O. drafted the manuscript; S.O., Y.O., M.T, A.P., O.Y., S.A., A.S., K.N. and K.O. edited and revised the manuscript; S.O., Y.O., M.T, A.P., O.Y., S.A., A.S., K.N. and K.O. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Ruth Austin for excellent technical assistance.
REFERENCES
- 1.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293: H1883–H1891, 2007. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 2.Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR, Meldrum KK. Toll-like receptor 4: a novel signaling pathway during renal fibrogenesis. J Surg Res 168: e61–e69, 2011. doi: 10.1016/j.jss.2009.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta 1833: 945–953, 2013. doi: 10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res 89: 201–210, 2001. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res 58: 88–111, 2008. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao XM, White DA, Dart AM, Du XJ. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther 134: 156–179, 2012. doi: 10.1016/j.pharmthera.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Hayashidani S, Tsutsui H, Ikeuchi M, Shiomi T, Matsusaka H, Kubota T, Imanaka-Yoshida K, Itoh T, Takeshita A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol 285: H1229–H1235, 2003. doi: 10.1152/ajpheart.00207.2003. [DOI] [PubMed] [Google Scholar]
- 8.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature 408: 740–745, 2000. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 9.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5: 1135–1142, 1999. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123: 594–604, 2011. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384, 2010. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 12.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther 30: 31–41, 2012. doi: 10.1111/j.1755-5922.2010.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest 115: 599–609, 2005. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of p53 prevents cardiac rupture after myocardial infarction in mice. Cardiovasc Res 70: 457–465, 2006. doi: 10.1016/j.cardiores.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Meneghin A, Choi ES, Evanoff HL, Kunkel SL, Martinez FJ, Flaherty KR, Toews GB, Hogaboam CM. TLR9 is expressed in idiopathic interstitial pneumonia and its activation promotes in vitro myofibroblast differentiation. Histochem Cell Biol 130: 979–992, 2008. doi: 10.1007/s00418-008-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merrell MA, Ilvesaro JM, Lehtonen N, Sorsa T, Gehrs B, Rosenthal E, Chen D, Shackley B, Harris KW, Selander KS. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res 4: 437–447, 2006. doi: 10.1158/1541-7786.MCR-06-0007. [DOI] [PubMed] [Google Scholar]
- 17.Mishina H, Watanabe K, Tamaru S, Watanabe Y, Fujioka D, Takahashi S, Suzuki K, Nakamura T, Obata JE, Kawabata K, Yokota Y, Inoue O, Murakami M, Hanasaki K, Kugiyama K. Lack of phospholipase A2 receptor increases susceptibility to cardiac rupture after myocardial infarction. Circ Res 114: 493–504, 2014. doi: 10.1161/CIRCRESAHA.114.302319. [DOI] [PubMed] [Google Scholar]
- 18.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624, 2007. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 19.Nevzorova YA, Hu W, Cubero FJ, Haas U, Freimuth J, Tacke F, Trautwein C, Liedtke C. Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis. Biochim Biophys Acta 1832: 1765–1775, 2013. doi: 10.1016/j.bbadis.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Ohm IK, Alfsnes K, Belland Olsen M, Ranheim T, Sandanger Ø, Dahl TB, Aukrust P, Finsen AV, Yndestad A, Vinge LE. Toll-like receptor 9 mediated responses in cardiac fibroblasts. PLoS One 9: e104398, 2014. doi: 10.1371/journal.pone.0104398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omiya S, Hikoso S, Imanishi Y, Saito A, Yamaguchi O, Takeda T, Mizote I, Oka T, Taneike M, Nakano Y, Matsumura Y, Nishida K, Sawa Y, Hori M, Otsu K. Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J Mol Cell Cardiol 46: 59–66, 2009. doi: 10.1016/j.yjmcc.2008.09.714. [DOI] [PubMed] [Google Scholar]
- 23.Pouleur AC, Barkoudah E, Uno H, Skali H, Finn PV, Zelenkofske SL, Belenkov YN, Mareev V, Velazquez EJ, Rouleau JL, Maggioni AP, Køber L, Califf RM, McMurray JJ, Pfeffer MA, Solomon SD; VALIANT Investigators . Pathogenesis of sudden unexpected death in a clinical trial of patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. Circulation 122: 597–602, 2010. doi: 10.1161/CIRCULATIONAHA.110.940619. [DOI] [PubMed] [Google Scholar]
- 24.Protti A, Dong X, Sirker A, Botnar R, Shah AM. MRI-based prediction of adverse cardiac remodeling after murine myocardial infarction. Am J Physiol Heart Circ Physiol 303: H309–H314, 2012. doi: 10.1152/ajpheart.00208.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Protti A, Sirker A, Shah AM, Botnar R. Late gadolinium enhancement of acute myocardial infarction in mice at 7T: cine-FLASH versus inversion recovery. J Magn Reson Imaging 32: 878–886, 2010. doi: 10.1002/jmri.22325. [DOI] [PubMed] [Google Scholar]
- 26.Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, Kass DA. Cardiomyocyte-specific transforming growth factor β suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res 114: 1246–1257, 2014. doi: 10.1161/CIRCRESAHA.114.302653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhee JW, Lee KW, Sohn WJ, Lee Y, Jeon OH, Kwon HJ, Kim DS. Regulation of matrix metalloproteinase-9 gene expression and cell migration by NF-kappa B in response to CpG-oligodeoxynucleotides in RAW 264.7 cells. Mol Immunol 44: 1393–1400, 2007. doi: 10.1016/j.molimm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Rohde D, Schön C, Boerries M, Didrihsone I, Ritterhoff J, Kubatzky KF, Völkers M, Herzog N, Mähler M, Tsoporis JN, Parker TG, Linke B, Giannitsis E, Gao E, Peppel K, Katus HA, Most P. S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via Toll-like receptor 4. EMBO Mol Med 6: 778–794, 2014. doi: 10.15252/emmm.201303498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 7: 30–37, 2010. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 30.Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol 163: 2433–2440, 2003. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Tu Q, Yan W, Xiao D, Zeng Z, Ouyang Y, Huang L, Cai J, Zeng X, Chen YJ, Liu A. CXC195 suppresses proliferation and inflammatory response in LPS-induced human hepatocellular carcinoma cells via regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem Biophys Res Commun 456: 373–379, 2015. doi: 10.1016/j.bbrc.2014.11.090. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe T, Otsu K, Takeda T, Yamaguchi O, Hikoso S, Kashiwase K, Higuchi Y, Taniike M, Nakai A, Matsumura Y, Nishida K, Ichijo H, Hori M. Apoptosis signal-regulating kinase 1 is involved not only in apoptosis but also in non-apoptotic cardiomyocyte death. Biochem Biophys Res Commun 333: 562–567, 2005. doi: 10.1016/j.bbrc.2005.05.151. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S, Takeda T, Watanabe T, Asahi M, Taniike M, Matsumura Y, Tsujimoto I, Hongo K, Kusakari Y, Kurihara S, Nishida K, Ichijo H, Hori M, Otsu K. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci USA 100: 15883–15888, 2003. doi: 10.1073/pnas.2136717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi AK, Krieg AM. CpG DNA rescue from anti-IgM-induced WEHI-231 B lymphoma apoptosis via modulation of I kappa B alpha and I kappa B beta and sustained activation of nuclear factor-kappa B/c-Rel. J Immunol 160: 1240–1245, 1998. [PubMed] [Google Scholar]