

Abstract
Long noncoding RNAs (lncRNAs) are emerging as crucial players in a myriad of biological processes. However, the precise mechanism and functions of most lncRNAs are poorly characterized. In this study, we presented genome-wide identification of lncRNAs in the patients with intervertebral disc degeneration (IDD) and spinal cord injury (control) using RNA sequencing (RNA-seq). A total of 124.6 million raw reads were yielded using Hiseq 2500 platform and approximately 88% clean reads could be aligned to human reference genome in both IDD and control groups. RNA-seq profiling indicated that 1,854 lncRNAs were differentially expressed (log2 fold change ≥ 1 or ≤−1, p < 0.05), in which 1,530 could potentially target 6,386 genes via cis-regulatory effects. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for these target genes suggested that lncRNAs were involved in diverse pathways, such as lysosome, focal adhesion, and MAPK signaling. In addition, a competing endogenous RNA (ceRNA) network was constructed for analyzing the function of lncRNAs. Further, quantitative real time PCR (qRT-PCR) was used to confirm the differentially expressed lncRNAs and ceRNA network. In conclusion, our results present the first global identification of lncRNAs in IDD and may provide candidate diagnostic biomarkers for IDD treatment.
1. Introduction
Intervertebral disc degeneration (IDD) is implicated as the major contributor of low back pain, which inflicts global burden with severe health care [1, 2]. Aetiology of IDD is complex, with both environmental and genetic factors playing roles in the disease process [3, 4]. For example, genetic polymorphisms in a number of genes, such as collagen I, collagen IX, collagen XI, aggrecan, extracellular matrix-degrading enzymes, inflammatory cytokines (IL-1, IL-6, and TNFα), Fas/FasL, and vitamin D receptors, have been associated with an increased risk of IDD [3]. Moreover, aberrant expression of noncoding RNAs (ncRNAs) has also been reported to be involved in the occurrence of IDD [5, 6].
NcRNAs are transcripts without protein-coding capacity, which are largely grouped into two major classes based on transcript size, small ncRNAs with length less than 200 nucleotides (nt), and long ncRNAs (lncRNAs, >200 nt) [7]. Small ncRNAs include many types, such as microRNAs (miRNAs), small interfering RNAs (siRNAs), and PIWI-interacting RNAs (piRNA), which have regulatory roles during diverse biological events [8]. Among these small ncRNAs, miRNAs are studied extensively in IDD, and we compared the expression of miRNAs between IDD and spinal cord injury specimens in previous study [9]. LncRNAs are initially considered as nonfunctional byproducts of RNA polymerase II transcripts and have been paid much attention recently because they may act as important cis- or trans-regulators in a wide variety of biological functions, such as gene expression, genome imprinting, chromatin modification, and epigenetic regulation as well [10–12]. Moreover, lncRNAs have been applied in the treatment of disease, such as cancer, neurodegenerative, and psychiatric diseases [13, 14].
Recently, Wan et al. have applied microarray method to identify 1,806 significantly differentially expressed lncRNAs in IDD compared to spinal cord injury group [15], whereas, unlike microarray method detecting known lncRNAs from an RNA pool [16], RNA sequencing (RNA-seq) is independent of currently available genome annotation or sequence and can be used to discover previously unknown lncRNAs in an unbiased manner [17].
In current study, we performed a comprehensive transcriptome analysis in IDD and control groups using RNA-seq and identified lncRNAs with differential expression between them. To explore the function of lncRNAs, we predicted their potential targets with cis-regulatory effects, which were then put into gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) for further analysis. In addition, it has been reported that lncRNA may function as competing endogenous RNA (ceRNA) in regulating gene expression. These lncRNAs could share miRNA response elements (MREs) with the transcripts of specific mRNAs and thus affect expression of these mRNAs [18, 19]. In combination with our previous miRNA profile [9], we constructed a ceRNA network to analyze the potential functions of lncRNAs. Furthermore, 10 differentially expressed lncRNAs and ceRNA network were confirmed by quantitative real time PCR (qRT-PCR). Taken together, our results may provide more candidate diagnostic biomarkers and treatment targets for IDD patients.
2. Materials and Methods
2.1. Sample Collection
The central nucleus pulposus (NP) was collected from patients with IDD (n = 6; average age 48.3, range 29–63 years) and spinal cord injury as control (n = 6; average age 44.5, range 29–57 years) (Table 1). The degree of disc degeneration was evaluated via magnetic resonance imaging (MRI) scan according to Pfirrmann grading classification [20].
Table 1.
Basic specimen's information.
| Sample | Age | Gender | MRI grade |
|---|---|---|---|
| IDD 1 | 55 | F | V |
| IDD 2 | 60 | F | IV |
| IDD 3 | 63 | F | V |
| IDD 4 | 29 | M | IV |
| IDD 5 | 34 | M | IV |
| IDD 6 | 49 | M | V |
| Control 1 | 36 | F | II |
| Control 2 | 47 | F | II |
| Control 3 | 29 | M | II |
| Control 4 | 43 | M | II |
| Control 5 | 55 | M | II |
| Control 6 | 57 | M | II |
F, female; M, male; MRI, magnetic resonance imaging.
This study was approved by the Human Ethics Committees Review Board at Xi'an Jiaotong University, Xi'an, China, and written informed consent was obtained from all participants.
2.2. RNA Extraction and Sequencing
Total RNA from six IDD and six spinal cord injury samples were extracted separately using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. Then, DNase I was added to remove contaminating genomic DNA. RNA quality and quantity were measured using a NanoDrop spectrophotometer (Thermo Scientific, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA). RNA integrity was determined by 1% gel electrophoresis. Equal amounts of total RNA from the IDD and spinal cord injury samples were pooled into experimental and control groups, respectively.
As for high throughout sequencing, ribosomal RNA (rRNA) was depleted from total RNA using the Ribo-Zero™ rRNA Removal Kit (Human/Mouse/Rat; Epicentre, USA) according to manufacturer's protocol. The cDNA libraries were prepared using an ScriptSeq™ v2 RNA-Seq Library Preparation Kit (Epicentre, USA) and were sequenced on Illumina HiSeq 2500 with 101 bp paired-end reads at the YingBio Tech, Shanghai, China.
2.3. RNA-Seq Reads Mapping and Transcriptome Assembly
The raw reads were first filtered to remove the adapter sequences and low-quality sequences using Trim Galore [21]. Next, clean reads were mapped to the human GRCh37 reference genome using TopHat [22]. To construct transcriptome, the mapped reads were assembled using Cufflinks [23]. The cuffcompare program was used to merge the RefSeq, Ensembl, Gencode, UCSC, Noncode, and Lncipedia human-known genes into one set of gene annotation for comparison with the assembled transcripts [23].
2.4. Pipeline for the Identification of lncRNA
To identify novel reliable lncRNAs from IDD, we employed a highly stringent pipeline to remove transcripts with evidence for protein-coding potential (Figure 1). Firstly, single-exon transcripts and the transcripts with length less than 200 nt were filtered. Next, three independent algorithms, coding potential calculator (CPC), coding potential assessment tool (CPAT), and phylogenetic codon substitution frequency (PhyloCSF), were applied to extract high reliable potential noncoding transcripts. A positive CPC score indicated a protein-coding potential transcript, whereas CPC value < 0 was considered as noncoding transcripts [24]. The CPAT coding probability score for protein determination varied from 0.364 to 0.44 for human [25], and negative PhyloCSF score indicated noncoding transcripts [26]. Here, we selected a quite stringent threshold for PhyloCSF score < −20 as ncRNA. Further, transcripts with CPC < 0, CPAT < 0.364, and PhyloCSF < −20 that encoded any protein domains cataloged in the Pfam database were filtered out utilizing HMMER software [27].
Figure 1.

Pipeline for the identification of lncRNAs based on RNA-seq.
2.5. LncRNA Classification
Depending on their relationships with the neighboring protein-coding genes, the identified lncRNAs can be classified to six categories: (1) sense or (2) antisense, the lncRNA transcript overlaps one or more exons of another transcript in the same or opposite DNA strand, respectively; (3) bidirectional, expression of lncRNAs is in the same direction as a neighboring coding transcript in the same chain; (4) intronic, lncRNAs derive wholly from within an intron of a second transcript; (5) intergenic, lncRNAs lie within the genomic interval between two genes; (6) small RNA (sRNA) host lncRNA [7, 28]. The differentially expressed lncRNAs were annotated with the following priority: sRNA host lncRNA > intronic lncRNA > sense lncRNA > antisense lncRNA > bidirectional lncRNA > intergenic lncRNA.
2.6. Differential Expression Analysis
The lncRNA and mRNA sequence reads of the IDD and control groups were normalized to fragments per kilobase of transcript per million mapped reads (FPKM) values [29]. Cuffdiff 2.0 was used to detect differentially expressed lncRNAs between the IDD and control groups (log 2 fold change (FC) ≥ 1 or ≤−1, p < 0.05) [23]. Those genes differentially expressed in IDD might be IDD specific genes.
2.7. GO and Pathway Analysis
For each differential expressed lncRNA, the nearest upstream and downstream (within 10 kb) protein-coding neighbors were identified as their cis-regulatory potential targets. To explore the roles of these target genes, we performed GO (http://www.geneontology.org) and pathway analysis. GO terms are comprised of biological process (BP), cellular component (CC), and molecular function (MF). Pathway analysis is a functional analysis that maps genes to the KEGG (http://www.genome.jp/kegg/) pathways. KEGG allowed us to determine the biological pathways that there is a significant enrichment of differential expressed mRNAs.
2.8. Construction of the ceRNA Network
In our previous study, 51 miRNAs were found to be differentially expressed in the IDD group compared with the spinal cord injury group [9]. To construct the ceRNA network, the interactions between differentially expressed lncRNAs and miRNAs were predicted by miRcode (http://www.mircode.org/) at first, and then, RNAs that were targeted by miRNAs with luciferase reporter assay support were from TarBase (http://www.microrna.gr/tarbase).
2.9. Validation by qRT-PCR
LncRNA and mRNA expression in IDD and control groups from the RNA-seq data analysis was validated by qRT-PCR by using SYBR-Green PCR Master Mix (Roche) in a ABI Q6 real time PCR instrument. Specific primers of each gene designed using Primer 5.0 were listed in Supplementary Table 1 in Supplementary Material available online at http://dx.doi.org/10.1155/2016/3684875. Thermal cycling conditions consisted of an initial denaturation step at 95°C for 10 min and then 40 cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. The lncRNAs and mRNAs expression levels were normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Expression fold change and expression level were calculated using 2−ΔΔCt method [30].
3. Results
3.1. RNA Sequencing and Transcriptome Reconstruction
Two cDNA preparations were sequenced from IDD and spinal cord injury groups. In total, 63,592,624 and 61,015,766 raw reads were generated from control and IDD RNA libraries, respectively. After discarding low-quality reads and adaptor sequences, we obtained approximately 59 million clean reads in both libraries. To identify putative lncRNAs from deep sequencing data, we developed a stringent filtering pipeline (Figure 1). At the beginning, clean reads were aligned to the human GRCh37 reference genome using TopHat [22]. Approximately 88% of the reads were aligned onto the human genome and 42.3% were uniquely mapped in both groups (Table 2). Then, transcripts were reconstructed using Cufflinks [29]. The reconstructed transcripts were annotated using the cuffcompare program from the Cufflinks package. Further, putative lncRNAs were screened from unknown transcripts according to the analysis work flow shown in Figure 1. Finally, we obtained 177,975 lncRNAs, of which 63 were novel. The average length of putative novel lncRNAs was 1,067 nt, varying from 205 (XLOC_001763) to 6217 nt (XLOC_037168) (Supplementary Table 2).
Table 2.
Summary of data from RNA-seq for IDD and control groups.
| Sample | Raw reads | Clean reads | Total mapped | Unique mapped |
|---|---|---|---|---|
| IDD | 61,015,766 | 58,911,634 (100%) | 51,929,341 (88.1%) | 24,934,606 (42.3%) |
| Control | 63,592,624 | 58,911,630 (100%) | 51,932,658 (88.2%) | 24,918,205 (42.3%) |
3.2. Differentially Expressed lncRNAs and mRNA Analysis
The transcript expression levels were normalized to FPKM values by the Cufflinks software. The volcano plots show the differential expression of transcripts in IDD and the control groups (Supplementary Figure 1). 1,854 lncRNAs (916 lncRNAs were upregulated and 938 lncRNAs downregulated) were significantly differentially expressed (Supplementary Table 3). Here, we presented the top 10 downregulated and upregulated mRNA and lncRNAs in Table 3. Among these, NONHSAT031859 (log2FC = 6.04) was the most significantly upregulated and NONHSAT006310 (log2FC = −7.58) was the most significantly downregulated. To classify the differentially expressed lncRNAs according to their position with protein-coding genes, we identified that vast majority were sense lncRNA (1302), followed by intergenic lncRNA (252), intronic lncRNA (134), sRNA host lncRNA (64), antisense lncRNA (59), and bidirectional lncRNA (35), as well as eight lncRNAs that could not be classified well (Supplementary Table 3).
Table 3.
The top 10 upregulated and downregulated lncRNAs between IDD and control groups.
| Upregulated lncRNAs | Downregulated lncRNAs | ||||
|---|---|---|---|---|---|
| Gene ID | Log2FC | Database | Gene ID | Log2FC | Database |
| NONHSAT031859 | 6.04 | Noncode | NONHSAT006310 | −7.58 | Noncode |
| ENSG00000103855 | 5.95 | Ensembl | XLOC_027116 | −6.328 | Predicted |
| CALHM2 | 5.72 | Refgene | IL10RA | −6.10 | Refgene |
| LOC100506325 | 5.52 | Refgene | ENSG00000113594 | −5.88 | Ensembl |
| ENSG00000242086 | 5.50 | Ensembl | ENSG00000071282 | −5.72 | Ensembl |
| NONHSAG009780 | 5.37 | Noncode | ENSG00000264187 | −5.58 | Ensembl |
| NONHSAT091589 | 5.03 | Noncode | PRR24 | −5.30 | Refgene |
| NONHSAT024268 | 4.68 | Noncode | NONHSAT024510 | −5.174 | Noncode |
| ENSG00000162576 | 4.42 | Ensembl | ENSG00000088035 | −4.54 | Ensembl |
| NONHSAT005499 | 4.21 | Noncode | ENSG00000014914 | −4.54 | Ensembl |
In total, 2,804 (1,444 upregulated and 1,360 mRNAs downregulated) mRNAs were significantly differentially expressed in IDD compared with the control group (Supplementary Table 4). The top 10 downregulated and upregulated mRNAs were presented in Table 4.
Table 4.
The top 10 upregulated and downregulated mRNAs between IDD and control groups.
| Upregulated mRNAs | Downregulated mRNAs | ||
|---|---|---|---|
| Gene symbol | Log2FC | Gene symbol | Log2FC |
| EPB41L2 | 8.87 | SIPA1L2 | −7.60 |
| SH3BP2 | 7.55 | KMT2E | −5.52 |
| ATM | 7.42 | PAAF1 | −5.69 |
| BTBD18 | 6.74 | ARMC10 | −5.07 |
| PRRC2C | 6.65 | RTN4 | −4.96 |
| LPP | 6.21 | MAP3K3 | −4.83 |
| CEP192 | 6.20 | MEF2A | −4.81 |
| NCSTN | 6.00 | DIP2A | −4.77 |
| PDZD2 | 5.49 | SLC5A3 | −4.74 |
| PRKCSH | 5.44 | POC5 | −4.68 |
3.3. Prediction of the Potential Target Genes of lncRNAs
It was considered that many lncRNAs function as cis-regulators, given that the expression of lncRNA is significantly correlated with their neighboring protein-coding genes as potential targets [31]. Here, we found that 1,530 differentially expressed lncRNAs were transcribed near (<10 kb) their protein-coding neighbor, and a total of 6,386 putative targets were collected (Supplementary Table 5). The number of putative targets for each lncRNA varied greatly. For example, ENST00000565580 had 30 target genes, maximum among these lncRNAs, followed by ENST00000523461 and ENST00000544639 with 27 and 26 target genes, respectively, which might indicate that these lncRNAs conferred to a broad-spectrum regulation. Simultaneously, 207 lncRNAs targeted only one gene, which might suggest a unique regulatory function played by these lncRNAs. To further understand the functions and associated pathways of these target genes, we performed GO and pathway analyses. The enriched GO terms of these potential targets involved many basic biological events, such as intracellular, protein binding, and cellular protein metabolic process (Figure 2(a)) (Supplementary Table 6). Based on our KEGG pathway analysis, the most enriched pathways corresponding to the differentially expressed lncRNAs were associated with lysosome, RNA transport, and aminoacyl-tRNA biosynthesis (Figure 2(b)) (Supplementary Table 7).
Figure 2.

Functional enrichment analysis of target genes of differentially expressed lncRNAs based on GO (a) and KEGG (b).
3.4. ceRNA Network Construction
A number of studies have demonstrated that some lncRNAs can serve as miRNA sponges to interact competitively with miRNAs to inhibit miRNA availability to mRNAs. Our previous results demonstrated that 25 miRNAs were upregulated and 26 were downregulated in the IDD group compared with the spinal cord injury group [9]. To examine the posttranscriptional regulatory function of differentially expressed lncRNAs in IDD, we constructed a lncRNA-miRNA-mRNA ceRNA network in combination with previous miRNA expression profile (Figure 3). In this ceRNA network, two downregulated miRNAs, has-miR-34a and has-miR-148a, were negatively correlated with their corresponding target genes E2F3 and VEGFA and ACVR1, respectively. Moreover, upregulated lncRNA PART1 harbor potential MRE for both has-miR-34a and has-miR-148a.
Figure 3.

ceRNA network in intervertebral disc degeneration. Round nodes represent mRNA, triangle nodes represent lncRNAs, diamond nodes represent miRNAs; violet edges indicate miRNA-target interactions; green represents upregulation, and red represents downregulation.
3.5. Verification of RNA-Seq Data by qRT-PCR
To confirm the expression patterns of lncRNAs identified by RNA-seq, 10 differentially expressed lncRNAs including five upregulated lncRNAs and five downregulated ones were selected for validation by qRT-PCR in 12 lumbar disc samples. Of these 10 lncRNAs, eight showed the same trends of up- and downregulation as the sequencing data (Figure 4(a)). In addition, expression level of mRNAs and lncRNA in ceRNAs was also verified by qRT-PCR (Figure 4(b)). Moreover, in the 12 lncRNAs and mRNAs, expression level of 9 genes showed significantly statistical differences between IDD group and control (Figure 5) (p < 0.05). Taken together, our these results showed good correlation between qRT-PCR and RNA-seq data and different expression level of genes.
Figure 4.
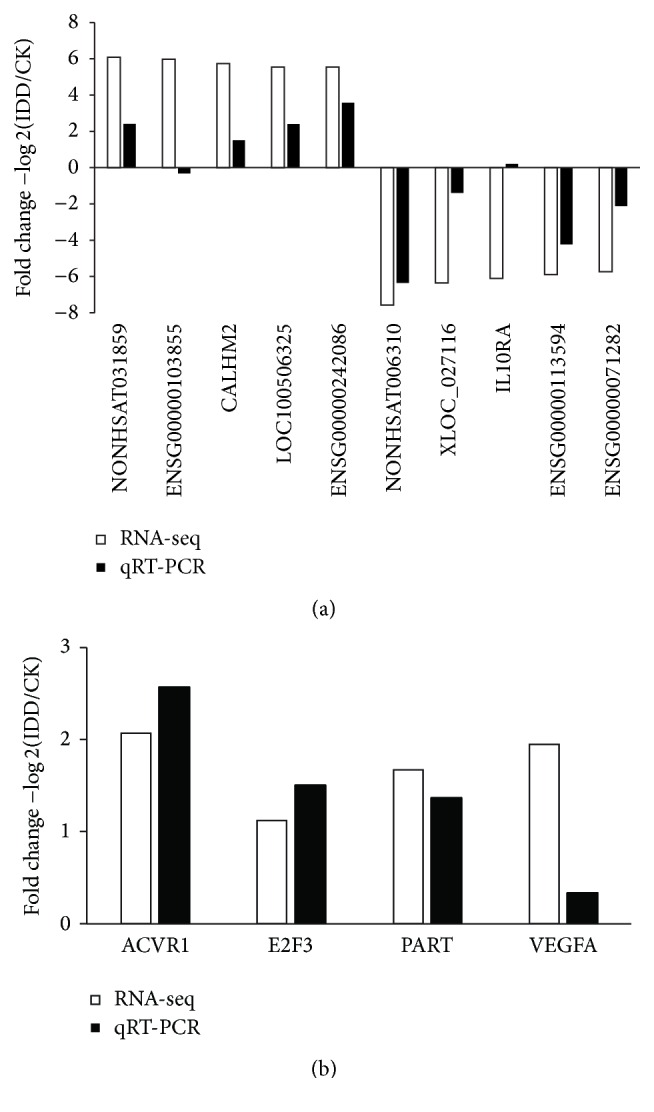
Validation of RNA-seq data by qRT-PCR. (a) 10 lncRNAs differentially expressed in IDD in comparison with control by RNA-seq were verified by qRT-PCR. (b) qRT-PCR analysis of expression levels of lncRNA (PART1) and mRNAs (ACVR1, E2F3, and VEGFA) in ceRNA network.
Figure 5.
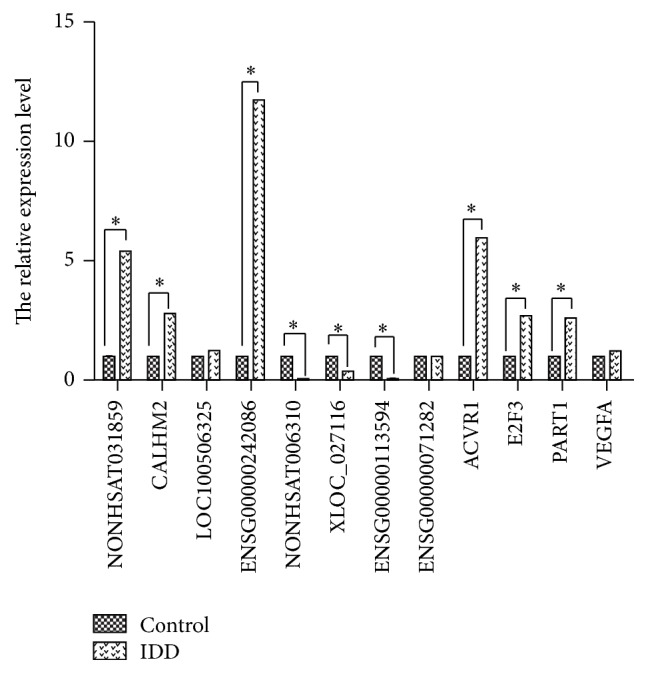
The relative expression level of 12 lncRNAs and mRNAs. The first four differentially expressed lncRNAs were upregulated in IDD, the second four differentially expressed lncRNAs were downregulated in IDD, and the third four differentially expressed genes were one lncRNA (PART1) and three mRNAs (ACVR1, E2F3, and VEGFA) in ceRNA network. The asterisk (∗) means significantly different expression between IDD group and control.
4. Discussion
In this study, we present the comprehensive identification and analysis of IDD specific lncRNAs using RNA-seq. Transcript predicted to have coding potential filtered by CPAT, CPC, and PhyloCSF, respectively. Thereby more reliable putative novel lncRNAs were attained. We identified 1,854 lncRNAs and 2,804 protein-coding genes which are differentially expressed in IDD group compared with the spinal cord injury group. In addition, the expression levels of 10 lncRNAs with significant differential expression in the IDD were verified by qRT-PCR.
Comparing our results with previous microarray study [20], the differentially expressed lncRNAs were totally different. However, we found that differentially expressed mRNAs showed partial overlap (Supplementary Table 8). For example, CHN1 and MAD1L1 exhibited quite similar upregulation and downregulation in two studies, respectively, while DGKZ and PTP4A3 were much more induced in RNA-seq data. Moreover, KEGG pathway analysis of the differentially expressed mRNAs was also partially overlaid, including lysosome, focal adhesion, and ubiquitin mediated proteolysis (Supplementary Figure 2) [15]. Samples taken from patients of different ages and high throughput technology may explain this big difference. Microarray is greatly dependent on designed probes and hence cannot comprehensively characterize dynamic and relatively low expression of lncRNAs. Also, lncRNAs have strong tissue specificity, and many lncRNAs are not identified at present [16, 32].
The functions of lncRNAs could be indirectly reflected by the functions of neighbor protein-coding genes. To better understand the regulatory roles of the putative lncRNA candidates, we also employed GO and KEGG to analyze the function of potential target genes of lncRNAs. Among the top 20 enriched KEGG terms, lysosome, endocytosis, and phagosome were relevant to autophagy which is a catabolic process involving the degradation of dysfunctional cellular components by lysosomal systems [33]. Ma et al. have reported that autophagy may play an important role in IDD, and silent information regulation 2 homolog-1 (SIRT1) protected degenerative human nucleus pulposus (NP) cells against apoptosis via promotion of autophagy [34]. An array of cardiovascular risk factors such as age, smoking, hypertension, high cholesterol, and diabetes has been reported to be related to IDD and back pain [35]. Cardiovascular risk related pathways were enriched in our results, including dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy (ARVC), and hypertrophic cardiomyopathy (HCM). Intervertebral disc is the largest avascular organ in the human body and has been identified as immune-privileged organ [36]. Exposure of NP to the immune system is able to evoke autoimmune reaction, which play an essential role in pathophysiology of IDD [37, 38]. Epithelial cell signaling in Helicobacter pylori infection, Staphylococcus aureus infection, and antigen processing and presentation might be associated with the breakdown of immune privilege resulting in IDD at last. Further, it has been reported that p38 MAPK gene expression is upregulated in senescent human annulus fibrosus (AF) cells compared to nonsenescent cells [39].
Both miRNA and lncRNA have been shown to be associated with IDD. In combination with our previous miRNA profile, we constructed a ceRNA network, of which upregulated lncRNA PART1 and downregulated has-miR-34a and has-miR-148a were the core elements.
In conclusion, the identified lncRNAs that are differentially expressed in IDD and control samples could have functional roles in IDD development. This study provides novel insights into the discovery and annotation of IDD development-related lncRNAs in mammalian genome.
Supplementary Material
Supplementary Figure 1. Differential expression of transcripts between IDD and control groups. Volcano plots of log10 (p-value) versus log2 fold change. Red dots indicated the differentially expressed transcripts between IDD and control groups. Supplementary Figure 2. Functional enrichment analysis of differentially expressed mRNAs between IDD and control groups based on KEGG. Supplementary Table 1. Prime list for qRT-PCR Supplementary Table 2. Information of novel lncRNA Supplementary Table 3. Information of differentially expressed lncRNAs Supplementary Table 4. Information of differentially expressed mRNAs Supplementary Table 5. Target genes of differentially expressed lncRNAs Supplementary Table 6. GO analysis of potential targets of lncRNAs Supplementary Table 7. KEGG analysis of potential targets of lncRNAs Supplementary Table 8. Differential expression of mRNAs between RNA-seq and microarray.
Acknowledgments
This study was supported by the grant from the National Health and Family Planning Commission (Grant no. 2011-1).
Competing Interests
All the authors declare that there are no competing interests in this study.
References
- 1.Gore M., Sadosky A., Stacey B. R., Tai K.-S., Leslie D. The burden of chronic low back pain: clinical comorbidities, treatment patterns, and health care costs in usual care settings. Spine. 2012;37(11):E668–E677. doi: 10.1097/brs.0b013e318241e5de. [DOI] [PubMed] [Google Scholar]
- 2.Katz J. N. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. The Journal of Bone & Joint Surgery—American Volume. 2006;88(supplement 2):21–24. doi: 10.2106/jbjs.e.01273. [DOI] [PubMed] [Google Scholar]
- 3.Kalb S., Martirosyan N. L., Kalani M. Y. S., Broc G. G., Theodore N. Genetics of the degenerated intervertebral disc. World Neurosurgery. 2012;77(3-4):491–501. doi: 10.1016/j.wneu.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Ikegawa S. The genetics of common degenerative skeletal disorders: osteoarthritis and degenerative disc disease. Annual Review of Genomics and Human Genetics. 2013;14:245–256. doi: 10.1146/annurev-genom-091212-153427. [DOI] [PubMed] [Google Scholar]
- 5.Li Z., Yu X., Shen J., Chan M. T. V., Wu W. K. K. MicroRNA in intervertebral disc degeneration. Cell Proliferation. 2015;48(3):278–283. doi: 10.1111/cpr.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C., Wang W. J., Yan Y. G., et al. MicroRNAs: new players in intervertebral disc degeneration. Clinica Chimica Acta. 2015;450:333–341. doi: 10.1016/j.cca.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Ponting C. P., Oliver P. L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Esteller M. Non-coding RNAs in human disease. Nature Reviews Genetics. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 9.Zhao B., Yu Q., Li H., Guo X., He X. Characterization of microRNA expression profiles in patients with intervertebral disc degeneration. International Journal of Molecular Medicine. 2014;33(1):43–50. doi: 10.3892/ijmm.2013.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fatica A., Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nature Reviews Genetics. 2014;15(1):7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 11.Wilusz J. E., Sunwoo H., Spector D. L. Long noncoding RNAs: functional surprises from the RNA world. Genes and Development. 2009;23(13):1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J. T. Epigenetic regulation by long noncoding RNAs. Science. 2012;338(6113):1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 13.Wapinski O., Chang H. Y. Long noncoding RNAs and human disease. Trends in Cell Biology. 2011;21(6):354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Harries L. W. Long non-coding RNAs and human disease. Biochemical Society Transactions. 2012;40(4):902–906. doi: 10.1042/BST20120020. [DOI] [PubMed] [Google Scholar]
- 15.Wan Z., Song F., Sun Z., et al. Aberrantly expressed long noncoding RNAs in human intervertebral disc degeneration: a microarray related study. Arthritis Research & Therapy. 2014;16, article 465 doi: 10.1186/s13075-014-0465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C., Kikyo N. Strategies to identify long noncoding RNAs involved in gene regulation. Cell and Bioscience. 2012;2(1, article no. 37) doi: 10.1186/2045-3701-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atkinson S. R., Marguerat S., Bähler J. Exploring long non-coding RNAs through sequencing. Seminars in Cell and Developmental Biology. 2012;23(2):200–205. doi: 10.1016/j.semcdb.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P. P. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tay Y., Rinn J., Pandolfi P. P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfirrmann C. W. A., Metzdorf A., Zanetti M., Hodler J., Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine. 2001;26(17):1873–1878. doi: 10.1097/00007632-200109010-00011. [DOI] [PubMed] [Google Scholar]
- 21.Krueger F. Trim Galore! http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- 22.Trapnell C., Pachter L., Salzberg S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C., Roberts A., Goff L., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong L., Zhang Y., Ye Z.-Q., et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research. 2007;35(2):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L., Park H. J., Dasari S., Wang S., Kocher J.-P., Li W. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Research. 2013;41(6, article e74) doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin M. F., Jungreis I., Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27(13):i275–i282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn R. D., Clements J., Eddy S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research. 2011;39(2):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alvarez-Dominguez J. R., Hu W., Yuan B., et al. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123(4):570–581. doi: 10.1182/blood-2013-10-530683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C., Williams B. A., Pertea G., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak K. J., Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Ebisuya M., Yamamoto T., Nakajima M., Nishida E. Ripples from neighbouring transcription. Nature Cell Biology. 2008;10(9):1106–1113. doi: 10.1038/ncb1771. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma K.-G., Shao Z.-W., Yang S.-H., et al. Autophagy is activated in compression-induced cell degeneration and is mediated by reactive oxygen species in nucleus pulposus cells exposed to compression. Osteoarthritis and Cartilage. 2013;21(12):2030–2038. doi: 10.1016/j.joca.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Jhawar B. S., Fuchs C. S., Colditz G. A., Stampfer M. J. Cardiovascular risk factors for physician-diagnosed lumbar disc herniation. Spine Journal. 2006;6(6):684–691. doi: 10.1016/j.spinee.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z., Zhang M., Zhao X.-H., et al. Immune cascades in human intervertebral disc: the pros and cons. International Journal of Clinical and Experimental Pathology. 2013;6(6):1009–1014. [PMC free article] [PubMed] [Google Scholar]
- 37.Sun Z., Wan Z.-Y., Liu Z.-H., et al. Expression of soluble Fas and soluble FasL in human nucleus pulposus cells. International Journal of Clinical and Experimental Pathology. 2013;6(8):1567–1573. [PMC free article] [PubMed] [Google Scholar]
- 38.Hangai M., Kaneoka K., Kuno S., et al. Factors associated with lumbar intervertebral disc degeneration in the elderly. Spine Journal. 2008;8(5):732–740. doi: 10.1016/j.spinee.2007.07.392. [DOI] [PubMed] [Google Scholar]
- 39.Gruber H. E., Ingram J. A., Norton H. J., Hanley E. N., Jr. Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence-associated β-galactosidase in human and sand rat discs. Spine. 2007;32(3):321–327. doi: 10.1097/01.brs.0000253960.57051.de. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Differential expression of transcripts between IDD and control groups. Volcano plots of log10 (p-value) versus log2 fold change. Red dots indicated the differentially expressed transcripts between IDD and control groups. Supplementary Figure 2. Functional enrichment analysis of differentially expressed mRNAs between IDD and control groups based on KEGG. Supplementary Table 1. Prime list for qRT-PCR Supplementary Table 2. Information of novel lncRNA Supplementary Table 3. Information of differentially expressed lncRNAs Supplementary Table 4. Information of differentially expressed mRNAs Supplementary Table 5. Target genes of differentially expressed lncRNAs Supplementary Table 6. GO analysis of potential targets of lncRNAs Supplementary Table 7. KEGG analysis of potential targets of lncRNAs Supplementary Table 8. Differential expression of mRNAs between RNA-seq and microarray.