

Abstract
Apert syndrome is the rare acrocephalosyndactyly syndrome type 1, characterized by craniosynostosis, severe syndactyly of hands and feet, and dysmorphic facial features. It demonstrates autosomal dominant inheritance assigned to mutations in the fibroblast growth factor receptor gene. Presently described is case of a 19-year-old female patient diagnosed on physical examination with Apert syndrome based on acrocephaly, prominent forehead, ocular hypertelorism, proptosis, short and broad nose, pseudoprognathism, dental crowding and ectopia, maxillar hypoplasia, low hairline, webbed neck, pectus excavatum, and severe, bilateral syndactyly of hands and feet. The multiple phenotypic signs of Apert syndrome make multidisciplinary team, including dentist, neurosurgeon, plastic surgeon, physiatrist, ophthalmologist, perinatalogist and geneticist, essential for successful management.
Keywords: Acrocephalosyndactyly, Apert syndrome, craniosynostosis
Apert syndrome was firstly described by a French physician, Eugene Apert [1]. This syndrome is a form of acrocephalodactyly (Type 1). It is a rarely seen congenital disorder characterized by a autosomal dominant inheritance which manifests itselt with craniosynostosis, midface hypoplasia, and symmetric syndactyly of hands, and feet [2].
Craniofacial deformities specific to Apert syndrome (AS) include craniofacial deformities, acrocephaly (cone-shaped calvarium), prominent forehead, proptosis, hyperteleorism, and flattened nose with a low bridge. Oral signs may be enumerated as pseudocleft, high-arched palate, transverse, and sagittal maxillary hypoplasia, dental crowding, delay in dentition, ectopic teeth, disarrayed teeth, and teeth crowding can be enumerated. Mandibula is generally normal in size, and pseudoprognatism can be seen. Rarely symptoms related to central nervous system, cardiac, gastrointestinal, and urogenital system, and vertebral anomalies have been reported [3].
Herein, we have aimed to present a 91-year-old female patient diagnosed as AS based on dysmorphic facial manifestations (prominent forehead, ocular hypertelorism, proptosis, lateral gaze, flattened nose with low bridge, hypoplasic maxilla), disarrayed teeth, webbed neck, short hairline, pectus excavatus, severe syndactyly of hands, and feet, in the light of the literature information.
CASE REPORT
A 19 year-old female patient applied to the medical board. At first examination, dysmorphic face, hand, and foot deformities, webbed neck were observed. The patient had normal height, and weight for her age. Family history of the patient was inquired. Her elder sister had also the same dysmorphic facial features. Her mental examination was unremarkable. She hadn’t had any symptoms related to cardiac, and respiratory system up to now. On physical examination acrocephaly, prominent forehead, hypertelorism, proptosis, lateral gaze of the left eye, broad nasal root, webbed neck, short hairline, pectus excavatum, cutaneous syndactyly of both hands, enlarged thumbs, subluxations of distal phalanges of hands, total syndactyly of toes of both feet, polydactyly (6 fingers), and brachydactyly were detected (Figure 1).
FIGURE 1.
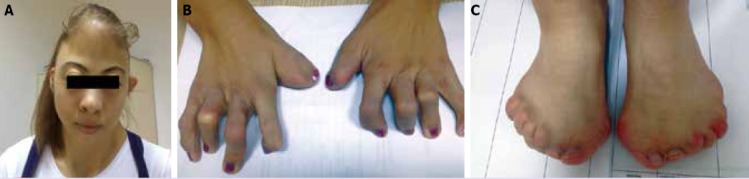
(A) Frontal view of the patient’s face, and neck. (B) Anteroposterior radiogram of patient’s both hands reveal widened thumbs, cutaneous syndactyly, and flexion deformities of all fingers. (C) Anteroposterior view of both feet, complete syndactyly.
Radiological evaluation revealed acroephaly, flattened occiput, increase in anterior opening of maxilla, and hypoplasia, teeth crowding, ectopic teeth, tapered inferior tip of the mandibula, and pseudoprognatism (Figure 2 A). Cutaneous syndactyly of both hands, flexion deformity of all fingers, widening of distal phalanx of the thumb, and occasional subluxation of distal phalanges were detected (Figure 2 B). Cervical, and lumbar vertebras were not abnormal. Anteroposterior radiograms of both feet demonstrated total syndactyly, polydactyly (6 toes), brachydactyly of 4. digits of both feet, clinodactyly of both big toes, and occasional subluxations (Figure 2 C).
FIGURE 2.
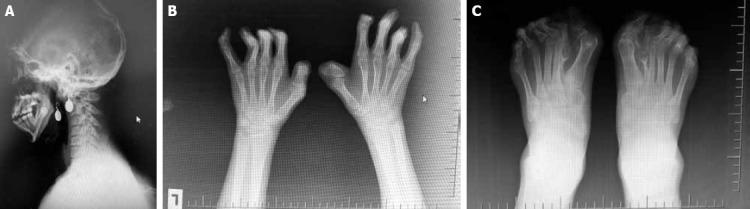
(A) Lateral radiogram of patient’s head, and neck. (B) Anteroposterior (AP) radiogram of patient’s both hands. (C) Anteroposterior (AP) radiogram of both feet of the patient.
DISCUSSION
AS is known as acrocephalosyndactyly type I which is a form of craniosynostosis. AS is characterized mainly by premature craniosynostosis, hypertelorism, syndactyly of hands, and feet, and many phenotypical symptoms. It has an autosomal dominant inheritance, and develops as a mutation of fibroblast growth factor receptor -2 gene (FGFR2) on 10q26 gene locus. FGFR2 gene enables coding of a protein called fibroblast growth factor receptor -2 gene. This protein is one of the four FGFRs responsible for the formation of blood vessels, wound healing, embryonic evolution, and regulation of cellular division, growth, and maturation. FGFR binds to fibroblast growth factors with higher affinity, and plays an important role in signal pathways which function in the fusion process of skull bones.
Molecular basis of Apert syndrome is very specific. Two different types of mutation have been demonstrated in the binding site between immunoglobulin like loop- 2, and immunoglobulin like loop -3 on fibroblast growth factor receptor -2 (FGFR2). Besides, intracellular signals oriented by FGFR play an important role in the embryogenesis, and in its deficiency, premature gastrulation, implantation anomalies, impairment of epithelial-mesencyhmal interaction, and defects in membranous, and endochondrial bone formation are seen. Most of the patients have a normal karyotype [4]. Although in our country specific gene mutation analysis is performed in private medical centers, in our province we haven’t this opportunity.
AS is especially seen in children of the parents (frequently advanced paternal age) of advanced age. It was learnt that her father was 42 years of age when she was born. Although generally paternal mutations are seen, most of the cases are sporadic, and develop because of new mutations. Its incidence is 9.9–15.5 per one million live births which do not differ between genders [5, 6].
Phenotypic manifestations of the disease are explained by premature fusion of cranial sutures Premature closure of coronal sutures before 3 months of age causes shorter anteroposterior diameter, high, and prominent forehead associated with acrocephalic (cone-shaped) head. The most prominent symptoms of this syndrome are syndactyly of hands, and feet [5, 6]. Our patient also had the most marked symptoms namely acrocephaly, syndactyly of fingers, and toes.
In these patients midface is hypoplasic. Eye manifestations include hypertelorism, proptosis, and downslanting palpebral fissures. Nose, and nasal root is short, and widened. Our patient carries all specific facial characteristics.
Rarely internal organ (renal, cardiac, gastrointestinal, genitourinary) involvement, elbow, shoulder, vertebral column (vertebral malsegmentation, fusion, hemivertebra) deformities, and frequently central nervous system defects (cerebellar, gyral, cortical defects, lysencephaly, hypogenesis or agenesis of corpus callosum, ventriculomegaly) which cause mental disorders have been reported. In patients with Apert syndrome, upper respiratory tract infections, sleep apnea, and malnutrition can be seen Respiratory distress can be severe requiring endotracheal intubation or tracheostomy. In our case apart from mild degrees of dyspnea, and snoring, any other symptom suggesting internal organ pathology was not found. In patients with AS, true megalencephaly is seen, however generally mental disorders are rarely encountered [2, 3, 4, 5, 6].
During physical examination of our patient webbed neck, and short hairline which are rarely seen in patients with AS were observed. These features are discriminating characteristics between Turner and Noonan syndrome. Normal height of our patient who had not mental disorder ruled out other diagnoses. Vertebral anomalies and most frequently cervical fusion which can be seen in 68% of the patients was not detected in our patient [7].
Anteroposterior diameter of anterior cranial fossa is relatively shorter, because of forward displacement of greater wings of sphenoid bone, forehead is steep, wide, and flat, temporal regions are protruded, and occiput is flattened. In our patient a prominent forehead, and a flat occipitus were observed. Because of forward displacement of the sphenoid bone, and blockade of the frontal bone, maxillary bone can not develop on all three planes. Consequently, maxillary height, width of the nasal cavity, and nasopharyngeal height decrease, This anatomic configuration severely prevents development of oropharyngeal, and nasopharyngeal cavity. In these patients, usually oral respiration is seen with resultant impairment of respiratory functions. The patient said that she hadn’t shortness of breath, however she stated that she had snored some nights Ophthalmic symptoms have been explained by enlargement of middle cranial fossa towards anteroinferior direction because of the position of the greater wings of sphenoid bone, and decrease in the anteroposterior diameter of the orbita secondary to impingement of the orbita on lateral walls of the orbita [8]. Down syndrome characterized by hypertelorism, flattened nose, short stature, and mental retardation may be considered in the differential diagnosis. Oral symptoms are explained by decrease in especially anteroposterior diameter of the maxilla with resultant crowding of teeth, and increase in anteror opening of the oral cavity [9].
In patients with AS, hand, and foot deformities which effect daily life can be seen [10]. Pedal deformities which can be named in the literature as ‘Apert’s foot’ were divided in 3 types by Baluth and von Torne [11]. Our patient was consistent with type 3 where syndactyly was seen in all toes. In AS patients with hand, and foot deformities, syndactyly should be corrected in order to increase quality of life of these patients. For this purpose surgical correction including partial amputation can be applied. Feet of the AS patients are not frequently suitable for wearing normal shoes because of pressure pain, and often surgery is required [12].
In the differential diagnosis other genetic disorders which can be seen apart from craniosynostosis include Crouzon, Carpenter (acrocephalosyndactyly type 2), Chotzen, and Pfeiffer syndromes. Specific association of craniosynostosis has been correlated with mutation in the FGFR gene [13]. In the Crouzon syndrome characterized by craniosynostosis, and dysmorphic face, acrocephaly, brachycephaly, exophthalmos, proptosis, hypertelorism, hooked nose, hypoplasic maxilla, ear, and palatine deformities occur. AS especially demonstrates similar characteristics with Crouzon syndrome. In the Crouzon syndrome, contrary to AS, extremities are not involved, and craniofacial deformities lead a milder course. However in AS, multiple sutures fuse prematurely. The face is asymmetric, forehead is more prominent, and exophthalmos is not so severe. Hand, and foot deformations, and especially extreme cases of syndactyly is its discriminative feature. We did not think of Crouzon syndrome in our patient with prominent extremity involvement. Some authors consider Apert-Crouzon syndrome as a separate entity. Pfeiffer syndrome is characterized by craniosynostosis, enlarged thumb, and toes. Interestingly, in our patient thumbs of both hands were enlarged just like Pfeiffer syndrome. However in Carpenter syndrome cloverleaf skull is a typical manifestation with with facial paralysis, and characteristic facial features [14, 15, 16]. The concomitancy between AS and toes with preaxial polydactyly in Carpenter syndrome has been reported in many cases. Our patient with polydactylic toes resembles a case with Carpenter syndrome. In the literature, it has been indicated that polydactylic form of AS may always accompany Carpenter syndrome [17].
In summary, although our patient had signs, and symptoms characteristic of AS, as seen in Pfeiffer syndrome, she also manifested a different mutation per se characterized by an enlarged thumb, and polydactylic form of AS associated with Carpenter syndrome Syndromes characterized by craniosynostosis can not be definitely discriminated from each other, and as was seen in our case, their concomitancy in rare cases has been reported. Besides, our case demonstrated differences from other cases with having webbed neck, and short hairline.
If family history of AS is detected, then fetal DNA analysis can detect specific mutations. Prenatal diagnosis can be made by demonstration of craniosynostosis, and syndactyly on prenatal ultrasound, and detection of FNGR gene mutation at 16. gestational week. AS is a rarely seen disorder without a complete cure which imposes material, and moral burden on the patient, his/her family, and health organizations. Therefore essential approach should be to establish the diagnosis by prenatal US, and termination of the pregnancy [18].
Since symptoms of AS demonstrate large variability, the diagnosis, and treatment of the disease require treatment by a multidisciplinary team in collaboration with neurosurgeon, plastic, and reconstructive surgeon, physiatrist, ophthalmologist, psychiatrist, neurologist, perinatologist, and genetician. Diagnosis is based on clinical, radiological, and genetic evaluation. Though its definitive treatment is not available, for anatomical deformities corrective surgery can provide cure. Cranial corrective operations, and fronto-orbital surgery are advised for infants aged 6–9 months, and generally reconstruction of synostosis is considered for patients older than 6 years. Besides these patients can manifest emotional, and behavioural disorders and because of severe craniofacial anomalies, they require psychiatric consultation [19].
Footnotes
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
REFERENCES
- 1.DeGiovanni CV, Jong C, Woollons A. What syndrome is this? Apert syndrome. Pediatr Dermatol. 2007;24:186–8. doi: 10.1111/j.1525-1470.2007.00372.x. [DOI] [PubMed] [Google Scholar]
- 2.Bhatia PV, Patel PS, Jani YV, Soni NC. Apert’s syndrome:Report of a rare case. J Oral Maxillofac Pathol. 2013;17:294–7. doi: 10.4103/0973-029X.119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar GR, Jyothsna M, Ahmed SB, Sree Lakshmi KR. Apert’s Syndrome. Int J Clin Pediatr Dent. 2014;7:69–72. doi: 10.5005/jp-journals-10005-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varoli FP, Santos KCP, Costa C, Oliveira JX. Apert syndrome:clinical and radiographic features and case report. Rev Odonto Cienc. 2011;26:96–9. [Google Scholar]
- 5.Cohen MM, Jr, Kreiborg S. Agenesis of the corpus callosum. Its associated anomalies and syndromes with special reference to the Apert syndrome. Neurosurg Clin N Am. 1991;2:565–8. [PubMed] [Google Scholar]
- 6.Cohen MM., Jr Craniosynostosis and syndromes with craniosynostosis:incidence, genetics, penetrance, variability, and new syndrome updating. Birth Defects Orig Artic Ser. 1979;15:13–63. [PubMed] [Google Scholar]
- 7.Kreiborg S, Barr M, Jr, Cohen MM., Jr Cervical spine in the Apert syndrome. Am J Med Genet. 1992;43:704–8. doi: 10.1002/ajmg.1320430411. [DOI] [PubMed] [Google Scholar]
- 8.Katzen JT, McCarthy JG. Syndromes involving craniosynostosis and midface hypoplasia. Otolaryngol Clin North Am. 2000;33:1257–84. doi: 10.1016/s0030-6665(05)70280-2. [DOI] [PubMed] [Google Scholar]
- 9.Açıkgöz Y, Nurşen B, Yalın T, İncesu L, Küçüködük Ş. Apert Syndrome:A Case Report and Review of the Literature O.M.Ü. Tıp Derg. 2006;23:59–64. [Google Scholar]
- 10.Mason WH, Wymore M, Berger E. Foot deformities in Apert’s syndrome. Review of the literature and case reports. J Am Podiatr Med Assoc. 1990;80:540–4. doi: 10.7547/87507315-80-10-540. [DOI] [PubMed] [Google Scholar]
- 11.Blauth W, von Törne O. “Apert’s foot”(in acrocephalo-syndactyly) (author’s transl). [Article in German] Z Orthop Ihre Grenzgeb. 1978;116:1–6. [Abstract] [PubMed] [Google Scholar]
- 12.Meyer JL. Apert’s syndrome:(acrocephalosyndactylism) J Foot Surg. 1981;20:210–3. [PubMed] [Google Scholar]
- 13.Cohen MM., Jr Craniosynostoses:phenotypic/molecular correlations. Am J Med Genet. 1995;56:334–9. doi: 10.1002/ajmg.1320560327. [DOI] [PubMed] [Google Scholar]
- 14.Jabs EW. Toward understanding the pathogenesis of craniosynostosis through clinical and molecular correlates. Clin Genet. 1998;53:79–86. doi: 10.1111/j.1399-0004.1998.tb02648.x. [DOI] [PubMed] [Google Scholar]
- 15.Benmiloud S, Chaouki S, Atmani S, Hida M. Apert syndrome. [Article in French] Pan Afr Med J. 2013;14:66. doi: 10.11604/pamj.2013.14.66.2178. [Abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krueger JL, Ide CH. Acrocephalosyndactyly. (Apert’s syndrome) Ann Ophthalmol. 1974;6:787–9. [PubMed] [Google Scholar]
- 17.Yonenobu K, Tada K, Tsuyuguchi Y. Apert’s syndrome-a report of five cases. Hand. 1982;14:317–25. doi: 10.1016/s0072-968x(82)80068-5. [DOI] [PubMed] [Google Scholar]
- 18.Hansen WF, Rijhsinghani A, Grant S, Yankowitz J. Prenatal diagnosis of Apert syndrome. Fetal Diagn Ther. 2004;19:127–30. doi: 10.1159/000075135. [DOI] [PubMed] [Google Scholar]
- 19.Gündüz S, Demirel N, Baş AY, Okumuş N, Zenciroğlu A. Apert Sendromu. Turkish J Pediatr Dis. 2013;2:94–5. [Google Scholar]