

Significance
The antimalarial artemisinin is a sesquiterpene lactone produced by glandular secretory trichomes on the leaves of Artemisia annua. Using a mutant impaired in artemisinin synthesis, we demonstrate the importance of nonenzymatic conversions in terpenoid metabolism and highlight the ability of A. annua glandular secretory trichomes to redirect flux into a sesquiterpene epoxide. The research presented offers insight into the mechanism of the final steps of artemisinin synthesis in A. annua, with significant implications for future production of secondary compounds in native versus heterologous host systems.
Keywords: artemisinin, p450 oxidase, terpenoid, sesquiterpene, Artemisia annua
Abstract
Artemisinin, a sesquiterpene lactone produced by Artemisia annua glandular secretory trichomes, is the active ingredient in the most effective treatment for malaria currently available. We identified a mutation that disrupts the amorpha-4,11-diene C-12 oxidase (CYP71AV1) enzyme, responsible for a series of oxidation reactions in the artemisinin biosynthetic pathway. Detailed metabolic studies of cyp71av1-1 revealed that the consequence of blocking the artemisinin biosynthetic pathway is the redirection of sesquiterpene metabolism to a sesquiterpene epoxide, which we designate arteannuin X. This sesquiterpene approaches half the concentration observed for artemisinin in wild-type plants, demonstrating high-flux plasticity in A. annua glandular trichomes and their potential as factories for the production of novel alternate sesquiterpenes at commercially viable levels. Detailed metabolite profiling of leaf maturation time-series and precursor-feeding experiments revealed that nonenzymatic conversion steps are central to both artemisinin and arteannuin X biosynthesis. In particular, feeding studies using 13C-labeled dihydroartemisinic acid (DHAA) provided strong evidence that the final steps in the synthesis of artemisinin are nonenzymatic in vivo. Our findings also suggest that the specialized subapical cavity of glandular secretory trichomes functions as a location for both the chemical conversion and the storage of phytotoxic compounds, including artemisinin. We conclude that metabolic engineering to produce high yields of novel secondary compounds such as sesquiterpenes is feasible in complex glandular trichomes. Such systems offer advantages over single-cell microbial hosts for production of toxic natural products.
The sesquiterpene lactone artemisinin is the active ingredient in artemisinin-combination therapies—the most effective treatment for malaria currently available. The production of artemisinin occurs in specialized 10-cell biseriate glandular trichomes present on the leaves, stems, and inflorescences of Artemisia annua (1–3). Artemisinin is phytotoxic (4) and is believed to accumulate in the subapical extracellular cavity of glandular trichomes (2). This ability of trichomes to transfer compounds into extracellular cavities (5, 6) overcomes the problem of cellular toxicity. Conveniently, natural products located in these cavities are readily extractable as exemplified by artemisinin. This natural product is extracted on a commercial scale by submerging intact dried A. annua leaf material in organic solvent with the active ingredient being directly crystallized from the condensed organic fraction (7). There has been much interest in determining the steps involved in the biosynthesis of artemisinin in recent years, largely driven by efforts to produce this compound through a completely biosynthetic microbial-based fermentation route (8, 9). Presently microbial production is at best semisynthetic, terminating at artemisinic acid (AA), which must then be extracted from culture and chemically converted to artemisinin by using photooxidation (8, 10). The lack of a low-cost, scalable conversion process is considered to be a major factor in the failure so far of the semisynthetic route to sustainably impact the market, making it uncompetitive with plant-based production (11).
Although the enzymatic steps involved in production of the nonphytotoxic precursors amorpha-4,11-diene (A-4,11-D) and dihydroartemisinic acid (DHAA) have been elucidated (12–15) and the associated genes have been shown to be highly expressed in both the apical and subapical cells of the glandular secretory trichomes (3, 16), the final steps in the conversion of DHAA to artemisinin are considered to be nonenzymatic and may be extracellular (17, 18). Therefore, microbial-based “complete” synthetic biology routes to artemisinin may never be achievable. Meanwhile, modern molecular breeding has succeeded in improving A. annua (19), creating hybrids reaching artemisinin yields of 1.4% dry leaf biomass in commercial field trials (20) (www.artemisiaf1seed.org/).
The glandular secretory trichomes of A. annua produce almost 600 secondary or specialized metabolites, many of which are terpenoids (21). These include a significant number of terpene allylic hydroperoxides and endoperoxides (21). This latter class, of which artemisinin is a member, are typically bioactive and therefore potential targets for development as pharmaceuticals (22). Consistent with their phytochemical complexity, it is known that glandular secretory trichomes express multiple members of gene families, encoding enzymes of specialized metabolism, including terpene synthases and cytochrome P450 oxidases (16, 19, 23). Many of these enzymes are considered to be promiscuous (24). We reasoned that this plasticity could be exploited by developing biochemical knockouts, redirecting flux to new high-value sesquiterpenes in a proven plant production system.
Recent attempts to knock down the A-4,11-D synthase by using RNAi in self-pollinating A. annua resulted in only a modest (30–50%) reduction in artemisinin levels (25). We have chosen to target amorpha-4,11-diene C-12 oxidase (CYP71AV1), which catalyzes the three-step conversion of A-4,11-D to AA (13, 26, 27). When we knocked out this enzyme, as expected, artemisinin was not produced; however, rather than A-4,11-D accumulating, it was instead readily converted to a sesquiterpene epoxide, arteannuin X. Detailed metabolite analysis revealed that production of this compound paralleled the production of artemisinin during leaf maturation, with early steps occurring in young leaves and later steps in older leaves. Our findings confirm the function of the CYP71AV1 enzyme in planta and also demonstrate the flexibility of glandular secretory trichome biochemistry, such that it is capable of redirecting the flux of A-4,11-D into a sesquiterpene epoxide at levels similar to artemisinin.
Results and Discussion
Disruption of CYP71AV1 Results in the Accumulation of a Sesquiterpene Epoxide at the Expense of Artemisinin.
We used the TILLING method (28) to screen for mutations in the single-copy (Fig. S1) CYP71AV1 gene in an F2 population of A. annua that had been derived from an ethyl methane sulfonate (EMS)-mutagenized population of the Artemis F1 hybrid, as described (19). This process resulted in an allelic series of 10 mutants, of which just 1 was nonsense because of a G-to-A transition in the second exon of CYP71AV1 (Fig. S2A). This mutation, which we designate cyp71av1-1, gives a predicted conversion of amino acid Trp-124 in the polypeptide to a stop codon, resulting in a major truncation of the enzyme and loss of most of the putative heme-binding sites, as well as Ser-473, which is crucial for catalyzing oxidation reactions (Fig. S2 B and C).
Fig. S1.

CYP71AV1 gene copy number for individual plants from three genotype classes relative to the squalene synthase (SS) gene. Gene copy number for 15 individual plants from three genotype classes derived from an F2 backcrossed segregating population was established by using qPCR as described in SI Materials and Methods.
Fig. S2.

Discovery and characterization of cyp71av1-1 mutation. (A) CYP71AV1 genomic DNA sequencing from segregating WT (i) heterozygous cyp71av1-1 (ii) and homozygous cyp71av1-1 (iii); position of G-A transition is indicated by arrow. (B) Alignment of deduced amino acids of CYP71AV1 closest homologs from the Artemisia genus. Amino acid sequences based on National Center for Biotechnology Information deposited cDNAs: A. annua (EF197889), A. absinthium (BAM68810), A. afra (BAM68809), A. martima (BAM68815), A. abrotanum (BAM68811), A. chemafolia (BAM68819), A. kumarensis (BAM68817), A. schmidtiana (BAM68816), A. campestris (BAM68812), A. japonica (BAM68814), and A. capillaris (BAM68821). Red arrow indicates the stop codon in cyp71av1-1 mutant; red dots represent potential heme binding sites based on homology with human CYP1A2; and the star identifies Ser-472, critical for the second oxidation reaction (12). (C, Upper) Position of the cyp71av1-1 mutation in the CYP71AV1 gene model. (C, Lower) CYP71AV1 protein structure modeled by I-TASSER (46) on the 10 most closely related structural analogs; parts of structure missing in the cyp71av1-1 mutant are highlighted in yellow, putative heme-binding residues are represented as wires, and Ser-472, crucial for the second oxidation step (27), is highlighted in red.
Previous work had shown that early stage intermediates in the artemisinin pathway accumulate in young A. annua leaves, and, as they mature, artemisinin accumulates (25, 29). To investigate the effects of the cyp71av1-1 mutation on artemisinin biosynthesis, we analyzed three leaf developmental stages: juvenile (leaves 1–5 as counted down from the apical meristem), expanding (leaves 7–9), and mature (leaves 11–13). To generate material for this analysis, we backcrossed cyp71av1-1 to Artemis parents, selfed the progeny, and performed DNA marker-based selection of wild-type (WT), heterozygous, and homozygous cyp71av1-1 individuals from the segregating backcrossed F2 population (Fig. S3). We did not detect any morphological differences between WT and cyp71av1-1 material (Fig. S4). We also extended the analysis to include oven-dried leaf material stripped from entire plants to investigate metabolite conversions occurring after harvest.
Fig. S3.

Overview of the cyp71av1-1 mutation breeding scheme from mutagenesis to the F2 backcross population. Bx, backcrossed; het, heterozygous cyp71av1-1; hom, homozygous cyp71av1-1; WT, segregating WT.
Fig. S4.

Morphological characterization of the cyp71av1-1 mutant segregating population. (A) Photographs show four representative 12-wk-old plants from each of three genotype groups. (B) Trait data recorded for 12-wk-old plants from each of the three genotype groups (A/A, homozygous cyp71av1-1; G/A, heterozygous cyp71av1-1; and G/G, segregating WT). Error bars, SEM (n = 15); letters represents one-way ANOVA Tukey’s range test results.
Compared with WT and heterozygous material, the juvenile leaves of cyp71av1-1 contain significantly elevated levels of the first committed metabolite in the artemisinin pathway, A-4,11-D (Fig. 1 A, i and Dataset S1), consistent with the reported in vitro activity of CYP71AV1 (13, 26, 27). Metabolite profiling further revealed a complete loss of all metabolites downstream of A-4,11-D including artemisinin, which typically accumulate in juvenile, expanding and mature WT leaves (Fig. 1 A, ii–viii and C and Datasets S2 and S3).
Fig. 1.

Effects of cyp71av1-1 mutation on selected sesquiterpene levels in fresh and dried leaves. (A) Level of selected sesquiterpenes were quantified by GC-MS (i) and UPLC-MS (ii–x) in fresh leaf (L) 1–5 (juvenile), 7–9 (expanding), and 11–13 (mature) as counted from the apical meristem, plus oven-dried whole plant-stripped leaves (dry) from 12-wk-old glasshouse-grown homozygous (hom), heterozygous (het) cyp71av1-1, and segregating WT as described in SI Materials and Methods. Error bars represent SEM (n = 15 for L1–5, L7–9, and L11–13; n = 6 for dry leaf). Letters represent Tukey’s range test results after one-way ANOVA or restricted maximum likelihood (see SI Materials and Methods for details). Groups not sharing letters indicate statistically significant differences. (B) Summary of the effects of cyp71av1-1 mutation on the level of selected sesquiterpenes. Red cross indicates steps of the pathway targeted by cyp71av1-1 mutation. Full arrows, known enzymatic steps; dotted arrows, potential nonenzymatic conversions; brown dotted arrows, novel pathway operating in the cyp71av1-1 mutant; full green arrows, metabolite changes (all types of leaves). AAA, artemsinic aldehyde; AAOH, artemsinic alcohol; ALDH1, aldehyde dehydrogenase; AMS, amorpha-4,11-diene synthase; ArtB, arteannuin B; CPR, cytochrome P450 reductase; DBR2, artemisinic aldehyde Δ 11(13) reductase; DeoxyArt, deoxyartemisinin; DHAAA, dihydroartemsinic aldehyde; DXR, 1-deoxy-d-xylulose-5-phosphate reductoisomerase; DXS, 1-deoxy-d-xylulose-5-phosphate synthase; FPP, farnesyl diphosphate; FPS, farnesyl diphosphate synthase; G-3-P, glyceraldehyde-3-phosphate; HDR, 4-hydroxy-3-methylbut-2-enyl diphosphate reductase; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; HMGR, 3-hydroxy-3-methylglutaryl CoA reductase; MEcPP, 2-C-methyl-d-erythritol-2,4-cyclopyrophosphate; MEP, 2-C-methylerythritol 4-phosphate; MVA, mevalonate.
Other classes of secondary metabolites, including aromatic alcohols and ketones, coumarins, monoterpenes, and other sesquiterpenes remained largely unchanged in cyp71av1-1 (Datasets S1 and S3). However, levels of some minor monoterpenes (eucalyptol, borneol, and sabinene) and sesquiterpenes (calarene, α-bisabolol, and cedrenol,) were reduced, and levels of two minor flavonoids (retusin and artemetin) were increased in cyp71av1-1 (Datasets S1 and S3). These and other changes were largely restricted to the juvenile leaves (Datasets S1 and S3 and Fig. S5), which are relatively dense in glandular trichomes (29) and exhibit high expression levels of terpene synthases (30). Recent attempts to silence AMORPHA-4,11-DIENE SYNTHASE expression in self-pollinating varieties of Artemisia annua resulted in increased levels of two nonamorphadiene sesquiterpenes, caryophyllene and copaene, which may be due to an elevated pool of farnesyl diphosphate acting as substrate for other sesquiterpene synthases in the glandular trichomes (25).
Fig. S5.

Principal component analysis (PCA) of UPLC-MS (A) and GC-MS (B) data of four leaf types from segregating cyp71av1-1 mutant populations. PCA of 55 UPLC-MS identified peaks (A) and 47 GC-MS identified metabolites (B). Leaf types, corresponding with Fig. 1 are represented by colors: green, leaf 1–5; purple, leaf 7–9; brown, leaf 11–13; gray, oven-dried leaf. Genotypes represented by shapes: circles, homozygous cyp71av1-1; triangles, heterozygous cyp71av1-1; crosshairs, segregating WTs. Only PC1 and PC2, which have explained maximal variance, are shown. PCA was performed on log-scaled data and mean-centered data; dotted ellipse, Hotelling’s 95% confidence interval.
Ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS) analysis revealed that cyp71av1-1 leaves accumulate large amounts of an oxygenated C14 metabolite (Dataset S3, Peak_ID M221.1535T43). One-dimensional (1D) and 2D NMR (spectroscopic techniques were used to elucidate the structure of this amorphadiene sesquiterpene as (2S,3R,6S)-3-methyl-6-(2R-methyloxiran-2-yl)-2-(3-oxobutyl) cyclohexanone, which we refer to as arteannuin X (Fig. 1 A, ix and B). At 0.4% leaf dry weight in mature leaves the concentration of arteannuin X in cyp71av1-1 is almost half that of artemisinin in WT leaves and the developmental profile for accumulation in expanding and mature leaves is similar for both compounds (Fig. 1 A, ix and Dataset S3). The fact that arteannuin X is present in trace amounts in WT and heterozygous cyp71av1-1 material (Fig. 1 A, ix and Dataset S3) suggests that it normally occurs as a by-product of A-4,11-D oxidation. In vivo and in vitro formation of low abundance by-products derived from other intermediates of artemisinin synthesis have been reported (12, 17, 18).
NMR analysis of cyp71av1-1 extracts identified a second major compound, A-4,11-D tertiary allylic hydroperoxide (A-4,11-DOOH; Fig. 1 A, x and B) that had not previously been reported in WT A. annua. The pattern of accumulation of A-4,11-DOOH (Fig. 1 A, x) preempted that of arteannuin X reaching a concentration of almost 0.15% leaf dry weight in juvenile and expanding leaves of cyp71av1-1 before decreasing in mature leaves (Fig. 1 A, x). Although A4,11-DOOH was sufficiently stable to survive the chromatographic isolation procedures that were required to obtain it in a pure state for analysis by NMR, it was found to be unstable under prolonged storage in deuterated chloroform, where it spontaneously converted to arteannuin X. This finding provided circumstantial evidence that arteannuin X might be biosynthesized from A-4,11-D via its tertiary hydroperoxide (A-4,11-DOOH) in vivo, in much the same way that DHAA has been shown to be transformed to artemisinin via the tertiary allylic hydroperoxide of DHAA, dihydroartemsinic acid tertiary hydroperoxide (DHAAOOH) (17).
In Planta Similarities Between the Synthesis of Arteannuin X and Artemisinin.
Using UPLC-MS, we compared the conversion profile of A-4,11-D to arteannuin X in leaves with that of DHAA to artemisinin. Specifically, we also monitored DHAAOOH, the final intermediate in the in vivo production of artemisinin (Fig. 1 A, iv and B). We found that DHAAOOH levels peak in expanding WT leaves at a concentration of 0.5% leaf dry weight (Fig. 1 A, iv and Dataset S3). Artemisinin levels increase gradually from juvenile to mature leaves, reaching a maximum concentration of 1.2% dry leaf weight and remaining stable during the postharvest drying process. (Fig. 1 A, iv and Dataset S3). Previous work has shown that the DHAAOOH intermediate can also give rise to both dihydro-epi-deoxyarteannuin B (DHEDB) (31), and (by Hock-cleavage) deoxyartemisinin (17). Although DHEDB remains at a concentration threefold lower than artemisinin throughout leaf maturation and after harvest (Fig. 1 A, vi and Dataset S3), the levels of deoxyartemisinin increase during dry leaf storage, accumulating to 0.1% leaf dry weight (Fig. 1 A, vii and Dataset S3). These data suggest that, after harvest, any remaining DHAAOOH is preferentially converted to deoxyartemisinin rather than artemisinin.
We next carried out a more detailed analysis of the progression during leaf maturation of A-4,11-D to either arteannuin X or artemisinin in cyp71av1-1 and WT, respectively, by performing 1H NMR analysis on individual extracts from a 24-leaf maturation series (Fig. 2). Determination of the relative amounts of the three most abundant sesquiterpene metabolites associated with cyp71av1-1 (A-4,11-D; A-4,11-DOOH; and arteannuin X) revealed a progressive decline in A-4,11-D, which was matched by an increase in arteannuin X. This analysis also demonstrated that the amount of A-4,11-DOOH reaches a maximum in leaves 7–8 (Fig. 2A). This pattern is entirely consistent with our hypothesis that A-4,11-D is converted to arteannuin X via the intermediate A-4,11-DOOH. 1H NMR analysis of WT material clearly demonstrated that a decline in DHAA inversely correlates with an increase in artemisinin, which reaches a maximum at leaves 14–15, whereas the DHAAOOH intermediate peaks around leaves 7–8 (Fig. 2B), as for A-4,11-DOOH.
Fig. 2.

Developmental patterns of artemisinin and arteannuin X biosynthesis. Leaves 1–24 (counting from apical meristem and shown below graphs) detached from the main stem of three cyp71av1-1 (A) and three WT (B) plants. Chloroform extracts were subjected to NMR analysis (see SI Materials and Methods for details), and abundance for selected metabolites calculated from the integration of distinctive resonances is shown as proportion of the total for each leaf. Error bars, SEM (n = 3).
The results of the above experiments revealed clear parallels in the conversion of A-4,11-D to arteannuin X via the hydroperoxide intermediate (A-4,11-DOOH) and the final steps in the conversion of DHAA to artemisinin via DHAAOOH. There is strong in vivo and in vitro evidence for a nonenzymatic autoxidation of DHAA to DHAAOOH and subsequent nonenzymatic rearrangement to artemisinin (17). Our data suggest that a similar autooxidation operates in cyp71av1-1 to convert A-4,11-D to A-4,11-DOOH and on to arteannuin X. The spontaneous conversion of A-4,11-DOOH to arteannuin X in deuterated chloroform noted above is entirely consistent with this hypothesis. A speculative pathway to account for this conversion is detailed in Fig. S6.
Fig. S6.

Theoretical scheme for the synthesis of arteannuin X from A-4,11-D (A) and comparison with the synthesis of artemisinin from DHAA (B). We propose that the first stages in the synthesis of arteannuin X proceed by the same mechanism that has been established for artemisinin (17) to effect oxidative cleavage at the C-4/C-5 position (A). Thus, A-4,11-DOOH, formed by an “ene-type” reaction of singlet oxygen with the 4,5-double bond in A-4,11-D, undergoes Hock cleavage to an enolic intermediate, which can then participate in a second oxidation to yield a hydroperoxy-aldehyde, similar to that produced from DHAOOH in the biogenesis of artemisinin (B). At this point in our proposed scheme, the synthetic routes to artemisinin and arteannuin X diverge. For artemisinin synthesis, the hydroperoxy-aldehyde intermediate from DHAA incorporates a carboxylic acid group at the 12 position which effectively “locks in place” the 3 new heterocyclic rings that are formed when the hydroperoxy-aldehyde “zips up” with the 4-keto and 5-aldehyde groups (B). For arteannuin X synthesis the hydroperoxy-aldehyde from A-4,11-D undergoes an alternative reaction, in which the terminal oxygen of the hydroperoxide is transferred to the 11,13-double bond, thereby producing the epoxide group found in arteannuin X (A). The final stages of the biogenesis of arteannuin X must then involve a second oxidative carbon–carbon bond cleavage reaction at C-5/C-6, in which C-5 is lost to form arteannuin X (A).
In Vivo Evidence for Nonenzymatic DHAA Conversion in cyp71av1-1 and WT Trichomes.
Previous reports have suggested that peroxidase and/or dioxygenase enzymes may be involved in the conversion of DHAA to artemisinin (16, 32). To further investigate DHAA conversion, we fed [U-13C15]-DHAA to boiled, intact, and dark-incubated trichomes that had been isolated from cyp71av1-1 (which lacks endogenous DHAA) and WT leaves (see SI Materials and Methods and Figs. S7–S9 for details). Chloroform extracts of [U-13C15]-DHAA–fed trichomes and no-trichome controls were subjected to UPLC-MS analysis, and the mass spectrum of each sesquiterpene 12C monoisotope metabolite was used to predict the mass spectra expected from the corresponding [U-13C15]-isotopomer. Metabolite concentrations were first normalized to the trichome density for a given sample. Because DHAA can slowly degrade and convert spontaneously to “downstream” products over time, labeled metabolite concentrations in trichome samples were also corrected by subtracting concentrations measured in matched time-equivalent buffer controls. (Fig. 3).
Fig. S7.

Microscopic evaluation of the glandular secretory trichome extracts from A. annua WT and cyp71av1-1 leaves before and after mechanical disruption. Images were taken under bright field with 8x magnification for the intact (A and B) and mechanically disrupted trichome extracts from the cyp71av1-1 mutant leaves (A and C) and WT (B and D) as described in SI Materials and Methods. Scale bars are depicted in micrometers.
Fig. S9.

Leaf-tagging experiment. The first expanded leaf was tagged on three 8-wk-old homozygous cyp71av1-1 i (AA) and segregating WT (GG) individuals. The number of fully expanded leaves above the tag was scored daily over 40 d. A linear regression curve was fitted through the data points to illustrate average time needed for emergence of the new leaf for each genotype. Error bars, SD; n = 3.
Fig. 3.

Feeding intact and protein-extracted glandular secretory trichomes (GSTs) with 13C-isotope–labeled DHAA. Intact GSTs isolated from young leaves of cyp71av1-1 (A) or segregating WT (B) and trichome protein extracts from cyp71av1-1 (C) or segregating WT (D) were fed with [U-13C15]-DHAA as described in SI Materials and Method. The concentration of selected [U-13C15]-labeled metabolites was first corrected for the different densities of the GST extracts and then calculated with subtraction of relevant feeding buffer controls, containing no trichomes or protein extracts. Metabolites are represented by shapes: artemisinin, triangles; DHEDB, squares. Treatments are represented by colors: white, intact, light-incubated GSTs; black, intact, dark-incubated GSTs; red, boiled, light-incubated GSTs. Level of metabolites was monitored by UPLC-MS. See SI Materials and Methods for details. Error bars, SEM (n = 3). Labeled substrate ([U-13C15]-DHAA) levels started at a concentration of 30–40 μM (off the scale of the graphs) and decreased as expected over the course of the experiments.
It was found that [U-13C15]-artemisinin did not accumulate substantially over the 5-d feeding period in extracts of [U-13C15]-DHAA–fed trichomes in either cyp71av1-1 or WT leaves (Fig. 3 A and B, white, red, and black triangles). Lack of artemisinin accumulation in trichomes fed with [U-13C15]-DHAA is perhaps not surprising, because previous results have indicated that DHAAOOH cannot efficiently undergo Hock cleavage into artemisinin in an aqueous environment (such as trichome extraction buffer) and that it may preferentially form DHEDB (17). We found an accumulation of [U-13C15]-DHEDB in extracts from both light-incubated boiled and intact trichomes from cyp71av1-1 and WT leaves (Fig. 3 A and B, white and red squares). Dark-incubated samples did not show an accumulation of the [U-13C15]-labeled DHEDB (Fig. 3 A and B, black squares), leading us to conclude that DHEDB formation is light-dependent. Both cyp71av1-1 and WT trichomes showed very similar patterns of [U-13C15]-DHEDB accumulation, which reached a plateau between 2 and 3 d after feeding commenced (Fig. 3 A and B, white and red squares). It is also evident that boiling accelerates the formation of DHEDB in both cyp71av1-1 and WT trichomes (Fig. 3 A and B, red vs.. white squares), consistent with the process being nonenzymatic. We did not detect labeled DHAAOOH itself, which suggests that the experimental conditions favored rapid conversion of this intermediate through to DHEDB.
To investigate whether uptake of [U-13C15]-DHAA into isolated trichomes is a limiting factor in the feeding experiment, we fed the labeled substrate to crude protein extracts from isolated trichomes. The products and temporal pattern of their accumulation was very similar to that obtained for the intact trichome feeding (Fig. 3 C and D). Notably, there was no accumulation of [U-13C15]-artemisinin over the 5-d feeding period (Fig. 3 C and D, red, black, and white triangles), whereas [U-13C15]-DHEDB accumulated in both light-incubated boiled and nonboiled protein extracts from cyp71av1-1 and WT trichomes (Fig. 3 C and D, red and white squares). It is also evident that boiling accelerates the formation of DHEDB in both cyp71av1-1 and WT trichome protein extracts (Fig. 3 C and D, red vs. white squares), consistent with the process being nonenzymatic.
The intact trichome-feeding and trichome protein extract-feeding experiments demonstrate that in cyp71av1-1, a block in artemisinin and DHEDB biosynthesis can be rescued by direct feeding of DHAA, which is expected, given the role of CYP71AV1 in the artemisinin biosynthetic pathway (Fig. 1B). The lack of further accumulation of artemisinin over a 5-d period in trichomes maintained in an aqueous medium contrasts with the gradual accumulation of artemisinin in trichomes during leaf maturation (Figs. 1 and 2). These observations lead us to suggest that the nonaqueous environment present in the intact subapical cavity of glandular secretory trichomes is essential for the efficient conversion of DHAA to artemisinin via DHAAOOH. In the absence of such an environment, DHAA is instead converted to DHEDB in a light-dependent nonenzymatic process.
The hydrophobic nature of A-4,11-D prevented us from preparing aqueous solutions of [U-13C15]-A-4,11-D for trichome-feeding experiments and performing a similar analysis of arteannuin X in the cyp71av1-1 mutant.
Given that the final steps in artemisinin biosynthesis appear to be nonenzymatic, the question arises as to how its production during leaf maturation is controlled. It is reasonable to assume that active transport system(s) will be responsible for pumping artemisinin precursors into the subapical cavity of glandular secretory trichomes. Transport of DHAA into the subapical cavity could be a limiting factor, with spatial and temporal expression patterns of relevant transporters controlling the increase in artemisinin during leaf maturation (Fig. 2B).
SI Materials and Methods
Plant Material.
Artemis is an F1 hybrid variety developed by Mediplant produced by crossing C4 and C1 parental material of East Asian origin (35). Its artemisinin content has been reported to reach 1.4% of the leaf dry weight when grown in the field (36). Plants were grown for 12 wk in the glasshouse, under long-day conditions (16-h day/8-h night) at 22 °C max/17 °C min in 4-in pots using Levington F2– seed and modular compost.
Establishment and Screening of an EMS-Mutagenized Population.
A Tilling population was generated from F1 Artemis seed, mutagenized by a 5-h treatment of 200 mM EMS. The seed was sown out, and the resulting plants were out-crossed to provide an M2 population. Young leaf DNA at a concentration of 5 ng/µL from individual M2 plants was pooled fourfold for screening. The full-length genomic DNA sequence of the Artemis CYP71AV1 gene for TILLING was obtained by PCR using gene-specific primers designed based on the GenBank-deposited sequence EF197889. A 1,990-bp fragment of CYP71AV1 was amplified in a two-step PCR. The first step was carried out with unlabeled primers: 5′-ATGGCACTCTCACTGACCACTTC-3′ (left) and 5′-CTAGAAACTTGGAACGAGTAACAACTC-3′ (right) on 12.5 ng of pooled gDNA in 10-μL volumes. Nested PCR and labeling with IR dyes was performed on a 1/10 dilution of the first PCR with a mixture of unlabeled M13-tailed primers [5′-TGTAAAACGACGGCCAGTCCACTTCCATTGCTCTTGCAACG-3′ (left) and 5′-AGGAAACAGCTATGACCATGCATCGTGGCTCCAGAGCTC-3′ (right)] and with M13 left primer (5′-TGTAAAACGACGGCCAGT-3′) labeled with IRDye700 and M13 right primer (5′-AGGAAACAGCTATGACCAT-3′) labeled with IRDye 800 (MWG). Heteroduplex formation, CEL I nuclease digestion, and analysis on the LI-COR 4300 DNA sequencer platform were carried out as described (37). All mutants found on the TILLING gels were verified by Sanger sequencing of both DNA strains of PCR-amplified fragments using the following primers: 5′-CGTAGAGAGTGTGTGTTACAGGCATG-3′ (left) and 5′-TTTGTGGAGTGCTGTCCAGAGGGTG-3′ (right).
Plant Crossing.
Cuttings from parental genotypes were maintained in 4-in pots under 16-h days for 12 wk. Plants were then transferred to 12-h days to induce flowering. Flowering was identified as the point at which the first ray florets were visible. Once flowering commenced, bags were placed over two plants to enable hybrid production. These bags were shaken every 2 d to encourage pollination. Once all flowers had died back, the bags were removed, and the flower heads were allowed to dry out under glass for a further 6 wk before harvesting.
DNA Preparation.
For DNA extraction, 30–50 mg of fresh leaf material was harvested from plants growing in the glasshouse. DNA was extracted by using Qiagen BioSprint 96. Extracted DNA was quantified spectrophotometrically by using NanoDrop-8000 (NanoDrop Products) and normalized to a concentration of 10 ng/μL for SSR and KASP assays or 1 ng/μL for quantitative PCR (qPCR) analysis.
KASP SNP Assay for cyp71av1-1 Mutation Status.
Twenty nanograms of leaf genomic DNA extracted from individual plants was used for 10-μL KASPar assay reaction containing: 1× KASP V4.0 low ROX master mix (LGC Genomics); a concentration of 167 nM each of the two allele-specific primers: cyp71av1-1_ForG 5′-GAAGGTGACCAAGTTCATGCTCTTGCACCTTATGGTGAATACTGG-3′ plus cyp71av1-1_ForA 5′-GAAGGTCGGAGTCAACGGATTCTTGCACCTTATGGTGAATACTGA-3′ and 414 nM universal primer cyp71av1-1_Rev 5′-CTTAACACTCAAAAGCTCCAATGTGCAAA-3′, according to the manufacturer’s recommendations. Allelic discrimination runs and allelic discrimination analysis were performed on Viia7 system (Life Technologies Ltd.) according to the manufacturer’s recommendations.
CYP71AV1 Gene Copy Determination by qPCR.
Three technical replications of 10-μL reactions containing 1 ng of leaf genomic DNA from single plants and 200 nM gene-specific primers in 1× Power SYBR Green PCR Master Mix (Life Technologies Ltd.), plus dilutions of cDNA were run on an ViiA7 Real-Time PCR System (Life Technologies Ltd.). Amplification conditions were as follows: 2 min at 50 °C; 10 min at 95 °C; 40 cycles each of 15 s at 95 °C, followed by 1 min at 60 °C; 15 s at 95 °C; 20 s at 60 °C; and 15 s at 95 °C. Melting curve was collected at the end of each run by using following thermal profile: 95 °C, 15 s, 1.6 °C per s ramp rate; followed by 60 °C, 1 min, 1.6 °C per s ramp rate; and 95 °C, 15 s, 0.05 °C per s ramp for dissociation step. Only reactions resulting in amplicons with predicted Tm were analyzed further. Background subtraction, average PCR efficiency for each amplicon, and N0 values were calculated by using LinRegPCR software (Version 2012) (38). Gene copy number was calculated by using squalene synthase (SS) as a single-copy reference and represented as N0 CYP71AV1/N0 SS. The following PCR primers were used for the analysis: CYP71AV1_For 5′-TCTCACTGACCACTTCCATTGC-3′; CYP71AV1_Rev 5′-TGTAAACGAACAAAAGGATCGTTG-3′; SS_For 5′-GAGGACTCTGAGAAAGCCGTTC-3′, and SS_Rev 5′- AGCATTTGTCACCATATCATTTAAGC-3′.
Trichome Density Measurements.
Trichome density was quantified on the abaxial surface of the terminal leaflets of leaves 14–16 (counting from the apical meristem). Trichomes were visualized by using a Zeiss fluorescent dissecting microscope (fitted with a 470/40-nm excitation filter/525/50-nm emission filter). Images were recorded by using AxioVision software (Version 4.7; Carl Zeiss Ltd.). Trichome number was counted manually across 3 × 0.5-mm2 per leaflet. Average (mean) trichome density was determined for the leaf.
Metabolite Analysis by UPLC-MS and -MS.
Fifteen plants from each of five genotype classes were grown from seeds in 4-inch pots under 16-h days for 12 wk. Metabolite profiles were generated from 50 mg fresh weight pooled samples of leaves at different developmental stages: 1–5 (counted from the apical meristem), representing the juvenile stage; leaves 7–9, representing the young, expanding stage; and leaves 11–13, representing the mature, expanded stage (Fig. 1B). Fresh leaf samples were stored at −80 °C. Dry leaf material was obtained from 14-wk-old plants, cut just above the zone of senescing leaves, and dried for 14 d at 40 °C. Leaves were stripped from the plants, and leaf material was sieved through 5-mm mesh to remove small stems. Trichome-specific metabolites were extracted as described (20) with minor modifications. Briefly, 50 mg of fresh material was extracted by gentle shaking in 500 µL of chloroform for 1 h. Dry leaf material (0.5 g) was ground to a fine powder by using a TissueLyser II ball mill fitted with stainless steel grinding jars (Qiagen) operated at 25 Hz for 1 min. Ten-milligram subsamples were extracted in 9:1 (vol/vol) chloroform:ethanol with gentle shaking for 1 h and then analyzed as per fresh material. For UPLC-MS analysis of sesquiterpenes, a diluted [1:5 (vol/vol) extract:ethanol] 2-µL aliquot was injected on an Acquity UPLC system (Waters) fitted with a Luna 50- × 2-mm 2.5-μm HST column (Phenomenex). Metabolites were eluted at 0.6 mL/min and 40 °C by using a linear gradient from 40% to 100% acetonitrile containing 0.1% formic acid over 2.5 min. For enhanced resolution runs, a Waters Acquity C18 BEH 100- × 2.1-mm 1.7-µm column was used with the same solvents, and the gradient was extended to 25 min. Pseudomolecular [M+H]+ ions were detected by using a Thermo Fisher LTQ-Orbitrap mass spectrometer fitted with an atmospheric pressure chemical ionization source operating in positive ionization mode under the control of Xcalibur software (Version 2.1). Data were acquired over the m/z range 100–1,000 in FTMS centroid mode with resolution set to 7,500 FWHM at m/z 400. Data extraction and analysis were performed by using packages and custom scripts in R (Version 3.2.2) (https://www.R-project.org/). XCMS (39) incorporating the centWave algorithm (40) was used for untargeted peak extraction. Deisotoping, fragment, and adduct removal was performed by using CAMERA (41). Artemisinin was quantified by using the standard curve of the response ratio of artemisinin (Sigma) to internal standard (β-artemether; Hallochem Pharmaceutical) that was previously added to extracts and standards. Metabolites were identified with reference to authentic standards or NMR-resolved structures, and empirical mass formulas were calculated by using the R package rcdk (42) within 10-ppm error and elemental constraints of: C = 1–100, H = 1–200, O = 0–20, and n = 0–1. Peak concentrations were calculated by using bracketed response curves, where standard curves were run every ∼30 samples. Metabolite concentrations were expressed as a proportion of the residual dry leaf material after extraction.
Under typical “short run” conditions, low levels of DHAA and AA appeared to be detected in homozygous cyp71av1-1 leaf extracts (Dataset S3). These were suspected to be misidentifications arising from by coeluting ion fragments of mutant-specific metabolites. A “long run” method used the same solvents and loading conditions, but with a Waters C18 BEH 1.7-µm 2.1- × 100-mm column over an extended 25-min elution gradient to maximize peak capacity and resolution. This method allowed coeluting peaks to be distinguished from genuine DHAA and AA peaks and confirmed the absence of both metabolites in homozygous cyp71av1-1 (Dataset S2).
For analysis of monoterpenes and volatile sesquiterpenes, an aliquot of chloroform extract (before dilution with ethanol for UPLC analysis) was taken for GC-MS analysis using an Agilent 6890 GC interfaced to a Leco Pegasus IV TOF MS (Leco). A 1-µL aliquot was injected into a CIS4 injector (Gerstel) fitted with a 2-mm ID glass liner containing deactivated glass wool at 10 °C. The injector was ramped from 10 °C to 300 °C at 12 °C/s and then held at 300 °C for 5 min. The carrier case was He at constant flow of 1 mL/min and the injection split ratio was 1:10. Peaks were eluted by using a Restek Rxi-5Sil MS column, 30-m × 0.25-mm ID × 0.25-µm film thickness (Thames Restek). The following temperature gradient was used: isothermal 40 °C 2 min, ramp at 20 °C per min to 320 °C, and then hold for 1 min; total run time ∼20 min. The transfer line was maintained at 250 °C, and the MS was used to collect −70 eV EI scans over the m/z range 20–450 at a scan rate of 20 spectra per s. Acquisition was controlled by ChromaTof software (Version 4.5; Leco). ChromaTof was used to identify peaks and deconvolute spectra from each run, assuming a peak width of 3 s and a minimum s/n of 10. Peak areas were reported as deconvoluted total ion traces (DTICs). Further analyses including annotation against authentic standards, between-sample peak alignment, grouping, consensus DTIC reporting, and missing value imputation were performed by using custom scripts in R.
R was used for all statistical data analysis using the stats base package, nlme (https://cran.r-project.org/web/packages/nlme/index.html), and pcaMethods (43).
NMR Structural Data for A-4,11-D, Amorphadiene Tertiary Hydroperoxide, and Arteannuin X.
Leaf material was extracted with chloroform, and the extract was separated by gradient-column chromatography, followed by preparative-scale isocratic HPLC to obtain purified fractions for identification by NMR spectroscopy. Both 1D experiments (1H and 13C) and 2D NMR techniques [heteronuclear single quantum coherence (HSQC), heteronuclear multiple bond correlation (HMBC), COSY, and NOESY] were used for structural characterization, which also generated full assignments for all 1H and 13C NMR resonances. The fully assigned NMR spectra for the known natural product A-411-D have not been reported previously in the literature, and the two metabolites, amorphadiene tertiary hydroperoxide and arteannuin X (Art X), are novel compounds.
Structural Characterizations by 1D and 2D NMR.
All spectra were acquired at a temperature of 298 K on an Avance III 700 spectrometer (Bruker Biospin) operating at a 1H frequency of 700.13 MHz using a 5-mm inverse (TCI) probe and standard Bruker pulse sequences.
The 1D NMR Techniques.
(i) 1H chemical shifts were established from a 1H NMR experiment, recorded by using 128 scans (repetitions) of the pulse sequence “zg30” (including 2 dummy scans) with 131,072 points over a 15-ppm sweep width. The time data were weighted by using a negative exponential function (line broadening of 0.2 Hz), followed by 1D Fourier transformation to produce a spectrum with 131,072 points.
(ii) 13C chemical shifts were established from a 13C NMR experiment, recorded using 3,000 scans (repetitions) of the pulse sequence “zgpg60” (including 2 dummy scans) with 131,072 points over a 270 ppm sweep width. The time data were weighted by using a negative exponential function (line broadening of 1 Hz), followed by 1D Fourier transformation to produce a spectrum with 131,072 points.
The 2D NMR Techniques.
(i) 1H-13C single-bond connections were derived from the gradient edited-HSQC, recorded by using 16 scans (repetitions) of the pulse sequence “hsqcedetgpsisp2.2” (including 32 dummy scans), with 2,048 data points and 256 T1 increments over a 10 ppm 1H and 160 ppm 13C sweep width, optimized for a 1JCH coupling of 145 Hz. The time data were weighted by using a Qsine function (SSB = 2) in both dimensions, followed by 2D Fourier transformation to produce a spectrum with 2,048 points in the normal frequency dimension and 1,024 points in the second dimension (F1).
(ii) 1H-13C two- and three-bond connections were derived from the gradient HMBC experiment (magnitude mode), recorded using 16 scans (repetitions) of the pulse sequence “hmbcetgpl3nd” (including 32 dummy scans), with 4,096 data points and 256 T1 increments over a 10 ppm 1H and 240 ppm 13C sweep width, optimized for 2JCH/3JCH couplings of 8 Hz. The time data were weighted by using a Qsine function (SSB = 2) in both dimensions, followed by 2D Fourier transformation to produce a spectrum with 4,096 points in the normal frequency dimension and 1,024 points in the second dimension (F1).
(iii) 1H-1H connections (predominantly through three-bonds) were derived from a phase-sensitive gradient-enhanced 1H-1H COSY experiment, recorded by using two scans (repetitions) of the pulse sequence “cosygpmfqf” (including eight dummy scans), with 2,048 data points and 256 T1 increments over a 10 ppm sweep width (both dimensions). The time data were weighted by using a sine function in both dimensions, followed by 2D Fourier transformation to produce a spectrum with 2,048 points (both dimensions).
(iv) Through-space proximities between 1H atoms, separated by a distance of up to ∼5 Å, were derived from a phase-sensitive gradient-enhanced NOESY experiment, recorded using 4 scans (repetitions) of the pulse sequence “noesygpph” (including 16 dummy scans), with 2,048 data points and 256 T1 increments over a 10 ppm sweep width (both dimensions), using a mixing time of 800 ms with a relaxation delay of 2 s. The time data were weighted by using a Qsine function (SSB = 2) in both dimensions, followed by 2D Fourier transformation to produce a spectrum with 2,048 points (both dimensions).
The 2D NMR Studies (CDCl3) on A-4,11-D.

HSQC revealed information on 1H and 13C chemical shifts for each of the methyl, methylene, and methine groups in A-4,11-D, as follows:
1-H (δH 1.32, dddd, J = 11, 3, 3, 3 Hz) correlated with 1-C (δC 41.8).
2-Hα (δH 1.56, dddd, J = 12, 12, 6, 2 Hz) correlated with 2-C (δC 25.8).
2-Hβ (δH 1.96, dddd, J = 12, 3, 3, 3 Hz) correlated with 2-C (δC 25.8).
3-Hα (δH 1.78, dddd, J = 16, 6 Hz) correlated with 3-C (δC 26.5).
3-Hβ (δH 1.89, dd, J = 16, 12 Hz) correlated with 3-C (δC 26.5).
5-H (δH 5.06, br s) correlated with 5-C (δC 120.9).
6-H (δH 2.55, br s) correlated with 6-C (δC 37.6).
7-H (δH 1.99, d, J = 11 Hz) correlated with 7-C (δC 47.6).
8-Hα (δH 1.51, dddd, J = 12, 3, 3, 3 Hz) correlated with 8-C (δC 26.0).
8-Hβ (δH 1.27, dddd, J = 12, 12, 12, 3 Hz) correlated with 8-C (δC 26.0).
9-Hα (δH 0.98, dddd, J = 12, 12, 12, 3 Hz) correlated with 9-C (δC 35.4).
9-Hβ (δH 1.68, dddd, J = 12, 3, 3, 3 Hz) correlated with 9-C (δC 35.4).
10-H (δH 1.41, dq, J = 10, 7 Hz) correlated with 10-C (δC 27.8).
12-H3 (δH 1.74, s) correlated with 12-C (δC 22.6).
13-Ha (δH 4.87, br s) correlated with 13-C (δC 109.8).
13-Hb (δH 4.65, br s) correlated with 13-C (δC 109.8).
14-H3 (δH 0.89, d, J = 7 Hz) correlated with 14-C (δC 19.8).
15-H3 (δH 1.60, s) correlated with 15-C (δC 23.7).
HMBC revealed information on 1H and 13C connectivities between the various methyl, methylene, and methine groups identified above.
1-C (δC 41.8) correlated with 9-Hβ (δH 1.68, dddd, J = 12, 3, 3, 3 Hz) and 14-H3 (δH 0.89, d, J = 7 Hz).
3-C (δC 26.5) correlated with 2-Hβ (δH 1.96, dddd, J = 12, 3, 3, 3 Hz) and 15-H3 (δH1.60, s).
4-C (δC 134.6) correlated with 2-Hβ (δH 1.96, dddd, J = 12, 3, 3, 3 Hz), 3-Hα (δH 1.78,dd, J = 16, 6 Hz) and 15-H3 (δH 1.60, s).
5-C (δC 120.9) correlated with 15-H3 (δH 1.60, s).
6-C (δC 37.6) correlated with 2-Hβ (δH 1.96, dddd, J = 12, 3, 3, 3 Hz) and 8-Hβ (δH1.27, dddd, J = 12, 12, 12, 3 Hz).
7-C (δC 47.6) correlated with 8-Hβ (δH 1.27, dddd, J = 12, 12, 12, 3 Hz), 9-Hβ (δH 1.68, dddd, J = 12, 3, 3, 3 Hz), 12-H3 (δH 1.74, s), 13-Ha (δH 4.87, br s) and 13-Hb (δH 4.65, br s).
9-C (δC 35.4) correlated with 8-Hα (δH 1.51, dddd, J = 12, 3, 3, 3 Hz), 8-Hβ (δH 1.27, dddd, J = 12, 12, 12, 3 Hz) and 14-H3 (δH 0.89, d, J = 7 Hz).
10-C (δC 27.8) correlated with 9-Hβ (δH 1.68, dddd, J = 12, 3, 3, 3 Hz) and 14-H3 (δH 0.89, d, J = 7 Hz).
11-C (δC 148.0) correlated with 8-Hα (δH 1.51, dddd, J = 12, 3, 3, 3 Hz), 8-Hβ (δH 1.27, dddd, J = 12, 12, 12, 3 Hz), 12-H3 (δH 1.74, s) and 13-Hb (δH 4.65, br s).
12-C (δC 22.6) correlated with 13-Ha (δH 4.87, br s) and 13-Hb (δH 4.65, br s).
13-C (δC 109.8) correlated with 12-H3 (δH 1.74, s).
Note that no HMBC correlation were observed for 2-C (δC 25.8), 8-C (δC 26.0), 14-C (δC 19.8), and 15-C (δC 23.7).
Strong, well-resolved COSY correlations observed are indicated by red arrows on the structure of A-4,11-D.
The 2D NMR Studies (CDCl3) on Amorphadiene Tertiary Hydroperoxide (A411DOOH).
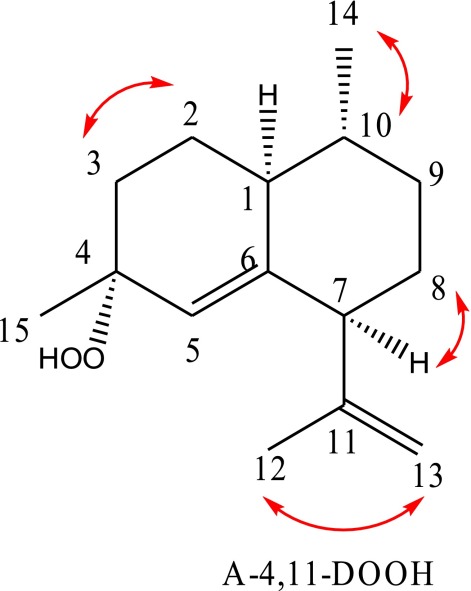
HSQC revealed information on 1H and 13C chemical shifts for each of the methyl, methylene, and methine groups in A-4,11-DOOH, as follows:
1-H (δH 1.64, m) correlated with 1-C (δC 44.5).
2-Hα (δH 1.38, dddd, J = 12, 12, 7, 3 Hz) correlated with 2-C (δC 24.0).
2-Hβ (δH 2.01, dddd, J = 12, 9, 6, 4 Hz) correlated with 2-C (δC 24.0).
3-Hα (δH 1.89, ddd, J = 13, 10, 4 Hz) correlated with 3-C (δC 29.8).
3-Hβ (δH 1.55, dd, J = 13, 8, 3 Hz) correlated with 3-C (δC 29.8).
5-H (δH 5.18, br s) correlated with 5-C (δC 121.4).
7-H (δH 2.57, d, J = 13 Hz) correlated with 7-C (δC 51.5).
8-Hα (δH 1.76, dddd, J = 12, 3, 3, 3 Hz) correlated with 8-C (δC 32.3).
8-Hβ (δH 1.45, dddd, J = 12, 12, 12, 3 Hz) correlated with 8-C (δC 32.3).
9-Hα (δH 1.22, dddd, J = 12, 12, 12, 3 Hz) correlated with 9-C (δC 35.3).
9-Hβ (δH 1.78, dddd, J = 12, 3, 3, 3 Hz) correlated with 9-C (δC 35.3).
10-H (δH 1.23, m) correlated with 10-C (δC 39.0).
12-H3 (δH 1.73, s) correlated with 12-C (δC 21.9).
13-Ha (δH 4.93, br s) correlated with 13-C (δC 112.2).
13-Hb (δH 4.75, br s) correlated with 13-C (δC 112.2).
14-H3 (δH 0.96, d, J = 7 Hz) correlated with 14-C (δC 20.1).
15-H3 (δH 1.28, s) correlated with 15-C (δC 24.2).
HMBC revealed information on 1H and 13C connectivities between the various methyl, methylene, and methine groups identified above.
1-C (δC 44.5) correlated with 14-H3 (δH 0.96, d, J = 6 Hz).
3-C (δC 29.8) correlated with 15-H3 (δH 1.28, s).
4-C (δC 81.5) correlated with 15-H3 (δH 1.28, s).
5-C (δC 121.4) correlated with 15-H3 (δH 1.28, s).
7-C (δC 51.5) correlated 5-H (δH 5.18, br s), 12-H3 (δH 1.73, s), 13-Ha (δH 4.93, br s) and 13-Hb (δH 4.75, br s).
9-C (δC 35.3) correlated with 14-H3 (δH 0.96, d, J = 6 Hz).
10-C (δC 39.0) correlated with 14-H3 (δH 0.96, d, J = 6 Hz).
11-C (δC 146.6) correlated with 12-H3 (δH 1.73, s).
12-C (δC 21.9) correlated with 13-Ha (δH 4.93, br s) and 13-Hb (δH 4.75, br s).
13-C (δC 112.2) correlated with 12-H3 (δH 1.73, s).
Note that no HMBC correlation were observed for 2-C (δC 24.0), 6-C (δC 146.8), 8-C (δC 32.3), 14-C (δC 20.1) and 15-C (δC 24.2).
Strong, well-resolved COSY correlations observed are indicated by red arrows on the structure of A-4,11-DOOH.
The 2D NMR Studies (CDCl3) on Arteannuin X.
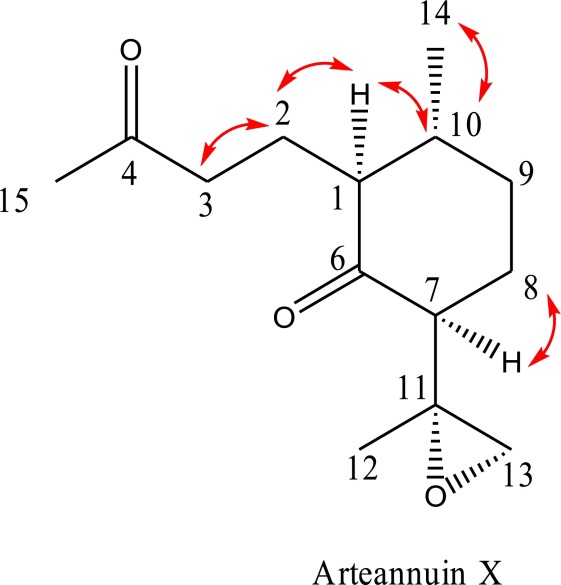
HSQC revealed information on 1H and 13C chemical shifts for each of the methyl, methylene, and methine groups in Arteannuin X, as follows:
1-H (δH 2.03, dd, J = 10, 1 Hz) correlated with 1-C (δC 56.8).
2-Ha (δH 1.83, dddd, J = 14, 9, 6, 2 Hz) correlated with 2-C (δC 20.0).
2-Hb (δH 1.76, ddd, J = 12, 9, 9, 6 Hz) correlated with 2-C (δC 20.0).
3-Ha (δH 2.60, ddd, J = 18, 9, 6 Hz) correlated with 3-C (δC 41.3).
3-Hb (δH 2.39, dd, J = 18, 9, 6 Hz) correlated with 3-C (δC 41.3).
7-H (δH 2.43, dd, J = 13, 5 Hz) correlated with 7-C (δC 56.3).
8-Hα (δH 2.06, dddd, J = 12, 5, 5, 3 Hz) correlated with 8-C (δC 28.7).
8-Hβ (δH 1.32, dddd, J = 12, 12, 12, 4 Hz) correlated with 8-C (δC 28.7).
9-Hα (δH 1.50, dddd, J = 12, 12, 12, 4 Hz) correlated with 9-C (δC 34.1).
9-Hβ (δH 1.89, dddd, J = 12, 3, 3, 3 Hz) correlated with 9-C (δC 34.1).
10-H (δH 1.56, m) correlated with 10-C (δC 40.1).
12-H3 (δH 1.38, s) correlated with 12-C (δC 20.9).
13-Ha (δH 2.62, d, J = 5 Hz) correlated with 13-C (δC 51.6).
13-Hb (δH 2.57, d, J = 5 Hz) correlated with 13-C (δC 51.6).
14-H3 (δH 1.09, d, J = 7 Hz) correlated with 14-C (δC 20.7).
15-H3 (δH 2.14, s) correlated with 15-C (δC 30.0).
HMBC revealed information on 1H and 13C connectivities between the various methyl, methylene, and methane groups identified above.
1-C (δC 56.8) correlated with 2-Hb (δH 1.76, ddd, J = 14, 9, 6 Hz) and 14-H3 (δH 1.09, d, J = 7 Hz).
3-C (δC 41.3) correlated with 2-Hb (δH 1.76, ddd, J = 14, 9, 6 Hz) and 15-H3 (δH 2.14, s).
4-C (δC 209.2) correlated with 2-Hb (δH 1.76, ddd, J = 14, 9, 6 Hz), 3-Ha (δH 2.60, ddd, J = 18, 9, 6 Hz), 3-Hb (δH 2.39, ddd, J = 18, 9, 6 Hz) and 15-H3 (δH 2.15, s).
6-C (δC 211.0) correlated with 2-Hb (δH 1.76, ddd, J = 14, 9, 6 Hz) and 7-H (δH 2.43, dd, J = 13, 5 Hz).
7-C (δC 56.3) correlated with 12-H3 (δH 1.38, s), 13-Ha (δH 2.62, d, J = 5 Hz) and 13- Hb (δH 2.57, d, J = 5 Hz).
8-C (δC 28.7) correlated with 7-H (δH 2.43, dd, J = 13, 5 Hz).
9-C (δC 34.1) correlated with 14-H3 (δH 1.09, d, J = 7 Hz).
10-C (δC 40.1) correlated with 14-H3 (δH 1.09, d, J = 7 Hz).
11-C (δC 55.8) correlated with 7-H (δH 2.43, dd, J = 13, 5 Hz), 12-H3 (δH 1.38, s), 13- Ha (δH 2.62, d, J = 5 Hz) and 13-Hb (δH 2.57, d, J = 5 Hz).
12-C (δC 20.7) correlated with 13-Ha (δH 2.62, d, J = 5 Hz).
13-C (δC 51.6) correlated with 7-H (δH 2.43, dd, J = 13, 5 Hz) and 12-H3 (δH 1.38, s).
Note that no HMBC correlation were observed for 2-C (δC 20.0), 14-C (δC 20.7), and 15-C (δC 30.0).
Strong well-resolved COSY correlations are indicated by red arrows on the structure of Arteannuin X.
NMR Analysis of Leaf-Tagging Experiments.
In this experiment, 24 individual leaves were sampled in three biological replicates from sequential nodes on the stem [i.e., from the top (tag 1, first expanded leaf) to the base of a single plant (tag 24, excluding senescing leaves)]. Each leaf was then extracted individually in deuterated chloroform as for UPLC- and GC-MS analysis. The chloroform extracts were analyzed by 1H NMR spectroscopy, and the relative concentrations of significant metabolites were quantified by integration of characteristic and well-resolved peaks appearing in the 1H NMR spectra.
1H-NMR spectra were acquired under automation at a temperature of 298 K on an Avance II+ 500 spectrometer (Bruker Biospin) operating at 500.13 MHz, using a 5-mm BBFO probe. A relaxation delay of 5 s was used for all 1H NMR experiments, which consisted of 128 scans of 65,536 data points with a spectral width of 15 ppm. Free induction decays were Fourier-transformed, after the application of an exponential window function with a line broadening of 0.3 Hz, and both phasing and baseline correction were carried out before performing accurate integration. All 1H NMR chemical shifts were referenced to the (CH3)3 signal of TMS at δH 0.00.
Well-resolved resonances in the 1H NMR spectra of each of the seven metabolites: (A-411-D; A-4,11-DOOH; and Arteannuin X from cyp71av1-1 mutant; and DHAA, DHAAOOH, artemisinin, and DHEDB from the segregating WT) are reported below. Although any of these resonances could in principle be used for integration, it is those that are underlined were selected to quantify the relative amounts for each of three metabolites in the cyp71av1-1 mutant leaf-tag experiments and the four metabolites in the WT leaf-tag experiments. These resonances were chosen because they were the best resolved from other confounding resonances in 1H NMR spectra of the crude extracts and were therefore judged the most suitable for performing accurate integrations.
cyp71av1-1.
A-4,11-D.
1H NMR (CDCl3, 500 MHz): δH 5.06 (1H, br s), 4.87 (1H, br s), 4.64 (1H, br s), 2.55 (1H, br s), 1.74 (3H, s), 1.60 (3H, s), 0.88 (3H, d, J = 6 Hz).
A-4,11-DOOH.
1H NMR (CDCl3, 500 MHz): δH 5.18 (1H, br s), 4.93 (1H, br s), 4.75 (1H, br s), 2.56 (1H, d, J = 13 Hz), 1.73 (3H, s), 1.28 (3H, s), 0.96 (3H, d, J = 6 Hz).
Arteannuin X.
1H NMR (CDCl3, 500 MHz): δH 2.61 (1H, d, J = 5 Hz), 2.59 (1H, ddd, J = 18. 9, 5 Hz), 2.57 (1H, d, J = 5 Hz), 2.13 (3H, s), 1.36 (3H, s), 1.08 (3H, d, J = 6 Hz).
WT.
DHAA.
1H NMR (CDCl3, 500 MHz): δH 5.11 (1H, br s), 2.50 (2H, m), 1.63 (3H, s), 1.18 (3H, d, J = 7 Hz), 0.87 (3H, d, J = 6 Hz).
DHAAOOH.
1H NMR (CDCl3, 500 MHz): δH 5.23 (1H, br s), 2.72 (1H, dq, J = 7, 7 Hz), 1.31 (3H, s), 1.28 (3H, d, J = 7 Hz), 0.93 (3H, d, J = 6 Hz).
Artemisinin.
1H NMR (CDCl3, 500 MHz): δH 5.86 (1H, s), 3.40 (1H, dq, J = 6, 7 Hz), 2.44 (1H, ddd, J = 14, 13, 4 Hz), 1.45 (3H, s), 1.21 (3H, d, J = 7 Hz), 1.00 (3H, d, J = 6 Hz).
DHEDB.
1H NMR (CDCl3, 500 MHz): δH 5.63 (1H, s), 3.15 (1H, dq, J = 7, 7 Hz), 1.69 (3H, s), 1.15 (3H, d, J = 7 Hz), 0.93 (3H, d, J = 6 Hz).
The three or four integrals from each metabolite in the +H NMR spectra of cyp71av1-1 mutant and WT extract were summed, and the relative abundance of each metabolite is expressed as the fraction of the summed total.
Trichome Isolation.
Trichomes were extracted from the top seven leaves pooled from 20 glasshouse-grown 12-wk-old individuals from each of the two genotype classes by using the method modified from ref. 44. Freshly harvested leaves were incubated in the extraction buffer (20 mL per 1 g of leaves) containing: 100 mM sorbitol, 1 mM sucrose, 25 mM KCl, 2.5 mM succinic acid, 2.5 mM DTT, 2.5 mM MgCl2, 0.5 mM EGTA, 0.25 mM Na2HPO4, 0.05 mM Na4P2O7, 12.5 mM Hepes (pH 7.3), and 0.5-mm glass beads (1 g per g of leaf) for 30 min at 4 °C. Leaves were vigorously shaken for 5 min, and extract was filtered through the series of copper sieves: 200, 50, and 20 µm. Purified trichomes were washed from the 20-µm sieve and kept on ice. The final flow through was returned to the container with the leaf material, and the procedure was repeated five times. Viability staining was performed by using 125 nM MitoTrackerRed CMX (Life Technologies Ltd.), a red-fluorescent dye that stains the mitochondria in live cells, and visualized on a Zeiss LSM 710 invert microscope (Carl Zeiss). Only extracts showing positive staining were used for the subsequent experiments. The number of trichomes in the final extract was determined by counting on a hemocytometer slide.
Total Protein Extraction of Trichomes Isolated from Young Leaves.
Trichomes were isolated as described above, except that the extraction buffer contained cOmplete protease inhibitor mixture mix (Roche Diagnostics GmbH). An aliquot was used to count trichomes using a hemocytometer slide. The trichome preparation was split into three aliquots, and these were centrifuged at 2,500 × g for 20 min at +4 °C, and the supernatant was removed. Trichome pellets were ground in a Tissue Lyser II (Qiagen GmbH) by using tungsten beads and a setting of four cycles of 30 s at 1/30-s frequency and freezing of sample in liquid nitrogen between cycles. Ground trichome samples were resuspended in extraction buffer containing cOmplete protease inhibitor mixture mix. Efficient disruption of the trichomes was confirmed by microscopy (Fig. S7 C and D). Total protein content in each sample was established by using the Bradford Protein Assay (Bio-Rad).
Semisynthetic Production of 13C15-Labeled DHAA.
A Saccharomyces cerevisiae strain overexpressing mevalonate pathway genes, multiple copies of integrated A-4,11-D synthase, and transcriptionally inhibited in ergosterol production (9) was grown in shake-flask cultures with radioactively labeled 4% d-glucose-13C6 as the sole carbon source and a hydrophobic overlay used to trap volatiles. A-4,11-D was distilled from the hydrophobic overlay to >95% purity, and fully labeled 13C15 was confirmed by GC-MS analysis. 13C15 A-4,11-D was used for chemical synthesis of 13C15 DHAA in a process as described (45).
Trichome Feeding Experiments.
Isolated trichomes were aliquoted, and one portion of each extract was boiled for 10 min to heat inactivated enzymes present in the glandular secretory trichomes (GSTs). It was clearly evident from microscopy that boiling destroyed the subcuticular sacs of the GSTs and had also caused the complete loss of chlorophyll from the two subapical cell pairs as shown in Fig. S8 B and D.
Fig. S8.

Microscopic evaluation of the enriched glandular secretory trichome extracts from A. annua WT leaves before and after boiling. Images were taken under bright field with 10× (A and B) or 40× (C and D) magnification for the intact trichome extracts (A and C) or 10-min boiled extracts (B and D). Intact subapical cavities are indicted by arrows (A and C). Scale bars are depicted in micrometers.
Boiled (10 min) and nonboiled samples were gently spun down, and the extraction buffer was replaced with 0.5 mg/mL 13C15 DHAA dissolved in trichome extraction buffer containing 1% EtOH to improve substrate solubility: Dark incubated nonboiled samples were immediately wrapped in tin foil. Controls were established that contained feeding substrates in the trichome extraction buffer incubated without trichomes (NTC).
Trichomes were incubated in glass vials over 16-h light/8-h dark cycles or in constant dark, with gentle rotation. Sampling was performed in triplicate from the same vial at 15 min and 24, 48, 72, 96, and 120 h. This time course was selected based on the leaf growth rates observed for the mutant and WT. Both genotypes were able to emerge one leaf per day on the main stem (Fig. S9). Therefore, a 5-d period represents a sufficient time needed for young leaves rich in DHAA and A-4,11-D to become mature leaves synthesizing DHAAOOH, A-4,11-DOOH, and also their final conversion products: artemisinin and arteannuin X to appear. Trichome samples were immediately extracted with 3 volumes of chloroform and subjected to UPLC-MS analysis as described above, but without the addition of β-artemether standards added.
Trichome Total Protein Extract Feeding Experiments.
Cofactors required for the activity of the enzymes from the artemisinin biosynthesis pathway (13–15) were added to the extraction buffer at the following concentrations: FAD (5 µM), FMN (5 µM), NAD (1 mM), NADP (1 mM), NADPH (1 mM), and l-ascorbate (1 mM). Feeding was started with the addition of 0.5 mg/mL 13C15 DHAA (1% ethanol final). The heat-inactivated sample was boiled for 10 min. Dark-incubated nonboiled samples were immediately wrapped in tin foil. Controls were established that contained cofactors and feeding substrate in the trichome extraction buffer incubated without protein extracts.
Protein extracts and nonextracted controls were incubated in glass vials over 16-h light/8-h dark cycles or in constant dark, with gentle rotation. The sampling time course included early time points compared with the intact trichome experiment to ensure any early conversion was monitored. Extraction and UPLC-MS product analysis was exactly as described for intact trichome feeding.
Conclusion
We have described an A. annua CYP71AV1 knockout mutant that provides in planta confirmation for the function of this enzyme. The cyp71av1-1 mutant accumulates high levels of A-4,11-D, which is converted to arteannuin X, a nor- seco-amorphane sesquiterpene epoxide. This work clearly demonstrates the plasticity of metabolism in the glandular secretory trichomes of A. annua; when one pathway is blocked, novel sesquiterpene alternatives are produced, highlighting the potential of trichomes as factories for production of new compounds with potential medicinal and industrial applications.
We found that the in vivo oxidation of A-4,11-D to arteannuin X parallels that of DHAA to artemisinin during the progression of leaf maturation. We were able to chemically complement cyp71av1-1 by externally feeding DHAA to intact trichome preparations and trichome protein extracts. This finding demonstrated that the conversion of DHAA to DHEDB, via a tertiary allylic hydroperoxide, is a nonenzymatic, light-requiring process. The lack of accumulation of artemisinin in these experiments supports the idea that a nonaqueous environment, as provided by the subapical cavity of glandular secretory trichomes, is essential for the nonenzymatic production of the endoperoxide containing artemisinin from DHAA. Together, these findings highlight the importance of nonenzymatic conversions in terpenoid metabolism of A. annua glandular secretory trichomes. These findings, together with the observation that artemisinin is known to be cytotoxic to various cell types (4, 33, 34), suggest a functional requirement for the specialized subapical cavity as a location for both chemical conversion and storage. It also highlights the challenges of producing certain types of plant natural products in microbial systems that lack this level of structural complexity and the need for more research into the compartmentation of metabolic processes in plant production systems.
Materials and Methods
Full details of plant material used, plant growth conditions, screening of EMS-mutagenized population, genotyping, metabolomic analyses, 2D NMR structural characterization, trichome extractions, and trichome feeding with 13C-labeled substrates are presented in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank C. Paddon and K. Monroe (Zagaya) for providing [U-13C15]-(uniformly) labeled A-4,11-D and DHAA; T. Winzer for preliminary experimental involvement; L. Doucet, D. Vyas, C. Whitehead, and B. Kowalik, for laboratory assistance; C. Abbot and A. Fenwick for horticulture assistance; P. Roberts for graphic design; C. Calvert for project management; and X. Simonnet and Médiplant for access to the Artemis variety. G.D.B. thanks the Chemical Analysis Facility at the University of Reading for provision of the 700-MHz NMR spectrometer used in these studies. This work was supported by The Bill and Melinda Gates Foundation and by Biotechnology and Biological Sciences Research Council Grant BB/G008744/1 (to G.D.B.), “The Biosynthesis of Artemisinin.”
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1611567113/-/DCSupplemental.
References
- 1.Duke SO, Paul RN. Development and fine structure of the glandular trichomes of Artemisia annua L. Int J Plant Sci. 1993;154(1):107–118. [Google Scholar]
- 2.Duke MV, Paul RN, Elsohly HN, Sturtz G, Duke SO. Localization of artemisinin and artemisitene in foliar tissues of glanded and glandless biotypes of Artemisia-Annua L. Int J Plant Sci. 1994;155(3):365–372. [Google Scholar]
- 3.Olsson ME, et al. Localization of enzymes of artemisinin biosynthesis to the apical cells of glandular secretory trichomes of Artemisia annua L. Phytochemistry. 2009;70(9):1123–1128. doi: 10.1016/j.phytochem.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Yan ZQ, et al. Mechanism of artemisinin phytotoxicity action: Induction of reactive oxygen species and cell death in lettuce seedlings. Plant Phys Biochem. 2015;88:53–59. doi: 10.1016/j.plaphy.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Lange BM, Ahkami A. Metabolic engineering of plant monoterpenes, sesquiterpenes and diterpenes—current status and future opportunities. Plant Biotechnol J. 2013;11(2):169–196. doi: 10.1111/pbi.12022. [DOI] [PubMed] [Google Scholar]
- 6.Lange BM, Turner GW. Terpenoid biosynthesis in trichomes—current status and future opportunities. Plant Biotechnol J. 2013;11(1):2–22. doi: 10.1111/j.1467-7652.2012.00737.x. [DOI] [PubMed] [Google Scholar]
- 7.Lapkin AA, Plucinski PK, Cutler M. Comparative assessment of technologies for extraction of artemisinin. J Nat Prod. 2006;69(11):1653–1664. doi: 10.1021/np060375j. [DOI] [PubMed] [Google Scholar]
- 8.Paddon CJ, Keasling JD. Semi-synthetic artemisinin: A model for the use of synthetic biology in pharmaceutical development. Nat Rev Microbiol. 2014;12(5):355–367. doi: 10.1038/nrmicro3240. [DOI] [PubMed] [Google Scholar]
- 9.Paddon CJ, et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496(7446):528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 10.Turconi J, et al. Semisynthetic artemisinin, the chemical path to industrial production. Org Process Res Dev. 2014;18(3):417–422. [Google Scholar]
- 11.Peplow M. Synthetic biology’s first malaria drug meets market resistance. Nature. 2016;530(7591):389–390. doi: 10.1038/530390a. [DOI] [PubMed] [Google Scholar]
- 12.Mercke P, Bengtsson M, Bouwmeester HJ, Posthumus MA, Brodelius PE. Molecular cloning, expression, and characterization of amorpha-4,11-diene synthase, a key enzyme of artemisinin biosynthesis in Artemisia annua L. Arch Biochem Biophys. 2000;381(2):173–180. doi: 10.1006/abbi.2000.1962. [DOI] [PubMed] [Google Scholar]
- 13.Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS. Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett. 2006;580(5):1411–1416. doi: 10.1016/j.febslet.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. The molecular cloning of artemisinic aldehyde Delta11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J Biol Chem. 2008;283(31):21501–21508. doi: 10.1074/jbc.M803090200. [DOI] [PubMed] [Google Scholar]
- 15.Teoh KH, Polichuk DR, Reed DW, Covello PS. Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. Botany. 2009;87(6):635–642. [Google Scholar]
- 16.Soetaert SS, et al. Differential transcriptome analysis of glandular and filamentous trichomes in Artemisia annua. BMC Plant Biol. 2013;13:220. doi: 10.1186/1471-2229-13-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown GD, Sy L-K. In vivo transformations of dihydroartemisinic acid in Artemisia annua plants. Tetrahedron. 2004;60(5):1139–1159. [Google Scholar]
- 18.Sy LK, Brown GD. The role of the 12-carboxyllic acid group in the spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron. 2002;58(5):909–923. [Google Scholar]
- 19.Graham IA, et al. The genetic map of Artemisia annua L. identifies loci affecting yield of the antimalarial drug artemisinin. Science. 2010;327(5963):328–331. doi: 10.1126/science.1182612. [DOI] [PubMed] [Google Scholar]
- 20.Larson TR, et al. A survey of artemisinic and dihydroartemisinic acid contents in glasshouse and global field-grown populations of the artemisinin-producing plant Artemisia annua L. Ind Crops Prod. 2013;45:1–6. [Google Scholar]
- 21.Brown GD. The biosynthesis of artemisinin (Qinghaosu) and the phytochemistry of Artemisia annua L. (Qinghao) Molecules. 2010;15(11):7603–7698. doi: 10.3390/molecules15117603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanescu B, Miron A, Corciova A. Sesquiterpene lactones from artemisia genus: Biological activities and methods of analysis. J Anal Methods Chem. 2015;2015:247685. doi: 10.1155/2015/247685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Wang Y, Zhang Q, Qi Y, Guo D. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genomics. 2009;10:465. doi: 10.1186/1471-2164-10-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weng JK, Philippe RN, Noel JP. The rise of chemodiversity in plants. Science. 2012;336(6089):1667–1670. doi: 10.1126/science.1217411. [DOI] [PubMed] [Google Scholar]
- 25.Ma DM, et al. A genome-wide scenario of terpene pathways in self-pollinated Artemisia annua. Mol Plant. 2015;8(11):1580–1598. doi: 10.1016/j.molp.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Ro DK, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440(7086):940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 27.Komori A, et al. Comparative functional analysis of CYP71AV1 natural variants reveals an important residue for the successive oxidation of amorpha-4,11-diene. FEBS Lett. 2013;587(3):278–284. doi: 10.1016/j.febslet.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 28.McCallum CM, Comai L, Greene EA, Henikoff S. Targeting induced local lesions IN genomes (TILLING) for plant functional genomics. Plant Physiol. 2000;123(2):439–442. doi: 10.1104/pp.123.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lommen WJ, Schenk E, Bouwmeester HJ, Verstappen FW. Trichome dynamics and artemisinin accumulation during development and senescence of Artemisia annua leaves. Planta Med. 2006;72(4):336–345. doi: 10.1055/s-2005-916202. [DOI] [PubMed] [Google Scholar]
- 30.Olofsson L, Engström A, Lundgren A, Brodelius PE. Relative expression of genes of terpene metabolism in different tissues of Artemisia annua L. BMC Plant Biol. 2011;11:45. doi: 10.1186/1471-2229-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sy LK, Zhu NY, Brown GD. Syntheses of dihydroartemisinic acid and dihydro-epi-deoxyarteannuin B incorporating a stable isotope labe at the 15-position for studies into the biosynthesis of artemisinin. Tetrahedron. 2001;57(40):8495–8510. [Google Scholar]
- 32.Bryant L, Flatley B, Patole C, Brown GD, Cramer R. Proteomic analysis of Artemisia annua—towards elucidating the biosynthetic pathways of the antimalarial pro-drug artemisinin. BMC Plant Biol. 2015;15:175. doi: 10.1186/s12870-015-0565-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsmark Jessing K, Duke SO, Cedergreeen N. Potential ecological roles of artemisinin produced by Artemisia annua L. J Chem Ecol. 2014;40(2):100–117. doi: 10.1007/s10886-014-0384-6. [DOI] [PubMed] [Google Scholar]
- 34.Li W, et al. Yeast model uncovers dual roles of mitochondria in action of artemisinin. PLoS Genet. 2005;1(3):e36. doi: 10.1371/journal.pgen.0010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delabays N, Simonnet X, Gaudin M. The genetics of artemisinin content in Artemisia annua L. and the breeding of high yielding cultivars. Curr Med Chem. 2001;8(15):1795–1801. doi: 10.2174/0929867013371635. [DOI] [PubMed] [Google Scholar]
- 36.Townsend T, et al. The use of combining ability analysis to identify elite parents for Artemisia annua F1 hybrid production. PLoS One. 2013;8(4):e61989. doi: 10.1371/journal.pone.0061989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Till BJ, Zerr T, Comai L, Henikoff S. A protocol for TILLING and Ecotilling in plants and animals. Nat Protoc. 2006;1(5):2465–2477. doi: 10.1038/nprot.2006.329. [DOI] [PubMed] [Google Scholar]
- 38.Ruijter JM, et al. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6):e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78(3):779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 40.Tautenhahn R, Böttcher C, Neumann S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhl C, Tautenhahn R, Böttcher C, Larson TR, Neumann S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal Chem. 2012;84(1):283–289. doi: 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guha R. Chemical informatics functionality in R. J Stat Softw. 2007;18(5) [Google Scholar]
- 43.Stacklies W, Redestig H, Scholz M, Walther D, Selbig J. pcaMethods—a bioconductor package providing PCA methods for incomplete data. Bioinformatics. 2007;23(9):1164–1167. doi: 10.1093/bioinformatics/btm069. [DOI] [PubMed] [Google Scholar]
- 44.McCaskill D, Croteau R. Monoterpene and sesquiterpene biosynthesis in glandular trichomes of peppermint (Mentha X Piperita) rely exclusively on plastid-derived isopentenyl diphosphate. Planta. 1995;197(1):49–56. [Google Scholar]
- 45.Westfall PJ, et al. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci USA. 2012;109(3):E111–E118. doi: 10.1073/pnas.1110740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, et al. The I-TASSER Suite: Protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.