

Significance
There exists an urgent need for minimally invasive molecular analysis tools for cancer assessment and management, particularly in advanced-stage lung cancer, when tissue procurement is challenging and gene mutation profiling is crucial to identify molecularly targeted agents for treatment. High-throughput compartmentalization and multigene profiling of individual circulating tumor cells (CTCs) from whole-blood samples using modular gene panels may facilitate highly sensitive, yet minimally invasive characterization of lung cancer for therapy prediction and monitoring. We envision this nanoplatform as a compelling research tool to investigate the dynamics of cancer disease processes, as well as a viable clinical platform for minimally invasive yet comprehensive cancer assessment.
Keywords: circulating tumor cells, microfluidics, rare-cell sorting, reverse transcription-PCR, single-cell analysis
Abstract
Circulating tumor cells (CTCs) are established cancer biomarkers for the “liquid biopsy” of tumors. Molecular analysis of single CTCs, which recapitulate primary and metastatic tumor biology, remains challenging because current platforms have limited throughput, are expensive, and are not easily translatable to the clinic. Here, we report a massively parallel, multigene-profiling nanoplatform to compartmentalize and analyze hundreds of single CTCs. After high-efficiency magnetic collection of CTC from blood, a single-cell nanowell array performs CTC mutation profiling using modular gene panels. Using this approach, we demonstrated multigene expression profiling of individual CTCs from non–small-cell lung cancer (NSCLC) patients with remarkable sensitivity. Thus, we report a high-throughput, multiplexed strategy for single-cell mutation profiling of individual lung cancer CTCs toward minimally invasive cancer therapy prediction and disease monitoring.
Blood-based biomarkers, including genomic, transcriptomic, proteomic, and other cellular components, are amenable to minimally invasive approaches for cancer detection and assessment (1). Circulating tumor cells (CTCs), which are currently defined as epithelial cells in a cancer patient’s bloodstream derived from tumor cell release, are an established prognostic blood biomarker, because their concentration in blood is associated with high tumor burden (2). CTC gene expression and gene mutation profiling may therefore noninvasively recapitulate the primary and metastatic tumor composition, potentially yielding clinically actionable information. Therefore, CTC single-cell mutation analysis for clinical application may be a practical approach for tumor progression monitoring and treatment selection.
CellSearch is the only US Food and Drug Administration-approved technology since its introduction in 1999 to identify CTCs in whole blood (3). Many other innovative platforms (2) leverage various physical and biochemical properties of CTCs for highly sensitive and efficient enrichment (2), encompassing improved epithelial cell adhesion molecule (EpCAM)-based capture [e.g., CTC Chip (4), HB-Chip (5), µNMR (6), MagSweeper (7), and MagSifter (8)], size-selective capture [e.g., Cluster-Chip (9), ISET (2, 10), and Vortex technologies (11)], inertial-based capture [e.g., iChip platform (12)], nanomaterials-based capture [e.g., nanofilms (13), NanoVelcro (14), and functionalized graphene oxide nanosheets (15)], and image-based cytometry [e.g., HD-CTC (16)].
These current platforms, unfortunately, are overwhelmingly reliant on the enumeration of CTCs by immunocytochemistry (ICC) to measure tumor burden, which clearly does not probe underlying CTC tumor biology. To overcome limitations of existing CTC identification strategies, downstream analysis of individual CTCs via highly sensitive and specific single-cell measurements of tumor-derived blood components is imperative. Investigators have detected CTCs via gene expression directly from unprocessed whole blood (17, 18), but these measurements are typically (i) obscured by background signals from circulating mRNA, white blood cells (WBCs), and other blood components (19) and (ii) performed using ensemble measurements that inadequately represent individual cell phenotypes. Because this analysis averages out signals across individual CTCs, the heterogeneity of different CTC populations is not detected, which could play a critical role in understanding a patient’s tumor biology, metastatic potential, and clinical course (20). Other reported methods for single-cell multigene expression CTC profiling of breast cancer (21) and metastatic prostate cancer (22) suffer from limited throughput—each captured cell is manually selected, lysed, and subjected to reverse transcription into cDNA before parallel multigene expression profiling, thus precluding their large-scale clinical application.
In particular, lung cancer presents an urgent need for a routine, minimally invasive clinical assessment tool, because (i) it is the leading cause of cancer deaths worldwide (23); (ii) many lung cancer patients are diagnosed at the most advanced stage; (iii) new molecularly targeted therapies are emerging to improve progression-free and overall survival for those patients with driver mutations such as epidermal growth factor receptor (EGFR) (24) and anaplastic lymphoma kinase (ALK) (25); (iv) new methods for diagnosis and treatment monitoring are imperative because accessing the primary tumor or metastatic lesions can be technically challenging, morbid, and costly; and (v) CTCs have been isolated and profiled for key driver mutations in non–small-cell lung cancer (NSCLC) (17).
With the above needs in mind, we report the development, validation, and demonstrated clinical utility of an integrated nanoplatform that features a magnetic sifter [MagSifter (8)] and single-cell nanowell array capable of sorting up to 25,600 cells [Nanowell (26, 27)] to capture, easily compartmentalize, and molecularly characterize single CTCs from blood (Fig. 1). We report here a proof-of-concept demonstration showing single-CTC multigene expression and mutation profiling for tumor interrogation using four-plex modular gene panels, including panels for EGFR mutation detection for therapy prediction and monitoring.
Fig. 1.

Overall workflow of the integrated nanoplatform. (A) First, whole-blood samples are processed for isolation of CTCs by the MagSifter. Streptavidinated 150-nm iron oxide magnetic nanoparticles (MNPs) are conjugated to biotinylated anti-EpCAM antibodies for EpCAM-positive selection of circulating tumor cells (CTCs), which are epithelial in origin. Magnetically labeled CTCs are captured on the sifter when flowed through it under an applied magnetic field and subsequently eluted in the absence of a magnetic field. (B) Subsequently, all eluent is directly loaded without staining onto a Nanowell device, and cells are seeded into individual compartments by centrifugation. After drying, single-cell RT-PCR mix is applied to the nanowells, which are then sealed with PCR tape. (C) The Nanowell device is then placed into a thermocycler for multiplex gene mutation and expression analysis via reverse transcription and PCR amplification. Multigene expression via up to four off-the-shelf hydrolysis probes with discrete emission spectra can be imaged in each nanowell. These fluorescence signals are then analyzed with custom MATLAB and R scripts for identification and characterization of putative CTCs based on modular panels of representative genes. These panels are flexible and can be adjusted to accommodate clinical needs such as therapy prediction and disease monitoring.
Results
Overall Workflow.
The Nanowell performs massively parallel sorting of individual CTCs into 25,600 separate compartments. First, the MagSifter technology (8) (Fig. 1 and Fig. S1) tags CTCs with magnetic nanoparticles for high-throughput, high-efficiency CTC enrichment from blood. After MagSifter enrichment from 2-mL whole-blood samples (see Fig. S2 for illustration of captured cells via conventional ICC), red blood cells (RBCs) are hemolyzed (28) and DNase I is added to remove circulating genomic DNA, obtaining ultrapure cellular eluent that is seeded into Nanowell by direct pipetting and centrifugation. Single-cell multigene RT-PCR is performed with our modular multigene panels: a CTC identification panel with telomerase reverse transcriptase (TERT) (29, 30) and mesenchymal–epithelial transition (MET) (31, 32) to identify putative CTCs in advanced-stage NSCLC patients, and a therapy prediction and monitoring panel with TERT, an EGFR mutation, and its corresponding wild-type gene for identification and mutation detection of putative CTCs in advanced-stage NSCLC. NSCLC and noncancer cell lines were used to validate the gene expression panels on the integrated nanoplatform. To demonstrate its potential for clinical translation, samples from both healthy human subjects and patients diagnosed with NSCLC with the CTC identification panel (TERT and MET) were assayed. Then, therapy prediction and monitoring (TERT, EGFR mutant, and EGFR wild-type) panels were assayed using advanced-stage NSCLC patient blood samples.
Fig. S1.

Photographic depiction of active device components of the integrated nanodiagnostic platform. (A) A single Magnetic Sifter (MagSifter) chip fabricated with standard microelectromechanical systems (MEMS) technology. (B) A MagSifter chip assembled in a custom-designed laser-cut acrylic holder and fastened by four steel screws and two rubber O-rings. (C) A Nanowell device fabricated with standard soft lithographic techniques, featuring 25,600 individual wells in a 1 × 1-cm2 area for single-cell compartmentalization and RT-PCR analysis. (D) A Nanowell device assembled on a standard-sized microscope slide with an accompanying gasket for reagent loading.
Fig. S2.

CTCs captured and identified by MagSifter through ICC with three stains (DAPI, a nuclear stain, in blue; CK, an epithelial marker, in green; and CD45, a leukocyte marker, in red). (A) Exemplary section of a fluorescence microscopic image of CTCs collected from a stage IV NSCLC patient. (Scale bar, 50 µm.) (B) After isolation, standard immunostaining with DAPI, CK, and CD45 visually identifies different cell populations with a variety of morphology and staining patterns. Cells that were positive for both DAPI and CK can be categorized into (i) pathological CTCs, (ii) cells with irregular morphology (Irregular CTC*), (iii) cells lacking definitive cellular integrity that are putatively apoptotic or necrotic (Apoptotic CTC*), and (iv) cancer-associated cells (CACs) (56) that are triple-positive for DAPI, CK, and CD45. For comparison, WBCs, which are DAPI-positive, CK-negative, and CD45-positive, are also shown.
Single-Cancer-Cell Detection Using the Nanowell Assay on Cell Lines.
We first identified mutations in HCC827 and H661 cell lines (Fig. 2A), which are mutant and wild type, respectively, for EGFR exon 19 deletion (del19) in bulk and then at the individual-cell level to demonstrate the utility of Nanowell. Bulk gene expression analysis of total mRNA extracted from 106 cells each of HCC827 and H661 indicated their mutually exclusive gene expression of EGFR E746-A750del [the most common del19 mutation (33)] and wild type, respectively (P < 0.0001, two-sample t test). Nanowell confirmed differential del19 expression between HCC827 and H661, with individual HCC827 cells expressing only EGFR E746-A750del (green) and individual H661 cells exhibiting only wild-type EGFR (orange) (P < 0.0001) (Fig. 2B).
Fig. 2.

Translating bulk gene expression to single-cell gene expression profiling on Nanowell using cell lines. (A) Two cell lines (HCC827 and H661) were selected according to their EGFR exon 19 deletion (del19) mutational status (mutant and wild type, respectively). Bulk expression analysis by PCR showed that HCC827 cells exhibited measured signal for only the EGFR E746-A750del (the most common EGFR del19 mutation shown in green), with undetectable wild-type EGFR. In contrast, H661 cells exhibited measured signal for only the wild-type EGFR shown in yellow, with undetectable E746-A750del. (B) The same two cell lines each underwent single-cell analysis on Nanowell using EGFR E746-A750del and wild-type probes in FAM (shown in green) and HEX (shown in orange) channels, respectively. HCC827 cells exhibited fluorescence predominantly in the FAM channel, indicating the presence of EGFR del19 mutation. (Scale bar, 200 µm.) In contrast, the assay of H661 cells exhibited fluorescence predominantly in the HEX channel, indicating the presence of wild-type EGFR. (Scale bar, 200 µm.) For each cell line, we observed a clear differentiation between the EGFR mutant and wild-type expression levels from single cells in Nanowell, and the bulk expression measurements matched the corresponding Nanowell signal, with high statistical significance (P < 0.0001 for both cases, t test). (C) Bulk gene expression among five NSCLC cell lines (A549, H661, H1650, H1975, and HCC827), a noncancerous fibroblast cell line (PCS-201), and WBCs are shown by quantitative RT-PCR. The five cancer cell lines exhibited high TERT and MET gene expression, whereas the fibroblast cells and WBCs exhibited no TERT and low MET expression. (D) Single-cell analysis using Nanowell on cancer and noncancer populations by RT-PCR expression analysis on H1650 and PCS-201 cell lines. (Scale bar, 50 µm.) Each cell line was stained with different CellTracker dyes (green for H1650; orange for PCS-201) and then imaged to illustrate the concordance between each cell’s Nanowell location and RT-PCR signal (TERT shown in blue). Nanowell showed a clear differentiation between H1650 (cancer; n = 16) and PCS-201 (noncancer; n = 14) cell lines based on TERT expression, with high statistical significance (P < 0.0001, t test).
We further examined the bulk gene expression of 106 cells each of five NSCLC cell lines (A549, H661, H1650, H1975, and HCC827), a noncancerous fibroblast (PCS-201) cell line, and WBCs from 2 mL of healthy human blood. Cancer cells expressed significantly higher TERT and MET than PCS-201 cells and WBCs (P < 0.0001, t test), both of which expressed no TERT and low MET (Fig. 2C), thus implying that TERT and MET are good CTC markers.
We then assessed Nanowell’s ability to distinguish between cancerous and noncancerous cells by differential gene expression profiling of H1650 and PCS-201 cell lines. H1650 and PCS-201 cells were stained with CellTracker Green CMFDA and Orange CMRA dyes (Life Technologies), respectively, and assayed together in a Nanowell for TERT. Nanowell images were acquired in three fluorescence channels corresponding to H1650 cells (as denoted in green color), PCS-201 cells (denoted as red color), and TERT gene expression (denoted as blue color) (Fig. 2D). TERT expression signals revealed a significant differentiation between H1650 and PCS-201 cells (P < 0.0001, t test).
Four-Plex Gene Expression of Single H1650 Cells Spiked into Healthy Blood.
To validate the suitability of a modular gene panel including vimentin (VIM) and aldehyde dehydrogenase (ALDH) in addition to TERT and MET for CTC identification, we examined the bulk gene expression of 106 cells from each of five NSCLC cell lines (A549, H661, H1650, H1975, and HCC827), a noncancerous fibroblast (PCS-201) cell line, and WBCs from 2 mL of healthy human blood. We noted heterogeneous VIM and ALDH expression across these cell types (Fig. S3).
Fig. S3.

The same six cell lines as in Fig. 2 and WBCs were also assayed in bulk for VIM and ALDH gene expression to demonstrate the heterogeneity of gene expression across different cell lines and to evaluate the capability for four-plex expression analysis using our primer–probe sets.
After performing these in vitro experiments, we assessed whether our platform could isolate cells spiked into healthy human blood using H1650 cells, which mimics the NSCLC patient condition (Fig. 3A). After demonstrating bulk gene expression for our four-plex assay TERT, MET, VIM, and ALDH, we observed distinct patterns of gene expression on individual cells isolated from doped blood (Fig. 3B). One cell population expressed high VIM only (Fig. 3 C, E, and F), which we attribute to WBCs’ mesenchymal cell origin (34), whereas the second cell population expressed high TERT, MET, and VIM, and variable ALDH (Fig. 3 D–F), corresponding to H1650s bulk expression. Importantly, these single-cell observations matched our bulk analysis from Fig. 2 and Fig. S3. Quantitative analysis of total gene expression, normalized by H1650 expression, indicated differential TERT and MET expression in WBCs and H1650 cells (P < 0.0001 for both cases, t test), indicating TERT and MET are both excellent markers to identify cancer cells compared with WBCs (Fig. 3E). Average VIM and ALDH expression was similar between H1650 cells and WBCs, and H1650 expressed ALDH with higher variance (Fig. 3F).
Fig. 3.

Validation of our integrated nanoplatform with blood samples. (A) Workflow schematic of validation experiments. H1650 cells were spiked into whole-blood samples from healthy human subjects and processed through the MagSifter and Nanowell. (B) Scanned fluorescence images on Nanowell after RT-PCR amplification of four genes on the spike-in cells recovered using MagSifter. (Scale bar, 50 µm.) These genes were selected to identify putative CTCs (TERT and MET) and to demonstrate the multiplex capability of the assay using biologically informative targets (VIM and ALDH). Two major populations were visually observed: one with a primarily red color (C) and another with a primarily yellow color (D). (C) This cell population primarily expressed VIM and none of the other three assayed genes, which is characteristic of leukocytes (WBCs). (D) In contrast, a second cell population expressed TERT, MET, and VIM, and partially expressed ALDH, which is characteristic of H1650 cancer cells. (E) Quantitative analysis of Nanowell after RT-PCR exhibited a clear differentiation of TERT and MET expression levels between the cell populations in C and D (nominally WBCs and H1650 cells), with high statistical significance (P < 0.0001 for both cases, t test). (F) Quantitative analysis of VIM and ALDH expression revealed similar VIM expression between the two cell populations, whereas the ALDH expression in the latter cell population exhibited higher variance. (G) A two-channel fluorescence composite scan of the entire Nanowell array (25,600 wells) after on-chip RT-PCR using TERT and MET probes shows a stage IV NSCLC patient sample (Left), with a substantial number of TERT and MET positive wells. In contrast, the similar scan of a healthy donor blood sample (Right) lacked TERT and MET signal. (Scale bar, 1,000 µm.) (H) The MagSifter and Nanowell were used for CTC identification in blood samples (defined as the population of EpCAM+ cells with expression of both TERT and MET in a nanowell). Our 55 assayed samples consisted of 35 advanced-stage NSCLC samples and 20 healthy control samples. (I) Multigene expression of CTCs from NSCLC patient blood. A representative portion of the fluorescence scan (superimposed in four channels) of a completed Nanowell multigene expression panel for a stage III NSCLC patient sample displayed heterogeneous single-cell gene expression across nanowells. (Scale bar, 50 µm.) (J) A quantitative representation of the four-plex data confirmed that putative CTCs exhibit high TERT and MET expression and variable VIM and ALDH expression. In contrast, WBCs exhibit high VIM expression, variable ALDH expression, and low TERT and MET expression.
Multiplex Gene Expression Analysis of CTCs in NSCLC Cancer Patients.
Following a successful demonstration of multiplexed gene profiling of individual cancer cells from cell lines in spiked blood, we proceeded to analyze whole-blood samples from 55 human subjects (from June 2014 to August 2016). We analyzed blood from 20 healthy individuals from the Stanford Blood Center and 35 patients with stage IV NSCLC of adenocarcinoma histology to first identify CTCs by single-cell TERT and MET expression profiling, and second to determine whether these putative CTCs were abundant in NSCLC and not in healthy patients (Fig. 3 G and H and Table S1).
Table S1.
Clinical information of patients enrolled in cohort who provided blood samples
| PTS | Nanowell | Diagnosis | Stage | Histology | Age | Sex | Tumor mutation |
| 1 | 0 | NSCLC | IV | AC | 63 | F | EGFR e18+ |
| 2 | 3 | NSCLC | IV | AC | 52 | F | NRAS |
| 3 | 3 | NSCLC | IV | AC | 54 | F | None |
| 4 | 3 | NSCLC | IV | AC | 60 | F | EGFR L858R |
| 5 | 7 | NSCLC | IV | AC | 66 | F | EGFR e19 Del |
| 6 | 8 | NSCLC | IV | AC | 55 | F | EGFR e19 Del |
| 7 | 8 | NSCLC | IV | AC | 79 | M | EGFR L858R |
| 8 | 9 | NSCLC | IV | AC | 79 | M | None |
| 9 | 10 | NSCLC | IV | AC | 63 | F | EFGR e19 Del |
| 10 | 11 | NSCLC | IV | AC | 73 | F | EFGR L858R |
| 11 | 12 | NSCLC | IV | AC | 35 | F | EGFR e19 Del |
| 12 | 13 | NSCLC | IV | AC | 45 | F | EGFR e19 Del |
| 13 | 13 | NSCLC | IV | AC | 66 | M | EGFR e19 Del |
| 14 | 14 | NSCLC | IV | AC | 67 | M | EGFR e19 Del |
| 15 | 15 | NSCLC | IV | AC | 35 | F | ERBB2 e20 insertion |
| 16 | 16 | NSCLC | IV | AC | 57 | F | EGFR L858R |
| 17 | 16 | NSCLC | IV | AC | 55 | M | EGFR e19, L858R, T790M |
| 18 | 19 | NSCLC | IV | AC | 55 | F | EGFR e19 Del |
| 19 | 22 | NSCLC | IV | AC | 67 | F | EGFR L858R |
| 20 | 23 | NSCLC | IV | AC | 73 | F | EGFR e19 Del |
| 21 | 24 | NSCLC | IV | AC | 74 | M | EGFR L858R |
| 22 | 25 | NSCLC | IV | AC | 46 | F | EGFR L858R |
| 23 | 25 | NSCLC | IV | AC | 74 | M | EGFR e19 Del |
| 24 | 25 | NSCLC | IV | AC | 59 | F | EGFR e19 Del |
| 25 | 26 | NSCLC | IV | AC | 69 | F | EGFR L858R |
| 26 | 27 | NSCLC | IV | AC | 50 | F | EGFR L858R |
| 27 | 30 | NSCLC | IV | AC | 52 | F | NRAS-182A > T, AA change Q61L |
| 28 | 32 | NSCLC | IV | AC | 60 | F | EGFR L858R |
| 29 | 45 | NSCLC | IV | AC | 66 | M | RAS c.34G > T, p.Gly12Cys |
| 30 | 50 | NSCLC | IV | AC | 66 | F | None |
| 31 | 54 | NSCLC | IV | AC | 75 | M | EGFR e21 Rearg |
| 32 | 68 | NSCLC | IV | AC | 66 | F | Short in-frame deletion in EGFR e19 Del |
| 33 | 69 | NSCLC | IV | AC | 58 | F | Short in-frame deletion in EGFR e19 Del |
| 34 | 71 | NSCLC | IV | AC | 65 | F | EGFR e19 Del |
| 35 | 181 | NSCLC | IV | AC | 75 | F | EGFR L858R |
AC, adenocarcinoma; F, female; M, male; NSCLC, non–small-cell lung cancer.
A representative stage IV Nanowell scan revealed a substantial number of nanowells with positive TERT and MET signal from single putative CTCs, whereas the healthy donor Nanowell exhibited negligible signals (Fig. 3G). Putative CTCs were detected in almost all advanced-stage NSCLC samples, from 0 to 181 [interquartile range (IQR), 10–30] (Fig. 3H). In contrast, up to 7 positive wells out of 25,600 were detected in healthy controls (IQR, 2–6). Considering 7 or more putative CTCs in 2 mL as a positive sample, we detected putative CTCs in 31 of 35 (88.6%) patients. Because CellSearch detects clinically meaningful levels of CTCs in just 53% of patients with advanced-stage NSCLC (35), our platform demonstrated higher capability to identify putative CTCs.
Interestingly, a stage III NSCLC patient blood sample revealed heterogeneity of expression across putative CTCs (Fig. 3I). From each cell’s multigene expression profile, a clear differentiation between putative CTCs and WBCs emerged, as predicted from the spike-in cell line experiments. Putative CTCs were identified by their high expression of both TERT and MET, whereas WBCs were identified by nanowells lacking TERT and MET expression and exhibiting high VIM expression. Variable VIM and ALDH expression was observed for putative CTCs (Fig. 3J), which may reflect the heterogeneity of CTC populations present in lung cancer patients (36, 37).
EGFR Mutation Detection in Single CTCs from NSCLC Patients.
We then used our integrated nanoplatform to detect the most common and clinically relevant EGFR mutations (del19, L858R, and T790M) in CTCs from seven stage IV NSCLC adenocarcinoma patients who had known EGFR mutation status confirmed by tumor biopsy. Putative CTCs were assessed for three genes: TERT, the EGFR mutation (E746-A750del, L858R, or T790M), and the corresponding EGFR wild type (Fig. 4). Because TERT gene exhibits superior distinction on cancer cell identification (Fig. 2), putative CTCs were identified by high TERT expression. Notably, we were able to accurately detect each patient’s known mutation from their putative CTCs (Fig. 4 B, D, and F), whereas the “bulk” RT-qPCR measurements from the same patient’s blood yielded undetectable levels of the assayed genes (Fig. 4 B and D). Putative CTCs from E746-A750del visually appeared green (indicative of the EGFR mutation), whereas yellow (EGFR wild type) was nearly absent (Fig. 4A). Quantitative analysis of putative CTCs confirmed that the majority of these were primarily EGFR mutant for del19, with very low wild-type EGFR expression (Fig. 4B). Both L858R and the T790M de novo mutations were also detected via our integrated nanoplatform, revealing the heterozygous expression of both mutant and wild-type alleles (Fig. 4 C–F). We further examined expression levels of EGFR mutations and their corresponding wild-type genes in CTCs from seven advanced NSCLC patients with known mutational statuses. Each putative CTC was identified by EGFR and TERT expression, and heterogeneous levels of EGFR expression across those CTCs were observed (Fig. 4G).
Fig. 4.

Multigene expression of CTCs from NSCLC patient blood samples (results from three of the seven patient samples that were analyzed for CTC gene mutation profiling are shown here). (A) Mutation detection assay on a stage IV NSCLC patient sample with EGFR del19 mutation previously confirmed by primary tumor biopsy and sequencing. Using a three-plex assay of TERT (Cy5, shown in blue), EGFR E746-A750del (FAM, shown in green), and EGFR wild type (HEX, shown in yellow), putative CTCs were identified by high TERT expression. (Scale bar, 50 µm.) (B) All TERT-positive wells (blue) exhibited EGFR mutant signal (green) and very low EGFR wild-type signal (yellow), indicating that putative CTCs were detected and identified as primarily EGFR del19 mutant. Subsequent quantitative analysis of TERT-positive wells confirmed the majority of identified putative CTCs were primarily EGFR del19 mutant. In addition, a few putative CTCs also exhibited some EGFR wild-type signal. (C) Nanowell scan of the EGFR L858R mutation assay on a stage IV NSCLC patient sample with EGFR L858R mutation. (D) Putative CTCs identified by their TERT expression exhibit heterozygous expression of both mutant and wild-type forms. (E) Nanowell scan of the EGFR T790M mutation assay of a stage IV NSCLC patient sample with de novo EGFR T790M mutation. (F) Putative CTCs demonstrate heterozygosity for the mutation. (G) Expression levels of EGFR mutations and their corresponding wild-type genes in CTCs from seven advanced NSCLC patients. Each CTC was identified by EGFR and TERT expression. Although heterogeneous levels of CTCs and patterns of EGFR expression across those CTCs were observed, follow-up work regarding these patients’ clinical outcomes is necessary for proper heterogeneity analysis.
Assay Performance Comparison.
As shown in Fig. S4, bulk PCR measurements performed directly on patients’ whole blood yielded undetectable signals for EGFR mutations (del19 and others). Although EGFR wild type was found to be widely expressed from bulk assays of advanced-stage patients’ blood, EGFR mutant genes were not. In addition, we assessed the analytical sensitivity of conventional PCR in vitro on NSCLC cell lines (HCC827 and H1650). Our data indicate that, although conventional PCR can reliably detect CTC-specific genes from a total of 100 cancer cells, this signal fluctuates significantly for 10 cells and is undetectable above background for <10 cells. Genes that were insufficiently expressed (e.g., ALDH, as in Fig. S5) were not detectable above threshold for samples of 10 or fewer cells. We also note that these experiments were performed on homogenized cell populations to extrapolate the number of cells in a sample, so this approach is expected to yield the most consistent and reliable serial dilution measurements. These observations confirm that conventional PCR cannot reliably assay “enriched” samples of cell populations of <10 or fewer cells, and hence a higher sensitivity approach is needed to reliably assay heterogeneous CTC populations for tumor-specific and therapy-relevant mutant gene panels.
Fig. S4.

Heat map representation of selected genes measured by bulk RT-PCR directly from patients’ whole-blood samples. We observed that conventional RT-PCR assays of mRNA extracted from patients’ blood yielded undetectable TERT expression for all samples and no MET expression for 76% of patient samples.
Fig. S5.

Assay performance comparison. (A) Cycle thresholds for PCR on 100 cells, 10 cells, and the theoretical expected value (based on 100 cells) for each of four genes assayed on NSCLC cells. Although the most highly expressed gene, VIM, could be reliably detected for samples of 10 cells, TERT and MET genes displayed deviations from their expected values. Furthermore, the least expressed gene, ALDH, was undetectable for samples of 10 cells. For all four genes, all assays on fewer than 10 cells yielded undetectable signals. These observations highlight the limitations of conventional PCR performed directly on cell eluent populations after enrichment. Assay performance of (B) “Bulk” gene expression directly from whole blood, (C) “CTC Enriched” gene expression after MagSifter processing, and (D) the Nanowell assay for “Single CTC” analysis. From an advanced-stage NSCLC patient with a known EGFR del19 mutation (patient A), assay measurements for putative CTC characterization among Bulk, CTC Enriched, and Single CTC are shown. Bulk and CTC Enriched PCR results indicated that no genes were detected except for constitutive GapDH, thereby showing insufficient sensitivity. The Nanowell assay identified eight CTCs, seven of which exhibited both del19 mutant and its wild-type gene together, with the lone remaining CTC exhibiting the wild type. Assay measurements on an NSCLC patient with a EGFR T790M mutation (patient B) indicated that, although GapDH and other wild-type genes were detected in “Bulk,” no genes were detected. The Nanowell assay identified 18 CTCs, 12 of which exhibited both T790M and its wild-type gene together. The remainders showed the EGFR wild-type gene only. Assay measurements on another NSCLC patient with EGFR del19 mutation (patient C) revealed the same result as patient A. No genes except for constitutive GapDH in Bulk were detected. The Nanowell assay identified 74 CTCs, 42 of which exhibited both del19 mutant and its wild-type gene together. The 32 remaining CTCs exhibited the EGFR wild-type gene only.
To investigate mutational detection directly from enriched CTC population, we performed (i) a conventional PCR on extracted mRNAs from whole-blood samples of three advanced stage NSCLC patients, (ii) a conventional PCR on enriched CTCs by the MagSifter technology, and 3) our proposed MagSifter–Nanowell assay in a parallel manner, to compare their sensitivity. mRNAs from whole blood and from enriched CTCs were extracted using the TRIzol protocol described above. Two advanced-stage NSCLC patients with the EGFR del19 mutation, and an advanced-stage NSCLC patient with the EGFR T790M mutation were recruited for assay comparison. In all cases of mRNA analysis from whole blood and enriched CTCs, neither mutant genes (EGFR del19, and EGFR T790M) nor TERT were detected (Fig. S5), whereas Nanowell could identify putative CTCs, which are either EGFR mutant, EGFR wild type, or both. This result agrees with our cell line experiment indicated above, thus showing the greater sensitivity of the Nanowell assay, especially when fewer numbers of CTCs are presented.
Additionally, to investigate Nanowell’s performance against ICC, we recruited 11 NSCLC patients and 1 healthy donor to directly compare the signal positivity for immunostaining (CK+, DAPI+, and CD45−) and Nanowell RT-PCR assay (TERT+ and MET+) (Table S2). Each patient’s sample was split into two 2-mL samples for CTC assessment by both conventional enumeration (immunostaining) and Nanowell assay. Note that each 2 mL of patient blood was processed with MagSifter in parallel. Representative immunostained CTCs are illustrated in Fig. S2. All resulting immunostained CTCs were confirmed by a Stanford Hospital pathologist. Enumeration results were positive for 3 of 11 cases (defined as CTC > 1), whereas the Nanowell assay measured positive signals above threshold for 8 of 11 cases, as shown in Table 1 (the full analysis is given in Table S2). These observations indicate the higher sensitivity of our approach, which is because of the signal amplification by PCR. We note that the 11 patients enrolled in this study spanned all lung cancer stages, representing early-stage tumors that are thought to release low numbers of CTCs and those more advanced tumors that are expected to release more CTCs.
Table S2.
Comparison of Nanowell and enumeration data measured by various parameters for validation purposes
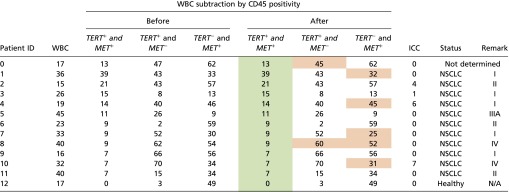 |
In particular, Nanowell signals were obtained both before and after WBC subtraction by CD45 positivity, as well as assessed with three different possible criteria (i.e., TERT+ and MET−, TERT− and MET+, or both). Notably, the measurements of putative CTCs (shown in green) remained completely unaffected after the exclusion of WBCs by CD45 positivity (although a few individual TERT+ and MET− or TERT− and MET+ signals were slightly affected; shown above in orange). These data indicate that the Nanowell assay’s analytical sensitivity exceeded that of conventional ICC (enumeration) for putative CTC detection in NSCLC patients (with relevant clinical information included).
Table 1.
Comparison of putative CTCs assayed by Nanowell (both TERT and MET positivity) and conventional ICC
| Patient ID | Nanowell | ICC | Types | Lung cancer stage |
| 1 | 39 | 0 | NSCLC | I |
| 2 | 21 | 4 | NSCLC | II |
| 3 | 15 | 1 | NSCLC | I |
| 4 | 14 | 6 | NSCLC | I |
| 5 | 11 | 0 | NSCLC | III |
| 6 | 9 | 0 | NSCLC | II |
| 7 | 9 | 0 | NSCLC | I |
| 8 | 9 | 0 | NSCLC | IV |
| 9 | 7 | 0 | NSCLC | I |
| 10 | 7 | 7 | NSCLC | IV |
| 11 | 7 | 0 | NSCLC | II |
| 12 | 0 | 0 | Healthy | N/A |
Markedly, the Nanowell assay’s analytical sensitivity exceeded that of conventional ICC for putative CTC detection in NSCLC patients. More information can be found in Table S2.
Discussion
Our integrated nanoplatform is a high-throughput, massively parallel CTC enrichment and subsequent molecular characterization tool for tumor biology and, potentially, for clinical use as a diagnostic/prognostic tool (38). It achieves accurate identification of hundreds to thousands of putative CTCs with multiplex gene mutation profiling capabilities. Each of the 25,600 nanowell compartments are 20 × 20 × 50 µm3, with 20-pL volumes that are 106 times smaller than those of typical PCR assays, thereby enabling higher sensitivity from higher mRNA concentrations. To this point, we observed that conventional RT-PCR assays of mRNA extracted from patients’ blood yielded undetectable TERT expression for all samples and no MET expression for 76% of patient samples (Fig. S4). Nanowell assays also achieve excellent specificity by using hydrolysis probes (e.g., TaqMan).
Detection of EGFR mutations, such as del19, L858R, and T790M, in lung cancer is now proving critical to inform clinical treatment decisions with EGFR tyrosine kinase inhibitor (TKI) therapies (39). Because 15% of lung adenocarcinomas harbor EGFR mutations—a number that is at least twice as high in the Asian population—many patients undergoing treatment in clinics for whom biopsy material is difficult to obtain may not be receiving optimal therapy. Furthermore, because resistance inevitably develops in patient with EGFR driver mutations that are treated over 12–24 months, and currently this can be monitored only by imaging and clinical gestalt, blood biomarkers for disease monitoring are desperately needed to guide management. Circulating tumor DNA (ctDNA) is a promising avenue to guide therapy, but recent data suggest that CTCs may be a complementary, rather than extraneous, approach (40). For example, ctDNA analysis from a del19 patient (Fig. 4A) using cancer personalized profiling by deep sequencing (41) (CaPP-Seq) detected del19 mutation at an allele frequency of 3.12%, indicating nontrivial tumor burden with this particular mutation.
Additionally, recent studies have demonstrated the promising potential of next-generation sequencing (NGS) profiling of CTCs (29, 42), especially to inform prognostic assessment and precise personalized cancer treatment (43). Unfortunately, widespread adoption of NGS for single-CTC sequencing is currently hindered by high costs and technical complexity for extracting clinically actionable information. Our integrated nanoplatform has the advantages of lower cost (with reagent costs of ∼25 US dollars per assay) and fast sample-to-answer time (Table S3) given its targeted strategy. Nonetheless, we believe NGS is a powerful tool that will become more cost-effective over time, and we envision using the Nanowell as a validation tool to confirm targets on CTCs discovered by whole-genome approaches.
Table S3.
Itemized summary of assay processing time including individual processing steps
| Processing step | Time required | Total time elapsed |
| Blood incubation with EpCAM antibody | 1:00 | 1:00 |
| Blood incubation with MNPs | 1:00 | 2:00 |
| MagSifter enrichment of CTCs (from 2 mL of whole blood) | 0:25 | 2:25 |
| RBC lysis and DNase I treatment | 0:15 | 2:40 |
| Nanowell prep (cell seeding) | 0:20 | 3:00 |
| Single-cell RT-PCR | 2:30 | 5:30 |
| Signal acquisition | 0:20 | 5:50 |
Note that the reverse transcription of the mRNA from putative CTCs is performed within 3 h of starting each sample’s assay. The total assay time including signal acquisition is less than 6 h per sample (unit, hh:mm).
The modular nature of our multiplex assay using “off-the-shelf” commercial reagents provides versatility for exploring research questions on individual CTCs and acts as a valuable tool for CTC analysis. As an example, we have demonstrated heterogeneous expression of ALDH and VIM on putative CTCs in this study. Cell immortality and plasticity defined by these types of expression markers are well established to be important in cancer progression. Exactly how this type of CTC heterogeneity affects patient outcome and relates to imaging and clinical parameters is an important, and incipient, avenue of inquiry that should be pursued by research laboratories in the field.
The technology reported here requires further optimization before widespread application in clinical settings. First, comprehensive enrichment of an entire population of heterogeneous CTCs requires the use of multiple capture antibodies. Although our platform currently relies on EpCAM enrichment, the Nanowell assay is highly sensitive and specific to tumor-derived cells neglected by ICC (Fig. S2). Moreover, MagSifter can be generalized to accommodate multiple cell capture antibodies instead of, or in conjunction with, EpCAM. This approach would broaden the platform’s utility to many other cancers that shed CTCs [e.g., human epidermal growth factor receptor (HER2) for breast cancer, neuron-glial antigen 2 (NG2) for melanoma, and carbonic anhydrase IX (CAIX) for renal cell carcinoma] and to EpCAM-low and EpCAM-negative populations. Second, our platform currently employs up to four fluorophores for multiplex single-cell gene expression profiling, because primer–probe sets are only commercially available in four discrete excitation–emission spectra that can be resolved by conventional fluorescence microscopy. However, further modifications, including laser excitation, sharper bandpass filters, and narrower-emission hydrolysis probes, may extend the platform’s multiplex capability to accommodate 12 or more genes (26). This expansion will allow comprehensive mutational profiling to develop along with clinical advances to capture a comprehensive panel of relevant “actionable” mutations for therapy selection and disease monitoring. We also envision that the Nanowell as a clinical research tool to study (i) the assessment of heterogeneity across CTCs with patient’s clinical outcomes, (ii) lineage mapping studies of biopsied primary tumor cells, (iii) correlation studies with cell-free and ctDNA, and (iv) discrimination between benign and malignant states for early cancer detection.
Materials and Methods
Patient Samples and Clinical Data.
All patient-related research was approved by the Stanford Institutional Review Board before study initiation and informed consent was obtained from the participants. For each patient, samples of whole blood were drawn into 10-mL Vacutainer K2-EDTA tubes (Becton Dickinson), and sample ID numbers were assigned to anonymize clinical identification. Healthy human blood samples were obtained from the Stanford Blood Center (Palo Alto, CA), and patient blood samples were drawn from those with NSCLC from Pulmonary, Medical Oncology, and Radiation Oncology Clinics at Stanford University Medical Center. Healthy donor samples were used for our spike-in experiments with cell lines and as negative controls, whereas patient blood samples were processed for CTC identification and mutation detection. Lung cancer diagnoses were extracted from the medical record, and we defined advanced-stage NSCLC here as stage IV by the American Joint Committee on Cancer (AJCC-7) staging criteria.
Some NSCLC patient samples were plasma depleted for other research purposes and then resuspended with an equal volume of 1× Gibco PBS buffer (Life Technologies). All blood samples were processed as soon as possible after blood draw (within 1–24 h). For each 7.5-mL blood sample, 2 mL was processed with the MagSifter and ICC for CTC enumeration, 2 mL was independently processed with the integrated nanoplatform for single-CTC gene expression and mutation analysis, and the remaining volume was allocated for total mRNA extraction via a TRIzol protocol for bulk gene expression analysis.
Nanowell Processing.
Following CTC enrichment by MagSifter and treatment of the eluent with lysis buffer and DNase, the entire eluent was loaded into an assembled Nanowell array (Fig. S1D), by centrifugation at 3,000 rpm for 10 min. Because each Nanowell array contains 25,600 wells and the CTC population from 2 mL of blood is expected to be no more than 2,000 cells, each individual well has a 99.7% chance of containing either a single cell or no cell, according to the Poisson distribution (Table S4), and only a 0.3% of containing two or more cells, thereby representing a high-throughput method of analyzing “single” cells. Identification of putative CTCs was then performed as follows. First, fluorescence microscope images of the entire Nanowell array were acquired for identification and exclusion of wells containing WBCs by CD45 signal. Next, the entire Nanowell array was completely dried at 70 °C for 10 min to fix seeded cells into the wells and to completely deactivate the DNase. Then, an RT-PCR master mix was applied, and the array was centrifuged for nanowell seeding at 3,000 rpm (Sorvale Legend RT Centrifuge; Thermo Fisher Scientific) for 10 min. The RT-PCR master mix consisted of 2× reaction mix (CellsDirect One-Step qRT-PCR; Life Technologies), polymerases (SuperScript III RT/Platinum Taq Mix; Life Technologies), TaqMan probes (Life Technologies; Bio-Rad) for targeting specific genes, and diethylpyrocarbonate (DEPC)-treated water (for detailed quantities, see Table S5). To prevent water evaporation during PCR thermocycling, the array was then capped with a small piece of adhesive PCR sealant film (Bio-Rad) and centrifuged at 3,000 rpm for 10 min, and white mineral oil (W. S. Dodge Oil) was applied around the array edges. The array was then loaded into a thermal cycler (PTC-200; Peltier Thermal Cycler; Bio-Rad) using the following cycle parameters: for the first thermal cycler step, cell lysis and subsequent reverse transcription, the array was incubated at 50 °C for 45 min. This was followed by 10 cycles of 60 s at 95 °C for denaturation and 90 s at 65 °C for an annealing and extension step. Amplification commenced after with 35 cycles of 60 s at 90 °C and 90 s at 60 °C.
Table S4.
Probability of a Nanowell (in an array of 25,600) containing at most one cell
| No. of cells seeded into single nanowell | Probability of a well containing at most one cell, % |
| 100 | 99.99 |
| 500 | 99.98 |
| 1,000 | 99.93 |
| 2,000 | 99.71 |
| 3,000 | 99.36 |
| 4,000 | 98.90 |
| 5,000 | 98.32 |
| 10,000 | 94.09 |
Table S5.
Reagent composition for each Nanowell assay
| Reagent | Quantity |
| 2× Reaction mix | 50 µL |
| Hydrolysis probe 1 | 3 µL |
| Hydrolysis probe 2 | 3 µL |
| Hydrolysis probe 3 (if needed) | 3 µL |
| Hydrolysis probe 4 (if needed) | 3 µL |
| SuperScript III RT Platinum Taq Mix | 3 µL |
| 50 mM Magnesium sulfate (MgSO4) (for 3- or 4-plex) | 3 µL |
| DEPC water | 32 µL (or amount to make 100 µL total) |
| Total | 100 µL |
Nanowell Signal Acquisition.
For all Nanowell assays, automated scanning of the full array was performed at Stanford University’s Cell Science Imaging Facility (CSIF) on a Zeiss Axio Imager fluorescence microscope (Carl Zeiss) with Zeiss ZEN Pro software. Each Nanowell was imaged for FAM, HEX, Texas Red, and Cy5 fluorescence after single-cell RT-PCR with a ZEISS Axiocam 503 monochrome camera. For each Nanowell, 100 regions of the array were imaged at 10× magnification, and images were stitched together with ZEN Pro software.
Marker Gene and Fluorophore Selection.
We reviewed the biomedical literature using the search terms “circulating tumor cell,” “lung cancer,” and “blood” in MEDLINE to identify potential biomarkers for single-cell expression analysis of CTCs. We identified about 400 articles pertaining to lung cancer CTCs, and from this literature, we identified TERT (29, 30, 44–48) and MET (31, 32, 49–52) as promising marker genes for the CTC identification panel. TERT is up-regulated by many oncogenes, and overexpression of TERT is well established as dysregulated in cancers. TERT is required for the perpetuation of the malignant clone during cancer progression as it elongates telomeres in each cell division, but is not expressed in normal somatic cells. Similarly, hepatocyte growth factor receptor (HGFR), or MET, is a proto-oncogene whose up-regulation is closely associated with NSCLC. Similarly, we reviewed the literature and identified VIM and ALDH as promising marker genes for the heterogeneity panel. VIM is an epithelial–mesenchymal transition (EMT) marker, whose expression indicates the increased potential for tumor metastasis (36). Multiple markers of cancer cell stemness exist, but we chose ALDH as a marker that indicates tumorigenicity and clonogenicity of lung cancer (37) based on commercially available primer–probe sets. Finally, for the therapy prediction and monitoring panel, we selected EGFR E746-A750del mutation, its associated wild-type gene, and TERT. Other clinically relevant EGFR mutations, including L858R and T790M, can also be included as needed. Each marker was assigned a specific fluorescence channel: FAM (492-nm excitation, 517-nm emission) to TERT, HEX (530-nm excitation, 556-nm emission) to MET, Texas Red (596-nm excitation, 615-nm emission) to VIM, and Cy5 (650-nm excitation, 670-nm emission) to ALDH.
Four-Gene Expression Development.
Simultaneous gene expression measurement of two genes in the Nanowell array was previously reported (26). To extend this capability to accommodate four-plex gene expression analysis on a single cell, the procedure was optimized to avoid issues of reagent depletion and weaker signal for lower-expressing genes (53). We selected probes with four different fluorophores and minimal spectral overlap for simultaneous four-plex gene expression capability. The four fluorophores, FAM, HEX, Texas Red, and Cy5, had excitation and emission peaks of 492 and 517 nm, 530 and 556 nm, 596 and 615 nm, and 650 and 670 nm, respectively. Primer–probe assays were obtained commercially: TERT (Life Technologies), MET (Life Technologies), VIM (Bio-Rad), and ALDH (Bio-Rad). The four-plex RT-PCR process was optimized on conventional bulk assay in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad).
Statistical Analysis.
After single-cell RT-PCR and fluorescence image acquisition were performed, custom-developed MATLAB and R scripts were used to analyze signals from the Nanowell arrays. For the diagnosis of NSCLC patients with Nanowell assays for TERT and MET, the following protocol was performed. Bright-field images acquired before and after single-cell RT-PCR were first manually inspected for the pixel locations of the Nanowell array at the top left, top right, and bottom left of the image area to be analyzed. These pixel locations were entered into a MATLAB script that extracted the pixel spacing information for the Nanowell array by using its natural periodicity. The script then used this periodicity to extrapolate the location of each nanowell within the image area of interest, and extracted the pixel intensities within each nanowell for every fluorescence channel of interest. As successful amplification in a nanowell results in an extremely bright spot within the nanowell, it was determined that a metric comparing the dispersion of pixel intensities within each nanowell could determine whether amplification had successfully occurred. Thus, a custom R script was used to run an outlier detection algorithm for each fluorescence image, whereby a z score for the variance in pixel intensity within each nanowell was computed to determine the probability of each individual point being an outlier from the entire population. Due to the large number of nanowells in each population (typically >5,000 per image analyzed), the central limit theorem dictates that the distribution should follow a normal distribution, thus allowing use of the Z test to identify outliers at a significance level of P < 0.05. Additionally, due to the large number of points tested per image, Bonferroni correction was performed when conducting the Z test to avoid excessive type I errors, with the Bonferroni correction commonly regarded as one of the most conservative ways to control familywise error rates. The R script then computed a count for the nanowells with z scores above this threshold and classifies these nanowells as positive for the particular measurement. The images in the individual fluorescence channels were analyzed separately, and the nanowell counts were then collated either individually (as in the PE-fluorescence image pre–RT-PCR when excluding nanowells with WBCs) or together (as per the use of both FAM and HEX channel images when enumerating potentially cancerous entities after RT-PCR). For all analysis of images acquired by the Zeiss Axio Imager, including the EGFR mutation assay and the four-marker gene expression panel, individual nanowell fluorescence intensities were extracted from ZEN Pro for each channel and collated manually after subtraction of background intensities from the empty nanowells.
Identification of Putative CTCs.
In this study, “putative CTCs” are mostly defined as cells with the multigene signature having high expression in both TERT and MET genes. This multigene approach yields the best assay performance (compared with a single-gene assay of either TERT or MET) by minimizing the frequency of “false positives” that are acquired because of intrinsic and extrinsic sources of background noise. In addition, for mutational detection purposes, we define putative CTCs as cells having either TERT and an EGFR mutation gene, or TERT and the corresponding EGFR wild-type gene. Transcriptomic profiles of putative CTCs are stable up to 12 hours from the blood draw (Fig. S6).
Fig. S6.

Putative CTC assay measurements for four advanced-stage NSCLC patients at three different time points (t = 0, 6, and 12 h) after blood draw. (A) Our putative CTC assay measurements indicate that mRNA degradation over these time points do not significantly affect our results. All data measured within the 12-h time window are shown to be significantly exceeding the threshold value set by results (from Fig. 3H). (B) To assess the intrasample experimental variation of our assay, we performed triplicate assays at t = 0 h. The blue band in the graph shows the expected uncertainty range within which assay results are expected to fall over the 12-h time window. (C–E) Individual patients’ assay results indicate that no significant degradation or dysregulation in assay measurements was observed over time.
SI Materials and Methods
Magnetic Sifter Fabrication.
Standard photolithographic techniques were used to fabricate the magnetic sifter device (MagSifter). The detailed fabrication process and assembly were previously described (8, 54). In brief, silicon nitride (Si3N4) membranes were deposited by low-pressure chemical vapor deposition onto standard double-polished silicon (Si) wafers, and subsequently patterned by photolithography to create arrays of 40-µm square pores on the top side and a complementary array of hexagonal holes (with side lengths of 100 µm) on the bottom side. The hexagonal holes were completely etched through the Si wafers by deep reactive ion etching (standard Bosch process; STS Deep RIE Etcher; Surface Technology Systems) to fully expose the Si3N4 membranes. A Perkin–Elmer high vacuum RF-sputtering system (Perkin–Elmer) was then used to deposit a 12-µm-thick permalloy (Ni81Fe19) film onto each Si3N4 membrane, followed by a 30-nm-thick silicon dioxide (SiO2) passivation layer.
MagSifter Processing.
The MagSifter processing of whole-blood samples drawn from patients was previously described (8), but this procedure has been modified and improved for compatibility with downstream analysis on the Nanowell. First, 2 mL of whole blood was mixed with 2 mL of 2× PlusCellect (R&D Systems) buffer, then incubated with 10 µL of biotinylated anti-EpCAM antibody (diluted 1:10 with PBS; BioLegend) for 1 h. Next, the sample was incubated with 20 µL of MagCellect Streptavidin Ferrofluid (MAG999; R&D Systems) for 1 h to label cells with magnetic nanoparticles. This sample was flowed through a MagSifter and syringe pump assembly at a rate of 10 mL/h. Next, the MagSifter was washed with 1 mL of PBS at a flow rate of 10 mL/h, and the captured cells were eluted into 0.5 mL of PBS. Because the removal of RBC contaminants significantly increases the PCR efficiency of single-cell gene expression (28), the cell eluent was incubated in 12.5 mL of ammonium chloride-based RBC hemolysis buffer for 10 min at room temperature and then centrifuged at 750 × g for 5 min. The resulting pellet was resuspended in 150 µL of PBS and incubated with 30 µL of CD45-PE stain (BioLegend) for 30 min to identify and exclude white blood cells (WBCs) after cell sorting on Nanowell. The sample was again centrifuged at 750 × g for 5 min, and finally, the pellet was incubated in 0.3 U per µL of DNase I (Life Technologies) for 5 min to remove noncellular DNA, followed by the addition of 5 mM EDTA for DNase deactivation.
Nanowell Array Fabrication.
The fabrication of the single-cell Nanowell arrays was reported elsewhere (26). Each nanowell array (Nanowell) contains 25,600 discrete nanowells, each 20 µm × 20 µm in size, arranged in a square array of 160 rows and 160 columns. The desired pattern of a dense array of wells was first designed into a mask. Then, this pattern was molded onto a silicon wafer using standard photolithography processes. A negative photoresist, SU-8 2035 (MicroChem), was used to define the master pillar structure with height of 50 µm. We prepared polydimethlysiloxane (PDMS) (Sylgard 184; Dow Corning) with a base-to-curing-agent ratio of 10:1, which was then cast onto the master wafer and cured at 90 °C for 10 min. The Nanowell device was assembled onto a commercially available glass slide (Thermo Fisher Scientific). Finally, the entire array was exposed to UV light for 30 min for sterilization.
Cell Line Preparation.
Six cell lines were used in this study: five of NSCLC type [A549 (ATCC CCL185), HCC827 (ATCC CRL2868), NCI-H661 (ATCC HTB183), NCI-H1650 (ATCC CRL5883), and NCI-H1975 (ATCC CRL5908)] and one of fibroblast lines (PCS-201), all of which were purchased from the American Type Culture Collection (ATCC), which authenticates tissues by genotype. These five cell lines are representative non–small-cell lung cancer (NSCLC) subtypes: adenocarcinoma (A549, H1650, H1975, and HCC827) and large-cell lung carcinoma (H661). Cells were grown in either 25- or 75-cm2 canted-neck flasks (Dow Corning) until reaching at least 80% confluence. After reaching sufficient confluency, cells were collected by trypsinization. For all five NCSLC cell lines, the cell culture medium used was Roswell Park Memorial Institute (RPMI) medium, supplemented with 10% (vol/vol) HyClone FBS, 1% sodium pyruvate (Life Technologies) as an additional source of energy, and 1% penicillin–streptomycin (Life Technologies) to prevent bacterial contamination, under standard growth conditions of 37 °C and 5% CO2. For the fibroblast cell line, DMEM (Life Technologies) supplemented with 10% (vol/vol) HyClone FBS and 1% penicillin–streptomycin was used under standard growth conditions. All cell lines were regularly tested and verified that they were free from mycoplasma contamination.
Total mRNA Extraction from Whole Blood.
Total mRNA strands from either (i) cell lines or (ii) whole-blood samples were extracted using a TRIzol protocol (Life Technologies). For the cell line total mRNA extraction, once cell line confluency in 75-cm2 canted-neck flasks reached 80%, cells were harvested via trypsinization. Then 106 cells were resuspended in 1 mL of PBS and centrifuged at 600 × g for 2 min. For each whole-blood sample, 3.5 mL of whole blood was diluted to reach a total of 50 mL of hemolysis buffer, which consists of a hypotonic solution with ammonium chloride. After removal of PBS, 1.2 mL of TRIzol and 200 µL of chloroform were added and vortexed vigorously. After centrifuging at 13,500 × g for 10 min, the upper phase was collected and added to equal volume of isopropyl alcohol. After centrifuging again at 13,500 × g for 10 min, the supernatant was aspirated, and the resulting pellet was washed with 1 mL of 75% (vol/vol) ethyl alcohol. After a final centrifugation at 13,500 × g for 10 min, the supernatant was aspirated, and the resulting pellet was dried in a biohood for 5 min. Finally, the dried pellet was resuspended in 20 µL of diethylpyrocarbonate (DEPC)-treated water to form the working stock of extracted mRNA for “bulk” gene expression analysis. The extracted mRNA underwent reverse transcription with Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) (Promega) to produce cDNA, which was used for gene expression analysis through PCR.
CTC Enumeration by ICC.
CellSearch employs (i) magnetic separation of CTCs from blood samples using anti-epithelial cell adhesion molecule (EpCAM) antibody conjugated with magnetic nanoparticles and (ii) three-channel fluorescence microscopy for identifying and enumerating CTCs, which are defined as positive for 4′,6-diamidino-2-phenylindole (DAPI) (a nuclear stain) and cytokeratin (an epithelial marker) and negative for CD45 (a leukocyte marker). Similarly, after MagSifter enrichment of CTCs from patient blood samples, the cell eluent was resuspended in 400 µL of PBS. To stain the CTCs, three staining solutions were added (8): 40 µL of CD45-PE-Cy5, 5 µL of DAPI, and 3 µL of anti-cytokeratin-FITC. After incubating the sample for 30 min, PBS was added to reach a total of 15-mL volume. After centrifuging the solution at 750 × g for 7 min, the supernatant was aspirated and the resulting pellet was resuspended in 500 µL of PBS. The entire volume was centrifuged via a Cytospin standard single funnel onto a SuperFrost Plus microscope slide using a Shandon Cytospin 3 cytocentrifuge (Thermo Fisher Scientific) at 500 rpm for 3 min. Finally, fluorescence microscopic images of the cytospin slide were acquired at 40× magnification in DAPI, FAM, and Cy3 channels. Enumeration of CTCs was performed by a cytopathologist according to pathological criteria (DAPI+, CK+, CD45−, size, and morphology).
WBC Exclusion by CD45 Positivity from Putative CTC Characterization.
Any impurities from leukocytes among the CTC population may affect the single-cell gene expression process and hence possibly interfere with signal acquisition and interpretation. To address this concern, we analyzed the same patient cohort using CD45 positivity to assess the signal overlap between the WBC and CTC populations. Notably, our measurements of putative CTCs (defined as both TERT+ and MET+) both before and after exclusion by CD45 positivity were identical for all samples studied (as shown in Table S5). Although a few samples showed slight deviation in the number of TERT+ signals or MET+ signals (but never both), these results demonstrate the high purity of CTCs enriched by the MagSifter. Quantitatively, both positive cases were unaffected by WBCs, whereas TERT+ and MET− and TERT− and MET+ were affected by WBCs in 4/477 = 0.8% and 12/536 = 2% of observed wells, respectively.
Investigation of Assay Reproducibility in Regard to Possible mRNA Variation over Time.
Because mRNA is degraded and produced in response to new cellular environments, we evaluated the impact of time on the change in mRNA profiles expected. We recruited four advanced-stage NSCLC patients and assessed the effect of time lapse on mRNA degradation by extrapolating from putative CTC assay measurements at time = 0, 6, and 12 h after blood draw. In addition, one of these patients was assayed in triplicate at time = 0 h to determine the putative CTC assay variation as a baseline comparison with time-lapsed assay results. In particular, after RT-PCR was performed, we measured the putative CTCs for all four patients at each of the three time points (as shown in Fig. S6). The triplicate results from patient 1 (n = 15, 28, 28; average = 23.7 and SD = 7.5; shown as blue background in the graph) were established as a baseline level of intrasample variation (as shown in Fig. S6B), and we observed that, for all 12 patient-time data points (as shown in Fig. S6A), the putative CTC assay measurements fell within the expected range of experimental variation, therefore demonstrating that mRNA degradation does not occur to any significant extent over the typical elapsed time frame of our assay workflow. As an additional note, because we are measuring genes that are highly overexpressed in cancer cells (e.g., TERT and MET), we do not expect significant loss of expression levels during our endpoint measurements. As a point of comparison with existing CTC studies, we considered the transcriptome sequencing of individual CTCs (42). In this work, all blood samples were assayed within 3 h following blood draw, after which CTCs were then stained with calcein dyes and manually picked individually, thus resulting in a significant time delay in the CTC transcriptomic analysis. As another example, the work by Yu et al. (55) indicated at least 3 h of processing time just before mRNA amplification, with a separation time of >2 h (3 mL at 1.5 mL/h) and subsequent RNA extraction and purification steps (∼1 h). According to our timeline, our method is comparable to or even faster than these and other similar studies.
Acknowledgments
We thank Dr. Pragya Tripathi for assistance with preparation of patient blood samples, Dr. Ivan K. Dimov and Prof. Aram J. Chung for helpful discussions, and Dr. Christopher M. Earhart for MagSifter fabrication expertise. We also thank the Stanford Nanofabrication Facility for use of nanofabrication tools, the Cell Science Imaging Facility (CSIF) at Stanford for use of a Zeiss Axio Imager fluorescence camera, and the Lurie Nanofabrication Facility at University of Michigan (Ann Arbor, MI) for contributing to the MagSifter fabrication. This research was supported by NIH Awards U54CA151459 (to the Center for Cancer Nanotechnology Excellence and Translation) and R21CA185804 (to S.S.G. and S.X.W.), the Canary Foundation (S.S.G. and V.S.N.), and the LUNGevity Foundation (V.S.N.).
Footnotes
Conflict of interest statement: S.X.W. holds two issued patents on MagSifter, which are owned by Stanford University and outlicensed for possible commercialization. S.-m.P., D.J.W., C.C.O., S.S.G., V.S.N., and S.X.W. are coinventors of a pending patent filed by Stanford University on the subject of this work.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1608461113/-/DCSupplemental.
References
- 1.Hanash SM, Baik CS, Kallioniemi O. Emerging molecular biomarkers—blood-based strategies to detect and monitor cancer. Nat Rev Clin Oncol. 2011;8(3):142–150. doi: 10.1038/nrclinonc.2010.220. [DOI] [PubMed] [Google Scholar]
- 2.Krebs MG, et al. Molecular analysis of circulating tumour cells—biology and biomarkers. Nat Rev Clin Oncol. 2014;11(3):129–144. doi: 10.1038/nrclinonc.2013.253. [DOI] [PubMed] [Google Scholar]
- 3.Tibbe AG, et al. Optical tracking and detection of immunomagnetically selected and aligned cells. Nat Biotechnol. 1999;17(12):1210–1213. doi: 10.1038/70761. [DOI] [PubMed] [Google Scholar]
- 4.Nagrath S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450(7173):1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stott SL, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci USA. 2010;107(43):18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castro CM, et al. Miniaturized nuclear magnetic resonance platform for detection and profiling of circulating tumor cells. Lab Chip. 2014;14(1):14–23. doi: 10.1039/c3lc50621e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talasaz AH, et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci USA. 2009;106(10):3970–3975. doi: 10.1073/pnas.0813188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earhart CM, et al. Isolation and mutational analysis of circulating tumor cells from lung cancer patients with magnetic sifters and biochips. Lab Chip. 2014;14(1):78–88. doi: 10.1039/c3lc50580d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarioglu AF, et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Methods. 2015;12(7):685–691. doi: 10.1038/nmeth.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs MG, et al. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J Thorac Oncol. 2012;7(2):306–315. doi: 10.1097/JTO.0b013e31823c5c16. [DOI] [PubMed] [Google Scholar]
- 11.Sollier E, et al. Size-selective collection of circulating tumor cells using Vortex technology. Lab Chip. 2014;14(1):63–77. doi: 10.1039/c3lc50689d. [DOI] [PubMed] [Google Scholar]
- 12.Ozkumur E, et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med. 2013;5(179):179ra47. doi: 10.1126/scitranslmed.3005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, et al. Biodegradable nano-films for capture and non-invasive release of circulating tumor cells. Biomaterials. 2015;65:93–102. doi: 10.1016/j.biomaterials.2015.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin M, et al. Nanostructure embedded microchips for detection, isolation, and characterization of circulating tumor cells. Acc Chem Res. 2014;47(10):2941–2950. doi: 10.1021/ar5001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon HJ, et al. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat Nanotechnol. 2013;8(10):735–741. doi: 10.1038/nnano.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marrinucci D, et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys Biol. 2012;9(1):016003. doi: 10.1088/1478-3975/9/1/016003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maheswaran S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359(4):366–377. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devriese LA, et al. Circulating tumor cell detection in advanced non-small cell lung cancer patients by multi-marker QPCR analysis. Lung Cancer. 2012;75(2):242–247. doi: 10.1016/j.lungcan.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Sieuwerts AM, et al. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real-time PCR. Breast Cancer Res Treat. 2009;118(3):455–468. doi: 10.1007/s10549-008-0290-0. [DOI] [PubMed] [Google Scholar]
- 20.Haber DA, Velculescu VE. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014;4(6):650–661. doi: 10.1158/2159-8290.CD-13-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell AA, et al. Single cell profiling of circulating tumor cells: Transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One. 2012;7(5):e33788. doi: 10.1371/journal.pone.0033788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CL, et al. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate. 2013;73(8):813–826. doi: 10.1002/pros.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jemal A, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 24.Shepherd FA, et al. National Cancer Institute of Canada Clinical Trials Group Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 25.Solomon BJ, et al. PROFILE 1014 Investigators First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 26.Dimov IK, et al. Discriminating cellular heterogeneity using microwell-based RNA cytometry. Nat Commun. 2014;5:3451. doi: 10.1038/ncomms4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park SM, et al. Dual transcript and protein quantification in a massive single cell array. Lab Chip. 2016;16(19):3682–3688. doi: 10.1039/c6lc00762g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Soud WA, Rådström P. Purification and characterization of PCR-inhibitory components in blood cells. J Clin Microbiol. 2001;39(2):485–493. doi: 10.1128/JCM.39.2.485-493.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni X, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci USA. 2013;110(52):21083–21088. doi: 10.1073/pnas.1320659110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sozzi G, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 2003;21(21):3902–3908. doi: 10.1200/JCO.2003.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Kubo T, et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124(8):1778–1784. doi: 10.1002/ijc.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsao M-S, et al. Hepatocyte growth factor/scatter factor is an autocrine factor for human normal bronchial epithelial and lung carcinoma cells. Cell Growth Differ. 1993;4(7):571–579. [PubMed] [Google Scholar]
- 33.Kato Y, et al. Novel epidermal growth factor receptor mutation-specific antibodies for non-small cell lung cancer: Immunohistochemistry as a possible screening method for epidermal growth factor receptor mutations. J Thorac Oncol. 2010;5(10):1551–1558. doi: 10.1097/JTO.0b013e3181e9da60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satelli A, et al. Universal marker and detection tool for human sarcoma circulating tumor cells. Cancer Res. 2014;74(6):1645–1650. doi: 10.1158/0008-5472.CAN-13-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krebs MG, et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J Clin Oncol. 2011;29(12):1556–1563. doi: 10.1200/JCO.2010.28.7045. [DOI] [PubMed] [Google Scholar]
- 36.Al-Saad S, et al. The prognostic impact of NF-kappaB p105, vimentin, E-cadherin and Par6 expression in epithelial and stromal compartment in non-small-cell lung cancer. Br J Cancer. 2008;99(9):1476–1483. doi: 10.1038/sj.bjc.6604713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang F, et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009;7(3):330–338. doi: 10.1158/1541-7786.MCR-08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SM, Sabour AF, Son JH, Lee SH, Lee LP. Toward integrated molecular diagnostic system (iMDx): Principles and applications. IEEE Trans Biomed Eng. 2014;61(5):1506–1521. doi: 10.1109/TBME.2014.2309119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 40.Sundaresan TK, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res. 2016;22(5):1103–1110. doi: 10.1158/1078-0432.CCR-15-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman AM, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramsköld D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lohr JG, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32(5):479–484. doi: 10.1038/nbt.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyazu YM, et al. Telomerase expression in noncancerous bronchial epithelia is a possible marker of early development of lung cancer. Cancer Res. 2005;65(21):9623–9627. doi: 10.1158/0008-5472.CAN-05-0976. [DOI] [PubMed] [Google Scholar]
- 45.Lantuejoul S, et al. Differential expression of telomerase reverse transcriptase (hTERT) in lung tumours. Br J Cancer. 2004;90(6):1222–1229. doi: 10.1038/sj.bjc.6601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu CQ, et al. Amplification of telomerase (hTERT) gene is a poor prognostic marker in non-small-cell lung cancer. Br J Cancer. 2006;94(10):1452–1459. doi: 10.1038/sj.bjc.6603110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujita Y, et al. The diagnostic and prognostic relevance of human telomerase reverse transcriptase mRNA expression detected in situ in patients with nonsmall cell lung carcinoma. Cancer. 2003;98(5):1008–1013. doi: 10.1002/cncr.11611. [DOI] [PubMed] [Google Scholar]
- 48.Miura N, et al. Clinical usefulness of serum telomerase reverse transcriptase (hTERT) mRNA and epidermal growth factor receptor (EGFR) mRNA as a novel tumor marker for lung cancer. Cancer Sci. 2006;97(12):1366–1373. doi: 10.1111/j.1349-7006.2006.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsao M-S, et al. Differential expression of Met/hepatocyte growth factor receptor in subtypes of non-small cell lung cancers. Lung Cancer. 1998;20(1):1–16. doi: 10.1016/s0169-5002(98)00007-5. [DOI] [PubMed] [Google Scholar]
- 50.Sierra JR, Tsao M-S. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3(1) Suppl:S21–S35. doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheu C-C, et al. Combined detection of CEA, CK-19 and c-met mRNAs in peripheral blood: A highly sensitive panel for potential molecular diagnosis of non-small cell lung cancer. Oncology. 2006;70(3):203–211. doi: 10.1159/000094321. [DOI] [PubMed] [Google Scholar]
- 52.Cheng T-L, et al. Overexpression of circulating c-met messenger RNA is significantly correlated with nodal stage and early recurrence in non-small cell lung cancer. Chest. 2005;128(3):1453–1460. doi: 10.1378/chest.128.3.1453. [DOI] [PubMed] [Google Scholar]
- 53.Henegariu O, Heerema NA, Dlouhy SR, Vance GH, Vogt PH. Multiplex PCR: Critical parameters and step-by-step protocol. Biotechniques. 1997;23(3):504–511. doi: 10.2144/97233rr01. [DOI] [PubMed] [Google Scholar]
- 54.Earhart CM, Wilson RJ, White RL, Pourmand N, Wang SX. Microfabricated magnetic sifter for high-throughput and high-gradient magnetic separation. J Magn Magn Mater. 2009;321(10):1436–1439. doi: 10.1016/j.jmmm.2009.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu M, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487(7408):510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lustberg M, Jatana KR, Zborowski M, Chalmers JJ. Emerging technologies for CTC detection based on depletion of normal cells. Recent Results Cancer Res. 2012;195:97–110. doi: 10.1007/978-3-642-28160-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]