

Abstract
Although pharmacologic therapies have provided gains in reducing the mortality of heart failure, the rising incidence of the disease requires new approaches to combat its health burden. Twenty-five years ago, abnormal calcium cycling was identified as a characteristic of failing human myocardium. Sarcoplasmic reticulum calcium ATPase (SERCA2a), the sarcoplasmic reticulum calcium pump, was found to be a key factor in the alteration of calcium cycling. With the advancement of gene vectors, SERCA2a emerged as an attractive clinical target for gene delivery purposes. Using adeno-associated virus constructs, SERCA2a upregulation has been found to improve myocardial function in animal models. The clinical benefits of overexpressing SERCA2a have been demonstrated in the phase I study Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID). This study has demonstrated that a persistent expression of the transgene SERCA2a is associated with a significant improvement in associated biochemical alterations and clinical symptoms of heart failure. In the coming years, additional targets will likely emerge that are amenable to genetic manipulations along with the development of more advanced vector systems with safer delivery approaches.
Introduction
Despite gains in preventative care and risk factor management, the incidence of heart failure continues to increase in the United States. This can be partially explained by the improvement in the treatment of myocardial infarction and coronary artery disease. As more patients are surviving their first myocardial infarction, a larger population goes on to develop progressive left ventricular dysfunction and end-stage heart failure. This increased incidence is also bolstered by an aging population, which experiences higher rates of dilated cardiomyopathy and small-vessel ischemic disease.
Current pharmacologic approaches have provided some benefits for mortality and symptom control, but have been unable to fundamentally change the natural history of the disease. Although patients may remain stable for years on optimal medical therapy, progressive myocardial remodeling leads to increasing rates of cardiac decompensation and arrhythmia. The median survival after diagnosis was only 1.7 years for men and 3.2 years for women in the Framingham Heart Study, with only 25% of men and 38% of women surviving 5 years (Ho et al., 1993).
As a result of the morbidity and mortality associated with congestive heart failure, research efforts have focused on elucidating the mechanisms of structural remodeling associated with end-stage disease. There has been particular focus on the molecular changes inherent in cardiomyocyte dysfunction, including differential gene expression of the proteins involved in the contraction–relaxation cycle. Because of the focus on changes in gene expression, gene therapy has become an attractive clinical intervention. Funding efforts by the National Heart Lung and Blood Institute have resulted in the identification of numerous possible targets. This review will discuss some of the research on the various targets, with specific emphasis on the sarcoplasmic reticulum calcium ATPase (SERCA2a) and its recent clinical applications.
Traditional Targets for Heart Failure
One of the blockbuster drug classes in the field of heart failure has been the beta-blockers (Swedberg et al., 1980; Rydén et al., 1983; Bristow et al., 1986; Bristow et al., 1994; Gwathmey et al., 1999; Washington et al., 2001). Since the seminal clinical trials of the late 1980s and early 1990s, beta-blockade has been the mainstay of heart failure therapy as it has provided significant mortality benefits (Brophy et al., 2001). Downregulation of adrenergic stimulation has been found to preserve heart sensitivity to sympathetic tone, and reduce downstream activation of gene-modifying transcription factors (Lymperopoulos et al., 2013). Changes at the level of the β-adrenergic receptor appear to be linked to upregulation of a critical G protein-coupled receptor kinase 2 (GRK2). GRK2, which is the most abundantly expressed G protein-coupled receptor kinase in the heart, has been implicated in the pathogenesis of a dysfunctional cardiac β-adrenergic system, thereby accounting for a decline in cardiac function in the failing heart (Raake et al., 2008).
Several gene-based experiments have tested the hypothesis that genetic manipulation of the myocardial β-adrenergic receptor system can restore/enhance cardiac function. Although detrimental clinical outcomes have been long recognized with the use of phosphodiesterase inhibitors in the treatment of heart failure, improvement in cardiac function has been reported with overexpression/activation of adenylate-cyclase 6 (ADCY6) (Lai et al., 2004). The favorable effects of ADCY6 and manipulation of the β-adrenergic signaling system in preclinical studies have resulted in the initiation of a clinical trial in patients with heart failure (Kawase et al., 2008).
Decreasing remodeling in heart failure has also been facilitated by the inhibition of the renin–angiotensin system. Angiotensin-converting enzyme (ACE) inhibitors have been found to prevent cardiac remodeling in postischemic patients (Konstam et al., 1992). Numerous mechanisms underlie the protective effect of ACE inhibition, including vasodilatation that reduces afterload, increased diuresis that reduces afterload, and reduction in adrenergic stimulation. The downstream effectors of angiotensin II receptors include the mitogen-activated protein kinase pathway, which is known to facilitate expression of genes resulting in cardiac hypertrophy and remodeling (Dulin et al., 1998; Aoki et al., 2000). Currently, treatment is focusing on downregulation of angiotensin receptors (AT1) using RNAi-mediated antisense strategies. Mouse models have shown decreased remodeling in the rat myocardium using knockout strategies with retrovirus vectors (Phillips and Kimura, 2005). Currently, no clinical trials are underway that utilize this mechanism of action.
SERCA2a: Targeting Calcium Cycling
More than 25 years ago, Gwathmey et al. (1987) first reported that calcium cycling was abnormal in the failing human heart. Regardless of the etiology for the observed heart failure, the abnormal calcium cycling was caused in part by a decrease in the SERCA2a activity (Hasenfuss et al., 1994).
The sarcoplasmic reticulum plays an important role in orchestrating the movement of calcium during contraction and relaxation. Excitation leads to the opening of voltage-gated L-type calcium channels, allowing the entry of a small amount of calcium, which then stimulates the release of a much larger amount of calcium from the sarcoplasmic reticulum (SR) and subsequent contraction. During relaxation, calcium is re-accumulated into the sarcoplasmic reticulum by the sarcoplasmic reticulum calcium-ATPase pump (SERCA2a) and extruded extracellularly by the sarcolemmal Na+/Ca2+ exchanger. The contribution of SERCA and the Na+/Ca2+ exchanger to the restoration of diastolic calcium concentrations varies among species. In humans, it accounts for a majority of calcium exchange, with ∼75% of the calcium removed by SERCA2a and ∼25% by the Na+/Ca2+ exchanger.
A decrease in SR calcium ATPase activity and calcium uptake has been shown to be responsible for abnormal calcium homeostasis in both experimental and human failing hearts (Gwathmey et al., 1993; Schmidt et al., 1998, 1999). Figure 1 reveals calcium cycling dynamics in the heart. Calcium signals recorded from failing human myocardial cells reveal significantly prolonged calcium transients with an elevated end-diastolic intracellular calcium concentration (Gwathmay et al., 1987; del Monte et al., 1999, 2002a,b). Overexpression of SERCA2a not only significantly shortened the time course of the calcium transient, but also significantly reduced the end-diastolic calcium concentration while restoring the force–frequency relationship (del Monte et al., 1999, 2002a,b). In an animal model of pressure-overload hypertrophy in transition to failure, where SERCA2a protein levels and activity are decreased and severe contractile dysfunction is present, overexpression of SERCA2a by gene transfer in vivo restored both systolic and diastolic function to normal levels (Miyamoto et al., 2000). The preclinical observations that the overexpression of SERCA2a in a rodent model of heart failure improves contractility and restored contractility in cardiomyocytes isolated from failing human hearts provided a strong foundation as well as scientific rationale for developing SERCA2a as a clinical therapy for heart failure (Miyamoto et al., 2000; del Monte et al., 2002a,b).
FIG. 1.

Calcium cycling dynamics in the heart. The sarcoplasmic calcium cycling interactome. SERCA2a serves as the focal point for numerous secondary and tertiary regulators of calcium cycling. Calcium influx into the sarcoplasmic reticulum is regulated either by direct posttranslational modification of SERCA or through alterations in phospholamban binding capacity. Membrane receptors for neuroendocrine hormones influence downstream kinases to phosphorylate SERCA directly or to alter affector function. SERCA2a, sarcoplasmic reticulum calcium ATPase. Patients experiencing either dilated or hypertropic cardiomyopathy exhibit abnormal calcium cycling with widening of the sarcoplasmic calcium loading curve. Slower rates of calcium reuptake lead to slower relaxation of the sarcomere and poorer pumping kinetics.
The SERCA2a Interactome
The activity of SERCA2a is influenced by numerous transcription factors and membrane proteins that direct structural changes in the calcium channel and influence calcium uptake. Figure 1 presents many of the membrane and cytoplasmic components of the SERCA interactome. The most direct effector of SERCA2a-mediated calcium cycling is a sarcoplasmic membrane protein known as phospholamban. The function of phospholamban is to inhibit calcium flow through the channel, and it is regulated by phosphorylation via various kinases (Kranias and Hajjar, 2012).
Published experimental results support the hypothesis that an abnormal ratio of phospholamban to SERCA2a contributes significantly to the observed abnormalities in calcium handling and contraction in failing ventricular myocardium. Associated with a defective calcium uptake, there is a decrease in the relative ratio of SERCA2a/phospholamban in failing hearts (Meyer et al., 1999). Using transgenic and gene transfer approaches, increasing levels of phospholamban relative to SERCA2a in isolated cardiomyocytes have been reported to significantly alter intracellular calcium handling by prolonging the relaxation phase of the calcium transient, decreasing calcium release, and increasing end-diastolic calcium concentration (Davia et al., 1999). Using antisense strategies to inhibit phospholamban results in improved sarcoplasmic reticulum function, calcium mobilization, as well as significantly improved cell shortening in cardiomyocytes isolated from failing human hearts (del Monte et al., 2002a).
Conversely, in vitro overexpression of SERCA2a in neonatal rat cardiomyocytes largely “rescued” the phenotype created by increasing the phospholamban-to-SERCA ratio (Hajjar et al., 1997). More importantly, in human cardiomyocytes isolated from the left ventricles of patients with end-stage heart failure, gene transfer of SERCA2a resulted in an increase in both protein expression and SERCA2a pump activity, and induced a faster contraction velocity and enhanced relaxation (del Monte et al., 1999). Furthermore, diastolic calcium was decreased in failing human cardiomyocytes overexpressing SERCA2a, while systolic calcium was increased and the frequency response was normalized.
The SERCA2a/phospholamban ratio can also be normalized by decreasing phospholamban (Minamisawa et al., 1999). This was achieved by adenoviral gene transfer using antisense strategies with phospholamban (He et al., 1999). Overexpression of a dominant negative mutant of phospholamban or decreasing the steady-state level of phospholamban has been reported to enhance SERCA2a activity (He et al., 1999). This is consistent with the observation that genetic ablation of phospholamban rescues the abnormalities in contractile function in a mouse model of dilated cardiomyopathy (Minamisawa et al., 1999).
More recently, direct posttranslational modifications of the SERCA pump have been found to affect calcium cycling as well. One such modifier is the small ubiquitin-related modifier 1 (SUMO-1). SUMO-1 is a unique polypeptide that covalently conjugates to target proteins. This binding, termed SUMOylation, regulates many aspects of protein function, such as subcellular localization, protein–protein interactions, and transcriptional activity (Herrmann et al., 2007). A number of target proteins that are regulated by SUMOylation have been implicated in human disease (Sarge and Park-Sarge, 2009). For example, a decrease in lamin A SUMOylation could be a causative factor in familial dilated cardiomyopathy; two different mutations in lamin A have been identified in patients with this disease (Sylvius et al., 2005; Zhang and Sarge, 2008). These mutations, which cause the amino-acid substitutions Glu203Gly and Glu203Lys, are likely to lead to defective lamin A SUMOylation, as well as alterations in nuclear morphology and the subcellular localization of lamin A (Zhang and Sarge, 2008). We have shown that the activity and stability of SERCA2a in cardiomyocytes is modulated by SUMO-1, and that cardiac expression of SUMO-1 is significantly reduced in humans and animals with heart failure. Cardiac-specific overexpression of SUMO-1 in transgenic mice restored SERCA2a function, improved hemodynamic performance, and significantly increased survival after thoracic aorta constriction.
Consistent with these findings, a long-term cardiac benefit of SUMO-1 overexpression (using adeno-associated virus serotype 9 [AAV9]-mediated SUMO1 gene delivery) has been shown in mouse models of heart failure (Kho et al., 2011). Two months after gene transfer (4 months after thoracic aorta constriction), transgene-mediated SUMO-1 overexpression rescued pressure-overload-induced cardiac dysfunction concomitantly with increased SERCA2a function and improved left ventricular dilation, functional deterioration, and hemodynamic performance. Depletion of cardiac SUMO-1 using AAV9-mediated delivery of a small hairpin RNA in normal mice accelerated the decline in SERCA2a function and expression resulting in a dramatic increase in mortality. Importantly, these beneficial effects of SUMO-1 overexpression when SERCA2a was downregulated suggest a SERCA2a-dependent role of SUMO-1 in cardiac function. Further studies are required to define the molecular mechanism of SERCA2a regulation by SUMOylation and to investigate the association between low levels of SUMO-1 expression and heart failure.
Secondary Effectors
Numerous secondary and tertiary effectors have been identified that influence calcium influx via SERCA2a. These effectors either impact phospholamban expression and function, or directly interact with calcium stores.
One example of a calcium-modulating protein that has been implicated in intracellular regulatory activities is S100. S100A1 is the most abundant S100 protein isoform found in the heart, and is found to be decreased in heart failure (Remppis et al., 1996). S100A1 promotes cardiac contraction and relaxation by enhancing the activity of ryanodine receptors and SERCA2a (Kettlewell et al., 2005). Preclinical studies using AAV6 long-term expression of S100A1 have demonstrated a sustained reversal of cardiac dysfunction in several animal models of heart failure (Pleger et al., 2007, 2011). These studies reinforce a rationale for a clinical trial of S100A1 gene therapy for human heart failure (Most and Koch, 2007).
Phospholamban has been found to have its own set of inhibitors. Protein-phosphatase 1 (PP1) is a common cytosolic protein that removes phosphate groups from numerous targets, including phospholamban (Kranias and Hajjar, 2012). PP1 levels have been found to be elevated in heart failure models (Neumann et al., 1997). The direct relationship of protein-phosphatases with phospholamban makes them an attractive target for indirectly altering SERCA2a function (Fish et al., 2013).
Antiapoptosis proteins have also been identified to influence calcium cycling. Heat shock protein 20 has been found to mediate cardiovascular proliferation in a post-ischemic model (Qian et al., 2009). Utilizing adenovirus, it has been found that upregulation of HSP20 can be cytoprotective (Zhu and Wang, 2005). HS-1-associated protein (HAX-1), a mitochondrial protein, has been found to interact directly with phospholamban (Vafiadaki et al., 2007). Mouse models using overexpression of HAX-1 have shown a decrease of calcium influx via SERCA2a because of increased phospholamban activity (Han et al., 2006).
Gene Delivery Systems: Vectors
A variety of approaches have been considered for somatic gene transfer to cardiomyocytes. Gene delivery systems can be divided into two categories: nonviral systems and recombinant viral systems. Each of these systems has unique expression profiles and advantages as well as disadvantages.
Nonviral vectors include naked plasmid DNA, liposomal DNA complexes, polymer-carried DNA, and oligonucleotides. Plasmids are double-stranded circular DNA-containing transgenes encoding the protein of interest and have enhancer and promoter sequences. Nonviral vectors, however, have had unacceptably low transfection efficiencies. More recently, RNA vectors consisting of nanoparticles coupled with modified messenger RNA have been used as gene therapy vehicles with more success (Zangi et al., 2013).
Viral vectors from the family Retroviridae include retroviruses and lentiviruses. Retroviruses contain single-stranded positive-sense RNAs that utilize a virally encoded reverse transcriptase to generate double-stranded DNA. In order for the virus's DNA to integrate into the host cell genome, the integrity of the nuclear cell membrane must be disrupted. Retroviral transfection is therefore limited to dividing cells (Miller et al., 1990). Because of this behavior, retroviral vectors have mediated dramatic physiological effects in vivo in developing animals, but its impact on adult cardiac gene transfer has been limited by a general requirement for ongoing replication. Lentiviral vectors are single-stranded RNA viruses that utilize reverse transcriptase and integrate into the genome, resulting in long-term expression of transgenes. Lentiviral vectors, however, are capable of transducing nondividing mitotically quiescent cells (Vodicka, 2001). This process of transduction involves random integration into the genome, however. Random integration increases the risk of oncogenic transformation, which has become apparent in previous clinical trials treating severe combined immunodeficiency with lentiviral vectors (Cavazzana-Calvo et al., 2000; Hacein-Bey-Abina et al., 2003).
Unlike the retrovirus family, Adenoviridae is a nonintegrating virus family containing double-stranded DNA. Adenoviral gene transfer has been successfully used in vivo to manipulate the expression of calcium-handling proteins (Hajjar et al., 1998; Miyamoto et al., 2000). Recombinant adenoviral vectors offer several significant advantages. The viruses can be prepared at extremely high titer, infect nonreplicating cells, and confer high-efficiency and high-level transduction of cardiomyocytes. The major disadvantages to adenoviral gene transfer have been its transient expression and the host immune/inflammatory response to the vectors themselves, specifically the first-generation vectors (Di Paolo et al., 2009). Gene transfer to adult myocardium in vivo mediates high-level expression for approximately 1 week. By 2–4 weeks, expression has usually declined dramatically (Vassalli et al., 2003). The E1–E4 proteins elicit a significant innate immune response, which is the major challenge for using them in a therapeutic setting in human populations. However, third-generation “gutless” adenoviruses with simultaneous E1 and E4 deletions have been successfully used (Alba et al., 2005; Jiang et al., 2011). Adenoviral gene transfer with E1–E4-deleted vectors results in prolonged expression and lower immunogenicity (Jiang et al., 2011). Adenoviruses have served as attractive vectors for delivering temporary gene expression to myocardium, particularly in the setting of angiogenesis and cell proliferation. Altering cardiac remodeling in heart failure, however, requires longer gene expression profiles than is offered by adenoviruses.
AAV, a nonpathogenic human parvovirus, is gaining attention for its potential use as a human gene therapy vector (Carter and Samulski, 2000; Manno et al., 2006; Asokan et al., 2012). One of the most attractive features of recombinant AAV vectors is the ability to remain stable in host cells as integrated parvoviruses. An additional advantage of using AAV vectors is its low immunogenicity. There are 13 different serotypes of AAVs (AAV1–13) (Wu et al., 2006). AAV1, 6, 8, and 9 are effective in transducing cardiomyocytes (Wang et al., 2005; Pacak et al., 2006; Zincarelli et al., 2008). The major disadvantage of the AAV is that its coding capacity is limited to 4.6–4.8 kb and it is difficult to produce and purify (Snyder and Moullier, 2011). Neutralizing antibodies to various AAV serotypes are present in approximately 20–80% of the human population, thereby limiting the potential therapeutic use of AAV vectors (Calcedo et al., 2009; Boutin et al., 2010). Although the presence of antibodies to AAV serotypes is known to reduce transfection efficiency, no evidence for cellular immunity has been identified. This is a promising finding as it shows that AAV-infected cells are not targeted by cytotoxic T cells, allowing transfected cells to remain viable without immune system interference (Zaiss, 2002). While seropositivity continues to remain a major exclusion criteria in AAV-based clinical trials, the evolution of chimeric strains that lack cellular immunity makes AAVs viable candidates for gene therapy strategies (Ying et al., 2010).
Tissue-specific promoters
The principal tools of transgene expression in vitro have traditionally utilized viral vectors with constitutive promoters; this guarantees high protein titers with few innate obstacles to expression (Tilemann et al., 2012). The major disadvantage of viruses using constitutively active promoters, such as cytomegalovirus or Rous sarcoma virus promoters, is the nonspecificity of expression. The lack of full cardiotropism in current viral vectors poses a challenge of inappropriate transgene expression in noncardiac tissue, the impact of which has not been well elucidated.
One way to improve the delivery capability of viral vectors would be inclusion of a cardiomyocyte-specific promoter. This would allow expression of the transgene only in cardiomyocytes, thus eliminating the problem of inappropriate transgene expression in other organs and thereby avoiding a more widespread inflammatory response. Furthermore, transgene expression can also be engineered with a trigger response to cardiac stress, providing both temporal and geographic localization.
The basic components of an expression cassette include promoter/enhancer elements, the genes of interest, and an appropriate mRNA-stabilizing polyadenylation signal. Other frequently employed cis-acting elements include internal ribosome entry site sequences to allow expression of two or more genes without the need for an additional promoter, and introns and post-transcriptional regulatory elements to improve transgene expression. Tissue-specific promoters can be used to restrict transgene expression to the desired target cell population and avoid unintended cells such as antigen-presenting cells. For example, cardiomyocyte-specific promoters such as alpha-myosin heavy and light chains have been employed to restrict gene expression to the myocardium (Boecker et al., 2004). Similarly, the smooth muscle-specific promoter SM22α has been demonstrated to restrict gene expression to cells of this type. A number of studies have recently examined the regulation of the human BNP promoter and identified some of the elements involved in cardiomyocyte-specific expression (He et al., 2001). In transgenic mice, studies have shown that the proximal promoter confers a high-level of cardiac-specific expression, primarily in the ventricles (LaPointe et al., 2002).
In a number of instances, it is desirable to have precise regulation of expression of a therapeutic gene in vivo. Natural and synthetic enhancer promoters can be utilized to drive gene transcription in a temporal, spatial, or environmental signal-inducible manner in response to heat shock, hypoxia, radiation, chemotherapy, or epigenetic agents (Ye et al., 1999). Hypoxia, intravascular shear stress, and left ventricular strain have all been used in models of this type of regulation. In the setting of heart failure, a responsive system would be important if the gene of interest needs to be turned on or off for a short period. This would include the use of genes driving angiogenesis or stem cell recruitment or expansion (Ruan et al., 2001; Su and Kan, 2007).
Meganucleases and zinc finger proteins can be engineered to induce double-strand breaks at specific DNA sequences (Paques and Duchateau, 2007; Klug, 2010). These breaks are repaired by homologous recombination or by nonhomologous end joining, which results in insertions or deletions of a few base pairs. They can then be used to restore the normal reading frame of a gene with a specific mutation. Similarly, zinc finger protein transcription factors that specifically activate or repress virtually any gene can be engineered. Meganucleases and zinc finger proteins have been used to target dystrophin mutations and vascular endothelial growth factor (VEGF) expression by gene transfer (Chapdelaine et al., 2010). With the ever-expanding genetic mutations in dilated and hypertrophic cardiomyopathies (Konno et al., 2010), these nucleases can be used in the future to repair DNA in the affected organs by gene therapy.
Gene Delivery Systems: Methods of Vector Delivery
The in vivo delivery of vectors to achieve transfection of myocardium requires careful considerations of the target physiology and the disease profile. The utilization of gene therapy in heart failure requires homogenous transfection of myocardial tissue (Fig. 2). Heterogeneous expression would result in dysynchronous contraction and worsening heart failure because of poor ejection fraction. Heterogeneous expression can also increase the risk of arrhythmias because of differences in contraction timing and altered transmission of voltage gradients across the ventricle.
FIG. 2.
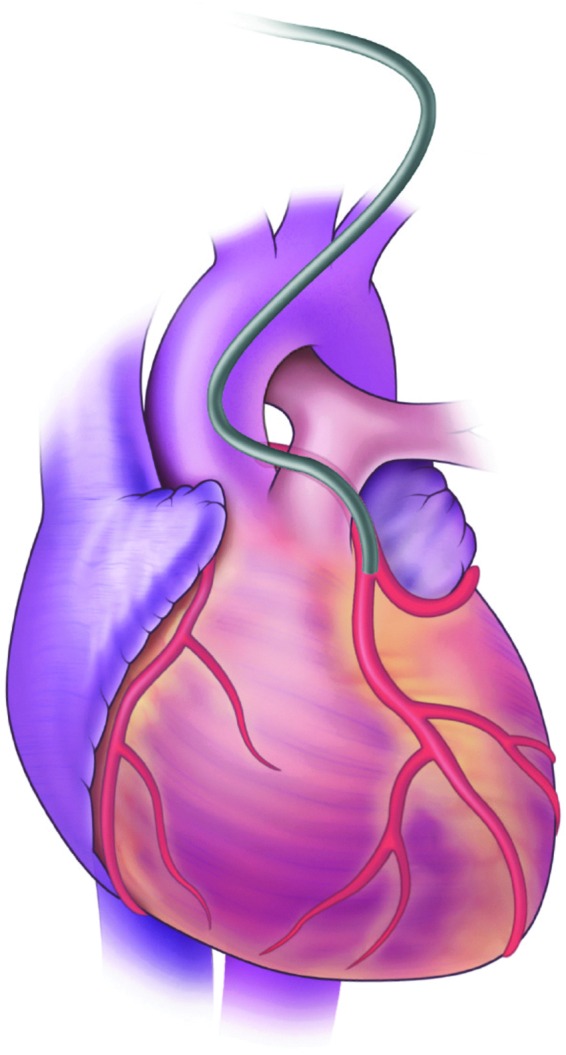
Percutaneous gene delivery into the circulation to the heart. Although vascular approaches to gene delivery in the heart circumvent the problem of localized transfection there can be problems with transfection kinetics. The amount of dwell time and pressure within the coronary circulation during interruption of coronary flow appears to be important for successful myocardial gene transfer.
Initial studies in myocardial gene transfer relied on direct injection of plasmid DNA, which resulted in excellent localized expression of the transgene restricted to the needle tracts (French et al., 1994a,b). This method is useful for delivery of transgenes, which code for soluble, secreted products, such as VEGF (Schalch et al., 2004). The vectors are injected either epicardially or endocardially. A primary advantage of direct injection is that it bypasses the endocardial barrier. In addition, in the case of coronary artery occlusion, the occluded area can be bypassed. Direct intramyocardal injection results in a high local concentration at the injection site and avoids exposure to the blood and possible deactivation of the vectors by circulating DNAases or neutralizing antibodies. In addition, this approach minimizes exposure of off target organs to the vector. Although invasive, vectors can be delivered using a surgical approach (e.g., thoracotomy), which allows visualization of the injection site, including the borders of myocardial infarctions. An endocardial injection can utilize a percutaneous approach with a catheter containing a retractable injection needle. Imaging is required and can consist of fluoroscopy, echocardiography, and/or magnetic resonance imaging (Lederman et al., 2002). The limitations associated with both the endocardial and epicardial approaches is that they cannot provide the global level of expression required in heart failure models. While localized expression may serve well for ischemic disease and angiogenesis, the more global altered pumping kinetics in cardiomyopathy would require larger expression profiles than offered by this approach.
Vascular approaches would circumvent the problem of localized transfection, with arterial delivery providing access to all areas of the myocardium (Fig. 2). The difficulty associated with vascular delivery involves transfection kinetics. The rapid flow of blood through the coronary vasculature prevents viral translocation into myocardial tissue. When adenovirus is delivered directly into the coronary arteries, it is rapidly washed out and results in significant nontarget tissue uptake in the liver and lung (Ishikawa et al., 2011, 2013). The endothelial lining of the vasculature also acts as a barrier that further reduces access.
In early studies in rats, an apical intraventricular injection of viral vectors during aortic and pulmonary artery cross-clamping to achieve homogenous gene delivery was used (Hajjar et al., 1998). Homogeneous gene delivery occurred as a result of increased net time spent within the cardiac circulation by the vectors. It is not well tolerated in large animals, however, because of the interruption of systemic perfusion and excessive left ventricle pressure overload. These features would make it poorly compatible in a clinical setting.
Efficient delivery of gene product has been accomplished through ex vivo intracoronary delivery during transient cardioplegic arrest or coronary recirculation. We and others have been effectively using cardiopulmonary bypass as a means to isolate the coronary circulation and inject gene products during cardioplegic arrest (Hayase et al., 2005). Davidson et al. (2001) demonstrated that by using cardiopulmonary bypass and cardioplegic arrest, intracoronary gene delivery of adenoviral vectors resulted in efficient myocardial uptake and expression with undetectable transgene expression in the liver or lung. Furthermore, while pharmacological pretreatment with vasodilatory and permeability enhancers has been shown to increase viral uptake in myocardial cells, the amount of dwell time and pressure within the coronary circulation during an interruption of flow appears to be the most important factors for successful lumen to myocardial gene transfer. Cardiopulmonary bypass provides the optimal conditions for such delivery as it maintains systemic perfusion while the heart remains arrested and protected from ischemic damage during perfusion with gene product (Davidson et al., 2001; White et al., 2011). Further optimization of the conditions of cardiopulmonary bypass-mediated gene delivery, including altering pressure by means of a retrograde cardioplegia balloon catheter, varying temperature and duration of vector dwell, as well as assessing various pharmacological enhancers of permeability, should improve delivery efficiency while reducing possible myocardial damage.
Studies have reported efficient and homogeneous transduction with retroinfusion into the coronary venous system as well as with antegrade coronary artery infusion (Kawase et al., 2008). A closed-loop recirculation retrograde venous approach is also feasible (Kaye et al., 2007; Byrne et al.,. 2008). Nevertheless, retrograde coronary infusion should be performed by a trained and experienced surgeon, as trauma to the coronary veins is a possibility and severe complications can result. Elevation of coronary venous pressures can result in myocardial edema and hemorrhage. More importantly, ischemia that might result from the use of any of the coronary infusion approaches might not be well tolerated in patients with advanced heart failure.
Intrapericardial delivery can be considered the least invasive delivery of vectors outside of simple intravenous infusion. Percutaneous pericardial puncture is feasible and relatively safe when using imaging techniques. A percutaneous approach can be minimally invasive via a substernal/xiphoid puncture. The pericardial cavity provides a large closed space, which can result in a longer duration of vector persistence, allows a high vector concentration, and minimizes leakage to other organs. However, the structural architecture of the visceral pericardium can limit transfection efficiency of superficial myocardial layers (Ladage et al., 2011). Addition of permeabilizing agents can increase delivery using a pericardial delivery approach; however, the toxicity and clinical feasibility of these agents has not been well elucidated (Roques et al., 2007; Karakikes et al., 2012). Mechanical methods utilizing microbubbles combined with echocardiography has been shown to provide efficient gene transduction of the myocardium as well (Beeri, 2002).
Clinical Trials
After over several decades of failures, gene therapy trials for specific monogenic diseases have shown some success, most notably for inherited blindness in pediatric patients with Leber congenital amaurosis (Jacobson et al., 2012). The earliest clinical trials in cardiovascular gene therapy focused on treating ischemic disease with angiogenesis as the target. Utilization of transient expression of angiogenic factors such as VEGF was hypothesized to increase collateral circulation in ischemic tissue. Numerous trials were conducted at the turn of the century utilizing naked DNA plasmids and adenoviral vectors (Giacca and Zacchigna, 2012). While phase I studies had shown safety in human disease, phase II trials did not identify a benefit in clinical outcomes (Gupta et al., 2009). Larger multicenter trials such as the NORTHERN trial did not find any difference in improvement of heart failure symptoms between VEGF administration and placebo groups (Stewart et al., 2009).
The elucidation of the mechanisms of calcium cycling, the established deficiencies of SERCA2a in heart failure models, and improved vector design made targeting heart failure an attractive option even in the context of gene therapy failures elsewhere. The Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) study was launched in 2007 to identify the clinical benefits of SERCA upregulation. As the first gene therapy trial in heart failure, it has shown promising results in patients with end-stage heart failure (Jessup et al., 2011).
CUPID was a two-part, phase 1/2, multicenter trial designed to evaluate safety and biological effects of delivery of SERCA2a cDNA by delivery of recombinant AAV1 (AAV1.SERCA) to patients with advanced heart failure. In part I of the study, patients were administered a single intracoronary artery infusion AAV1.SERCA2a in an open-label design. Four cohorts with three patients each received a single escalating dose of AAV1.SERCA. At the 12-month follow-up time point, the cohort receiving AAV1.SERCA demonstrated an acceptable safety profile. In this part I study, several patients demonstrated improvement in their cardiac parameters, including clinical symptoms, functional class, BNP levels, and echocardiographic findings of remodeling. New arrhythmias, which were a concern at the beginning of the study, did not occur during the phase I study.
In phase 2 of the study, 39 patients with advanced heart failure were randomized to receive intracoronary artery AAV.SERCA2a in one of three doses or placebo. New York Heart Association functional class, 6-min walk test, MVO2 max, NT-proBNP levels, and echocardiographic parameters were evaluated over 6 months. The cohort that received the high dose of AAV1.SERCA2a met criteria for success both at the individual level and at the group level. Compared with patients who received placebo, the group receiving AAV1.SERCA2a demonstrated improvement or stabilization in NYHA class, improvement on the Minnesota Living with Heart Failure questionnaire, 6 min walk test, MVO2 max, NT-proBNP levels, and left ventricle end-systolic volumes at the 12-month postinjection time point. The low and mid-range doses had reduced recurrent cardiovascular events for the first 6-month time point but were similar to placebo from 6-month to 1-year time points. However, the high-dose group continued to do significantly better than the placebo group at the 12-month time point with respect to both clinical and biochemical parameters. Figure 3 represents the event-curves showing differences in cumulative rate of events between the placebo and treatment groups (Jessup et al., 2011).
FIG. 3.

Event data for Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) trial. In the initial 6-month period, all treatment groups revealed decreased cardiovascular events, including hospitalizations, as compared with placebo. At the 12-month evaluation point, there was no difference between placebo and low-dose and medium-dose treatment groups. The high-dose treatment group, however, continued to show a significant difference in events compared with placebo.
The CUPID results support the possibility that AAV1.SERCA2a therapy may be an important addition to the heart failure treatment paradigm, particularly in patients with advanced disease and those who are poorly responsive to pharmacologic approaches. Further clinical studies are underway, including an international study of 200 patients that tests whether the high dose of AAV1.SERCA2a versus placebo, randomized 1:1, is an effective therapy to reduce the occurrence of clinical events in patients with advanced heart failure.
Forward Thinking
Gene therapy has experienced its share of trials and tribulations in the past two decades. Criticisms have focused particularly on the complexity of the diseases being targeted, and how monogenetic manipulations will not provide the therapeutic efficacy demanded from such high-investment interventions. As our understanding of the molecular mechanisms associated with heart failure have improved, the concept of single-gene manipulation has been shown to be both relevant and efficacious in treating disease. SERCA2a and its effectors serve as flagship examples of the success in targeting fundamental pathways of disease. In the coming years, the identification of more molecular targets and further advancement in vector technology will undoubtedly lead to safer and more effective clinical trials in gene therapy for heart failure.
Author Disclosure Statement
No competing financial interests exist.
References
- Alba R. Bosch A. Chillon M. Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther. 2005;12:S18–S27. doi: 10.1038/sj.gt.3302612. [DOI] [PubMed] [Google Scholar]
- Aoki H. Richmond M. Izumo S. Sadoshima J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem. J. 2000;347(Pt 1):275–284. [PMC free article] [PubMed] [Google Scholar]
- Asokan A. Schaffer D.V. Samulski R.J. The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeri R. New efficient catheter-based system for myocardial gene delivery. Circulation. 2002;106:1756–1759. doi: 10.1161/01.cir.0000035240.92015.e4. [DOI] [PubMed] [Google Scholar]
- Boecker W. Bernecker O.Y. Wu J.C., et al. Cardiac-specific gene expression facilitated by an enhanced myosin light chain promoter. Mol. Imaging. 2004;3:69–75. doi: 10.1162/15353500200404103. [DOI] [PubMed] [Google Scholar]
- Boutin S. Monteilhet V. Veron P., et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- Brophy J.M. Joseph L. Rouleau J.L. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann. Intern. Med. 2001;134:550. doi: 10.7326/0003-4819-134-7-200104030-00008. [DOI] [PubMed] [Google Scholar]
- Bristow M.R. Ginsburg R. Umans V., et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ. Res. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- Bristow M.R. O'Connell J.B. Gilbert E.M., et al. Dose-response of chronic beta-blocker treatment in heart failure from either idiopathic dilated or ischemic cardiomyopathy. Bucindolol Investigators. Circulation. 1994;89:1632–1642. doi: 10.1161/01.cir.89.4.1632. [DOI] [PubMed] [Google Scholar]
- Byrne M.J. Power J.M. Preovolos A., et al. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008;15:1550–1557. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- Calcedo R. Vandenberghe L.H. Gao G., et al. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P.J. Samulski R.J. Adeno-associated viral vectors as gene delivery vehicles. Int. J. Mol. Med. 2000;6:17–27. doi: 10.3892/ijmm.6.1.17. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M. Hacein-Bey S. de Saint Basile G., et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- Chapdelaine P. Pichavant C. Rousseau J., et al. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Ther. 2010;17:846–858. doi: 10.1038/gt.2010.26. [DOI] [PubMed] [Google Scholar]
- Davia K. Hajjar R.J. Terracciano C.M., et al. Functional alterations in adult rat myocytes after overexpression of phospholamban with use of adenovirus. Physiol. Genomics. 1999;1:41–50. doi: 10.1152/physiolgenomics.1999.1.2.41. [DOI] [PubMed] [Google Scholar]
- Davidson M.J. Jones J.M. Emani S.M., et al. Cardiac gene delivery with cardiopulmonary bypass. Circulation. 2001;104:131–133. doi: 10.1161/01.cir.104.2.131. [DOI] [PubMed] [Google Scholar]
- del Monte F. Harding S.E. Schmidt U., et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Monte F. Harding S.E. Dec G.W., et al. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002a;105:904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Monte F. Johnson C.M. Stepanek A.C., et al. Defects in calcium control. J. Card. Fail. 2002b;8:S421–S431. doi: 10.1054/jcaf.2002.129285. [DOI] [PubMed] [Google Scholar]
- Di Paolo N.C., et al. Virus binding to a plasma membrane receptor triggers interleukin-1 alpha-mediated proinflammatory macrophage response in vivo. Immunity. 2009;31:110–121. doi: 10.1016/j.immuni.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulin N.O. Alexander L.D. Harwalkar S., et al. Phospholipase A2-mediated activation of mitogen-activated protein kinase by angiotensin II. Proc. Natl. Acad. Sci. USA. 1998;95:8098–8102. doi: 10.1073/pnas.95.14.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish K.M. Ladage D. Kawase Y., et al. AAV9.I-1c delivered via direct coronary infusion in a porcine model of heart failure improves contractility and mitigates adverse remodeling. Circ. Heart Fail. 2013;6:310–317. doi: 10.1161/CIRCHEARTFAILURE.112.971325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French B.A. Mazur W. Ali N.M., et al. Percutaneous transluminal in vivo gene transfer by recombinant adenovirus in normal porcine coronary arteries, atherosclerotic arteries, and two models of coronary restenosis. Circulation. 1994a;90:2402–2413. doi: 10.1161/01.cir.90.5.2402. [DOI] [PubMed] [Google Scholar]
- French B.A. Mazur W. Geske R.S. Bolli R. Direct in vivo gene transfer into porcine myocardium using replication-deficient adenoviral vectors. Circulation. 1994b;90:2414–2424. doi: 10.1161/01.cir.90.5.2414. [DOI] [PubMed] [Google Scholar]
- Giacca M. Zacchigna S. VEGF gene therapy: therapeutic angiogenesis in the clinic and beyond. Gene Ther. 2012;19:622–629. doi: 10.1038/gt.2012.17. [DOI] [PubMed] [Google Scholar]
- Gupta R. Tongers J. Losordo D.W. Human studies of angiogenic gene therapy. Circ. Res. 2009;105:724–736. doi: 10.1161/CIRCRESAHA.109.200386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwathmey J.K. Copelas L. MacKinnon R., et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- Gwathmey J.K. Bentivegna L.A. Ransil B.J., et al. Relationship of abnormal intracellular calcium mobilisation to myocyte hypertrophy in human ventricular myocardium. Cardiovasc. Res. 1993;27:199–203. doi: 10.1093/cvr/27.2.199. [DOI] [PubMed] [Google Scholar]
- Gwathmey J.K. Kim C.S. Hajjar R.J., et al. Cellular and molecular remodeling in a heart failure model treated with the β-blocker Carteolol. Am. J. Physiol. 1999;276:H1678–H1690. doi: 10.1152/ajpheart.1999.276.5.H1678. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S. Von Kalle C. Schmidt M., et al. LMO2- associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Hajjar R.J. Schmidt U. Kang J.X., et al. Adenoviral gene transfer of phospholamban in isolated rat cardiomyocytes. Rescue effects by concomitant gene transfer of sarcoplasmic reticulum Ca2+-ATPase. Circ. Res. 1997;81:145–153. doi: 10.1161/01.res.81.2.145. [DOI] [PubMed] [Google Scholar]
- Hajjar R.J. Schmidt U. Matsui T. Guerrero J.L. Lee K.H. Gwathmey J.K. Dec G.W. Semigran M.J. Rosenzweig A. Modulation of ventricular function through gene transfer in vivo. Proc. Natl. Acad. Sci. USA. 1998;95:5251–5256. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y. Chen Y.S. Liu Z., et al. Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ. Res. 2006;99:415–423. doi: 10.1161/01.RES.0000237387.05259.a5. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G. Reinecke H. Studer R., et al. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- Hayase M. del Monte F. Kawase Y. MacNeill B.D. McGregor J. Yoneyama R. Hoshino K. Tsuji T. De Grand A.M. Gwathmey J.K. Frangioni J.V. Hajjar R.J. Cather-based antegrade intracoronary viral gene delivery with coronary venous blockade. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H2295–H3000. doi: 10.1152/ajpheart.00703.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H. Meyer M. Martin J.L., et al. Effects of mutant and antisense RNA of phospholamban on SR Ca2+-ATPase activity and cardiac myocyte contractility. Circulation. 1999;100:974–980. doi: 10.1161/01.cir.100.9.974. [DOI] [PubMed] [Google Scholar]
- He Q. Wang D. Yang X.P., et al. Inducible regulation of human brain natriuretic peptide promoter in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H368–H376. doi: 10.1152/ajpheart.2001.280.1.H368. [DOI] [PubMed] [Google Scholar]
- Herrmann J. Lerman L.O. Lerman A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 2007;100:1276–1291. doi: 10.1161/01.RES.0000264500.11888.f0. [DOI] [PubMed] [Google Scholar]
- Ho K.K. Anderson K.M. Kannel W.B., et al. Survival after the onset of congestive heart failure in Framingham Heart Study subjects. Circulation. 1993;88:107–115. doi: 10.1161/01.cir.88.1.107. [DOI] [PubMed] [Google Scholar]
- Ishikawa K. Tilemann L. Fish K. Hajjar R.J. Gene delivery methods in cardiac gene therapy. J. Gene Med. 2011;13:566–572. doi: 10.1002/jgm.1609. [DOI] [PubMed] [Google Scholar]
- Ishikawa K. Aguero J. Naim C., et al. Percutaneous approaches for efficient cardiac gene delivery. J. Cardiovasc. Transl. Res. 2013;6:649–659. doi: 10.1007/s12265-013-9479-7. [DOI] [PubMed] [Google Scholar]
- Jacobson S.G. Cideciyan A.V. Ratnakaram R., et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessup M. Greenberg B. Mancini D., et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B. Qian K. Du L., et al. Helper-dependent adenovirus is superior to first-generation adenovirus for expressing transgenes in atherosclerosis-prone arteries. Arterioscler. Thromb. Vasc. Biol. 2011;31:1317–1325. doi: 10.1161/ATVBAHA.111.225516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakikes I. Hadri L. Rapti K., et al. Concomitant intravenous nitroglycerin with intracoronary delivery of AAV1.SERCA2a enhances gene transfer inporcine hearts. Mol. Ther. 2012;20:565–571. doi: 10.1038/mt.2011.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase Y. Ly H.Q. Prunier F., et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008;51:1112–1119. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Kaye D.M. Preovolos A. Marshall T., et al. Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J. Am. Coll. Cardiol. 2007;50:253–260. doi: 10.1016/j.jacc.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Kettlewell S. Most P. Currie S., et al. S100A1 increases the gain of excitation-contraction coupling in isolated rabbit ventricular cardiomyocytes. J. Mol. Cell Cardiol. 2005;39:900–910. doi: 10.1016/j.yjmcc.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Kho C. Lee A. Jeong D., et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010;79:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- Konno T. Chang S. Seidman J.G. Seidman C.E. Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2010;25:205–209. doi: 10.1097/HCO.0b013e3283375698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstam M.A. Rousseau M.F. Kronenberg M.W., et al. Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure. SOLVD Investigators. Circulation. 1992;86:431–438. doi: 10.1161/01.cir.86.2.431. [DOI] [PubMed] [Google Scholar]
- Kranias E.G. Hajjar R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladage D. Turnbull I.C. Ishikawa K., et al. Delivery of gelfoam-enabled cells and vectors into the pericardial space using a percutaneous approach in a porcine model. Gene Ther. 2011;18:979–985. doi: 10.1038/gt.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai N.C. Roth D.M. Gao M.H., et al. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation. 2004;110:330–336. doi: 10.1161/01.CIR.0000136033.21777.4D. [DOI] [PubMed] [Google Scholar]
- LaPointe M.C. Yang X.P. Carretero O.A. He Q. Left ventricular targeting of reporter gene expression in vivo by human BNP promoter in an adenoviral vector. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1439–H1445. doi: 10.1152/ajpheart.01090.2001. [DOI] [PubMed] [Google Scholar]
- Lederman R.J. Guttman M.A. Peteres D.C. Thompson R.B. Sorger J.M. Dick A.J. Raman V.K. McVeigh E.R. Catheter-based endomyocardial injection with real-time magnetic resonance imaging. Circulation. 2002;105:1282–1284. doi: 10.1161/01.CIR.0000012425.71261.FC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A. Rengo G. Koch W.J. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ. Res. 2013;113:739–753. doi: 10.1161/CIRCRESAHA.113.300308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno C.S. Pierce G.F. Arruda V.R., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Meyer M. Bluhm W.F. He H., et al. Phospholamban-to-SERCA2 ratio controls the force-frequency relationship. Am. J. Physiol. 1999;276:H779–H785. doi: 10.1152/ajpheart.1999.276.3.H779. [DOI] [PubMed] [Google Scholar]
- Miller D.G. Adam M.A. Miller A.D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamisawa S. Hoshijima M. Chu G., et al. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- Miyamoto M.I. del Monte F. Schmidt U., et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc. Natl. Acad. Sci. USA. 2000;97:793–798. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Most P. Koch W.J. S100A1: a calcium-modulating inotropic prototype for future clinical heart failure therapy. Future Cardiol. 2007;3:5–11. doi: 10.2217/14796678.3.1.5. [DOI] [PubMed] [Google Scholar]
- Neumann J. Eschenhagen T. Jones L.R., et al. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell Cardiol. 1997;29:265–272. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- Pacak C.A. Mah C.S. Thattaliyath B.D., et al. Recombinant adenoassociated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- Paques F. Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr. Gene Ther. 2007;7:49–66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- Phillips M.I. Kimura B. Gene therapy for hypertension: antisense inhibition of the renin-angiotensin system. Methods Mol. Med. 2005;108:363–379. doi: 10.1385/1-59259-850-1:363. [DOI] [PubMed] [Google Scholar]
- Pleger S.T. Most P. Boucher M., et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- Pleger S.T. Shan C. Ksienzyk J., et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci. Transl. Med. 2011;3:92ra64. doi: 10.1126/scitranslmed.3002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J. Ren X. Wang X., et al. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 2009;105:1223–1231. doi: 10.1161/CIRCRESAHA.109.200378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raake P.W. Vinge L.E. Gao E., et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ. Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remppis A. Greten T. Schafer B.W., et al. Altered expression of the ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochim. Biophys. Acta. 1996;1313:253–257. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- Roques C. Salmon A. Fiszman M.Y., et al. Intrapericardial administration of novel DNA formulations based on thermosensitive Poloxamer 407 gel. Int. J. Pharm. 2007;331:220–223. doi: 10.1016/j.ijpharm.2006.11.056. [DOI] [PubMed] [Google Scholar]
- Ruan H. Su H. Hu L., et al. A hypoxia-regulated adeno-associated virus vector for cancer-specific gene therapy. Neoplasia. 2001;3:255–263. doi: 10.1038/sj.neo.7900157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydén L. Ariniego R. Arnman K., et al. A double-blind trial of metoprolol in acute myocardial infarction. Effects on ventricular tachyarrhythmias. N. Engl. J. Med. 1983;308:614–618. doi: 10.1056/NEJM198303173081102. [DOI] [PubMed] [Google Scholar]
- Sarge K.D. Park-Sarge O.K. Sumoylation and human disease pathogenesis. Trends Biochem. Sci. 2009;34:200–205. doi: 10.1016/j.tibs.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch P. Rahman G.F. Patejunas G., et al. Adenoviral-mediated transfer of vascular endothelial growth factor 121 cDNA enhances myocardial perfusion and exercise performance in the nonischemic state. J. Thorac. Cardiovasc. Surg. 2004;127:535–540. doi: 10.1016/j.jtcvs.2003.06.015. [DOI] [PubMed] [Google Scholar]
- Schmidt U. Hajjar R.J. Helm P.A., et al. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J. Mol. Cell Cardiol. 1998;30:1929–1937. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- Schmidt U. Hajjar R.J. Kim C.S., et al. Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am. J. Physiol. 1999;277:H474–H480. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- Snyder R. Moullier P. Methods in Molecular Biology Series. Vol. 807. Humana Press; New York, NY: 2011. Adeno-Associated Virus: Methods and Protocols. [Google Scholar]
- Stewart D.J. Kutryk M.J. Fitchett D., et al. NORTHERN Trial Investigators. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial. Mol. Ther. 2009;17:1109–1115. doi: 10.1038/mt.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H. Kan Y.W. Adeno-associated viral vector-delivered hypoxia- inducible gene expression in ischemic hearts. Methods Mol. Biol. 2007;366:331–342. doi: 10.1007/978-1-59745-030-0_19. [DOI] [PubMed] [Google Scholar]
- Swedberg K. Hjalmarson A. Waagstein F., et al. Beneficial effects of long-term beta-blockade in congestive cardiomyopathy. Br. Heart. J. 1980;44,117 doi: 10.1136/hrt.44.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvius N. Bilinska Z.T. Veinot J.P., et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 2005;42:639–647. doi: 10.1136/jmg.2004.023283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilemann L. Ishikawa K. Weber T. Hajjar R.J. Gene therapy for heart failure. Circ. Res. 2012;110:777–793. doi: 10.1161/CIRCRESAHA.111.252981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafiadaki E. Sanoudou D. Arvanitis D.A., et al. Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J. Mol. Biol. 2007;367:65–79. doi: 10.1016/j.jmb.2006.10.057. [DOI] [PubMed] [Google Scholar]
- Vassalli G. Bueler H. Dudler J., et al. Adeno-associated virus (AAV) vectors achieve prolonged transgene expression in mouse myocardium and arteries in vivo: a comparative study with adenovirus vectors. Int. J. Cardiol. 2003;90:229–238. doi: 10.1016/s0167-5273(02)00554-5. [DOI] [PubMed] [Google Scholar]
- Vodicka M.A. Determinants for lentiviral infection of non-dividing 764 cells. Somat. Cell. Mol. Genet. 2001;26:35–49. doi: 10.1023/a:1021022629126. [DOI] [PubMed] [Google Scholar]
- Wang Z. Zhu T. Qiao C. Zhou L. Wang B. Zhang J. Chen C. Li J. Xiao X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- Washington B. Butler K. Doye A.A., et al. Heart function challenged with β-receptor agonism and antagonism in a heart failure model. Cardiovasc. Drugs Ther. 2001;15:479–486. doi: 10.1023/a:1013755402109. [DOI] [PubMed] [Google Scholar]
- White J.D. Thesier D.M. Swain J.B., et al. Myocardial gene delivery using molecular cardiac surgery with recombinant adeno-associated virus vectors in vivo. Gene Ther. 2011;18:546–552. doi: 10.1038/gt.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. Asokan A. Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Ye X. Rivera V.M. Zoltick P., et al. Regulated delivery of therapeutic proteins after in vivo somatic cell gene transfer. Science. 1999;283:88–91. doi: 10.1126/science.283.5398.88. [DOI] [PubMed] [Google Scholar]
- Ying Y. Muller O.J. Goehringer C., et al. Heart-targeted adeno-associated viral vectors selected by in vivo biopanning of a random viral display peptide library. Gene Ther. 2010;17:980–990. doi: 10.1038/gt.2010.44. [DOI] [PubMed] [Google Scholar]
- Zaiss A.K. Liu Q. Bowen G.P. Wong N.C.W. Bartlett J.S. Muruve D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002;76:4580–4590. doi: 10.1128/JVI.76.9.4580-4590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangi L. Lui K.O. von Gise A., et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013;31:898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.Q. Sarge K.D. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J. Cell Biol. 2008;182:35–39. doi: 10.1083/jcb.200712124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.H. Wang X. Overexpression of heat-shock protein 20 in rat heart myogenic cells confers protection against simulated ischemia/reperfusion injury. Acta Pharmacol. Sin. 2005;26:1076–1080. doi: 10.1111/j.1745-7254.2005.00137.x. [DOI] [PubMed] [Google Scholar]
- Zincarelli C. Soltys S. Rengo G. Rabinowitz J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]