

Abstract
Our understanding of the genetic mechanisms that underlie biological processes has relied extensively on loss-of-function (LOF) analyses. LOF methods target DNA, RNA or protein to reduce or to ablate gene function. By analysing the phenotypes that are caused by these perturbations the wild-type function of genes can be elucidated. Although all LOF methods reduce gene activity the choice of approach (for example, mutagenesis, CRISPR-based gene editing, RNA interference, morpholinos or pharmacological inhibition) can have a major effect on phenotypic outcomes. Interpretation of the LOF phenotype must take into account the biological process that is targeted by each method. The practicality and efficiency of LOF methods also vary considerably between model systems. We describe parameters for choosing the optimal combination of method and system, and for interpreting phenotypes within the constraints of each method.
Loss-of-function (LOF) approaches have been applied to many fields and their substantial positive impact on biological and biomedical research is unquestionable. First pioneered in microorganisms, genetic screens in Caenorhabditis elegans1 and Drosophila melanogaster2,3 demonstrated how large groups of genes that are involved in a specific biological process could be identified through saturation mutagenesis, an approach that was later also applied to zebrafish (Danio rerio)4,5 and mice6,7. More recently, LOF screens based on RNA interference (RNAi) and genome-editing methods have been used in tissue culture systems to interrogate various cellular processes8,9. An important feature of cell-based assays is that they are amenable to large-scale combinatorial screens, which has important implications for understanding network structures and for identifying drug targets10,11.
LOF experiments can be divided into three main applications on the basis of the complexity of the gene sets that are interrogated. First, large-scale screens can be carried out using untargeted mutagenesis, knockout approaches such as CRISPR, or knockdown approaches such as RNAi, to systematically identify new factors that are involved in normal and pathological processes. Second, studies on focused sets of genes can be used to explore gene function for small numbers of known components, to study disease mechanisms, or to further evaluate candidate therapeutic targets that have been identified from large-scale screens. Third, as many genetic diseases involve at least a partial loss of gene function, generation of LOF alleles of these genes in cell culture or in vivo can be used to model these diseases in experimentally tractable systems for further investigation.
Although LOF approaches may initially all seem to achieve the same effect, that is, disruption of gene function, there are considerable conceptual differences among them that can lead to substantially different outcomes (TABLE 1). One key difference is that approaches such as RNAi, morpholinos, chemical inhibitors and hypomorphic mutations most often lead to the partial suppression of gene function, whereas null mutations ablate function (BOX 1). For pleiotropic genes, the partial suppression of activity may affect some functions, whereas a complete LOF mutation would remove all functions. Experimentally, there are several differences between LOF approaches that should be taken into account when designing assays. For example, approaches vary in the extent to which LOF is inducible and reversible (TABLE 1). These properties allow the assessment of gene function after an essential developmental role, which if perturbed would result in lethality. The temporal nature of a perturbation is also important in another way. Cells adapt to changes in genetic state12, and there may be differences in the effects of a short-term disruption, such as through RNAi or drug treatment, compared with the long-term LOF effects of a mutation. Finally, there are differences in the side effects. Methods such as RNAi can be associated with off-targets that may mask the true function of a gene and may lead to false-positive observations13,14. The disruption of one or more non-target genes can also be an issue with genome engineering, although the specificity of tools such as CRISPR is still under investigation and development9,15–18. Similarly, long-term knockdown or mutant alleles may be affected by secondary mutations or other forms of phenotypic compensation that suppress the effects of the primary LOF event and are fixed owing to selective pressure or by chance.
Table 1. Summary of loss-of-function approaches.
| Loss-of-function approach | Level of action | Duration | Specificity | Inducible | Reversible |
|---|---|---|---|---|---|
| Chemical mutagens | DNA | Sustained | None | No | No |
| Transposons | DNA | Sustained | None | Partially | In some cases |
| RNAi | mRNA | System dependent | Variable | Yes | System specific |
| Morpholinos | mRNA | Transient | Variable | Yes | No |
| Genome editing | DNA | Sustained | High | Partially | No |
| CRISPRa and CRISPRi | Transcription | System dependent | High | Yes | Yes |
| Degrons | Protein | Transient | High | Yes | Yes |
| Small-molecule inhibitors | Protein | Transient | Variable | Yes | Yes |
The information in this table represents the most widely used versions of each technology. There are, however, exceptions to many of the listed classifications. CRISPRa, CRISPR activation (transcriptional activation); CRISPRi, CRISPR interference (transcriptional inhibition); RNAi, RNA interference.
Box 1. The classification of mutations based on their behaviour in various genetic situations.
Mutations are associated with either loss of function (LOF) or gain of function (GOF) depending on the nature of the molecular lesion230. LOF mutations can be complete null (amorphic) or can be associated with reduced activity (hypomorphic). GOF mutations can be associated with increased activity relative to the wild-type gene (hypermorphic), new and abnormal expression patterns or functional gene products (neomorphic), or altered gene products that act antagonistically to the wild-type allele (antimorphic). LOF mutations are usually recessive (and hence require all alleles to be mutated for phenotypic effects to be observed), except in cases in which the gene is haploinsufficient. By contrast, GOF mutations are usually associated with dominant phenotypes. Except in cases in which the coding sequence (CDS) is deleted, leading to null mutations, molecular lesions do not immediately predict the nature of a mutation. A point mutation could either reduce or increase wild-type activity, leading to hypomorphic or hypermorphic activities, or could modify the product such that it acts as an antimorph. Furthermore, a single base change that introduces a stop codon in the CDS can lead to a shorter protein, a novel polypeptide (if downstream translational re-initiation occurs), a hypomorph (if sporadic translational read-though occurs) or a null (if nonsense-mediated decay efficiently targets the RNA for degradation). Careful genetic tests, such as the comparison of the homozygous mutant phenotype to the phenotype of the mutation over a deletion of the locus, need to be used to characterize the nature of the mutation. This is particularly important for truncated proteins or point mutations in which key protein domains may retain function or if an autoinhibitory region is affected. These types of tests should be easier to carry out now that whole-gene deletions can be generated using CRISPR–Cas9.
The selection of the most appropriate LOF approach and experimental system and careful interpretation of results with the caveats of the chosen approach in mind are essential in order to accurately annotate gene function. In this Review, we discuss the approaches and tools that are available for LOF studies in four animal models (Caenorhabditis elegans, Drosophila melanogaster, zebrafish and mice) as well as in cultured cells. We also discuss specific examples in which the outcome of approaches may differ, and why this is the case. Finally, we attempt to extract basic guidelines for the selection of appropriate LOF methods and highlight common pitfalls in the interpretation of results.
LOF approaches across organisms
Non-targeted DNA mutagenesis approaches
Several options exist for the non-targeted induction of mutations in genomic DNA (FIG. 1a). Historically, irradiation and chemical mutagens have been used in many model systems. For example, X-rays, ethyl methane sulfonate (EMS) and N-ethyl-N-nitrosourea (ENU) can be used to introduce widespread alterations throughout the genome19. X-Rays result in double-strand breaks (DSBs), which can be repaired to produce small insertions or deletions (indels), often resulting in frameshift mutations that lead either to no protein being made or to the production of truncated proteins19. Large deletions and inversions are also frequently induced by X-rays. EMS and ENU treatments generally result in the induction of single-nucleotide changes, leading to a wide range of mutation types, including gain-of-function (GOF) and LOF mutations, as well as temperature-sensitive mutations (BOX 1). The range of possible outcomes, together with a high mutation rate, has made these mutagens favourites for unbiased screens. They have been widely used in C. elegans20, D. melanogaster21,22 and fish (particularly D. rerio (zebrafish) and Oryzias latipes (known as medaka))4,5,23,24, and in more focused ways in mice7,25.
Figure 1. Overview of loss-of-function approaches.
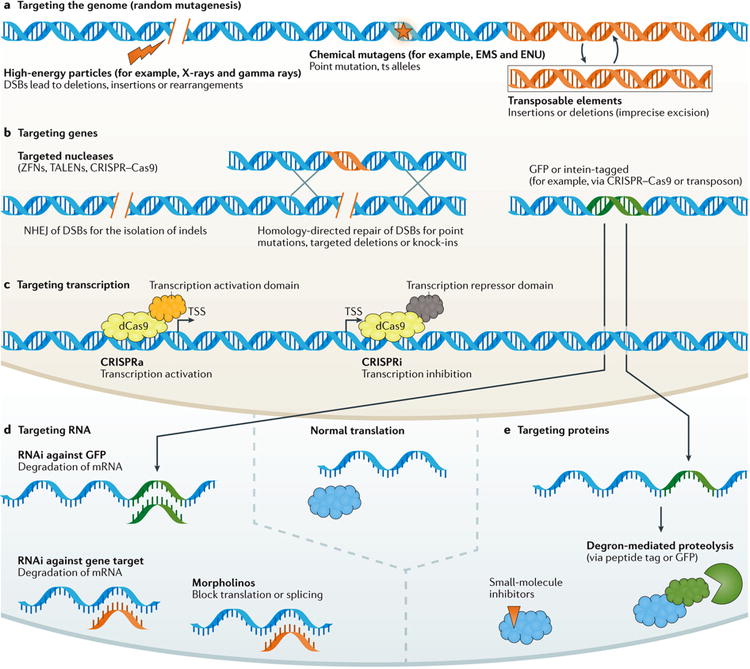
a | Targeting the genome. The modification of the genes themselves can result in loss-of-function (LOF) mutations. These mutations can be induced at random, for example, by using mutagens such as high-energy particles (such as, X-rays and gamma rays), which tend to induce double-strand breaks (DSBs), resulting in some cases in large deletions or complex rearrangements; by using chemical mutagens (such as, ethyl methane sulfonate (EMS) and N-ethyl-N-nitrosourea (ENU)), which tend to result in single base-pair changes; and by using transposons. The possible functional outcomes of classical mutations are many (BOX 1). b | Targeting genes. Zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), CRISPR–Cas9 and other gene-editing approaches can be used to induce DSBs. When repaired through non-homologous end joining (NHEJ), this can result in small insertions or deletions (indels). Combining one of these targeted approaches to generating DSBs with a homologous donor construct, these methods can be used to induce specific changes (for example, specific single base-pair changes) or deletions (for example, of an entire coding region or functional domain), or can be used to replace genes with marker or other constructs (knock-in). Gene-targeted approaches or transposons can also be used to insert fusion tags such as GFP into specific or random locations, respectively. In addition to (or instead of) disrupting genes, the insertion of tags such as GFP or inteins can be useful for RNA and protein-targeting approaches (parts d and e). c | Targeting transcription. A nuclease-dead version of the Cas9 protein (dCas9) fused to appropriate effector domains can be used in combination with guide RNAs (gRNAs) to target appropriate regions to bring about transcriptional activation (CRISPRa) or transcriptional inhibition (CRISPRi), leading to increased or decreased levels, respectively, of the transcript and hence in most cases, leading to increased or decreased levels of the wild-type protein. d | Targeting RNA. The introduction of RNA interference (RNAi) reagents that target a specific gene (or isoform), or targeting GFP (for GFP-tagged genes) can be used to reduce mRNA levels, leading to a reduction in protein levels. The introduction of morpholinos can lead to a block of translation or splicing of the target mRNA. In either case, some population of mRNA might evade RNAi or morpholino targeting, such that the approaches are likely to result in partial and incomplete LOF. e | Targeting proteins. When available, small-molecule inhibitors can be introduced, resulting in the disruption of protein function, for example, through the occupancy of a substrate-binding site or other disruption of function. Genetically tagging a protein of interest through knock-in can allow for its inducible degradation by recruitment of an effector protein that binds the introduced tag and earmarks the protein for proteolysis. ts, temperature-sensitive; TSS, transcription start site.
An important factor for random mutagenesis screens is the need to generate homozygous mutations. All non-targeted mutagenesis approaches initially result in the disruption of a single allele and, therefore, generally do not exhibit a phenotype, with the exception of dominant alleles and haploinsufficient loci that are sensitive to the loss of a single gene copy. Crossing schemes are thus required in order to generate homozygous animals in which the phenotypes of interest can be analysed. C. elegans is particularly well suited to the establishment and maintenance of large centralized collections of mutagenized animals that can be screened for phenotypes by the community. As hermaphrodites are self-fertile, newly created alleles can be propagated and made homozygous without mating. C. elegans larvae can survive freezing and so mutant lines can be maintained as frozen stocks that can be stored indefinitely in liquid nitrogen. The C. elegans knockout consortium has generated more than 8,000 lines, including 2,000 lines with fully sequenced genomes, which together provide putative LOF alleles for more than 50% of the 20,000 protein-coding genes26. Each line, however, carries many mutations (∼400 in the case of the sequenced Million Mutation Project (MMP) lines), which can complicate phenotypic analyses. The maintenance of large mutant collections is more challenging in other animal models, although in the case of zebrafish and mice, it is possible to store mutants in the form of frozen sperm. In addition to the thousands of mutants that have been identified in large-scale genetic screens, ongoing tilling projects in zebrafish have identified potentially disruptive mutations in more than 60% of all known zebrafish protein-coding genes27,28, and continuing work aims to mutate any additional genes of interest using the CRISPR–Cas9 gene-editing technology (E. Busch-Nentwich, personal communication).
In C. elegans, D. melanogaster, zebrafish, and to some extent in mice, animals can also be mutagenized de novo for screens in which only animals with a specific phenotype are kept for further study. Mutant lines that are generated by such screens are generally ‘cleaned up’ by out-crossing to wild-type animals, and a large number of strains harbouring mutations generated in this way by the D. melanogaster, C. elegans and zebrafish communities are available from public repositories (TABLE 2). It should be noted, however, that a considerable mutation load remains even after out-crossing29.
Table 2. Key features of loss-f-function approaches across organisms.
| Organism or system | Key advantages for LOF studies | Key resources |
|---|---|---|
| Caenorhabditis elegans |
|
|
| Drosophila melanogaster |
|
|
| D. melanogaster cells |
|
|
| Zebrafish |
|
|
| Mouse |
|
|
| Mammalian cells |
|
|
AAV, adeno-associated virus; DSBs, double-strand breaks; dsRNA, double-stranded RNA; LOF, loss-of-function; RNAi, RNA interference; shRNA, short hairpin RNA; siRNA, small interfering RNA; UAS, upstream activation sequence.
Second-generation mutagenesis approaches have focused on transposon-mediated gene disruption30–33. Transposons are genetic elements that move within genomes using an RNA intermediate or through the excision of a DNA element from one region and integration into another. When transposons integrate near or into a coding sequence, gene function can be disrupted. One advantage of transposon-mediated gene disruption is that mutation rates are lower than for chemical mutagens and so single genes are generally disrupted as a result of the element mobilization. However, owing to the insertional nature of this approach and biases in integration sites, the range of mutations that is generated is more limited than that from chemical-based approaches. Another key advantage of transposon-based mutagenesis lies in the possibility of rapidly mapping disrupted genes using inverse PCR amplification of the inserted transposon together with flanking genomic DNA. Transposon-based mutagenesis has been broadly used in C. elegans with the Tc1 transposon32,34 and Mos35 transposon, and in D. melanogaster with P-elements and the Piggybac transposon22,33,36,37. However, some transposable elements exhibit insertional bias, which limits the range of gene disruptions that can be generated. For example, P-elements, which are widely used in D. melanogaster, preferentially insert at the 5′ end of genes, thus biasing the genomic search space. In addition, Sleeping Beauty (a Tc1 family transposon) has an insertional bias towards heterochromatin, thus limiting its use for the disruption of many genes38. Another approach that has been applied in D. melanogaster is to use transposable elements such as Minos, which have less insertion bias, to introduce MiMIC cassettes into the introns of genes39. These cassettes, which include splice acceptor sequences followed by stop codons, resulting in premature translational termination and efficient inhibition of the target gene, can be subsequently modified within their new genomic location to easily introduce various DNA fragments at the location of the MiMIC. However, owing to low mutation rates, these approaches are not typically used for de novo screening. Instead, they have been used to generate libraries of mutant animals, which are then used for low-throughput and high-throughput studies (TABLE 2). In zebrafish, large collections of enhancer-trap mutants and gene-trap mutants have also been generated using retroviral vectors and transposable elements40–45, and the technology to generate conditional alleles is being refined45.
Insertional mutagenesis has found limited application in mammals owing to relatively high genomic complexity and the lower throughput of in vivo studies. Efforts to generate repositories of mutant mouse stocks initially took advantage of gene-trapping methods to generate large collections of knockout embryonic stem cell (ESC) lines46,47, which can subsequently be used to generate mutant mice, which can then be backcrossed and characterized. For experimentation in cell culture, a major limitation of non-targeted mutagenesis is an inability to generate homozygous mutations, which is achieved in vivo using crossing schemes. To overcome this limitation, one strategy has been to use cell lines that are deficient for Bloom's syndrome protein (BLM). These cells exhibit a high rate of mitotic recombination, resulting in gene conversion and the homozygosis of induced mutations48. In addition, several near-haploid and haploid mammalian cell lines have been derived that enable homozygous gene disruption through random insertional mutagenesis49–51. The resulting mutagenized cell pools can be used for phenotypic screening and deconvolution for the generation of mutant cell line collections52.
Targeting DNA with genome-editing technologies
The most recent approach for generating LOF alleles is targeted genome editing (FIG. 1b). In contrast to other methods, genome editing facilitates the precise modification of genome sequences, including the introduction of targeted knockouts, knock-ins, hypermorphic or hypomorphic variants, disease-associated mutations and modifications to non-coding genomic sequences53. In general, genome editing is enabled by the introduction of DSBs at a specific location by a targeted nuclease54. In the presence of a homologous donor DNA template, the DSB is repaired by homology-directed repair (HDR) using sequences on the template to insert or to exchange DNA at the target site. In the absence of a donor template, DSBs are repaired by direct re-ligation through non-homologous end joining (NHEJ). Because NHEJ is often error prone, indels might be introduced during repair. Indels in the coding sequence that disrupt the reading frame or that remove crucial amino acids can lead to a gene knockout, although other kinds of alleles may also be generated (BOX 1). By targeting nucleases to two different positions along a chromosome, it is also possible to excise the region between a pair of DSBs to generate a deletion55. Two DSBs can also be used to generate megabase-scale deletions, thereby removing many genes at once56.
Several sequence-specific nucleases have been developed to introduce DSBs at specific sites in the genome. These include programmable nucleases that target specific sites, such as meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and CRISPR–Cas9. Each of these has unique properties and advantages54.
Owing to their ease of use, efficiency and scalability, CRISPR-based systems have become the method of choice for most applications and organisms. Unlike the protein-encoded targeting specificity of meganucleases, ZFNs and TALENs, CRISPR uses a guide RNA (gRNA) to target the Cas9 nuclease to a specific sequence and to create a DSB at the target site. CRISPR–Cas9 has enabled the generation of cell lines and organisms with targeted gene disruptions57–63 and the template-mediated introuction of precise mutations57,58,64–67. In addition, by controlling Cas9 expression, mutations can be induced with temporal and spatial control68–73. However, as CRISPR targets genomic DNA, the persistence of both mRNA and protein limits the extent of the temporal control of this system.
CRISPR has been transformative, allowing the efficient genome editing of many organisms. This system works in a similar manner in all organisms, with the major differences focused on reagent delivery. Differences in the relative efficiency of NHEJ and HDR also influence the way in which CRISPR genome editing is used in each organism. For example, in C. elegans, in which HDR is robust74–76, precise LOF mutations are generated using oligonucleotides to template frame-shifts or whole-gene deletions. By contrast, in zebrafish, in which NHEJ is dominant, LOF alleles are generated by screening for Cas9-induced indels77.
CRISPR represents a major advance for LOF studies in mammalian cell culture systems. Previous methods, such as insertional mutagenesis, could only be used effectively in specialized (BLM-deficient or haploid) cell lines owing to the need to generate homozygous mutations. By contrast, CRISPR-based mutagenesis is sufficiently potent that multiple alleles can be generated simultaneously. Therefore, this technology allows targeted mutagenesis screens to be carried out for the first time in diploid or polyploid mammalian cell lines and primary cell cultures78. However, one disadvantage of CRISPR compared with chemical mutagenesis is that the range of alleles produced using this method is narrow. CRISPR generates mutations at a single targeted position, whereas chemical mutagens have the potential to generate sequence alterations at any location and, therefore, may provide information on the function of specific protein domains. Nevertheless, shortly after the development of CRISPR as a genome-engineering tool, a wide range of applications were pioneered in mammalian cell culture systems, including the engineering of complex and compound mutations57,58, transcriptional repression and activation systems that are based on a nuclease-dead mutant of Cas9 (dCas9) fused to transcriptional regulatory domains79,80 (further details below), and genome-wide screening81,82. The delivery of CRISPR–Cas9-based tools can be achieved through transfection57,58, viral delivery79 and microinjection60,83, making nearly all cell culture systems and tissues amenable to genome editing. However, one limitation of CRISPR is that the induction of DSBs can result in genotoxic stress, especially limiting its use for targeting highly amplified loci, as commonly found in cancer genomes84,85. An additional important issue for CRISPR-engineered cell lines is adaptive evolution over time, an issue that is not relevant to transient knockdown models such as RNAi or drug treatments. Adaptive changes have been well documented in yeast, for which the accumulation of specific secondary compensatory mutations seems to increase fitness12. In contrast to in vivo systems, in which secondary mutations can be removed or addressed via genetic crosses, this issue is important for cell culture. Thus, as a cautionary note, samples of early isolates should be preserved, and careful attention should be paid to the number of passages carried out before cell screening or other assays.
A major hurdle for gene editing in vertebrate somatic tissues is the delivery of the nuclease itself. This can be overcome by transgenic expression of Cas9, as has been done in mice86, zebrafish73 and C. elegans87, and so only necessitating the delivery of gRNAs and, if required, repair templates. Alternatively, to avoid the difficulties of Cas9 and gRNA delivery, CRISPR can be applied in cultured cells, which are then injected into mice, allowing genome-wide in vivo screens88,89. Nonetheless, complete abrogation of gene function in a somatic tissue is still most efficiently done through the generation of germline knockout alleles, a process that can be accelerated through CRISPR–Cas9-based editing in zygotes60,64,90. Many of the reagents necessary for implementing CRISPR–Cas9-based genome engineering are commercially available or are available through the Addgene non-profit plasmid repository. The large number of available mammalian cell lines provides a rich resource for CRISPR–Cas9-based studies of basic processes and human diseases. Moreover, recently developed organoid culture systems that closely mimic the composition of mammalian tissues will enable scalable cell culture studies in complex and increasingly relevant cellular contexts91.
A major limitation that is associated with most mutant alleles is the lack of stage- or tissue-specific inducibility and reversibility. This is a particular problem for the study of essential and pleiotropic genes, for which lethality at an early developmental stage might prevent the analysis of LOF effects at later stages. One solution is to use temperature-sensitive mutant alleles (BOX 2), in which the protein is functional in one temperature range but is not functional at other, usually higher, temperatures. Remarkably, ∼50% of temperature-sensitive alleles in C. elegans have been found to be ‘fast-acting’, that is, LOF phenotypes can be observed within seconds of exposure to the non-permissive temperature92. Temperature-sensitive alleles can provide precise information on gene function. For example, using fast-acting temperature-sensitive alleles and a device that is designed to induce rapid temperature changes, Davies et al.93 mapped the requirement for six cytoskeletal regulators to precise stages of cytokinesis, creating a functional timeline for cell division. In D. melanogaster and especially in mice, an alternative and widely used solution for low-throughput studies has been to generate conditional mutant animals using inducible recombination systems such as FLP–FRT in D. melanogaster94 and mice95, and Cre–lox in mice96,97. By controlling the temporal and spatial expression of the recombinase, mutations can be induced at specific developmental stages and in certain tissues98–101. In cultured cells, inducible mutations can be achieved using artificial introns that disrupt gene function only when inverted using the Cre–lox system102. In most cases, these inducible systems are not reversible, although some specialized gene- trap elements have been developed that allow both the induction and the reversion of mutations103.
Box 2. The generation of temperature-sensitive alleles.
Temperature-sensitive alleles are valuable tools for the study of genes with pleiotropic functions, for which an early function may mask the effect of later functions. Using temperature-sensitive alleles, it is possible to perturb protein function at a specific developmental stage using a temperature shift. In Caenorhabditis elegans, it is possible to carry out screens that enrich for reversible temperature-sensitive mutations92, and efforts are ongoing to systematically clone all available temperature-sensitive embryonic lethal mutants (B. Bowerman, personal communication). These efforts are predicted to identify temperature-sensitive alleles for nearly 25% of the ∼2,500 essential genes in C. elegans. For other organisms, most temperature-sensitive mutations were identified by chance in large-scale mutagenesis screens, and reliable methods to generate temperature-sensitive alleles at high frequency are needed. Recent advances in CRISPR genome-editing technologies offer a potential solution to this issue. For example, a new approach that uses CRISPR to introduce point mutations at high efficiency227 could be used to generate temperature-sensitive alleles in specific loci as done previously for the sevenless and MEK genes231,232. Alternatively, heat-inducible degrons can be used to generate transgenic temperature-sensitive alleles233. Another approach is to use conditionally active protein inteins whereby a temperature-sensitive splicing variant of the yeast Saccharomyces cerevisiae vacuolar ATPase subunit (VMA) intein is inserted into a target gene, such that it inhibits protein function. At the permissive temperature, the intein is ‘spliced’ out of the protein, restoring normal function234.
Targeting transcription
dCas9 lacks the ability to induce DSBs and provides a programmable DNA-binding protein that can be fused to effectors to illicit transcriptional changes and epigenetic modifications80 (FIG. 1c). For example, dCas9 has been fused to various transcriptional activators (known collectively as CRISPRa)79,104,109 and repressors such as the KRAB domain (known collectively as CRISPRi)79. Additionally, the direct modification of epigenetic marks by the fusion of dCas9 to histoneor DNA-modifying enzymes can be used to probe the function of both genes and non-coding regulatory elements110,112. In particular, these tools provide the opportunity to target the non-coding regulatory elements of the genome for both GOF and LOF assays with high specificity111,113. Although these tools have only been tested to a limited extent, initial results are promising. For example, these tools have been used in D. melanogaster114,115, C. elegans116 and zebrafish116,117 for single-gene studies and also genome-wide in mammalian cell culture118,119.
Targeting RNA
A commonly used LOF approach is the targeting of mRNAs using RNAi or morpholino reagents (FIG. 1d). A major difference between these methods and the direct mutation of the genome (FIG. 1) is that the inhibition of mRNA rarely causes complete LOF, with some level of the wild-type mRNA and protein still detectable, and high protein stability can delay the onset of effects, leading to weaker phenotypes.
RNAi reagents induce the sequence-specific elimination of targeted mRNA molecules. The RNAi pathway is triggered when double-stranded RNA (dsRNA) that is introduced into a cell interacts with the endogenous cellular machinery to illicit the cleavage of the target mRNA120. In general, RNAi has several advantages over other methods for perturbing gene expression. These include the simplicity of delivery, the capacity to generate hypomorphic phenotypes by partially knocking down gene function, and the ability to target specific exons, allowing the repression of specific isoforms121. A disadvantage of RNAi is the substantial possibility of re agent-specific off-targets. Knockdown efficiency also typically varies for different reagents, even for those that target the same gene. Differences in knockdown efficiency and off-targets can lead to considerable variability in the phenotypes observed with different reagents that target the same gene, often confounding the interpretation of results. Owing to divergence in RNAi pathways and different modes of delivery, the appropriate design of RNAi experiments can vary considerably for different organisms.
C. elegans is particularly amenable to RNAi because, in this system, silencing is heritable122. dsRNA that is introduced into hermaphrodites depletes both maternal and zygotic RNAs in all F1 progeny, generating large populations of animals that can be examined for phenotypes120,123. The delivery of RNAi reagents to worms is also straightforward: dsRNA can be delivered by injection, soaking or feeding of Escherichia coli strains that are engineered to express dsRNA against specific genes122,124. The effects spread from tissue to tissue (known as ‘systemic RNAi’)120,125. However, it should be noted that some tissues are refractory to RNAi126, and the degree of inactivation is sensitive to the experimental conditions122. Despite these caveats, the experimental ease of RNAi in C. elegans quickly made it the method of choice for large-scale screens127.
For systems in which RNAi is not systemic, it is possible to limit RNAi to particular tissues or developmental stages using inducible RNAi systems, for example, Gal4–UAS (upstream activation sequence) in D. melanogaster128–131 and doxycycline-inducible systems in mice132. Indeed, in large part owing to this inducible control, RNAi has become the tool of choice for in vivo large-scale screens and single-gene studies in D. melanogaster. This is facilitated by the availability of extensive collections of transgenic RNAi lines, as well as the ability to tightly control spatial and temporal expression of the RNAi reagent in vivo using the Gal4-UAS system130,131. Finally, D. melanogaster cell culture has been a productive platform for high-throughput screens owing to the straightforward design of potent RNAi triggers and their easily scalable delivery by simply bathing cells in long dsRNAs in an arrayed format (that is, a different single gene is targeted by dsRNAs in each well of a multi-well plate), and the less complex genome relative to those of mammals simplifies the interpretation of results133. However, a major limitation of this system is an inability to screen in a pooled format (in which complex mixtures of RNAi reagents are delivered to a population of cells), owing to the lack of technologies allowing the delivery of a single RNAi reagent to each cell, which limits the scale and number of screens that can be carried out.
At the time it was developed, RNAi was a transformative advance for the functional exploration of mammalian gene function, which for decades had lagged behind other, more ‘genetically tractable’ systems. Following pioneering work in C. elegans and the first successful small interfering RNA (siRNA) transfection experiments in mammalian cells134,135, a plethora of mammalian RNAi tools were established and later optimized on the basis of a growing understanding of endogenous microRNA (miRNA) pathways. The development of viral vectors for the integration of short hairpin RNA (shRNA) expression cassettes into genomic DNA has enabled stable gene silencing in cultured cells136, as well as pool-based genetic screens137,138. A common system is based on RNA polymerase III (Pol III)-driven expression of stem-loop structures (resembling pre-mi RNAs). These can be very effective but are prone to triggering unspecific effects owing to imprecise processing139 or to interference with endogenous miRNA pathways140,141. To mitigate such effects, synthetic shRNAs can be embedded in endogenous miRNA backbones142,143. Such ‘shRNAmirs’ can be expressed like natural mi RNAs in polycistronic transcripts from Pol II promoters, facilitating regulatable and tissue-specific RNAi144,145, direct coupling to reporter transgenes146,147 and combinatorial LOF studies11. Conditional shRNAmir cassettes have also been implemented in transgenic mice, enabling inducible and reversible gene suppression in disease models and normal tissues132,148–150. Over the past few years, a growing understanding of sequence determinants in miRNA biogenesis139,151,152 has led to substantial improvements in shRNAmir design, facilitating the construction of shRNAmir libraries that are functional under single-copy conditions153,154. Although all basic RNAi systems (siRNAs, shRNAs and shRNAmirs) have led to major discoveries in high-throughput genetic screens, they differ substantially in their on-target efficacy and the type and severity of associated off-targets, which should be considered in the interpretation of RNAi-based results155.
Although RNAi works efficiently in most common model systems, it has had very limited success so far in zebrafish and frogs156. Instead, communities working on zebrafish and frogs have focused on the use of morpholinos, which are nucleic acid analogues that bind an RNA target sequence and that prevent translation or splicing157. Morpholinos were first used in zebrafish158 in 2000, and since then more than 2,000 zebrafish papers using morpholinos have been published.
The specificity of morpholinos is a concern that has recently been revived following the direct comparison of phenotypes in morpholino-injected animals versus mutants generated using TALENs or CRISPR117,159–161. In many cases, there is a clear discrepancy between these phenotypes, leading to vigorous debate within the fish and frog communities162–165 (potential molecular explanations are discussed further below). The current recommendation for identifying a morpholino with minimal off-targets is to test it in a null mutant background, as it should not induce any additional phenotypes117. Such an approach of course relies on the ability to generate and validate null alleles, for example, by whole-gene deletion166, without disrupting other important genetic elements in the process.
Targeting proteins
The techniques discussed above involve the targeting of genomic DNA or mRNA with the goal of reducing or eliminating the gene product, or reducing protein function through amino acid substitution or truncation (FIG. 1). Although useful, these approaches also have disadvantages. In particular, they are dependent on the half-life of wild-type mRNA and protein, and so effects may be slow or incomplete owing to mRNA and/or protein persistence. LOF methods that directly target proteins overcome such problems.
There are two main options for disrupting function at the protein level: the inhibition of protein activity and the induction of protein degradation (FIG. 1e). Small-molecule inhibitors (SMIs) are the most widely used tools to modulate protein function and are amenable to high-throughput screening (HTS). For example, a growing collection of small molecules can be used to inhibit defined protein functions167. Despite the steady expansion of this resource (for example, Chemical Probes, SGC and DrugBank; see Further information), such compounds only cover a fraction of proteins. Moreover, although these inhibitors act quickly and can have a range of knockdown effects, they rarely achieve inhibition levels of a knockout and often suffer from a lack of specificity, which can complicate the interpretation of results. In addition, the targets of many small molecules remain unknown, resulting in the need for extensive follow-up assays to identify the relevant mechanism that leads to the observed phenotype.
Combining protein engineering with chemical modifiers of protein function is generally useful for both GOF and LOF studies. For example, proteolysis-targeting chimaeras (PROTACs) are molecules that bind to a specific protein and that recruit the ubiquitin-ligase machinery, resulting in the rapid degradation of the target protein168.
As a specific and generalizable approach for rapid protein suppression, several genetic strategies for rendering proteins the targets of ligand-induced decay have been established in mammalian cells169–171, D. melanogaster172, zebrafish173,174 and C. elegans175,176, which have been reviewed elsewhere177. One example of this approach, known as deGradFP, involves targeting GFP-tagged proteins with a GFP-specific nanobody fused to a domain that targets the protein for proteasome-mediated degradation. By combining the deGradFP transgene with an endogenously GFP-tagged protein, the nanobody directs the degradation signal sequence to the protein of interest, resulting in the degradation of the target172. Key advantages of deGradFP are that it allows inducible and reversible degradation178 and can be applied to any endogenously GFP-tagged genes. Such collections are already available for D. melanogaster178–181. Another broadly applicable approach is the auxin-inducible degron (AID) system, which exploits the small-molecule-induced ubiquitylation of proteins that carry a short peptide tag by an exogenous plant F-box protein175,182,183. This system has been used in yeast183, mammalian cells183,184, C. elegans175 and D. melanogaster185 and presents a favourable alternative to GFP-based systems owing to a very short minimal degron length and target degradation in as little as 15 minutes. Finally, several alternative approaches have been developed to selectively inhibit aspects of protein function without the need to generate fusion proteins. For example, peptide aptamer interference (PAPTi) allows interference with specific protein–protein interactions186. This approach is likely to be particularly useful for studying specific aspects of pleiotropic protein function.
Comparing LOF approaches for single genes
Results that are obtained using different LOF approaches are often inconsistent. In some cases, this can be attributed to easily defined, addressable issues such as differences in knockdown efficiency, temporal aspects of the LOF approach (for example, fast-acting or not) or off-targets. In other cases, the differences reflect important aspects of biology. A recent study in zebrafish has highlighted the need for caution when interpreting results that are obtained using different LOF approaches. An investigation into the egfl7 gene, which encodes an extracellular matrix (ECM) protein, included the direct comparison of the phenotypes that are caused by a strong functional mutation created with TALENs and two knockdown approaches, morpholinos and CRISPRi, with CRISPRi causing a substantial reduction in mRNA levels117. The knockdown approaches led to a much stronger vascular phenotype than that observed in the mutant. Rather than assuming that this was an off- target of the knockdown approaches, the authors further investigated the inconsistency. Proteomic and transcriptomic profiling of informative samples led to the identification of several ECM protein-coding genes that were upregulated in mutant but not in knockdown embryos. These genes were shown to be able to compensate, at least partially, for the loss of egfl7 function. Similar observations were made for the vegfaa gene: vegfab was upregulated in vegfaa mutants but not in morphants117. Such observations are reminiscent of studies in yeast that showed that the partial inhibition of Bem1 via light-induced sequestration away from its normal site of action produced severe defects that were not observed using a bem1 deletion allele187, as well as studies in mice188,189 and Arabidopsis thaliana190.
In addition to these examples, several cases have been reported in which the characteristics of LOF disease models differ depending on the approach. For example, mir-122 is a liver-specific miRNA that promotes hepatitis C virus (HCV) replication191 and that is under development as a therapeutic target for this disease192. However, whereas antisense inhibition of mir-122 may be an effective treatment for HCV, later studies of sustained LOF showed oncogenic effects, inflammation and liver damage193,194. In addition, inconsistencies have been reported between different mutant mouse models of the necroptosis regulators RIPK1 and RIPK3, which have been implicated as candidate therapeutic targets for several human diseases195. In this case, knockout mutations of RIPK1 are not viable but kinase catalytic mutants of the same gene are viable; by contrast, knockout mutants of RIPK3 are viable but kinase catalytic mutants are not viable. These results illustrate the need to consider multiple LOF approaches in the development of accurate disease models and therapeutic strategies, especially when studying potentially pleiotropic proteins.
Similar inconsistencies have been documented between RNAi and SMI-based approaches196. For example, Aurora B is a kinase that is required during mitosis, the functions of which include the regulation of interactions between the kinetochore and microtubules and the initiation of the spindle checkpoint when microtubules are not properly attached197,198. When Aurora B is knocked down using RNAi, the spindle checkpoint is no longer activated when microtubules are disrupted with either paclitaxel or nocodazole SMIs199,200. By contrast, when Aurora B is pharmacologically inhibited, the spindle checkpoint was activated following nocodazole treatment but not following paclitaxel treatment199,201. Further investigation revealed that this difference is unlikely to be due to off-targets. Instead, it has been hypothesized that the differential inhibition of pleiotropic Aurora B functions is responsible199,200. Aurora B is part of the chromosome passenger complex, and removal of Aurora B by RNAi is thought to disrupt this complex. By contrast, pharmacological inhibition prevents Aurora B kinase activity but does not remove it from this complex, thus maintaining other complex functions199,202,203.
In addition to discrepancies between LOF methods, differences have also been observed using a single LOF method but with differing timing of gene disruption. One example of this occurred when comparing an inducible deletion of the retinoblastoma (Rb) gene to a stable LOF mutant204. Specifically, an inducible mutation in Rb was found to be sufficient to drive quiescent primary mouse embryonic fibroblasts (MEFs) to re-enter the cell cycle. By contrast, primary MEFs carrying a constitutive mutant allele of Rb were considerably less able to re-enter the cell cycle under the same conditions. This difference was found to be due to the partial compensation of Rb function by the Rb family member p107, which was upregulated in the c ons tit utive Rb-mutant cells.
Although there are numerous cases in which there is a clear difference between the results from multiple LOF approaches, it is not clear how widespread a phenomenon this is. These cases are probably viewed as either negative results or a failure of validation, and, as such, are rarely investigated deeply or reported in the literature. The recent development of the CRISPR system has resulted in an expansion of the LOF toolbox and has made the generation of null mutants relatively straightforward. Thus, there is likely to be increased pressure to ‘validate’ the results that are obtained using knockdown approaches with CRISPR-generated mutants. However, the examples provided above highlight the need for great care when interpreting results from different LOF approaches. Importantly, rather than representing a failure to validate, on detailed analysis, some inconsistencies may lead to fundamental discoveries regarding underlying compensatory mechanisms, protein complex formation and other biologically relevant processes.
Comparing LOF approaches in screens
A major application of LOF methods in the past 10 years has been HTS in cell lines, for example, using CRISPR, RNAi or SMIs in pooled or arrayed formats. The most recent addition to this toolbox is CRISPR, which was rapidly repurposed for HTS205. Past experience with RNAi is streamlining the implementation of CRISPR screens as many best practices for RNAi screens, such as reagent specificity and efficacy, pooled versus arraybased methods and reagent delivery (for example, lentiviral transduction versus DNA or RNA transfection) are applicable to CRISPR screening. However, there are fundamental differences among the various screening approaches that need to be fully appreciated. This will become increasingly important as results obtained from screens with new technologies such as CRISPR are compared with established methods.
Screens for essential genes have been carried out in many different model organisms and human cell lines206. By comparing results across multiple screens, attempts have been made to identify a core set of essential genes common to all cell types. Three studies consistently reported approximately 250–300 such generally essential genes based on RNAi screen results207–210. By contrast, more recent studies using knockout approaches have reported many more essential genes (approximately 2,000), and although these approaches have been used in far fewer cell lines, the majority of the identified essential genes were common between those tested211,212. For example, Wang et al.211 screened four cell types and identified only 95 genes that were specifically essential in only one cell line. Furthermore, 31 of these genes were later found to be false positives owing to cytotoxicity caused by repeated CRISPR-mediated cleavage of an amplified genomic region.
The implication of the difference in the number of generally essential genes identified by knockdown and knockout screening methods is likely to become clear as additional cell lines are tested using knockout approaches. One possibility is that the low number of common essential genes identified by RNAi is due to variability in knockdown efficiency, which could result in inconsistent results between screens. Conversely, the ‘maximal’ phenotypes that are caused by knockout methods may result in an inability to detect quantitative differences in viability phenotypes between cell lines. For example, a gene may be identified as essential in multiple cell lines when completely ablated but may show differential effects on viability when partially inhibited. Such a gene would be detected as a universal essential gene using knockout methods but as a context-dependent essential gene using knockdown methods.
Two recent studies directly compared CRISPR and RNAi screens that were designed to identify essential genes in mammalian cells213,214. Both studies carried out side-by-side comparisons of CRISPR and RNAi screens and used sets of known essential and non-essential genes to assess the relative quality of output from each technology. Interestingly, the two studies resulted in contrasting conclusions, with one study finding that CRISPR outperformed RNAi213 and the other study finding that the two technologies performed equally well214. Possible reasons for this discrepancy could be differences in the scale of screens and the design of shRNA libraries. Although Evers et al.213 used a conventional stem–loop library with ∼five shRNAs per gene, Morgens et al.214 used an optimized shRNAmir-based library containing 25 shRNA mirs per gene153. Further work will be required to determine performance differences of available RNAi and CRISPR libraries, and to evaluate their utility and complementarity in different screening scenarios. However, one conclusion from these two studies is that reagent and experimental design can have a greater effect on screen output than the choice of technology.
Another interesting observation by Morgens et al.214 is that shRNAmir and CRISPR screens identified similar numbers of positive control genes but had a modest overlap in results overall. Combining the results from the two screens increased the recovery of known essential genes from >60% to >85%214. Although the reasons for the poor concordance between RNAi and CRISPR screens remain unclear, these findings highlight the limitations of each method and the potential value of their combined use.
The value of combining results from parallel CRISPR and RNAi screens was recently highlighted in a study investigating the mechanism of action of the antiviral drug GSK983 (REF. 215). In this study, parallel CRISPR and RNAi screens were carried out in the presence and absence of GSK983 to identify the genes that are involved in the cytotoxicity that is caused by the drug. The rationale for this approach was that RNAi knockdown of a direct target of GSK983 would behave as a hypomorph and thus would lead to a stronger phenotype in the presence of the drug (owing to the combined downreglation and inhibition of the drug target) than either the RNAi or the drug treatment alone. By contrast, a CRISPR knockout of the same target would be expected to behave as a null and thus would have a similar effect in the presence or absence of the drug. On the basis of this logic, genes that scored positively in the RNAi screen but not in the CRISPR screen were retained as candidate drug targets. In addition, some hits were detected only using CRISPR, probably because they required a near-complete knockout in order to exhibit a phenotype. Therefore, the combination of these two screening approaches resulted in a more complete representation of the underlying biology than either would alone. This elegant study illustrates the power of cross-comparison between LOF approaches in large-scale screens.
Another major application of HTS is the identification of synthetic lethal interactions216–219. A synthetic lethal interaction is defined as a genetic interaction in which the simultaneous disruption of two genes results in lethality but the disruption of either alone does not. Synthetic lethal interaction screens have a number of applications, including determining the structure of signalling networks, assigning functions to redundant network components and identifying candidate drug targets for human disease. For example, synthetic lethal interactions with specific driver mutations in cancer provide promising candidates for the development of targeted therapies that specifically kill tumour cells while having no major toxicities in normal tissues. Both RNAi and mutagenesis screens have been used to identify putative synthetic lethal interactions219. However, these two technologies are likely to identify different classes of interactions. The first class of interactions follows the classical definition, in which knockout of the candidate gene alone has no viability effect. CRISPR is more likely to identify this type of relationship because RNAi is unlikely to completely ablate protein function. A second class of synthetic lethality occurs when complete knockout of the candidate is lethal in both wild-type cells and tumour cells, but partial inhibition is only lethal in the presence of the other disruption, such as in tumour cells. These interactions are unlikely to be identified using CRISPR but can be identified using RNAi screens. In addition, two recent studies found that CRISPR screens result in considerable nonspecific anti-proliferative effects when targeting amplified genomic regions84,85. Such effects will probably limit the ability of CRISPR screens to identify differentially essential genes between cancer cell lines in which genomic amplifications are common. Notably, the complete inhibition of protein function is unlikely to be achieved clinically using a drug. Many targeted cancer therapies already used in the clinic are based on the partial inhibition of essential genes216, emphasizing the importance of identifying this second class of synthetic lethal interaction. Moreover, to obtain a more complete understanding of the genetic relationships between network components, it will probably be important to combine results from both knockout screens (or unbiased mutagenesis) and complementary knockdown approaches.
Approaches to increase confidence in LOF results
LOF methods have been applied to a wide range of biological questions. However, relatively few examples have been reported in which different LOF methods have been applied to the same question in a manner that allows for cross-comparison. This is partly historical, with newer methods replacing older methods in common practice, and is also due to the likelihood that phenotypes that do not correlate are often attributed to false discovery and are excluded from further analysis. Whereas in many cases this may be the correct interpretation, such discrepancies can be due to differences in timing, duration, strength and mechanism of LOF (FIG. 2a; TABLE 1), and the differences in phenotype between approaches may be informative of the underlying biology and/or relevant to the development of therapeutic approaches.
Figure 2. Effects of different LOF approaches and potential for compensation.
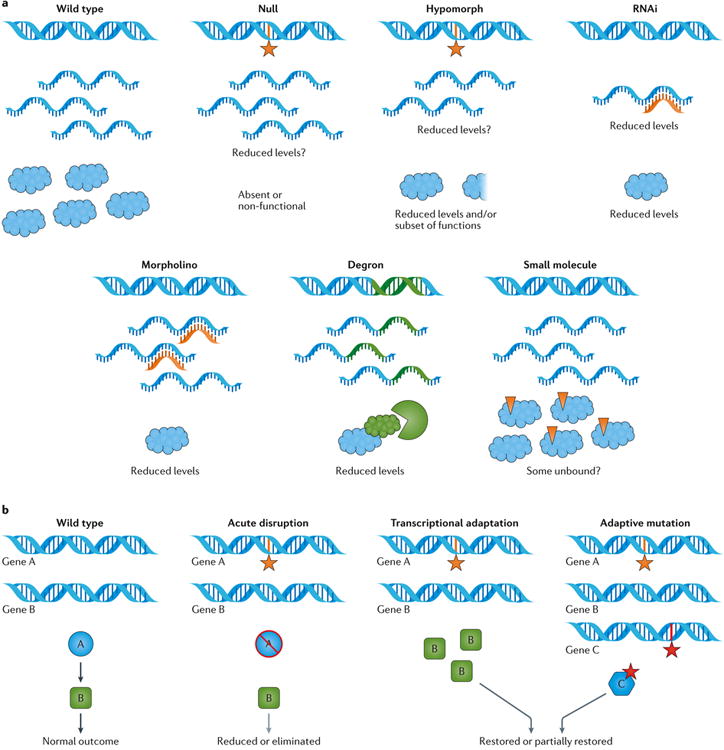
a | Different loss-of-function (LOF) reagents have different effects on RNA and proteins. Null mutations can result in no RNA, RNA of a different length (for example, shorter, as in a small deletion), or a full-length RNA with a nonsense or missense mutation, which may or may not be as stable as the wild-type transcript. In a gene deletion that results in a null mutation, the protein would be absent; however, nonsense or missense mutations can result in the production of a truncated or full-length but non-functional protein, which would also behave genetically as null. Similarly, for hypomorphs, the nature of the allele alone does not tell us the effect on RNA or protein levels, stability, or length. For some hypomorphic alleles, RNA levels are reduced; in others, the protein is wild-type in function but is reduced in abundance, has weak activity compared with wild-type, or has only a subset of the full set of wild-type activities (for example, when one but not all functional domains are disrupted or when a key residue of a protein with both catalytic and structural roles is disrupted). With effective RNA interference (RNAi) reagents, RNA sequences are wild-type but levels are reduced, leading to reduced levels of the protein. With morpholinos, RNA is present but translation or splicing is blocked, leading to reduced levels of the protein. For RNAi and morpholinos, the effectiveness of the reagent (for example, percentage knockdown), as well as the initial abundance and/or the half-life of the protein can affect the protein levels and thus, the timing and severity of a LOF phenotype. With a degron approach, in which proteins are targeted for proteolysis, RNA is present and protein levels are reduced. With a small-molecule approach, protein levels are typically not affected. In this case, some reduced level or subset of wild-type activities could result, for example, from a structural contribution made by protein bound to a small molecule that only affects a catalytic domain, and/or by a population of unbound protein. b | Compensation following acute or long-term LOF disruption. With acute disruption of gene A, the pathway, complex or other activity in which protein A participates is disrupted, leading to the reduction or elimination of the outcome. When cells have time to adapt to a disruption, transcriptional changes such as the upregulation of positively acting factors or the downregulation of negatively acting factors might partially or fully restore the outcome. In addition, cells might accumulate one or more mutations in genes controlling the same activity (for example, activating mutations in positive regulators or inactivating mutations in negative regulators), and/or mutations in alternative pathways or activities that also affect the outcome, leading to partial or full restoration of the outcome.
The selection of the most appropriate LOF method will depend on both the biological question to be answered and the gene under investigation. However, there are some common experimental themes to guide the selection of appropriate methods (BOX 3). In practice, the choice of LOF method is often based on practical limitations rather than on biological factors. For example, reagents for one LOF approach may already be available from stock centres but others would need to be generated to enable the use of a more appropriate approach. Alternatively, inherent limitations of an experimental system may limit choice. For example, the delivery of SMIs in vivo can be problematic owing to solubility issues. Additionally, when studying an essential gene, knockdown or inducible knockout reagents are required, which may prevent the use of more focused tools such as point mutations to perturb a specific aspect of protein function. In cases for which an ideal LOF method is unavailable or not possible, results should be interpreted with the chosen method in mind. For example, the lack of a phenotype with a knockout may be attributable to genetic compensation or secondary mutations (FIG. 2b). Furthermore, factors such as the type of genetic mutant used (that is, whether it is a complete null, a hypomorph or other (BOX 1)), the nature of any residual protein (FIG. 2a) and whether all or a subset of gene functions are likely to be disrupted should be considered in the interpretation and comparison of results.
Box 3. Considerations for the selection of LOF approaches for specific applications.
Differences in the outcome of loss-of-function (LOF) approaches are likely to vary on a gene-by-gene and a question-by-question basis (see the table). However, there are some common principles to help with the selection of the most appropriate method. For example, the selection of a LOF method may depend on the level of functional gene annotation available. For uncharacterized genes, there is little basis upon which to select the most appropriate LOF method. In this case, a knockout approach is likely to be the most appropriate as this provides the greatest chance of effectively disrupting all gene functions and in most cases results in the strongest possible phenotype, facilitating the initial characterization of the gene.
For cases in which a gene is partially characterized, more focused questions can be asked using LOF approaches with more specific properties. For example, to answer the question ‘what is the function of the kinase activity of gene X?’, pharmacological inhibition or a precise point mutation in the kinase domain of the protein may be most appropriate. Additionally, for cases in which quantitative differences in phenotypes must be assessed (that is, for genetic interactions), a knockdown approach may be appropriate because the ‘maximal’ phenotype caused by knockout could prevent accurate comparison (see the synthetic lethality and GSK983 examples in the main text). By contrast, pleiotropic genes may have a different dose dependence for each individual function and so, in this case, knockout may lead to more consistent effects than knockdown, which would result in differing phenotypes depending on the level of residual protein.
In addition, differences between LOF approaches may vary depending on the time between the LOF intervention and the phenotypic observation. For example, mutagenic methods such as CRISPR that target DNA may not reduce protein activity as quickly as pharmacological methods, and may therefore result in weaker phenotypes over short timescales. Over longer timescales, however, a CRISPR knockout could yield a stronger phenotype than pharmacological inhibition, which may only partially inactivate protein activity. At even later time points, genetic or transcriptional adaptation may suppress the phenotype of the knockout mutation, again leading to a weaker phenotype.
Finally, homeostatic regulation of a target gene may compensate for LOF at different levels. For example, large amounts of inactive CDK9 bound by 7SK small nuclear ribonucleoprotein (snRNP) form a cellular reservoir, from which it is released when CDK9 kinase activity is blocked by a small-molecule inhibitor (SMI). This results in a compensatory induction of crucial target genes such as MYC. Conversely, knockdown of CDK9 or overexpression of a dominant-negative mutant form do not induce compensatory MYC expression and can even counteract the effects of the SMI235.
| Application | LOF approach | Comments |
|---|---|---|
| General characterization of a gene with unknown function | Multiple approaches | Genetic knockout (for example, deletion of the open reading frame) will disrupt all gene functions and is most likely to provide the strongest phenotype |
| Study of essential genes | Knockdown or inducible knockout | Methods to perturb a subset of protein functions may also be useful (for example, SMIs) |
| Genes for which low levels of protein are sufficient to maintain function (for example, non-rate-limiting metabolic enzymes) | Knockout methods | Knockdown approaches result in residual wild-type protein and may not cause a phenotype in this case |
| Modelling drug action | Specific alleles or acute knockdown methods | Drugs may inhibit a subset of protein functions, requiring the generation of specific alleles to mimic the effect. Alternatively, all protein functions may be reduced similar to knockdown methods |
| Modelling disease-associated mutations | Specific alleles or non-coding mutations | Exact disease-associated mutations can be generated using genome-editing technologies |
Altogether, different LOF methods are better suited to different applications or goals, and in many cases, studies would benefit from the application of more than one approach to the same question. Moreover, questions should be formulated and results should be interpreted with the relevant mechanisms of LOF in mind. For singlegene studies, the use of multiple LOF approaches is generally feasible. However, for time-consuming and expensive high-throughput studies, such as the identification of drug targets by synthetic lethal screening, using multiple parallel approaches is not usually viable, with the possible exception of parallel, pooled screening with both CRISPR and RNAi libraries. In addition to the use of multiple LOF approaches, there is often value in carrying out LOF experiments across organisms. Conserved hits that are identified based on LOF approaches applied to more than one system can be considered with higher confidence220,221, and we predict that hits that are identified in cross-species studies will validate at higher rates across mammalian systems.
Concluding remarks and future directions
In conclusion, although on the surface LOF approaches seem straightforward, in practice they are complex, and the simple interpretation of the resulting phenotypes in the absence of robust approaches and detailed analyses can lead to false discovery and inaccurate conclusions. Therefore, great care must be taken when selecting the most appropriate LOF approach or approaches for any given application and organism, and careful comparison between LOF approaches is likely to be essential for accurately annotating gene function.
The tools and resources currently available for LOF studies in the worm, fly, fish and mammalian systems, although impressive, are set for major expansions in the next few years. In all systems, systematic efforts using CRISPR technologies are underway to mutate large numbers of genes. In addition, genome engineering will facilitate the generation of temperature-sensitive alleles (BOX 2). Despite the value of these approaches, it is important to keep in mind the power of unbiased genetic dissection of biological processes through random mutagenesis, such as using chemical mutagens. The ability to screen millions of chemically mutagenized genomes means that even mutations in non-annotated genes, non-coding genes, small genes and even regulatory sequences can be recovered222–224. New mutagens that yield a different spectrum of mutations promise to expand the range of mutable targets even further225. A remaining challenge, however, is the identification of the causative allele against a sea of mutations in a heavily mutagenized genome. Whole-genome mapping and sequencing techniques have reduced this burden, but mapping still often fails to narrow intervals down to single candidate genes226. Genome editing offers a solution, as candidate mutations can be recreated in a standardized genetic background and tested for the phenotype of interest. Recent advances in mammalian cells are already improving the efficiency of introducing single-base changes that precisely target one or both alleles227,228. Moreover, as the ease and efficiency of Cas9 combined with HDR is increased in animals76,229, genome editing may eventually replace genetic crosses to create in vivo models with multiple targeted genetic changes. The marriage of unbiased genetic screening and precision genome editing promises to herald a new era of genetic discovery.
Acknowledgments
This work was supported by the US National Institutes of Health (NIH) R01HD37047 (G.S.) and 5P01CA120964 (N.P.). Research in D.Y.R.S.' laboratory is funded in part by the DFG, EU and the Max Planck Society. Research in J.Z.'s laboratory is funded by ERC Starting Grant 336860, SFB grant F4710 of the Austrian Science Fund (FWF) and generous institutional support from Boehringer Ingelheim. M.M. is a recipient of a DOC Fellowship of the Austrian Academy of Sciences. S.E.M. is supported in part by NIH NIGMS R01 GM067761 (N.P., PI) and by NCI Cancer Center Support Grant NIH 5 P30 CA06516 (E. Benz, PI). C.A.G. and M.G. are supported in part by NIH DP2OD008586, R01DA036865, R21DA041878, and R21AR065956. N.P and G.S. are HHMI Investigators. The authors apologize to the authors of the many relevant papers that they were not able to cite owing to length limitations.
Glossary
- Knockout
A genetic perturbation that completely ablates gene function
- Knockdown
A perturbation at the DNA, RNA or protein level that reduces the amount of functional protein
- Pleiotropic
A gene that has roles in more than one stage, tissue or process
- Off-targets
Disruptive effects on gene function at unintended targets
- Enhancer-trap mutants
An exogenous DNA cassette or construct that is used to provide a minimal promoter and reporter such as Gal4 or GFP that, when inserted near a DNA element (such as an enhancer), becomes expressed under the control of that element. It is distinct from a gene trap in that an enhancer trap is not necessarily inserted within a gene
- Gene-trap mutants
An exogenous DNA cassette or construct that is used to provide a reporter such as Gal4 or GFP (for example, as an artificial exon) that, when inserted into a gene (for example, into an intron separating coding exons), becomes expressed under the control of the endogenous promoter of that gene
- Knock-ins
ntroduction of a specific exogenous sequence — such as a reporter, selectable marker or engineered mutation — into a specific genomic region, typically a gene region
- Arrayed format
A high-throughput screen format in which each reagent or set of gene-specific reagents (such as a small interfering RNA (siRNA) ‘mini-pool’) is in a separate well of a micro-well format plate. Positive results of a cell-based assay carried out in an arrayed format can be matched back with reagents (and thus the targeted gene) using a look-up table
- Pooled format
A high-throughput screen format in which gene-specific reagents are introduced into cultured cells en masse and at random, such that the identity of the reagent introduced into any given cell is not known. Positive results of a cell-based assay in a pooled format are typically identified through the sequencing of the starting and final reagent population, for example, reagent sequences extracted from cells at the start of a selection versus sequences remaining following a selection
- Morphants
Organisms that have been treated with a morpholino
Footnotes
Competing interests statement: The authors declare competing interests: see Web version for details.
References
- 1.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 3.Gans M, Audit C, Masson M. Isolation and characterization of sex-linked female-sterile mutants in Drosophila melanogaster. Genetics. 1975;81:683–704. doi: 10.1093/genetics/81.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haffter P, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Driever W, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. References 4 and 5 are leading papers that describe the first large-scale ENU screens conducted in zebrafish. [DOI] [PubMed] [Google Scholar]
- 6.Nolan PM, et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- 7.Kasarskis A, Manova K, Anderson KV. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci USA. 1998;95:7485–7490. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohr SE, Smith JA, Shamu CE, Neumüller RA, Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartenian E, Doench JG. Genetic screens and functional genomics using CRISPR/Cas9 technology. FEBS J. 2015;282:1383–1393. doi: 10.1111/febs.13248. [DOI] [PubMed] [Google Scholar]
- 10.Horn T, et al. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods. 2011;8:341–346. doi: 10.1038/nmeth.1581. Describes a large-scale dual-RNAi approach to genetic interaction mapping (>70,000 pairwise combinations) [DOI] [PubMed] [Google Scholar]
- 11.Bassik MC, et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152:909–922. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teng X, et al. Genome-wide consequences of deleting any single gene. Mol Cell. 2013;52:485–494. doi: 10.1016/j.molcel.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 2006;443:359–363. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni MM, et al. Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat Methods. 2006;3:833–838. doi: 10.1038/nmeth935. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Kriz AJ, Sharp PA. Target specificity of the CRISPR-Cas9 system. Quant Biol. 2014;2:59–70. doi: 10.1007/s40484-014-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng R, Lin G, Li J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2015;283:1218–1231. doi: 10.1111/febs.13586. [DOI] [PubMed] [Google Scholar]
- 17.Hendel A, Fine EJ, Bao G, Porteus MH. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015;33:132–140. doi: 10.1016/j.tibtech.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR–Cas9 nucleases. Nat Rev Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson P. Mutagenesis. Methods Cell Biol. 1995;48:31–58. [PubMed] [Google Scholar]
- 20.Corsi AK, Wightman B, Chalfie MA. Transparent window into biology: a primer on Caenorhabditis elegans. Genetics. 2015;200:387–407. doi: 10.1534/genetics.115.176099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.St Johnston D. The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet. 2002;3:176–188. doi: 10.1038/nrg751. Seminal review that describes successful approaches to genetic screening in D. melanogaster and key elements of the ‘genetic toolbox’ for flies. [DOI] [PubMed] [Google Scholar]
- 22.Venken KJT, Bellen HJ. Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods. 2014;68:15–28. doi: 10.1016/j.ymeth.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin SW, et al. A transgene-assisted genetic screen identifies essential regulators of vascular development in vertebrate embryos. Dev Biol. 2007;307:29–42. doi: 10.1016/j.ydbio.2007.03.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furutani-Seiki M, et al. A systematic genome-wide screen for mutations affecting organogenesis in Medaka, Oryzias latipes. Mech Dev. 2004;121:647–658. doi: 10.1016/j.mod.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520–524. doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutter H, Moerman D. Big Data in Caenorhabditis elegans: quo vadis? Mol Biol Cell. 2015;26:3909–3914. doi: 10.1091/mbc.E15-05-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kettleborough RNW, et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496:494–497. doi: 10.1038/nature11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan L, et al. Rapid identification and recovery of ENU-induced mutations with next-generation sequencing and Paired-End Low-Error analysis. BMC Genomics. 2015;16:83. doi: 10.1186/s12864-015-1263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarin S, et al. Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing. Genetics. 2010;185:417–430. doi: 10.1534/genetics.110.116319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miskey C, Izsvák Z, Kawakami K, Ivics Z. DNA transposons in vertebrate functional genomics. Cell Mol Life Sci. 2005;62:629–641. doi: 10.1007/s00018-004-4232-7. [DOI] [PubMed] [Google Scholar]
- 31.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 32.Greenwald I. lin-12, a nematode homeotic gene, is homologous to a set of mammalian proteins that includes epidermal growth factor. Cell. 1985;43:583–590. doi: 10.1016/0092-8674(85)90230-2. [DOI] [PubMed] [Google Scholar]
- 33.Spradling AC, Rubin GM. Transposition of cloned P elements into Drosophila germ line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- 34.Moerman DG, Benian GM, Waterston RH. Molecular cloning of the muscle gene unc-22 in Caenorhabditis elegans by Tc1 transposon tagging. Proc Natl Acad Sci USA. 1986;83:2579–2583. doi: 10.1073/pnas.83.8.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vallin E, et al. A genome-wide collection of Mos1 transposon insertion mutants for the C. elegans research community. PLoS ONE. 2012;7:e30482. doi: 10.1371/journal.pone.0030482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hacker U, Nystedt S, Barmchi MP, Horn C, Wimmer EA. piggyBac-based insertional mutagenesis in the presence of stably integrated P elements in Drosophila. Proc Natl Acad Sci USA. 2003;100:7720–7725. doi: 10.1073/pnas.1230526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hummel T, Klämbt C. P-Element mutagenesis. Methods Mol Biol. 2008;420:97–117. doi: 10.1007/978-1-59745-583-1_6. [DOI] [PubMed] [Google Scholar]
- 38.Ikeda R, et al. Sleeping beauty transposase has an affinity for heterochromatin conformation. Mol Cell Biol. 2007;27:1665–1676. doi: 10.1128/MCB.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venken KJT, et al. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods. 2011;8:737–743. doi: 10.1038/nmeth.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amsterdam A, et al. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golling G, et al. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet. 2002;31:135–140. doi: 10.1038/ng896. [DOI] [PubMed] [Google Scholar]
- 42.Clark KJ, et al. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat Methods. 2011;8:506–515. doi: 10.1038/nmeth.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trinh LA, et al. A versatile gene trap to visualize and interrogate the function of the vertebrate proteome. Genes Dev. 2011;25:2306–2320. doi: 10.1101/gad.174037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varshney GK, et al. A large-scale zebrafish gene knockout resource for the genome-wide study of gene function. Genome Res. 2013;23:727–735. doi: 10.1101/gr.151464.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quach HNB, et al. A multifunctional mutagenesis system for analysis of gene function in zebrafish. G3 (Bethesda) 2015;5:1283–1299. doi: 10.1534/g3.114.015842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zambrowicz BP, et al. Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- 47.Skarnes WC, et al. A public gene trap resource for mouse functional genomics. Nat Genet. 2004;36:543–544. doi: 10.1038/ng0604-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo G, Wang W, Bradley A. Mismatch repair genes identified using genetic screens in Blm- deficient embryonic stem cells. Nature. 2004;429:891–895. doi: 10.1038/nature02653. [DOI] [PubMed] [Google Scholar]
- 49.Carette JE, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. This study introduced genome-scale gene-trap mutagenesis screening to mammalian genetics by exploiting a near-haploid human cancer cell line. [DOI] [PubMed] [Google Scholar]
- 50.Leeb M, Wutz A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011;479:131–134. doi: 10.1038/nature10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elling U, et al. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Stem Cell. 2011;9:563–574. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bürckstümmer T, et al. A reversible gene trap collection empowers haploid genetics in human cells. Nat Methods. 2013;10:965–971. doi: 10.1038/nmeth.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilton IB, Gersbach CA. Enabling functional genomics with genome engineering. Genome Res. 2015;25:1442–1455. doi: 10.1101/gr.190124.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee HJ, Kim E, Kim JS. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Essletzbichler P, et al. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014;24:2059–2065. doi: 10.1101/gr.177220.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jinek M, et al. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. References 57–59 were the first demonstration of CRISPR–Cas9 repurposed for genome editing in eukaryotic cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang WY, et al. Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedland AE, et al. Heritable genome editing in C. elegans via a CRISPR–Cas9 system. Nat Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gratz SJ, et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang H, et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang N, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465–472. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–1034. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bassett AR, Tibbit C, Ponting CP, Liu JL. Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open. 2014;3:42–49. doi: 10.1242/bio.20137120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen Z, et al. Conditional knockouts generated by engineered CRISPR–Cas9 endonuclease reveal the roles of Coronin in C. elegans neural development. Dev Cell. 2014;30:625–636. doi: 10.1016/j.devcel.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 69.Li W, Yi P, Ou G. Somatic CRISPR–Cas9-induced mutations reveal roles of embryonically essential dynein chains in Caenorhabditis elegans cilia. J Cell Biol. 2015;208:683–692. doi: 10.1083/jcb.201411041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tian D, et al. Anillin regulates neuronal migration and neurite growth by linking RhoG to the actin cytoskeleton. Curr Biol. 2015;25:1135–1145. doi: 10.1016/j.cub.2015.02.072. [DOI] [PubMed] [Google Scholar]
- 71.Xue Z, et al. CRISPR/Cas9 mediates efficient conditional mutagenesis in Drosophila. G3 (Bethesda) 2014;4:2167–2173. doi: 10.1534/g3.114.014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dow LE, et al. Inducible in vivo genome editing with CRISPR–Cas9. Nat Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ablain J, Durand EM, Yang S, Zhou Y, Zon LIA. CRISPR/Cas9 vector system for tissue--specific gene disruption in zebrafish. Dev Cell. 2015;32:756–764. doi: 10.1016/j.devcel.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arribere JA, et al. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014;198:837–846. doi: 10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paix A, et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics. 2014;198:1347–1356. doi: 10.1534/genetics.114.170423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paix A, Folkmann A, Rasoloson D, Seydoux G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR–Cas9 ribonucleoprotein complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brocal I, et al. Efficient identification of CRISPR/Cas9-induced insertions/deletions by direct germline screening in zebrafish. BMC Genomics. 2016;17:259. doi: 10.1186/s12864-016-2563-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parnas O, et al. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell. 2015;162:675–686. doi: 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. This paper first demonstrated the broad utility of CRISPR–Cas9 targeting transcriptional regulators to specific genomic sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods. 2016;13:127–137. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shalem O, et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR–Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. References 81 and 82 are the first examples of the use of pooled gRNA libraries and the CRISPR–Cas9 platform to carry out whole genome knockout screens in human cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang H, et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 2012;149:605–617. doi: 10.1016/j.cell.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 84.Aguirre AJ, et al. Genomic copy number dictates a gene-independent cell response to CRISPR–Cas9 targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Munoz DM, et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 2016;6:900–913. doi: 10.1158/2159-8290.CD-16-0178. [DOI] [PubMed] [Google Scholar]
- 86.Platt RJ, et al. CRISPR–Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li W, Ou G. The application of somatic CRISPR–Cas9 to conditional genome editing in Caenorhabditis elegans. Genesis. 2016;54:170–181. doi: 10.1002/dvg.22932. [DOI] [PubMed] [Google Scholar]
- 88.Braun CJ, et al. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc Natl Acad Sci USA. 2016;113:E3892–E3900. doi: 10.1073/pnas.1600582113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen S, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burger A, et al. Maximizing mutagenesis with solubilized CRISPR–Cas9 ribonucleoprotein complexes. Development. 2016;143:2025–2037. doi: 10.1242/dev.134809. [DOI] [PubMed] [Google Scholar]
- 91.Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 2016;18:246–254. doi: 10.1038/ncb3312. [DOI] [PubMed] [Google Scholar]
- 92.O'Rourke SM, et al. A survey of new temperature-sensitive, embryonic-lethal mutations in C elegans: 24 alleles of thirteen genes. PLoS ONE. 2011;6:e16644. doi: 10.1371/journal.pone.0016644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davies T, et al. High-resolution temporal analysis reveals a functional timeline for the molecular regulation of cytokinesis. Dev Cell. 2014;30:209–223. doi: 10.1016/j.devcel.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Golic KG, Lindquist S. The FLP recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell. 1989;59:499–509. doi: 10.1016/0092-8674(89)90033-0. [DOI] [PubMed] [Google Scholar]
- 95.Lewandoski M. Conditional control of gene expression in the mouse. Nat Rev Genet. 2001;2:743–755. doi: 10.1038/35093537. [DOI] [PubMed] [Google Scholar]
- 96.Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6861–6865. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Duffy JB, Harrison DA, Perrimon N. Identifying loci required for follicular patterning using directed mosaics. Development. 1998;125:2263–2271. doi: 10.1242/dev.125.12.2263. [DOI] [PubMed] [Google Scholar]
- 99.Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development. 1995;121:1053–1063. doi: 10.1242/dev.121.4.1053. [DOI] [PubMed] [Google Scholar]
- 100.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 101.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 102.Koo BK, et al. Rapid, one-step generation of biallelic conditional gene knockouts. 2016 Preprint at bioRxiv http://dx.doi.org/10.1101/056549.
- 103.Robles-Oteiza C, et al. Recombinase-based conditional and reversible gene regulation via XTR alleles. Nat Commun. 2015;6:8783. doi: 10.1038/ncomms9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR–Cas9-based transcription factors. Nat Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chavez A, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RDA. Protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chavez A, et al. Comparison of Cas9 activators in multiple species. Nat Methods. 2016;13:563–567. doi: 10.1038/nmeth.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vojta A, et al. Repurposing the CRISPR–Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hilton IB, et al. Epigenome editing by a CRISPR–Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kearns NA, et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015;12:401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thakore PI, et al. Highly specific epigenome editing by CRISPR–Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lin S, Ewen-Campen B, Ni X, Housden BE, Perrimon N. In vivo transcriptional activation using CRISPR/Cas9 in Drosophila. Genetics. 2015;201:433–442. doi: 10.1534/genetics.115.181065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ghosh S, Tibbit C, Liu JL. Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic Acids Res. 2016;44:e84. doi: 10.1093/nar/gkw063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Long L, et al. Regulation of transcriptionally active genes via the catalytically inactive Cas9 in Celegans and D rerio. Cell Res. 2015;25:638–641. doi: 10.1038/cr.2015.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rossi A, et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. Reports that for two genes, zebrafish mutants and morphants differ in their adaptive transcriptional response. [DOI] [PubMed] [Google Scholar]
- 118.Gilbert LA, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR–Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. Seminal paper describing the first evidence that RNAi could function at substoichiometric levels. [DOI] [PubMed] [Google Scholar]
- 121.Alló M, et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol. 2009;16:717–724. doi: 10.1038/nsmb.1620. [DOI] [PubMed] [Google Scholar]
- 122.Conte D, MacNeil LT, Walhout AJM, Mello CC. RNA Interference in Caenorhabditis elegans. Curr Protoc Mol Biol. 2015;109:26.3.1–26.3.30. doi: 10.1002/0471142727.mb2603s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grishok A, Tabara H, Mello CC. Genetic requirements for inheritance of RNAi in C. elegans. Science. 2000;287:2494–2497. doi: 10.1126/science.287.5462.2494. [DOI] [PubMed] [Google Scholar]
- 124.Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. First description of gene knockdown upon feeding dsRNA-expressing E. coli to C. elegans. [DOI] [PubMed] [Google Scholar]
- 125.Jose AM, Hunter CP. Transport of sequence- specific RNA interference information between cells. Annu Rev Genet. 2007;41:305–330. doi: 10.1146/annurev.genet.41.110306.130216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tavernarakis N, Wang SL, Dorovkov M, Ryazanov A, Driscoll M. Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat Genet. 2000;24:180–183. doi: 10.1038/72850. [DOI] [PubMed] [Google Scholar]
- 127.Sugimoto A. High-throughput RNAi in Caenorhabditis elegans: genome-wide screens and functional genomics. Differentiation. 2004;72:81–91. doi: 10.1111/j.1432-0436.2004.07202004.x. [DOI] [PubMed] [Google Scholar]
- 128.Piccin A, et al. Efficient and heritable functional knock-out of an adult phenotype in Drosophila using a GAL4-driven hairpin RNA incorporating a heterologous spacer. Nucleic Acids Res. 2001;29:E55–E55. doi: 10.1093/nar/29.12.e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Giordano E, Rendina R, Peluso I, Furia M. RNAi triggered by symmetrically transcribed transgenes in Drosophila melanogaster. Genetics. 2002;160:637–648. doi: 10.1093/genetics/160.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dietzl G, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. Demonstrates the first example of tissue-specific RNAi in D. melanogaster enabling the genome-wide interrogation of gene functions. [DOI] [PubMed] [Google Scholar]
- 131.Perrimon N, Ni JQ, Perkins L. In vivo RNAi: today and tomorrow. Cold Spring Harb Perspect Biol. 2010;2:a003640. doi: 10.1101/cshperspect.a003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dow LE, et al. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell. 2015;161:1539–1552. doi: 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mohr SE. RNAi screening in Drosophila cells and in vivo. Methods. 2014;68:82–88. doi: 10.1016/j.ymeth.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 135.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 137.Berns K, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. This study reports the first targeted LOF screen in mammalian cells using shRNAs. [DOI] [PubMed] [Google Scholar]
- 138.Paddison PJ, et al. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 139.Gu S, et al. The loop position of shRNAs and pre-mi RNAs is critical for the accuracy of Dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McBride JL, et al. Artificial mi RNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 142.Zeng Y, Wagner EJ, Cullen BR, Carolina N. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- 143.Chung KH, et al. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006;34:e53. doi: 10.1093/nar/gkl143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hinterberger M, et al. Autonomous role of medullary thymic epithelial cells in central CD4+ T cell tolerance. Nat Immunol. 2010;11:512–519. doi: 10.1038/ni.1874. [DOI] [PubMed] [Google Scholar]
- 145.Dickins RA, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- 146.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zuber J, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dickins RA, et al. Tissue-specific and reversible RNA interference in transgenic mice. Nat Genet. 2007;39:914–921. doi: 10.1038/ng2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Premsrirut PK, et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145:145–158. doi: 10.1016/j.cell.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McJunkin K, et al. Reversible suppression of an essential gene in adult mice using transgenic RNA interference. Proc Natl Acad Sci USA. 2011;108:7113–7118. doi: 10.1073/pnas.1104097108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fellmann C, et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. 2011;41:733–746. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013;152:844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kampmann M, et al. Next-generation libraries for robust RNA interference-based genome-wide screens. Proc Natl Acad Sci USA. 2015;112:E3384–E3391. doi: 10.1073/pnas.1508821112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fellmann C, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 155.Fellmann C, Lowe SW. Stable RNA interference rules for silencing. Nat Cell Biol. 2014;16:10–18. doi: 10.1038/ncb2895. This review summarizes the development of RNAi reagents for mammalian genetics from first siRNA-mediated knockdowns to miRNA-based RNAi triggers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kelly A, Hurlstone AF. The use of RNAi technologies for gene knockdown in zebrafish. Brief Funct Genomics. 2011;10:189–196. doi: 10.1093/bfgp/elr014. [DOI] [PubMed] [Google Scholar]
- 157.Summerton JE. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007;7:651–660. doi: 10.2174/156802607780487740. [DOI] [PubMed] [Google Scholar]
- 158.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. Introduces the morpholino technology in zebrafish. [DOI] [PubMed] [Google Scholar]
- 159.Law SHW, Sargent TD. The serine–threonine protein kinase PAK4 is dispensable in zebrafish: identification of a morpholino-generated pseudophenotype. PLoS ONE. 2014;9:e100268. doi: 10.1371/journal.pone.0100268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kok FO, et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev Cell. 2015;32:97–108. doi: 10.1016/j.devcel.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Novodvorsky P, et al. klf2ash317 mutant zebrafish do not recapitulate morpholino-induced vascular and haematopoietic phenotypes. PLoS ONE. 2015;10:e0141611. doi: 10.1371/journal.pone.0141611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Blum M, De Robertis EM, Wallingford JB, Niehrs C. Morpholinos: antisense and sensibility. Dev Cell. 2015;35:145–149. doi: 10.1016/j.devcel.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 163.Lawson ND. Reverse genetics in zebrafish: mutants, morphants, and moving forward. Trends Cell Biol. 2016;26:77–79. doi: 10.1016/j.tcb.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 164.Schulte-Merker S, Stainier DYR. Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology. Development. 2014;141:3103–3104. doi: 10.1242/dev.112003. [DOI] [PubMed] [Google Scholar]
- 165.Stainier DYR, Kontarakis Z, Rossi A. Making sense of anti-sense data. Dev Cell. 2015;32:7–8. doi: 10.1016/j.devcel.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 166.Lim S, et al. A simple strategy for heritable chromosomal deletions in zebrafish via the combinatorial action of targeting nucleases. Genome Biol. 2013;14:R69. doi: 10.1186/gb-2013-14-7-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Arrowsmith CH, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bonger KM, Chen L, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat Chem Biol. 2011;7:531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Schneekloth JS, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 172.Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol. 2012;19:117–121. doi: 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- 173.Shin YJ, et al. Nanobody-targeted E3-ubiquitin ligase complex degrades nuclear proteins. Sci Rep. 2015;5:14269. doi: 10.1038/srep14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hermann A, Liewald JF, Gottschalk A. A photosensitive degron enables acute light-induced protein degradation in the nervous system. Curr Biol. 2015;25:R749–R750. doi: 10.1016/j.cub.2015.07.040. [DOI] [PubMed] [Google Scholar]
- 175.Zhang L, Ward JD, Cheng Z, Dernburg AF. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development. 2015;142:4374–4384. doi: 10.1242/dev.129635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Armenti ST, Lohmer LL, Sherwood DR, Nance J. Repurposing an endogenous degradation system for rapid and targeted depletion of C. elegansproteins. Development. 2014;141:4640–4647. doi: 10.1242/dev.115048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rakhit R, Navarro R, Wandless TJ. Chemical biology strategies for posttranslational control of protein function. Chem Biol. 2014;21:1238–1252. doi: 10.1016/j.chembiol.2014.08.011. This review provides a comprehensive overview of chemical-genetic tools. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nagarkar-Jaiswal S, et al. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. eLife. 2015;4:e05338. doi: 10.7554/eLife.05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lowe N, et al. Analysis of the expression patterns, subcellular localisations and interaction partners of Drosophila proteins using a pigP protein trap library. Development. 2014;141:3994–4005. doi: 10.1242/dev.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci USA. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sarov M, et al. A genome-wide resource for the analysis of protein localisation in Drosophila. eLife. 2016;5:e12068. doi: 10.7554/eLife.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Holland AJ, Fachinetti D, Han JS, Cleveland DW. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci USA. 2012;109:E3350–E3357. doi: 10.1073/pnas.1216880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 184.Natsume T, Kiyomitsu T, Saga Y, Kanemaki MT. Rapid protein depletion in human cells by auxin- inducible degron tagging with short homology donors. Cell Rep. 2016;15:210–218. doi: 10.1016/j.celrep.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 185.Trost M, Blattner AC, Lehner CF. Regulated protein depletion by the auxin-inducible degradation system in Drosophila melanogaster. Fly (Austin) 2016;10:35–46. doi: 10.1080/19336934.2016.1168552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yeh JTH, Binari R, Gocha T, Dasgupta R, Perrimon N. PAPTi: a peptide aptamer interference toolkit for perturbation of protein–protein interaction networks. Sci Rep. 2013;3:1156. doi: 10.1038/srep01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jost APT, Weiner OD. Probing yeast polarity with acute, reversible, optogenetic inhibition of protein function. ACS Synth Biol. 2015;4:1077–1085. doi: 10.1021/acssynbio.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Daude N, et al. Knockout of the prion protein (PrP)- like Sprn gene does not produce embryonic lethality in combination with PrPC-deficiency. Proc Natl Acad Sci USA. 2012;109:9035–9040. doi: 10.1073/pnas.1202130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.De Souza AT, et al. Transcriptional and phenotypic comparisons of Ppara knockout and siRNA knockdown mice. Nucleic Acids Res. 2006;34:4486–4494. doi: 10.1093/nar/gkl609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gao Y, et al. Auxin binding protein 1 (ABP1) is not required for either auxin signaling or Arabidopsis development. Proc Natl Acad Sci USA. 2015;112:2275–2280. doi: 10.1073/pnas.1500365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 192.Janssen HLA, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 193.Hsu SH, et al. Essential metabolic, anti- inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122:2871–2883. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tsai WC, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–2897. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–353. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 196.Weiss WA, Taylor SS, Shokat KM. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat Chem Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene. 2005;24:5005–5015. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- 198.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 199.Ditchfield C, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–936. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 201.Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore- microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 203.Girdler F, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006;119:3664–3675. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]
- 204.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 205.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR–Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhan T, Boutros M. Towards a compendium of essential genes — from model organisms to synthetic lethality in cancer cells. Crit Rev Biochem Mol Biol. 51:74–85. doi: 10.3109/10409238.2015.1117053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Marcotte R, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci USA. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Luo B, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cowley GS, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang T, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Blomen VA, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 213.Evers B, et al. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol. 2016;34:631–633. doi: 10.1038/nbt.3536. [DOI] [PubMed] [Google Scholar]
- 214.Morgens DW, Deans RM, Li A, Bassik MC. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol. 2016;34:634–636. doi: 10.1038/nbt.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Deans RM, et al. Parallel shRNA and CRISPR–Cas9 screens enable antiviral drug target identification. Nat Chem Biol. 2016;12:361–366. doi: 10.1038/nchembio.2050. This study compares state-of-the-art CRISPR and RNAi reagents in a systematic large-scale screen and demonstrates the complementarity of these two key technologies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nijman SMB. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585:1–6. doi: 10.1016/j.febslet.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 218.Westbrook TF, Stegmeier F, Elledge SJ. Dissecting cancer pathways and vulnerabilities with RNAi. Cold Spring Harb Symp Quant Biol. 2005;70:435–444. doi: 10.1101/sqb.2005.70.031. [DOI] [PubMed] [Google Scholar]
- 219.Thompson JM, Nguyen QH, Singh M, Razorenova OV. Approaches to identifying synthetic lethal interactions in cancer. Yale J Biol Med. 2015;88:145–155. [PMC free article] [PubMed] [Google Scholar]
- 220.Neumüller RA, et al. Conserved regulators of nucleolar size revealed by global phenotypic analyses. Sci Signal. 2013;6:ra70. doi: 10.1126/scisignal.2004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Housden BE, et al. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal. 2015;8:rs9. doi: 10.1126/scisignal.aab3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 223.Garbe D, Doto JB, Sundaram MV. Caenorhabditis elegans lin-35/Rb, efl-1/E2F and other synthetic multivulva genes negatively regulate the anaphase-promoting complex gene mat-3/APC8. Genetics. 2004;167:663–672. doi: 10.1534/genetics.103.026021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Doitsidou M, et al. A combinatorial regulatory signature controls terminal differentiation of the dopaminergic nervous system in C. elegans. Genes Dev. 2013;27:1391–1405. doi: 10.1101/gad.217224.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Noma K, Jin Y. Optogenetic mutagenesis in Caenorhabditis elegans. Nat Commun. 2015;6:8868. doi: 10.1038/ncomms9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zuryn S, Jarriault S. Deep sequencing strategies for mapping and identifying mutations from genetic screens. Worm. 2013;2:e25081. doi: 10.4161/worm.25081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Paquet D, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 229.Paix A, Schmidt H, Seydoux G. Cas9-assisted recombineering in C. elegans: genome editing using in vivo assembly of linear DNAs. Nucleic Acids Res. 2016;44:e128. doi: 10.1093/nar/gkw502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Muller HJ. In: Proceedings of the 6th International Congress of Genetics. Jones DF, editor. Banta: 1932. pp. 213–255. Influential and useful categorization of mutations (‘Muller's morphs’) [Google Scholar]
- 231.Simon MA, Bowtell DD, Dodson GS, Laverty TR, Rubin GM. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell. 1991;67:701–716. doi: 10.1016/0092-8674(91)90065-7. [DOI] [PubMed] [Google Scholar]
- 232.Hsu JC, Perrimon N. A temperature-sensitive MEK mutation demonstrates the conservation of the signaling pathways activated by receptor tyrosine kinases. Genes Dev. 1994;8:2176–2187. doi: 10.1101/gad.8.18.2176. [DOI] [PubMed] [Google Scholar]
- 233.Dohmen RJ, Wu P, Varshavsky A. Heat- inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- 234.Zeidler MP, et al. Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat Biotechnol. 2004;22:871–876. doi: 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- 235.Lu H, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. eLife. 2015;4:e06535. doi: 10.7554/eLife.06535. erratum 4, e09993 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Thompson O, et al. The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res. 2013;23:1749–1762. doi: 10.1101/gr.157651.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.C. elegans Deletion Mutant Consortium. Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 (Bethesda) 2012;2:1415–1425. doi: 10.1534/g3.112.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 239.Mohr SE, Hu Y, Kim K, Housden BE, Perrimon N. Resources for functional genomics studies in Drosophila melanogaster. Genetics. 2014;197:1–18. doi: 10.1534/genetics.113.154344. Comprehensive review of LOF tools for D. melanogaster. [DOI] [PMC free article] [PubMed] [Google Scholar]