

Abstract
This brief overview of premature senescence of dysfunctional endothelial and endothelial progenitor cells provides information on endothelial cell differentiation and specialization, their ontogeny, and controversies related to endothelial stem and progenitor cells. Stressors responsible for the dysfunction of endothelial and endothelial progenitor cells, as well as cellular mechanisms and consequences of endothelial cell dysfunction are presented. Metabolic signatures of dysfunctional endothelial cells and senescence pathways are described. Emerging strategies to rejuvenate endothelial and endothelial progenitor cells conclude the review.
1. INTRODUCTION
A balance between microvascular density and the surrounding tissues in the basin of vascular supply is one of the highly regulated parameters. It is kept constant to insure that the diffusional distances for oxygen supply of the cells do not exceed 500 μm. This constancy of relationship between vascular supply and tissue volume is a cornerstone of the concept developed by J. Folkman attributing tumor shrinkage to cutting the vascular endowment, but it has a much broader applicability to virtually all blood-supplied organs. Proper vascular density is most probably maintained by the long-lived endothelial cells, endothelial progenitor cells, and vascular endothelium stem cells, on the one hand, and proliferative capacity of cells within the basin of such vessels. And yet, despite this tight regulation, microvascular rarefaction is a constant companion of diverse chronic pathological states, thus leading to the loss of differentiated cells, their substitution with myofibroblasts, fibrosis, and organ failure. Understanding the causes and mechanisms of vascular drop-out in chronic diseases is paramount for reducing their deleterious consequences.
2. VASCULAR MORPHOGENESIS
Blood vessels are predominantly composed of endothelial cells that form the inner, luminal layer, and smooth muscle cells that form the surrounding vessel wall. During blood vessel development, endothelial cells are formed first, and undergo rapid expansion and coalescence into capillary plexi that are then remodeled into arterial–venous networks capable of sustaining systemic circulation. Vascular remodeling and maturation involves coordinated migration, cell cycle inhibition, and specification of endothelial subtypes (arterial, venous), as well as smooth muscle cell recruitment. Later in development, a subset of venous endothelial cells bud off to form the lymphatic vasculature.
2.1 Endothelial Cell Differentiation
The de novo emergence of primordial, unspecialized endothelial cells is referred to as vasculogenesis, which begins in the mammal within the extraembryonic yolk sac shortly after gastrulation. Herein, endothelial cells are formed from newly generated mesodermal progenitors, in response to signals from the adjacent visceral endoderm (Belaoussoff, Farrington, & Baron, 1998; Vokes & Krieg, 2002). Later stages of vasculogenesis include the formation of vascular channels and plexi that are remodeled into circulatory networks via the process of angiogenesis.
The signaling pathways that direct the differentiation of endothelial cells from mesodermal progenitors are still not entirely clear and under intense investigation. Murine gene deletion studies revealed that fibroblast growth factor 2 (FGF2 or bFGF) and bone morphogenetic protein 4 (BMP4) are not only critical for mesoderm formation, but also play an important role in endothelial cell specification there from (Marom, Levy, Pillemer, & Fainsod, 2005; Winnier, Blessing, Labosky, & Hogan, 1995; Yamaguchi, Harpal, Henkemeyer, & Rossant, 1994). Mouse embryonic stem (mES) cell differentiation studies suggest that BMP4 promotes mesoderm formation and initiates a program requiring FGF2 to promote the specification of angioblasts, or endothelial progenitors (Park et al., 2004; Pearson, Sroczynska, Lacaud, & Kouskoff, 2008).
Further commitment to an endothelial cell lineage is promoted by signals from the adjacent visceral endoderm, including Indian hedgehog (IHH), which is sufficient to induce the formation of endothelial cells in mouse embryo explants that lack endoderm (Byrd et al., 2002; Vokes & Krieg, 2002). Such effects by IHH may also be mediated by BMP4, as they are during the differentiation of endothelial cells from human ES cells (Kelly & Hirschi, 2009). Vascular endothelial growth factor (VEGF), also produced by the visceral endoderm early in vascular development, is another key regulator of vasculogenesis (Carmeliet et al., 1996; Ferrara et al., 1996; Miquerol, Langille, & Nagy, 2000). VEGF-A predominantly signals through two receptors, VEGFR1 (Flt-1) and VEGFR2 (Flk-1 or Kdr), and mice lacking Flk-1 are embryonic lethal and lack vascular plexus development, despite normal formation of angioblasts (Schuh, Faloon, Hu, Bhimani, & Choi, 1999; Shalaby et al., 1995). Consistent with this, Flk-1 −/− mES cells generate endothelial cells, but they fail to propagate in vitro (Schuh et al., 1999); thus, VEGF-A may regulate the survival and/or propagation of endothelial cells, not necessarily their differentiation.
Transcriptional regulators in the ETS family are also known to play important roles in endothelial cell development. There are ~30 mammalian ETS factors and several, such as Ets1, Erg, Fli-1, and Etv2, have been demonstrated to play important roles in vascular development (De Val & Black, 2009; Dejana, Taddei, & Randi, 2007). In fact, regulatory regions for almost all endothelial genes contain ETS binding sites, suggesting that ETS factors could potentially regulate most, if not all, endothelial-specific gene expression (De Val & Black, 2009; Dejana et al., 2007). Etv2 (ER71/etsrp), or ETS variant 2, appears to play a critical role in the differentiation of mesodermal progenitors toward an endothelial cell fate. Etv2 is initially broadly expressed within the primitive streak mesoderm, but then restricted to developing vascular endothelial cells (Kataoka et al., 2011; Lee et al., 2008; Salanga, Meadows, Myers, & Krieg, 2010). Mice deficient for Etv2 are embryonic lethal and lack a yolk sac primitive vascular plexus, dorsal aortae, and endocardium, despite normal mesoderm formation (De Val et al., 2008). Overexpression of Etv2 leads to ectopic expression of endothelial markers in multiple cell types in vivo, suggesting Etv2 is necessary and sufficient for vasculogenesis. Etv2 expression is restricted to Flk-1+ mesoderm in mES cells and its induction within embryoid bodies results in de novo production of Flk-1+ cells (De Val et al., 2008). Thus, Etv2 appears to regulate expression of VEGF receptor Flk-1, which is associated with the specification of an endothelial cell fate. Much remains to be learned about the regulation of this important process.
2.2 Endothelial Cell Specialization
Once primordial endothelial cells are formed, they must be further specialized to perform unique functions. In a properly remodeled circulatory system, the blood vasculature is subdivided into two interconnected yet structurally and functionally distinct networks of arteries and veins. Unlike veins, arteries withstand high blood pressure and are surrounded by several layers of smooth muscle cells and connective tissues. Endothelial cell identity is known to be modulated by hemodynamic flow forces (le Noble et al., 2004); however, the downstream molecular mechanisms that regulate phenotypic changes are not clear.
Nonetheless, signaling pathways that have been implicated in earlier aspects of vascular development are thought to also play significant roles in arterial–venous specification. For example, VEGF-A binds to Flk-1 and coreceptor neuropilin-1 (NP-1) (Gu et al., 2003), activating Notch signaling (Lawson et al., 2001; Lawson, Vogel, & Weinstein, 2002) and promoting EphrinB2 expression, while suppressing EphB4; thus, establishing an arterial identity while suppressing a venous fate in endothelial cells (Wang, Chen, & Anderson, 1998). Studies in mouse ES cell also demonstrate that high VEGF-A levels induce arterial endothelial cells whereas low to intermediate concentrations induce venous differentiation, and inhibition of Notch signaling results in an arterial to venous fate switch (Lanner, Sohl, & Farnebo, 2007). Wnt signaling is also involved in the specification of an arterial fate; β-catenin (β-cat), a transcriptional coactivator of Wnt signaling pathway, upregulates Notch ligand Dll4 and promotes arterial specification (Corada et al., 2010; Yamamizu et al., 2010). In addition, Hh signaling also acts upstream of VEGF-A via smoothened receptor to promote arterial endothelial cell specification and repress venous fates (Wilkinson et al., 2012; Williams et al., 2010).
Venous endothelial cell specification is thought to be mediated by COUP-TFII (chicken ovalbumin upstream promoter-transcription factor II), a member of the orphan nuclear receptor superfamily. Endothelial-specific deletion of COUP-TFII leads to arterialization of veins, whereas ectopic expression results in fusion of veins and arteries (You et al., 2005). COUP-TFII is thought to suppress NP-1 function and Notch signaling to maintain vein identity.
The lymphatic vasculature is thought to develop primarily from the venous system. It is essential for returning extravasated proteins and cells to the blood stream and for proper immune system surveillance. These roles are clearly distinct from those of blood vessels; thus, generation of lymphatic endothelial cells requires the further specification of a subpopulation of the venous endothelial cells via distinct regulatory pathways. Lymphatic endothelial cell development is thought to be induced by expression of Prospero homeodomain transcription factor (Prox1), which is expressed by the subpopulation of venous endothelial cells that sprout, migrate, and form the lymphatic system (Srinivasan et al., 2007; Wigle et al., 2002; Wigle & Oliver, 1999). Genetic ablation of Prox1 leads to a general failure of lymphatic endothelial cell specification from venous endothelium (Francois et al., 2012; Johnson et al., 2008). In addition, lymphatic endothelial cells were shown to dedifferentiate into blood vessel endothelial cells upon inactivation of Prox1 during postnatal stages (Johnson et al., 2008). Prox1 is therefore not only necessary for specification of lymphatic lineage, but also for maintenance of lymphatic endothelial cell identity. Both the SRY-related gene Sox18 and COUP-TFII, which is necessary for specifying venous endothelial cell fate, were shown to regulate Prox1 expression (Francois et al., 2008; Lee et al., 2009; Lin et al., 2010; Pennisi et al., 2000; Srinivasan et al., 2010). Sox18 is expressed by cardinal vein endothelial cells that eventually express Prox1 and form the lymphatic vasculature. Ectopic Sox18 expression in blood vessel endothelial cells induces Prox1 and lymphatic endothelial cell marker expression.
Following specification, the lymphatic endothelial cells migrate out of veins to form lymph sacs and then lymphatic vessels (Francois et al., 2012). VEGF-C signaling, via Flt4 receptor (VEGFR3), is necessary for lymphangiogenic development. Homozygous and heterozygous deletion of VEGF-C in mouse embryos results in a failure of Prox1-positive lymphatic endothelial cell progenitors to sprout (Karkkainen et al., 2004), and inhibition of Flt4 function leads to impaired development of the lymphatic system (Makinen et al., 2001). Thus, analogous to the role VEGF-A plays in primordial endothelial cell development, VEGF-C signaling appears to regulate the survival and propagation of lymphatic endothelial cells, and not necessarily their specification.
Hemogenic endothelial cells are also specialized from a small subset of the primordial endothelium within the yolk sac and AGM during embryonic development. These cells acquire hematopoietic potential and give rise to hematopoietic stem and progenitor cells, and their specification and function has recently been reviewed elsewhere (Gritz & Hirschi, 2016).
2.3 Controversial EPC Identity
Although it is not difficult to identify cells that function as endothelial progenitor cells (angioblasts, primordial endothelial cells) based on origin, location, and marker expression, or lack thereof, during embryonic development, their identification in postnatal tissues is far less clear. Cells that can be isolated from blood, bone marrow, and other adult tissues, have been shown to participate in vessel regeneration in injured tissues; however, their origin, phenotype, lineage potential and function in situ, have been heavily debated (as previously reviewed in Hirschi, Ingram, & Yoder, 2008). In addition, not all populations referred to as adult endothelial progenitor cells are the same; thus, leading to confusion and controversy in this field.
3. ONTOGENY OF ENDOTHELIAL AND ENDOTHELIAL PROGENITOR CELLS
Endothelial cell turnover is slow under physiological conditions, averaging from 2 months to 3 years (rev. in Brown, 2010). Damaged endothelial cells are eliminated as a result of patrolling function of the noninvading subset of Ly6Clow monocytes. Intravital microscopy studies showed these monocytes crawling on the luminal surface of glomerular and peritubular capillaries and scavenging microparticles (Carlin et al., 2013). In response to nucleic acid “danger” signaling, this subset of cells exhibit longer dwell times in glomerular and peritubular capillaries, attach to the damaged endothelial cells, and recruit neutrophils, which ultimately induce focal necrosis and disposal of cellular debris. Endothelial cells escaping this type of in situ elimination, detach from the basement membrane and appear in the circulation.
The number of colony-forming EPC is declining with age, as determined in healthy aged subjects (Hill et al., 2003), while EPC numbers are inversely correlated with cardiovascular risk (Hill et al., 2003; Werner et al., 2005). The role played by telomere attrition as a driver of replicative cell senescence in aging is well-established. Chen, Brodsky, and Goligorsky (2002) showed that, contrary to the replicative senescence, stress-induced premature senescence (SIPS) of endothelial cells, occurs even in the presence of relatively unaffected telomeres. A diverse group of stress signals, such as pro-oxidants, asymmetric dimethylarginine (ADMA), nonenzymatically glycation-modified proteins induce cell cycle arrest, SIPS and eventual apoptosis in low-passage cultured cells and in young mice. The fact that SIPS can be reversed after the withdrawal of the offending stressor, is of therapeutic importance; however, if the stressor persists, SIPS becomes irreversible, endothelial cells undergo apoptosis, and the process culminates in microvascular rarefaction. What accounts for this point-of-no-return remains unknown.
In aging animals, endothelium-dependent vasorelaxation and angiogenic competence are reduced compared to young ones (Reed, Karres, Eyman, & Vernon, 2007). The same is true for prematurely senescent Klotho-mice (Shimada et al., 2004). Notably, caloric restriction rescues angiogenic competence (Facchetti, Monzani, Cavallini, Bergamini, & La Porta, 2007). Intriguingly, clearance of p16Ink4a-positive senescent cells delays age-associated disorders, but does not lead to extended lifespan (Baker et al., 2011).
One of the downstream targets of caloric restriction, sirtuin-1, is robustly expressed in EPC and EC; however, its expression declines with age and following application of cardiovascular stressors (Chen, Qin, et al., 2012; Chen, Xavier, et al., 2012). Sirtuin-1 deficiency leading to premature senescence of EPC and EC occurs as a result of stress-induced loss of lysosomal membrane integrity and leakage of cathepsin B, which is capable of directly degrading this deacetylase, as shown in in vitro studies. EPC isolated from the bone marrow of mice genetically engineered to lack endothelial sirtuin-1 exhibit higher rate of premature senescence and apoptosis and lower stress tolerance even at a young age (Chen, Qin, et al., 2012; Chen, Xavier, et al., 2012).
4. iPSC TO EPC TRANSITION
The identity of EPC is rather nebulous. The confusion is explainable by the fact that several consensus markers of EPC, either bone marrow-derived, or circulating, or tissue-resident, are also expressed by circulating endothelial cells, endothelial stem cells, or cells of monocytic lineage; these include CD31, CD34, VEGF receptor-2 (Khakoo & Finkel, 2005; Richardson & Yoder, 2011). Two distinct populations of EPC exist in human blood: early and late outgrowth EPC, which are distinguishable on the basis of their proliferative capacity: 20-fold vs 1000-fold expansion, respectively. EPC, and especially the endothelial colony-forming subset, which is enriched in the cord blood (ECFC), have a distinct therapeutic potential (Alev, Li, & Asahara, 2011). Thus, the search for adult autologous sources for such cells is of great value. One of the most remarkable technical accomplishments of the past decade was the establishment and optimization of protocols for generating EPC from iPSC. Yoder and coworkers (Prasain et al., 2014) have successfully achieved this goal by first revisiting the earlier approach by James et al. (2010) based on application of TGF-beta inhibitor to CD144+ cells. However, expression of CD144+ is preceded by the expression of NP-1 in human pluripotent stem cells committed toward endothelial lineage. This earlier finding of Cimato et al. (2009) was explored by exposing pluripotent stem cells to mesodermal differentiation factors with endothelial specification: activin-A, BMP-4, FGF-2, and VEGF (Prasain et al., 2014) which resulted in formation of NP-1+CD31+ cells highly resembling those obtained from the cord blood. It is important to emphasize that NP-1+CD31+ cells did not form teratomas during 6 months of follow-up observation after their inoculation. Remarkably, endothelial cells obtained from patients with peripheral artery disease expressed low levels of NP-1 compared to control cells. Using an NP-1 agonist Fc-NP-1, Prasain et al. (2014) were able to overcome this deficiency and restore proliferative potential while reducing senescence of these cells.
Intriguingly, c-Kit+ cells exhibit somewhat similar robustness in angiogenesis and proliferation. Fang et al. (Fang, Wei, Pentinmikko, Leinonen, & Salven, 2012; Sedwick, 2012) described solitary cells or small clusters of c-Kit+ cells present in all three layers of the vascular wall: adventitial, medial, and intimal. These c-Kit+/VEGFR2+/CD45− cells are clonogenic and differentiate toward EC, SMC, and fibroblasts (Bearzi et al., 2009). Only 0.4% of all vascular wall cells are lin−CD31+CD105 +Sca1+CD117/c-Kit+, referred to as vascular endothelial stem cells (VESCs). Adaptive transfer of isolated VESCs demonstrated that a single c-kit+ VESC can generate in vivo functional blood vessels that incorporate into host circulation. VESCs also display long-term self-renewal capacity as shown by repeated rounds of cell isolation and in vivo serial transplantation. Most recent publication on the pulmonary c-Kit+ cells assigns to them a role of an exclusively endothelial precursor (Liu et al., 2015). Genetic lineage tracing studies of c-Kit+ cells revealed that they are present in lung vasculature and coexpress endothelial markers CD31 and VE-cadherin (but not epithelial markers) under basal conditions or during repair after two different lung injury models. It would be necessary in the future to compare VESC (lin−CD31+CD105+Sca1+CD117/c-Kit+) with ECFC (NP-1+CD31+ cells) to address the question: are these overlapping populations or distinct ones? To put this question differently: Is there a single or multiple populations of EPC? Considering the many ways to isolate EPCs, one might deduce that there are multiple populations; however, presently such conclusion has no solid support.
5. METABOLIC SIGNATURES OF EPC, ENDOTHELIAL CELLS, AND THEIR DYSFUNCTIONAL PROFILE
Dysfunctional endothelial cells and their progenitors are a harbinger of diverse cardiovascular and renal diseases. Such cells lose some or all of their highly differentiated functions including production of vasoconstrictors and vasodilators controlling the blood pressure; blood coagulation and fibrinolysis; formation of new blood vessels; regulation of inflammatory processes and the traffic of leukocytes into and out of the vascular lumen; formation of a selective barrier between the blood and interstitium and controlling the transcapillary passage of different molecules; as well as maintenance and regeneration of surrounding tissues. In clinical settings, flow-dependent relaxation of conduit vessels, a function of endothelium-derived relaxing factors, mainly nitric oxide (NO), is suppressed in patients and animals with CKD. Different modifications of testing endothelium-dependent vasorelaxation, together with other surrogate biomarkers such as markers of oxidative stress (8-iso-PGF2α and oxidized LDL), pro-inflammatory markers (high-sensitivity CRP, lipoprotein-associated PLA2, soluble ICAM-1, IL-6, von Willebrand factor), inhibitor of endothelial nitric oxide synthase (eNOS) ADMA, circulating procoagulants and others have become the standard of diagnosing endothelial cell dysfunction (ECD) (Goligorsky, 2005, 2006; Ludmer et al., 1986). Fig. 1 puts forth diverse traditional and nontraditional risk factors, such as ADMA, advanced glycation end-products, and pro-oxidants, all leading to ECD via its most reliable attribute, uncoupling of eNOS. These metabolic perturbations reviewed previously (Goligorsky, 2005) result in a metabolic shift toward Warburg-type energetics and perturbations in cell cycle. The latter can be reversible or irreversible resulting in apoptosis and vascular rarefaction.
Fig. 1.
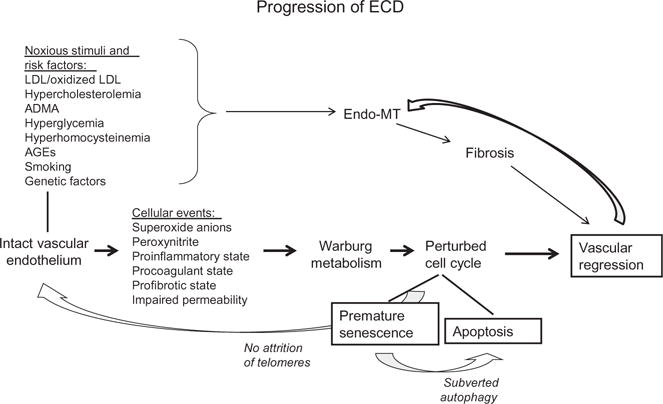
A schematic overview of progression of endothelial cell dysfunction, instigating noxious, and risk factors leading to altered cell metabolism and cell cycle.
To get broad unbiased insight into endothelial metabolism in health and disease, we isolated renal microvessels from mice with chronically inhibited eNOS screened using 2D electrophoresis, in-gel digestion, and mass-spectrometry analysis revealed at least 13 nonredundant differentially expressed proteins with high level of confidence. Five of those are specific for mitochondria, and two down-regulated proteins are the known components of the Krebs cycle: aconitase-2 and enoyl-coA-hydratase-1 (Addabbo et al., 2009). This deficiency of key enzymes is accompanied by reduced mitochondrial mass, mitochondrial oxidative stress, and switch to the normoxic glycolysis (Warburg type of metabolic hypoxia seen in chronic uncoupling of eNOS) to support energy metabolism. Moreover, by supplying cultured cells with the metabolic intermediate downstream of the deficient aconitase-2—α-ketoglutarate, which bypasses enzymatic bottleneck, we were able to not only restore energy metabolism but also prevent cell death or premature senescence.
It must be interjected here that endothelial cells are thought to heavily rely on normoxic glycolysis, at least in vitro (Verdegem, Moens, Stapor, & Carmeliet, 2014). However, it is proposed that glycolysis is enhanced specifically in “tip” cells. Yet, genetic targeting of cyclophilin D, a component of mitochondrial permeability transition pore, results in enhanced angiogenesis in vivo, suggesting that normally mitochondrial respiration contributes to the phenotype of endothelial cells (Marcu et al., 2015). It is possible that there exists an internal “rheostat” dynamically regulating OXPHOS and glycolytic contribution to energy metabolism and cell phenotype.
Therefore, in our particular case of endothelial dysfunction induced by chronic inhibition of NOS, we next supplemented animals with glutamine. This treatment results in improved vasculopathy (as judged by restored endothelium-dependent vasorelaxation) and decreased proteinuria (Addabbo et al., 2013). In addition, metabolomic studies conducted using liquid chromatography–mass spectrometry analyses disclose multiple metabolite abnormalities developing in ECD and restored by glutamine supplementation. Among those are several lysophospholipids, hippuric acid (all elevated), and glutamine/glutamate itself (reduced levels most probably due to consumption), which become normalized after glutamine supplementation (Addabbo et al., 2013). Hence, metabolic abnormalities affect EC functions and correction of these abnormalities leads to amelioration of ECD and vasculopathy, which contribute to progression of CKD.
Most recently, high-resolution NMR disclosed 44 metabolites associated with stem cell differentiation (Moussaieff et al., 2015). The main metabolic signature of pluripotent embryonic stem cells was found to be represented by normoxic glycolysis and generation of lactate and acetyl-CoA, which supports histone acetylation and blocks its deacetylation. Upon differentiation, cells rapidly lose acetyl-CoA and pluripotency, while showing increased histone deacetylation. Addition of acetate alone can halt differentiation by inducing histone pan-acetylation. Thus, Moussaieff and coauthors conclude that pluripotent state is maintained by normoxic glycolysis and generation of acetyl-CoA as an end-product, whereas metabolic switch depleting acetyl-CoA heralds early differentiation. This remarkable study is somewhat reminiscent of our work in dysfunctional endothelial cells. Analyzing our data in the light of the study by Moussaieff et al. (2015), one wonders whether the dysfunctional state of endothelial cells could be associated with acquisition or readiness to acquire pluripotency ground state. Such a scenario, if confirmed, would create a feedback mechanism for metabolically induced self-renewal.
In addition to that, the above findings of glutamine consumption and depletion in dysfunctional endothelia displaying Warburg-type metabolic profile may shed light on additional biochemical aberrations, some confirmed, others speculative. For instance, impaired expression and activity of sirtuin 1 (SIRT1) and uncoupling of eNOS, lysosomal dysfunction and impaired autophagy, all leading to premature vascular senescence and cardiovascular disease have been previously documented by our laboratory (Brodsky et al., 2004; Chen, Qin, et al., 2012; Chen, Xavier, et al., 2012; Lin et al., 2014; Maizel et al., 2014; Patschan et al., 2008). The hypothetical schema ascribing to Warburg metabolism premature senescence and dysfunction of EPC and EC illustrates its role in reverse Krebs cycle, increased production of acetyl-CoA, and consumption of nicotinamide dinucleotide (NAD) with all the functional sequelae for cells and organisms (Fig. 2).
Fig. 2.
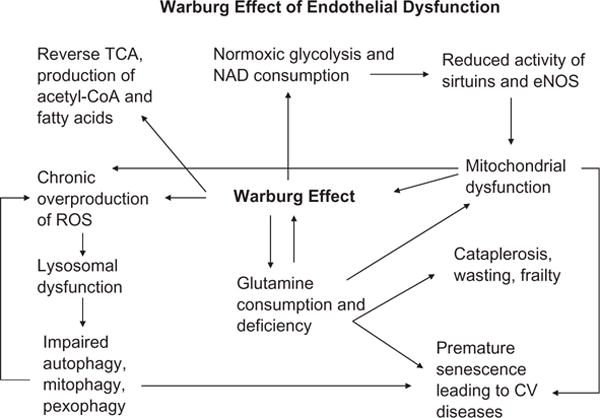
A schematic depiction of endothelial dysfunction through metabolic aberrations, mitochondrial and lysosomal dysfunction, impaired autophagy, mitophagy and pexophagy leading to normoxic glycolysis (Warburg-type respiration), consumption of glutamine and NAD, and defective SIRT expression/activity. These abnormalities of endothelial metabolism and function result in premature vascular senescence.
Several additional pathways from the dysfunctional endothelium to fibrosis and progressive organ dysfunction are summarized in Fig. 3. It proposes three major processes of premature cell senescence, endothelial–mesenchymal transition (Endo-MT) and the loss of endothelial surface glycocalyx (ESG) as main perpetrators of lost endothelial functions and their consequences leading to accumulation of myofibroblasts and matrix deposition. All three will be discussed later.
Fig. 3.
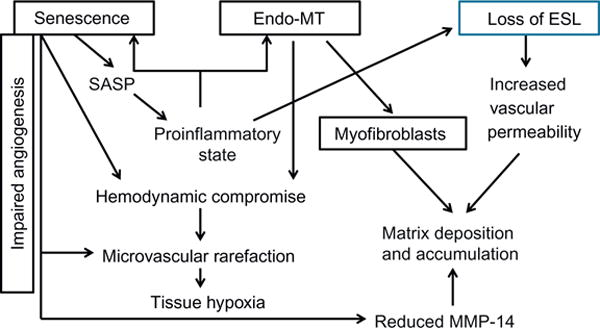
Endothelium-dependent pathways of fibrosis and progressive organ dysfunction.
Various noxious stimuli lead to SIPS of EPC and vascular endothelium and contribute to development of vasculopathy. We have recently demonstrated that cell stress induces SIPS by depleting SIRT1, a member of the family of NAD-dependent deacetylases, which has been implicated in cellular and organismal longevity. We were able to outline a novel pathway triggered by stress-induced lysosomal membrane permeabilization leading to release of cathepsins and cleavage of SIRT1. As a follow-up, we characterized mice deficient in endothelial SIRT1 and demonstrated that it is dispensable under normal conditions, but leads to exaggerated fibrotic response after injury. MMP-14 deficiency developing in endothelial SIRT1 knockout mice was responsible in part for the observed fibrosis and pharmacological maneuvers restoring MMP-14 prevented it (Chen, Qin, et al., 2012; Chen, Xavier, et al., 2012; Vasko et al., 2014). In fact, Potente et al. (2007) were the first to demonstrate that inhibition of SIRT1 results in modulation of multiple genes, including downregulation of MMP-14, in endothelial cells. This master metalloproteinase, MMP-14, in turn, is crucial for diverse physiological functions such as cell motility, adhesion to extracellular matrix proteins, cell differentiation and signaling. Below, we offer a simplified schema of diverse MMP-14 targets pertinent to functions of EPC and endothelial cells that could be affected by its deficiency (Fig. 4). Among the targets of MMP-14, apart from the matrix proteins, are several epitopes potentially involved in fibrosis, such as endoglin (an extracellular domain of the TGF-β auxiliary receptor richly expressed on endothelial cells), tissue transglutaminase (a multifunctional protein richly expressed on the surface of vascular endothelium and in the extracellular matrix, where it cross-links fibronectin, collagen, vitronectin, and osteopontin), delta-like-1 (a Notch ligand), and Dickkopf-1 (a natural antagonist of Wnt-frizzled interaction through binding to LRP6 leading to the inhibition of Wnt/β-catenin pathway). It is not known whether and how the fragments released by MMP-14 (sheddome) affect signaling.
Fig. 4.
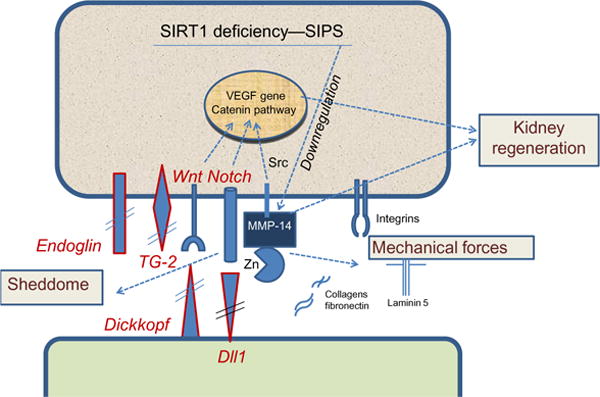
Cartoon summarizing the distinct potential pathways involved in SIRT1 deficiency- and SIPS-induced down-regulation of MMP-14 consequently leading to perturbations in the sheddome (endoglin, tissue transglutaminase-2 (TG2), Dickkopf, delta-like-1), mechanome (cell and matrix rigidity), and regenerative ability of the kidney (or other organs).
6. SENESCENCE-ASSOCIATED SECRETORY PRODUCTS
Senescent endothelial cells not only compromise the function of the endothelial lining of the vessels, but also affect the neighboring cells by their secretome referred to as senescence-associated secretory products (SASPs). SASP is composed of TGF-α, galectin-3, IGFBP-3, -4, and -6, MIC-1 (rev. in Suzuki & Boothman, 2008), among other site-specific components. Dysfunctional senescent endothelial cells produce excessive amounts of collagen XVIII and its C-terminal antiangiogenic fragment, endostatin (O’Riordan, Mendelev, Patschan, Chander, & Goligorsky, 2007). High-resolution mass-spectrometry analysis of the secretome of endothelial progenitor cells disclosed 133 proteins, some membrane-bound, others secretory (Hemmen et al., 2011). Specifically, soluble forms of VEGF receptors, adhesion molecules, semaphorin 3F, and TGF-β, CD109, members of Roundabout family, as well as endothelial markers were detected. Mass spectrometry screen of the secretome of colony-forming units, precursors of mature endothelial cells, identified 272 nonredundant proteins of which 124 were also found in cultured EPC (Pula, Mayr, Evans, et al., 2009). Secretory products included MMP-9, IL-8, MIF, various cathepsins and protease inhibitors, S100 proteins A11, A8, and A4, PAI-2 and apolipoprotein E, as well as a proangiogenic and pro-survival factor, thymidine phosphorylase. In many ways, the panoply of functional effects probably afforded by normal and abnormal secretomes remains unresolved.
7. ENDOTHELIAL–MESENCHYMAL TRANSITION
Endo-MT is defined as a process of acquisition by endothelial cells of mesenchymal and stem cell-like phenotypic characteristics. Endo-MT is a normal developmental stage taking place during embryonic formation of heart valves and septa. In adulthood, Endo-MT may turn to be a contributor to vascular drop-out and development of fibrosis, as detected in multiple organs, including heart and kidney (Li, Qu, & Bertram, 2009; O’Riordan et al., 2007; Zeisberg et al., 2007). It has recently been demonstrated that glomerulosclerosis in tensin-2-deficient mice is associated with Endo-MT of glomerular endothelial cells (Kato, Mizuno, & Ito, 2014). Some estimates show that from 30 to 50% of interstitial fibroblasts originate from the endothelium, although other investigations dramatically reduce this figure (LeBlue et al., 2013). Dejana’s group has demonstrated Endo-MT in the brain contributing to the onset and progression of cerebral cavernous malformation (Madedaluno et al., 2013). Xu et al. (2015) demonstrated that aberrant Endo-MT causes endocardial fibroelastosis. In fibrodysplasia ossificans progressiva Endo-MT is responsible for excessive generation of osteoblasts and chondrocytes from endothelial cells, thus illustrating yet another end-result of this process (Medici et al., 2010). TGF-β is considered to be a major mediator of Endo-MT, whereas BMP-7—a factor counteracting Endo-MT, as demonstrated using endothelial cell fate-tracing technique (rev. in Medici & Kalluri, 2012). Actions of TGF-β are mediated via activin receptor-like kinases 1 and 5 (Alk1 and Alk5). Activation of Alk1 uniquely expressed on endothelial cells results in cell migration, proliferation and angiogenesis, whereas stimulation of Alk5 induces via Smads 2/3 phosphorylation the transcription of SM22α, fibronectin, and PAI-1 which mediate differentiation along the smooth muscle/mesenchymal phenotype and leads to formation of myofibroblasts. Endoglin, another specific endothelial TGF coreceptor, regulates balance between activation of these two Alk pathways. Notably, prolonged activation with TGF-β results in the escape of Alk1 signaling and predominant signaling via Alk 5, thus promoting Endo-MT (rev. in Gaengel, Genove, Armulik, & Betsholtz, 2009). Ubil et al. (2014) described the possibility of reversing cardiac fibrosis by generating endothelial cells from mesenchymal cells.
Gene microarray analysis of cultured endothelial cells treated with an inhibitor of NOS, the most common pathogenic step toward endothelial dysfunction, revealed upregulation of collagen XVIII and its antiangiogenic fragment endostatin, a finding confirmed in vivo in mice chronically treated with NOS inhibitor (O’Riordan et al., 2007). Enhanced generation of endostatin in these animals leads to the development of Endo-MT and eventual rarefaction of renal microvasculature, thus further compounding vascular and parenchymal pathology. In fact, young mice overexpressing endostatin or mice chronically infused with it show signs of premature senescence, microvascular rarefaction, and renal fibrosis (Lin et al., 2014). Endo-MT can be induced not only by TGF-β, but also by activation of Notch which could serve as a regulator of partial or complete Endo-MT (rev. by Welch-Reardon, Wu, & Hughes, 2015). FGF and its downstream target let-7 miRNA suppress TGF-β-induced Endo-MT, thus preventing fibrosis (Chen, Qin, et al., 2012; Chen, Xavier, et al., 2012).
An intriguing relationship between ciliated endothelium and development of Endo-MT has recently been recognized. Endothelial cells located at the sites of low or oscillatory blood flow are endowed with primary cilia, which appear to be involved in the induction of shear-sensitive transcription factor Kruppel-like factor 2 (KLF2) (Nauli et al., 2008). In mice lacking endothelial primary cilia KLF2 and KLF4 fail to increase in response to shear stress and are prone to Endo-MT, the situation that can be rescued by overexpression of KLF4 (Egorova et al., 2011).
8. PERTURBATIONS OF ENDOTHELIAL SURFACE GLYCOCALYX
An intact ESG determines most of parameters characterizing normally functioning and dysfunctional endothelial cells, such as regulation of vascular permeability, mediation of blood cell–vessel wall interactions, participation in coagulation and modulation of vasodilation in blood vessels. When it is perturbed, the homeostatic and vasoprotective functions of the glycocalyx become errant and partially contribute to the pathogenesis of several diseases. In particular, diabetes, congestive heart failure, sepsis, and cancer have shown alterations in the endothelial glycocalyx prior to the development of symptoms (Nieuwdorp et al., 2006; Reitsma, Slaaf, Vink, van Zandvoort, & oude Egbrink, 2007; Weinbaum, Tarbell, & Damiano, 2007).
9. MICROVASCULAR ENDOWMENT, MICROVASCULAR PATENCY, AND MICROVASCULAR DROP-OUT
It is conceivable that Endo-MT is at the cross-section of several pathophysiological conditions. Capillary endothelium in the adipose tissue can undergo Endo-MT and give rise to preferential adipogenic differentiation of mesenchymal cells (Gupta et al., 2012; Tran et al., 2012), whereas in patients with fibrodysplasia ossificans progressiva generation of osteoblasts and chondrocytes occurs via Endo-MT (Medici et al., 2010). This could account for the expansion of fat cells or chondrocytes and reduction in capillary endowment. It is quite likely that similar processes occur in other tissues. The process of microvascular drop-out has distinct intermediate steps. We surmise that the earlier stage is represented by the loss of vessel patency: endothelial cells are present in usual numbers on immunohistochemical examination of tissues, but the number of actually perfused capillaries is reduced. We observed this dissociation between the vascularity detected through counting CD31-labeled vascular structures and patency, as detected using intravenous injection of tomato lectin specifically labeling endothelia of perfused vessels in murine kidneys undergoing fibrogenesis and in mice exposed to elevated levels of endostatin (Xavier et al., 2015; Lin et al., 2014). It is not excluded that initially microvasculature is nonpatent due to the excessive vasoconstriction, but the total reserve of microvascular profiles remains unchanged, as it occurs in the skin microvasculature even under normal conditions—a large proportion of capillaries in the ambient temperature is nonperfused; however, when the temperature increases, the blood flow across the recruited capillaries jumps nearly 200-fold. Later on, with ensuing stagnation of blood flow, the loss of the shear stress and damage to endothelial cells induce local platelet adhesion, attract macrophages, which in turn recruit other leukocytes, eventually disposing of cell debris (Brown, 2010). Thus, the following stage is likely presented by the obliteration of the capillary lumen, which leaves behind the deendothelialized basement membranes, variably referred to as a “string vessels” or “empty basement membrane tubes”, or “empty sleeves” as remnants of preexisting vessels. This remaining scaffold of the basement membrane, rich in endothelial and pericyte growth factors, such as VEGF, bFGF, and PDGF, facilitates the repopulation of string vessels with endothelial cells, leading to deposition of a new layer of the basement membrane material, renewed patency of vessels, and ability to restore blood flow to the tissue. Restoration of endothelial lining of string vessels occurs by proliferation of EC and is reportedly assisted by endothelial progenitor cells (Otani et al., 2002). Sequential rounds of de-and recellularization of string vessels may be responsible for the appearance of thickened basement membranes.
10. ENDOTHELIUM-TARGETING REJUVENATION THERAPEUTICS
Emerging principles of rejuvenation pharmacology are based on interfering with one of three major pathways of cell aging: sirtuins, target of rapamycin (mTOR), and insulin-like growth factor. Pioneering studies of heterochronic parabiosis (sharing circulatory system between young and old mice) have revealed restoration of stem cell competence in aged animals and opened the possibility of rejuvenating therapies (Conboy et al., 2005). A member of the activating/TGF-β superfamily, growth differentiation factor 11 (GDF11) has been identified using modified aptamer-based proteomic technology as one of the factors present in the serum of young mice, reduced in aged animals, and capable of restoring cardiac function upon supplementation of old mice (Loffredo et al., 2013). The issue has been questioned, however, in follow-up studies (Egerman et al., 2015).
Based on the earlier studies of SIRT1 and its activation by resveratrol, a number of small molecule SIRT1 activators has been synthesized and are currently tested (Hubbard & Sinclair, 2014). Sirtuin-activating compounds (STACs) exert their effect by allosteric activation of this deacetylase. Three generations of STACs include, in addition to resveratrol, quercetin, and butein (first generation), SRT 1720, 1460, and 2183 (second generation), and STAC-5, -9, and -10 (third generation), all extending life-span and/or health-span. These compounds are presently undergoing clinical trials. In fact, dietary restriction, which possibly induces SIRT1, acts via mTOR signaling and NAD-dependent pathways accompanied by a shift toward oxidative metabolism (Moroz et al., 2014), both representing novel venues of rejuvenation therapy.
NAD+ is a cofactor necessary for activation of several sirtuins. NAD+ bioavailability is reduced in disease states and aging (Verdin, 2015). A precursor of NAD+, nicotinamide, is paving ways as a therapy to correct NAD+ deficiency.
Another venue for rejuvenation therapy is based on the series of findings implicating mTOR activation and the resulting defect in autophagy in senescence. Application of mTOR inhibitor, rapamycin, has been shown to act as a rejuvenating therapy, even when applied late in life (Harrison et al., 2009).
11. CONCLUSION
This brief overview of the origins of endothelial and endothelial progenitor cells, their function and dysfunction in maintaining (micro)vascular integrity in health and disease, and development of premature senescence of these cells in the course of chronic diseases seeks to paint in broad brushstrokes the picture of abnormal metabolism of dysfunctional endothelium, transition of endothelial cells towards mesenchymal phenotype with vascular drop-out and formation of myofibroblasts and induction of fibrogenesis, culminating in organ fibrosis and functional failure. We also sketch some emerging rejuvenation therapies that may be applicable to the endothelium. However, a long and tortuous path remains before we arrive to a better understanding, prophylaxis, and treatment of ECD.
Acknowledgments
Studies in the authors’ labs were supported in part by NIH grants DK54602, DK052783, and DK45462; Westchester Artificial Kidney Foundation/The New York Community Trust (M.S.G.); and NIH grants UH3 EB017103, EB016629, and HL128064 (K.H.).
Footnotes
CONFLICT OF INTEREST
Authors declare no conflicts of interest.
References
- Addabbo F, Chen Q, Patel DP, Rabadi M, Ratliff B, Zhang F, et al. Glutamine supplementation alleviates vasculopathy and corrects metabolic profile in an in vivo model of endothelial cell dysfunction. PLoS One. 2013;8(6):e65458. doi: 10.1371/journal.pone.0065458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addabbo F, Ratliff B, Park HC, Kuo MC, Ungvari Z, Csiszar A, et al. The Krebs cycle and mitochondrial mass are early victims of endothelial dysfunction. American Journal of Pathology. 2009;174:34–43. doi: 10.2353/ajpath.2009.080650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alev C, Li M, Asahara T. Endothelial progenitor cells: A novel tool for the therapy of ischemic diseases. Antioxidants & Redox Signaling. 2011;15:949–965. doi: 10.1089/ars.2010.3872. [DOI] [PubMed] [Google Scholar]
- Baker D, Wijshake J, Tchykonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearzi C, Leri A, Lo Monaco F, Rota M, Gonzalez A, Hosoda T, et al. Identification of coronary vascular progenitor cell in the human heart. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15885–15890. doi: 10.1073/pnas.0907622106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Belaoussoff M, Farrington SM, Baron MH. Hematopoietic induction and respecification of a-p identity by visceral endoderm signaling in the mouse embryo. Development (Cambridge, England) 1998;125:5009–5018. doi: 10.1242/dev.125.24.5009. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, et al. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circulation Research. 2004;94(3):377–384. doi: 10.1161/01.RES.0000111802.09964.EF. [DOI] [PubMed] [Google Scholar]
- Brown W. A review of string vessels or collapsed, empty basement membrane tubes. Journal of Alzheimer’s Disease. 2010;21:725–739. doi: 10.3233/JAD-2010-100219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd N, Becker S, Maye P, Narasimhaiah R, St-Jacques B, Zhang X, et al. Hedgehog is required for murine yolk sac angiogenesis. Development (Cambridge, England) 2002;129:361–372. doi: 10.1242/dev.129.2.361. [DOI] [PubMed] [Google Scholar]
- Carlin L, Stamatiades E, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153:362–375. doi: 10.1016/j.cell.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single vegf allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Chen J, Brodsky SV, Goligorsky MS. Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circulation Research. 2002;90:1290–1298. doi: 10.1161/01.res.0000022161.42655.98. [DOI] [PubMed] [Google Scholar]
- Chen P, Qin L, Barnes C, Charisse K, Zhang S, Ali R, et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Reports. 2012;2:1684–1696. doi: 10.1016/j.celrep.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. American Journal of Pathology. 2012;180:973–983. doi: 10.1016/j.ajpath.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimato T, Beers J, Ding S, Ma M, McCoy JP, Boehm M, et al. Neuropilin-1 identifies endothelial precursors in human and murine embryonic stem cells before CD34 expression. Circulation. 2009;119:2170–2178. doi: 10.1161/CIRCULATIONAHA.109.849596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Conboy M, Wagers A, Girma E, Weissman I, Rando T. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, et al. The wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating dll4/notch signaling. Developmental Cell. 2010;18:938–949. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochimica et Biophysica Acta. 2007;1775:298–312. doi: 10.1016/j.bbcan.2007.05.003. [DOI] [PubMed] [Google Scholar]
- De Val S, Black BL. Transcriptional control of endothelial cell development. Developmental Cell. 2009;16:180–195. doi: 10.1016/j.devcel.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val S, Chi NC, Meadows SM, Minovitsky S, Anderson JP, Harris IS, et al. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell. 2008;135:1053–1064. doi: 10.1016/j.cell.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerman M, Cadena S, Gilbert J, Meyer A, Nelson H, Swalley S, et al. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metabolism. 2015;22:164–174. doi: 10.1016/j.cmet.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorova A, Khedoe P, Goumans M, Yoder B, Nauli S, Dijke P, et al. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circulation Research. 2011;108:1093–1101. doi: 10.1161/CIRCRESAHA.110.231860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchetti F, Monzani E, Cavallini G, Bergamini E, La Porta CA. Effect of caloric restriction regimen on the angiogenic capacity of aorta and on the expression of endothelin-1 during aging. Experimental Gerontology. 2007;42:662–667. doi: 10.1016/j.exger.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Fang S, Wei J, Pentinmikko N, Leinonen H, Salven P. Generation of functional blood vessels from a single c-kit+ adult vascular endothelial stem cell. PLoS Biology. 2012;10:1001407. doi: 10.1371/journal.pbio.1001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the vegf gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Francois M, Caprini A, Hosking B, Orsenigo F, Wilhelm D, Browne C, et al. Sox18 induces development of the lymphatic vasculature in mice. Nature. 2008;456:643–647. doi: 10.1038/nature07391. [DOI] [PubMed] [Google Scholar]
- Francois M, Short K, Secker GA, Combes A, Schwarz Q, Davidson TL, et al. Segmental territories along the cardinal veins generate lymph sacs via a ballooning mechanism during embryonic lymphangiogenesis in mice. Developmental Biology. 2012;364:89–98. doi: 10.1016/j.ydbio.2011.12.032. [DOI] [PubMed] [Google Scholar]
- Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- Goligorsky MS. Endothelial cell dysfunction: Can’t live with it, how to live without it. American Journal of Physiology. Renal Physiology. 2005;288:F871–F880. doi: 10.1152/ajprenal.00333.2004. [DOI] [PubMed] [Google Scholar]
- Goligorsky MS. Clinical assessment of endothelial dysfunction: Combine and rule. Current Opinion in Nephrology and Hypertension. 2006;15:617–624. doi: 10.1097/01.mnh.0000247497.62505.72. [DOI] [PubMed] [Google Scholar]
- Gritz E, Hirschi KK. Specification and function of hemogenic endothelium during embryogenesis. Cellular and Molecular Life Sciences. 2016;73(8):1547–1567. doi: 10.1007/s00018-016-2134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, et al. Neuropilin-1 conveys semaphorin and vegf signaling during neural and cardiovascular development. Developmental Cell. 2003;5:45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Mepani R, Kleiner S, Lo JC, Khandekar MJ, Cohen P, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metabolism. 2012;15:230–239. doi: 10.1016/j.cmet.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison D, Strong R, Sharp Z, Nelson J, Astle C, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmen K, Reinl T, Buttler K, Behler F, Dieken H, Jänsch L, et al. High-resolution mass spectrometric analysis of the secretome from mouse lung endothelial progenitor cells. Angiogenesis. 2011;14:163–172. doi: 10.1007/s10456-011-9200-x. [DOI] [PubMed] [Google Scholar]
- Hill J, Zalos G, Halcox J, Schenke W, Waclawiw M, Quyyumi A, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. New England Journal of Medicine. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard B, Sinclair D. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends in Pharmacological Sciences. 2014;35:146–154. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James D, Nam HS, Seandel M, Nolan D, Janovitz T, Tomishima M, et al. Expansion and maintenance of human embryonic stem cell-derived endothelial cells by TGFbeta inhibition is Id1 dependent. Nature Biotechnology. 2010;28(2):161–166. doi: 10.1038/nbt.1605. http://dx.doi.org/10.1038/nbt.1605. Epub 2010 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, Frase SL, et al. Lymphatic endothelial cell identity is reversible and its maintenance requires prox1 activity. Genes & Development. 2008;22:3282–3291. doi: 10.1101/gad.1727208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor c is required for sprouting of the first lymphatic vessels from embryonic veins. Nature Immunology. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Hayashi M, Nakagawa R, Tanaka Y, Izumi N, Nishikawa S, et al. Etv2/er71 induces vascular mesoderm from flk1+pdgfralpha+ primitive mesoderm. Blood. 2011;118:6975–6986. doi: 10.1182/blood-2011-05-352658. [DOI] [PubMed] [Google Scholar]
- Kato T, Mizuno S, Ito A. A decrease in glomerular endothelial cells and endothelial-mesenchymal transition during glomerulosclerosis in the tensin2-deficient mice (ICGN strain) Acta Histochemica et Cytochemica. 2014;47:265–271. doi: 10.1267/ahc.14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Hirschi KK. Signaling hierarchy regulating human endothelial cell development. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:718–724. doi: 10.1161/ATVBAHA.109.184200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakoo A, Finkel T. Endothelial progenitor cells. Annual Review of Medicine. 2005;56:79–101. doi: 10.1146/annurev.med.56.090203.104149. [DOI] [PubMed] [Google Scholar]
- Lanner F, Sohl M, Farnebo F. Functional arterial and venous fate is determined by graded vegf signaling and notch status during embryonic stem cell differentiation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:487–493. doi: 10.1161/01.ATV.0000255990.91805.6d. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, et al. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development (Cambridge, England) 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Vogel AM, Weinstein BM. Sonic hedgehog and vascular endothelial growth factor act upstream of the notch pathway during arterial endothelial differentiation. Developmental Cell. 2002;3:127–136. doi: 10.1016/s1534-5807(02)00198-3. [DOI] [PubMed] [Google Scholar]
- LeBlue V, Taduri G, O’Connell J, Teng Y, Cooke V, Sugimoto H, et al. Origin and function of myofibroblasts in kidney fibrosis. Nature Medicine. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kang J, Yoo J, Ganesan SK, Cook SC, Aguilar B, et al. Prox1 physically and functionally interacts with coup-tfii to specify lymphatic endothelial cell fate. Blood. 2009;113:1856–1859. doi: 10.1182/blood-2008-03-145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, et al. Er71 acts downstream of bmp, notch, and wnt signaling in blood and vessel progenitor specification. Cell Stem Cell. 2008;2:497–507. doi: 10.1016/j.stem.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, et al. Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development (Cambridge, England) 2004;131:361–375. doi: 10.1242/dev.00929. [DOI] [PubMed] [Google Scholar]
- Li J, Qu X, Bertram J. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. American Journal of Pathology. 2009;175:1380–1388. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FJ, Chen X, Qin J, Hong YK, Tsai MJ, Tsai SY. Direct transcriptional regulation of neuropilin-2 by coup-tfii modulates multiple steps in murine lymphatic vessel development. The Journal of Clinical Investigation. 2010;120:1694–1707. doi: 10.1172/JCI40101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Chen J, Ziman BD, Marshall S, Maizel J, Goligorsky MS. Endostatin and kidney fibrosis in aging: A case for antagonistic pleiotropy? American Journal of Physiology. Heart and Circulatory Physiology. 2014;306(12):H1692–H1699. doi: 10.1152/ajpheart.00064.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Huang X, Zhang H, Tian X, He L, Yang R, et al. c-Kit+ cells adopt vascular endothelial but not epithelial cell fates during lung maintenance and repair. Nature Medicine. 2015;21:866–868. doi: 10.1038/nm.3888. [DOI] [PubMed] [Google Scholar]
- Loffredo F, Steinhauser M, Jay S, Gannon J, Pancoast J, Yalamanchi P, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludmer P, Selwyn A, Shook T, Wayne RR, Mudge GH, Alexander RW, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. New England Journal of Medicine. 1986;315:1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- Madedaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, et al. EndMT contributes to the onset and progression of cerebral cavernous malformation. Nature. 2013;498:492–496. doi: 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- Maizel J, Xavier S, Chen J, Lin C, Vasko R, Goligorsky MS. Sirtuin 1 ablation in endothelial cells is associated with impaired angiogenesis and diastolic dysfunction. American Journal of Physiology. Heart and Circulatory Physiology. 2014;307(12):H1691–H1704. doi: 10.1152/ajpheart.00281.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble vegf receptor-3. Nature Medicine. 2001;7:199–205. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- Marcu R, Kotha S, Zhi Z, Qin W, Neeley C, Wang R, et al. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circulation Research. 2015;116:1336–1345. doi: 10.1161/CIRCRESAHA.116.304881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marom K, Levy V, Pillemer G, Fainsod A. Temporal analysis of the early bmp functions identifies distinct anti-organizer and mesoderm patterning phases. Developmental Biology. 2005;282:442–454. doi: 10.1016/j.ydbio.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Medici D, Kalluri R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Seminars in Cancer Biology. 2012;22:379–384. doi: 10.1016/j.semcancer.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D, Shore E, Lounev V, Kaplan S, Kalluri R, Olsen B. Conversion of vascular endothelial cells into multipotent stem-like cells. Nature Medicine. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development (Cambridge, England) 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- Moroz N, Carmona J, Anderson E, Hart A, Sinclair D, Blackwell T. Dietary restriction involves NAD-dependent mechanisms and a shift toward oxidative metabolism. Aging Cell. 2014;13:1075–1085. doi: 10.1111/acel.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metabolism. 2015;21:392–402. doi: 10.1016/j.cmet.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Nauli S, Kawanabe Y, Kaminski J, Pearce W, Ingber D, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwdorp M, Mooij HL, Kroon J, Atasever B, Spaan JA, Ince C, et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55:1127–1132. doi: 10.2337/diabetes.55.04.06.db05-1619. [DOI] [PubMed] [Google Scholar]
- O’Riordan E, Mendelev N, Patschan S, Chander P, Goligorsky MS. Chronic NOS inhibition actuates endothelial-mesenchymal transformation. American Journal of Physiology. 2007;292:H285–H294. doi: 10.1152/ajpheart.00560.2006. [DOI] [PubMed] [Google Scholar]
- Otani A, Kinder K, Ewalt K, Otero F, Schimmel P, Friedlander M. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nature Medicine. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- Park C, Afrikanova I, Chung YS, Zhang WJ, Arentson E, Fong Gh G, et al. A hierarchical order of factors in the generation of flk1- and scl-expressing hematopoietic and endothelial progenitors from embryonic stem cells. Development (Cambridge, England) 2004;131:2749–2762. doi: 10.1242/dev.01130. [DOI] [PubMed] [Google Scholar]
- Patschan S, Chen J, Gealekman O, Krupincza K, Wang M, Shu L, et al. Mapping mechanisms and charting the time course of premature cell senescence and apoptosis: Lysosomal dysfunction and ganglioside accumulation in endothelial cells. American Journal of Physiology. Renal Physiology. 2008;294(1):F100–F109. doi: 10.1152/ajprenal.00261.2007. [DOI] [PubMed] [Google Scholar]
- Pearson S, Sroczynska P, Lacaud G, Kouskoff V. The stepwise specification of embryonic stem cells to hematopoietic fate is driven by sequential exposure to Bmp4, activin A, bFGF and VEGF. Development (Cambridge, England) 2008;135:1525–1535. doi: 10.1242/dev.011767. [DOI] [PubMed] [Google Scholar]
- Pennisi D, Gardner J, Chambers D, Hosking B, Peters J, Muscat G, et al. Mutations in sox18 underlie cardiovascular and hair follicle defects in ragged mice. Nature Genetics. 2000;24:434–437. doi: 10.1038/74301. [DOI] [PubMed] [Google Scholar]
- Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes & Development. 2007;21(20):2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasain N, Lee M, Vemula S, Meador JL, Yoshimoto M, Ferkowicz MJ, et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nature Biotechnology. 2014;32:1151–1157. doi: 10.1038/nbt.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pula G, Mayr U, Evans C, et al. Proteomics identifies thymidine phosphorylase as a key regulator of the angiogenic potential of colony-forming units and endothelial progenitor cell cultures. Circulation Research. 2009;104:32–40. doi: 10.1161/CIRCRESAHA.108.182261. [DOI] [PubMed] [Google Scholar]
- Reed M, Karres N, Eyman D, Vernon RB. Culture of murine aortic explants in 3-dimensional matrix: A novel, miniaturized assay of angiogenesis in vitro. Microvascular Research. 2007;73:248–252. doi: 10.1016/j.mvr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: Composition, functions, and visualization. Pflügers Archiv-European Journal of Physiology. 2007;454:345–359. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson M, Yoder M. Endothelial progenitor cell: Quo Vadis? Journal of Molecular and Cellular Cardiology. 2011;50:266–272. doi: 10.1016/j.yjmcc.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salanga MC, Meadows SM, Myers CT, Krieg PA. Ets family protein etv2 is required for initiation of the endothelial lineage but not the hematopoietic lineage in the Xenopus embryo. Developmental Dynamics: An Official Publication of the American Association of Anatomists. 2010;239:1178–1187. doi: 10.1002/dvdy.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh AC, Faloon P, Hu QL, Bhimani M, Choi K. In vitro hematopoietic and endothelial potential of flk-1(−/−) embryonic stem cells and embryos. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2159–2164. doi: 10.1073/pnas.96.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedwick C. On the hunt for vascular endothelial stem cells. PLoS Biology. 2012;10(10):e1001408. doi: 10.1371/journal.pbio.1001408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Shimada T, Takeshita T, Murohara T, Sasaki K, Egami K, Shintani S, et al. Angiogenesis and vasculogenesis are impaired in the precocious-aging klotho mouse. Circulation. 2004;110:1148–1155. doi: 10.1161/01.CIR.0000139854.74847.99. [DOI] [PubMed] [Google Scholar]
- Srinivasan RS, Dillard ME, Lagutin OV, Lin FJ, Tsai S, Tsai MJ, et al. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes & Development. 2007;21:2422–2432. doi: 10.1101/gad.1588407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan RS, Geng X, Yang Y, Wang Y, Mukatira S, Studer M, et al. The nuclear hormone receptor coup-tfii is required for the initiation and early maintenance of prox1 expression in lymphatic endothelial cells. Genes & Development. 2010;24:696–707. doi: 10.1101/gad.1859310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Boothman D. Stress-induced premature senescence (SIPS)—Influence of SIPS on radiotherapy. Journal of Radiation Research. 2008;49:105–112. doi: 10.1269/jrr.07081. [DOI] [PubMed] [Google Scholar]
- Tran KV, Gealekman O, Frontini A, Zingaretti MC, Morroni M, Giordano A, et al. The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metabolism. 2012;15:222–229. doi: 10.1016/j.cmet.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubil E, Duan J, Pillai I, Rosa-Garrido M, Wu Y, Bargiacchi F, et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–590. doi: 10.1038/nature13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko R, Xavier S, Chen J, Lin CH, Ratliff B, Rabadi M, et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: Relevance to fibrosis of vascular senescence. Journal of the American Society of Nephrology. 2014;25(2):276–291. doi: 10.1681/ASN.2013010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdegem D, Moens S, Stapor P, Carmeliet P. Endothelial cell metabolism: Parallels and divergences with cancer cell metabolism. Cancer & Metabolism. 2014;2:19. doi: 10.1186/2049-3002-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Vokes SA, Krieg PA. Endoderm is required for vascular endothelial tube formation, but not for angioblast specification. Development (Cambridge, England) 2002;129:775–785. doi: 10.1242/dev.129.3.775. [DOI] [PubMed] [Google Scholar]
- Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-b2 and its receptor eph-b4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annual Review of Biomedical Engineering. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Welch-Reardon K, Wu N, Hughes C. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35:303–308. doi: 10.1161/ATVBAHA.114.303220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. New England Journal of Medicine. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, et al. An essential role for prox1 in the induction of the lymphatic endothelial cell phenotype. The EMBO Journal. 2002;21:1505–1513. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98:769–778. doi: 10.1016/s0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- Wilkinson RN, Koudijs MJ, Patient RK, Ingham PW, Schulte-Merker S, van Eeden FJ. Hedgehog signaling via a calcitonin receptor-like receptor can induce arterial differentiation independently of vegf signaling in zebrafish. Blood. 2012;120:477–488. doi: 10.1182/blood-2011-10-383729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C, Kim SH, Ni TT, Mitchell L, Ro H, Penn JS, et al. Hedgehog signaling induces arterial endothelial cell formation by repressing venous cell fate. Developmental Biology. 2010;341:196–204. doi: 10.1016/j.ydbio.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes & Development. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Xavier S, Vasko R, Matsumoto K, Zullo J, Chen R, Maizel J, et al. Curtailing endothelial TGFb signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. Journal of the American Society of Nephrology. 2015;26(4):817–829. doi: 10.1681/ASN.2013101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Friehs I, Zhong H, Melnychenko I, Tampe B, Alnour F, et al. Endocardial fibroelastosis is caused by aberrant endothelial-to-mesenchymal transition. Circulation Research. 2015;116:857–866. doi: 10.1161/CIRCRESAHA.116.305629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. Fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes & Development. 1994;8:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]
- Yamamizu K, Matsunaga T, Uosaki H, Fukushima H, Katayama S, Hiraoka-Kanie M, et al. Convergence of notch and beta-catenin signaling induces arterial fate in vascular progenitors. The Journal of Cell Biology. 2010;189:325–338. doi: 10.1083/jcb.200904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY. Suppression of notch signalling by the coup-tfii transcription factor regulates vein identity. Nature. 2005;435:98–104. doi: 10.1038/nature03511. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature Medicine. 2007;13(8):952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]