

Summary
Bisphenol A (BPA) is a key monomer in the production of plastics. It has been shown to be hepatotoxic. Inflammation and oxidative stress are closely linked with liver fibrosis, the major contributing factor to hepatic failure. Therefore, the aim of this study was to evaluate the impact of chronic exposure to BPA on the development of hepatic fibrosis in male rats and to determine the cross‐talk between the hepatic cytokine network, oxidative stress and apoptosis. For this purpose, 30 male Wistar albino rats were divided into three equal groups as follows: the first group was given no treatment (normal control group); the second group was given corn oil once daily by oral gavage for 8 weeks (vehicle control group); and the third group received BPA (50 mg/kg body weight/day, p.o.) for 8 weeks. BPA administration induced liver fibrosis as reflected in an increase in serum hepatic enzymes activities, hepatic hydroxyproline content and histopathological changes particularly increased collagen fibre deposition around the portal tract. In addition, there was inflammation (as reflected in increase in interleukin‐1beta ‘IL‐1β’, decrease in interleukin‐10 ‘IL‐10′ serum levels and increase in IL‐1β/IL‐10 ratio), oxidative stress (as reflected in increase in malondialdehyde (MDA) level, reduction in reduced glutathione (GSH) content and inhibition of catalase (CAT) activity) and apoptosis [as reflected in an increase in caspase‐3 level and a decrease in numbers of B‐cell lymphoma 2 (BCL2)‐immunopositive hepatocytes]. Interestingly, BPA had an upregulating effect on an extracellular matrix turnover gene [as reflected in matrix metalloproteinase‐9 (MMP‐9)] and a downregulating effect on its inhibitor gene [as reflected in tissue inhibitor of matrix metalloproteinase‐2 (TIMP‐2)] expression. Thus, the mechanism by which BPA induced liver fibrosis seems to be related to stimulation of the inflammatory response, along with oxidative stress, the apoptotic pathway and activation of extracellular matrix turnover.
Keywords: apoptosis, IL‐1β/IL‐10, liver fibrosis, matrix metalloproteinase‐9, oxidative stress, tissue inhibitor of matrix metalloproteinase‐2
Bisphenol A (BPA), 4,4′‐isopropylidenediphenol, is a ubiquitous environmental endocrine‐disrupting chemical. It has been used extensively in the production of polycarbonate plastics and epoxy resins that are then used in reusable drinking and baby bottles, food storage containers, inner‐lining of food cans, dental sealants, currency papers, toys and computer units (Diamanti‐Kandarakis et al. 2009). BPA can be released from these products in air, water and food under high temperature and acidic or basic conditions (Welshons et al. 2006). Moreover, people are exposed to this contaminant during handling of thermal papers used in cash register receipts through dermal contact (Biedermann et al. 2010). Thus, human exposure to BPA is unavoidable in daily life. Neonates and infants are exposed to BPA during maternal pregnancy and/or lactation through the placenta (Wan et al. 2010) and breast milk (Sun et al. 2004). Such exposure can induce deleterious effects on reproductive organ development (Suzuki et al. 2002) and disturbances in metabolic (Liu et al. 2013), cardiovascular, immune and neuroendocrine functions as well as cancer (De Coster & van Larebeke 2012; Rochester 2013).
The liver is an essential metabolic organ responsible for keeping homeostasis all over the body. It has several major functions such as metabolism, detoxification of xenobiotics, storage of glycogen and production of bile, cholesterol and proteins (Grijalva & Vakili 2013). As BPA is primarily metabolized by the liver through glucuronic acid conjugation, this organ is more vulnerable to lower doses of BPA than other organs (Pottenger et al. 2000; Moon et al. 2012). BPA induces oxidative stress and partial fat infiltration and inhibits cytochrome P450 isoforms in the liver of rats (Hanioka et al. 1998; Bindhumol et al. 2003; Rönn et al. 2013). Other in vivo experimental studies have shown that developmental exposure to BPA causes liver diseases, including hepatic steatosis (Jiang et al. 2014), liver tumours (Weinhouse et al. 2014) and metabolic syndrome (Wei et al. 2011; van Esterik et al. 2014). Moon et al. (2012) have reported that BPA may upregulate the expression of pro‐inflammatory cytokines, including interleukin‐6 (IL‐6) and tumour necrosis factor‐alpha (TNF‐α), inducing lipid peroxidation in the liver and impairing hepatic mitochondrial function. Chronic liver injury is one of the major causes of progressive liver fibrosis, bringing about cirrhosis, liver failure and carcinoma (Thompson & Patel 2010). A plethora of evidence has shown that an uncontrolled inflammatory cascade and generation of reactive oxygen species (ROS) are involved in hepatic tissue remodelling and fibrosis (Steib et al. 2007; Friedman 2008). On these bases, hepatic fibrosis is a likely consequence of BPA‐induced liver injury. Therefore, this study aimed to explore the hepatotoxic effect of BPA after long‐term exposure in male rats and to underline molecular mechanisms.
Materials and methods
Chemicals
Bisphenol A (BPA) (purity > 99%), hydroxyproline (Hyp), chloramine‐T and p‐dimethylaminobenzaldehyde were purchased from Sigma Chemical Co. (St Louis, MO, USA). Corn oil was supplied from ARMA Oils Company (10th of Ramadan City, Egypt). All other chemicals were of the commercially available highest grade.
Animals
Thirty male Wistar albino rats (2–3 months old) weighing 180–220 g were obtained from the Egyptian Organization for Biological Products and Vaccines (Cairo, Egypt). Rats were housed in an air‐conditioned atmosphere, at a temperature of 25°C with alternatively 12‐h light and dark cycles. They were acclimatized for 1 week in stainless steel rodent cages prior to starting the experiment. Animals were kept on a standard rodent chow (El‐Nasr, Abu Zaabal, Egypt) and water ad libitum. The diet pellets contained 20% protein, 13% fiber and ash, 5% total fat (including 2% unsaturated fat) and 62% carbohydrate.
Experimental design
Animals were divided randomly into three groups (10 animals per group) as follows: the first group was given no treatment (normal control group); the second group was given 1 ml vehicle (corn oil) once daily by oral gavage for 8 weeks (vehicle control group); and the third group (BPA group) received BPA dissolved in corn oil (50 mg/kg body weight/day, p.o.) for 8 weeks (Hassan et al. 2012). At the end of the experimental period, rats were fasted overnight and anesthetized with urethane (1.3 mg/kg) and blood samples were collected from the retro‐orbital plexus and allowed to clot. Serum was separated by centrifugation at 1000 g for 20 min and used immediately for the assessment of liver function enzymes (alanine transaminase (ALT) and aspartate transaminase (AST) activities. The remaining aliquot of serum was stored at −20°C until use for the determination of IL‐1β and IL‐10 levels. Then, rats were sacrificed by decapitation and liver tissues were dissected, washed with ice‐cold saline, one portion (0.5 g from the right lobe) was frozen in liquid nitrogen (−170°C), kept at −80°C and subjected to determination of Hyp content, and matrix metalloproteinase‐9 (MMP‐9) and tissue inhibitor of matrix metalloproteinase‐2 (TIMP‐2) gene expressions. About 0.3 g of each liver tissue was homogenized in cold phosphate buffer (pH = 7.5). Tissue homogenates were centrifuged at 1000 g for 15 min at 4°C, and then supernatants were further used for the assessment of oxidative stress marker (malondialdehyde (MDA) and antioxidants (reduced glutathione (GSH) contents and catalase (CAT) activity)), as well as pro‐apoptotic maker (caspase‐3) level. A 4‐mm slice of hepatic tissues (from the right lobe) was fixed with 10% neutral‐buffered formaldehyde for histopathological and immunohistochemical examinations.
Biochemical analysis of serum ALT and AST enzymatic activities
These enzyme levels were determined spectrophotometrically by commercial diagnostic kits (Diamond Co., Cairo, Egypt) (Reitman & Frankel 1957) according to the manufacturer's instruction.
Quantification of Hepatic Hydroxyproline (Hyp) Content
The hepatic Hyp content was determined as previously described using the method of Fujita et al. (2003). Briefly, liver samples were hydrolysed in 6 N HCl at 120°C for 18 h. After filtration of the hydrolysate, 5 μl was added to 5 μl citric/acetate buffer (pH = 6) and 100 μl chloramine‐T solution in an ELISA plate. The mixture was incubated at room temperature for 20 min and then treated with 100 μl p‐dimethylaminobenzaldehyde solution and incubated at 65°C for 15 min. After cooling to room temperature for 10 min, optical densities of samples were read spectrophotometrically using a microplate reader (Sunrise, Austria) at 550 nm against a reagent blank containing no tissue and compared with a standard curve of known amounts of Hyp. Results were expressed as μg/g of wet tissue.
Histopathological examination
After proper fixation, liver specimens were dehydrated in ascending grades of ethyl alcohol (70%, 90%, 100%), cleared in xylol, impregnated and then embedded in paraffin wax. Five‐micrometre‐thick sections were cut using rotatory microtome (Leica RM 2155, England). Liver sections were stained with Mallory trichrome stain to study collagen fibers in relation to hepatic fibrosis (Drury & Wallington 1980). Sections were examined under a light microscope, and representative images were obtained. Area percentage of collagen fibers/(μm)2 surface area in liver tissue was measured morphometrically by Leica Qwin 500 image analyser (England) which was connected to a Leica microscope in five high‐power microscopic fields/section within total six sections from ten rats/group (Mohamad et al. 2011).
Real‐time quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR)
Total RNA was extracted from frozen liver using SV Total RNA Isolation System (Promega, Madison, WI, USA) and quantified spectrophotometrically at 260/280 nm. Residual genomic DNA was removed by incubating RNA with Rnase‐free DNase I. The total extracted RNA was reverse‐transcribed into complementary DNA (cDNA) using RT‐PCR kit (Stratagene, USA). Random hexamers (primers) for MMP‐9 (GenBank accession number NM_009660.3), TIMP‐2 (GenBank accession number NM_029482.1) and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH, housekeeping gene) (GenBank accession number NM _071085.1) genes were used for reverse transcription of RNA. The primer sequences used, product size and annealing temperature are listed in Table 1. Total volume of the master mixture was 19 μl for each sample. This mixture was added to the 13 μl RNA‐primer mixture resulting in 32 μl of cDNA. The last mixture was incubated in the programmed thermal cycler for 1 h at 37°C followed by inactivation of enzymes at 95°C for 10 min and finally cooled at 4°C. RNA was then changed into cDNA. The amplification reactions were performed in a 50 μl final volume, with thermal cycling conditions of 2 min at 50°C, 10 min at 95°C and 40 cycles of 15 s at 95°C, 30 s at 60°C, 30 s at 72°C and 10 min at 72°C. A mixture of 25 μl SYBR Green I qPCR mixture (2x), 0.5 μl liver cDNA, 2 μl primer pair mix and 22.5 μl H2O was prepared in each optical tube. After finishing PCR, the tubes were removed from the machine. The real‐time PCR result was analysed with qPCR equipment, step one applied biosystem software (Qiagen). The cycle number at which the transcripts were detectable (cycle threshold, Ct) was normalized to the cycle number of GAPDH, referred to as ΔCt. Relative changes in gene expression levels of samples to control were determined using the comparative (2−ΔΔCt) method (Schmittgen & Livak 2008).
Table 1.
The primers used for real‐time PCR
| Gene | Primer sequence | Annealing temperature | Product size |
|---|---|---|---|
| MMP‐9 | Forward primer: 5′‐CACAGACAGCCTTCTGCAAC‐3′ | 60°C | 141 bp |
| Reverse primer: 5′‐CATTTCCCACAGCCTTGAAT‐3′ | |||
| TIMP‐2 | Forward primer: 5′‐AATGACATCTATGGCAACCCC‐3′ | 58°C | 426 bp |
| Reverse primer: 5′‐AAGAACCATCACTTCTCTTG‐3′ | |||
| GAPDH | Forward primer: 5′‐AGC CAC ATC GCT GAG ACA C‐3′ | 55°C | 148 bp |
| Reverse primer: 5′‐GCC CAA TAC GACCAA ATCC‐3′ |
The ECM turnover marker was calculated, as previously described by Song et al. (2004), using the following equation: ECM turnover marker = MMP‐9 mRNA expression/TIMP‐2 mRNA expression.
Assessment of serum of pro‐inflammatory and anti‐fibrotic cytokines
Both IL‐1β and IL‐10 levels in serum were measured using RayBio® rat enzyme‐linked immunosorbent assay (ELISA) kits (RayBiotech Inc., Norcross, GA, USA) using a microtitre plate reader (Sunrise, Austria) at 450 nm following the manufacturers’ recommendations. The concentrations of these cytokines were expressed as pg/ml.
Determination of lipid peroxidation product (malondialdehyde “MDA”) content
Malondialdehyde (MDA) was estimated spectrophotometrically by measuring the concentration of thiobarbituric acid‐reactive substances (TBARS) in the liver using Biodiagnostic kit (Biodiagnostic, Dokki, Giza, Egypt) (Ohkawa et al. 1979). Briefly, 1 ml TBA reagent was added to 0.2 ml of tissue homogenate. Samples were incubated at 100°C for 30 min and cooled. The supernatant absorbance was read at 534 nm. Lipid peroxide level was expressed as nmol of TBARS/g wet tissue using 1,1,3,3‐tetraethoxypropane as a standard.
Measurement of hepatic antioxidants
To determine the non‐enzymatic antioxidant (GSH) content, we used the commercially available kit (Biodiagnostic, Dokki, Giza, Egypt) according to the method of Beutler et al. (1963). About 0.5 ml of tissue homogenate was added to a tube with 0.5 ml of 10% trichloroacetic acid (TCA). The tubes were shaken gently and allowed to stand for 5 min, followed by centrifugation at 700 g for 15 min. An aliquot of the resulting supernatant (0.5 ml) was added to a test tube containing 1 ml phosphate buffer and 0.1 ml Ellman's reagent (5,5′‐dithiobis (2‐nitrobenzoic acid)), and absorbance was read at 412 nm after 10 min. Results were expressed as mmol/g wet tissue. Estimation of enzymatic antioxidant (CAT) activity was made using kits provided by Biodiagnostic, Dokki, Giza, Egypt. CAT activity was assessed colorimetrically, where each unit of CAT decomposes 1 μM of hydrogen peroxide (H2O2) per min at 25°C and pH = 7 according to the method of Aebi (1984). CAT reacts with a known quantity of H2O2. The reaction was stopped after exactly one min with CAT inhibitor. In the presence of peroxidase, remaining H2O2 reacts with 3,5‐dichloro‐2‐hydroxybenzene sulphonic acid and 4‐aminophenazone to form a chromophore with a colour intensity inversely proportional to the amount of CAT in the original sample. Absorbance was read off at 510 nm. Enzyme activity was expressed as unit/g wet tissue.
Estimation of caspase‐3 level in hepatic tissues
Caspase‐3 level was determined using ELISA Kit (CUSABIO BIOTECH CO., LTD, China) and a microtitre plate reader (Sunrise, Austria) at 450 nm according to the instructions of the manufacturers. Levels were expressed as nmol/g tissue.
B‐cell lymphoma 2 (BCL2) Immunohistochemical staining
Paraffin‐embedded liver sections were deparaffinized and rehydrated. To block endogenous peroxidase activity, sections were incubated with 5% H2O2 for 10 min at room temperature. After rinsing the slides with phosphate‐buffered saline (PBS), they were incubated with primary antibodies against BCL2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Biotinylated secondary antibodies (DakoCytomation, Carpinteria, CA, USA) directed against immunoglobulin were then added and incubated for 15 min. Protein expression level was evaluated using a streptavidin–biotin–peroxidase kit. Sections were stained with diaminobenzidine (DAB) solution as a chromogen for BCL2 detection and then counterstained with haematoxylin (Xiaohui et al. 2007). A negative control was generated using normal tissues but omitting the primary antibody. Slides were examined under a light microscope, and representative images were taken. The number of BCL2‐positive hepatocytes was counted morphometrically in five randomly selected high‐power microscopic fields within total six liver sections from ten rats/group using a computerized image system composed of a Leica Qwin 500 image analyser (England) which is connected to a Leica microscope and was expressed as cell number per μm² (Salama et al. 2013).
Statistical analysis
Data were presented as mean ± SD, and statistical analysis was performed by GraphPad Prism software. Differences between the means of data were compared by one‐way analysis of variance (anova) test and Tukey–Kramer post hoc test for analysis of group differences. The correlations between the studied parameters were assessed using the Pearson's correlation coefficient (r). A P value of <0.05 was considered to be statistically significant.
Ethical approval statement
This study was performed according to ethical guidelines of the local ethics committee of animal handling (ECAHZU) (Zagazig University, Egypt) and the recommendations of Weatherall report.
Results
Liver function indices
Liver enzymes activities (ALT and AST) were decreased significantly in the control group receiving corn oil as compared to the normal group (P < 0.05). On the other hand, BPA administration significantly increased them by 3.44‐fold and 2.84‐fold, respectively, with respect to control values (P < 0.001) (Table 2).
Table 2.
Effect of daily oral administration of bisphenol A (50 mg/kg body weight) for 8 weeks on serum liver function biomarkers and cytokines as well as hepatic oxidative stress markers in male rats
| Groups | Parameters | |||||||
|---|---|---|---|---|---|---|---|---|
| ALT (U/l) | AST (U/l) | IL‐1β (pg/ml) | IL‐10 (pg/ml) | IL‐1β/IL‐10 | MDA (nmol/g tissue) | GSH (mmol/g tissue) | CAT (U/g tissue) | |
| Normal group | 20.09 ± 1.07 | 41.3 ± 3.09 | 96.8 ± 6.03 | 95 ± 4.43 | 1.03 ± 0.11 | 129.7 ± 3.79 | 2.68 ± 0.18 | 5.72 ± 0.11 |
| Control group | 16.68 ± 1.72* | 34.17 ± 4.4* | 78.88 ± 2.61* | 109 ± 6.29* | 0.73 ± 0.06 | 81.6 ± 2.67* | 3.47 ± 0.29* | 6.09 ± 0.07* |
| BPA group | 57.43 ± 1.84$ | 97.04 ± 4.98$ | 238.1 ± 19.33$ | 15.18 ± 1.82$ | 16.01 ± 3.34$ | 307 ± 10.09$ | 0.71 ± 0.04$ | 1.72 ± 0.04$ |
Values were expressed as mean ± SD (n = 6/group). *Significantly different from normal group at P < 0.05, $significantly different from control group at P < 0.001 using anova followed by Tukey–Kramer as a post hoc test. BPA, bisphenol A; ALT, alanine aminotransferase; AST, aspartate aminotransferase; IL‐1β, interleukin‐1beta; IL‐10, interleukin‐10; MDA, malondialdehyde; GSH, reduced glutathione; CAT, catalase.
Liver fibrosis markers
Liver fibrosis was evaluated biochemically by measuring Hyp content as an index of collagen accumulation and histologically by morphometric analysis of the area percentage of blue colour (collagen fibers) using Mallory trichrome stain. Biochemical measurement revealed a significant increase in the liver Hyp content in BPA‐intoxicated group reaching 6.36‐fold as matched to the control group (P < 0.001) (Figure 1). Liver sections from normal and control groups stained with Mallory trichrome revealed normal distribution of collagen fibers (blue stain) around central vein (CV) and portal tract (PT). However, BPA administration for 8 weeks resulted in a marked increase in the density of collagen fibers around portal tract (PT) (Figure 2A–C). Histomorphometric analysis supported these results where BPA induced a significant increase in fibrotic area around portal tract/μm2 surface area of liver tissue by 6.58‐fold compared to the control group (P < 0.001). In contrast, although corn oil alone significantly reduced hepatic Hyp content (P < 0.001), it produced a non‐significant reduction in fibrotic area% with respect to the normal group (Figures 1 and 2D).
Figure 1.
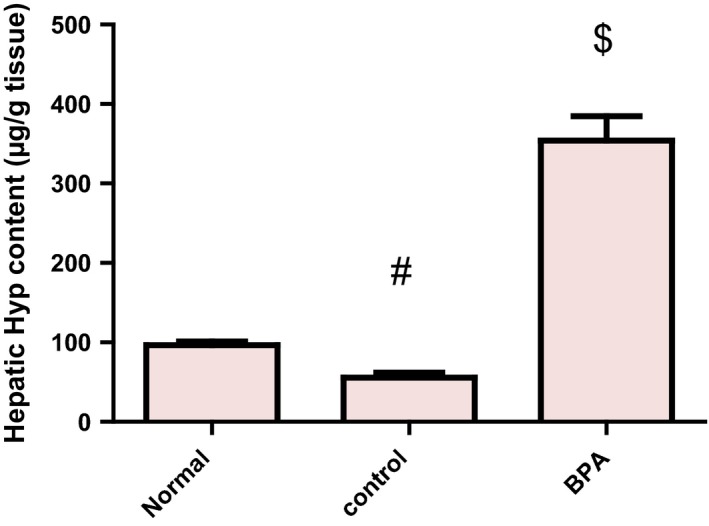
Effect of daily oral administration of bisphenol A (50 mg/kg body weight) for 8 weeks on hepatic Hyp content in male rats. Values were expressed as mean ± SD (n = 6/group). #Significantly different from normal group at P < 0.001, $Significantly different from control group at P < 0.001 using anova followed by Tukey–Kramer as a post hoc test. BPA, bisphenol A; Hyp, hydroxyproline.
Figure 2.
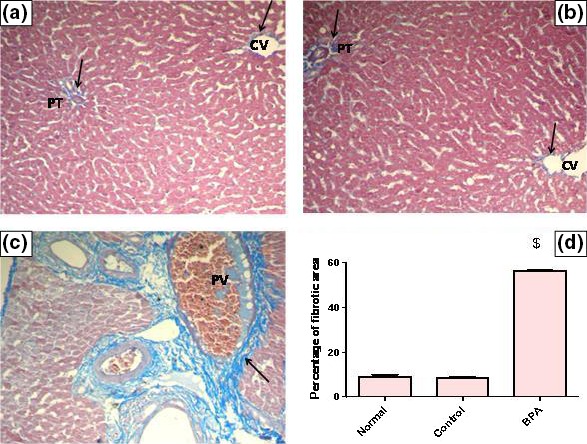
Representative photomicrographs of liver sections stained by Mallory trichrome (×200): (A) A liver section of an adult male albino rat of normal group, showing the normal distribution of collagen fibers around central vein (CV) and portal tract (PT) (arrows). N.B. The collagen fibers are stained blue. (B) A liver section obtained from an adult male albino rat of control group receiving vehicle (corn oil), showing normal distribution of collagen fibers around central vein (CV) and portal tract (PT) (arrows). (C) A liver section taken from an adult male albino rat receiving BPA, showing a marked increase in collagen fibers deposition around portal tract (arrow) and congested portal vein (PV) [Mallory trichrome × 200]. (D) The mean fibrosis (collagen) area percentage/μm2 surface area of liver tissue in the studied groups of adult male albino rats. Values were expressed as mean ± SD (n = 6/group). $ Significantly different from control group at P < 0.001 using anova followed by Tukey–Kramer as a post hoc test. BPA, bisphenol A.
Extracellular matrix turnover (ECM) markers
BPA‐administered rats exhibited a significant increase in hepatic MMP‐9 mRNA expression in comparison with the control group (P < 0.001). On the other hand, they displayed a significant decrease in hepatic TIMP‐2 mRNA expression relative to their control counterparts (P < 0.001) (Figure 3). ECM turnover marker (MMP‐9/TIMP‐2 mRNA ratio) was significantly increased in BPA group compared to the control group (84.72 ± 19.12 vs. 0.79 ± 0.2) (P < 0.001), implying an imbalance between MMP‐9 and TIMP‐2. The reverse pattern was observed in corn oil‐treated control group when compared to the normal group (P < 0.001) (Figure 3) except for ECM turnover marker which showed a non‐significant decrease from the normal value (0.79 ± 0.2 vs. 1.97 ± 0.22).
Figure 3.
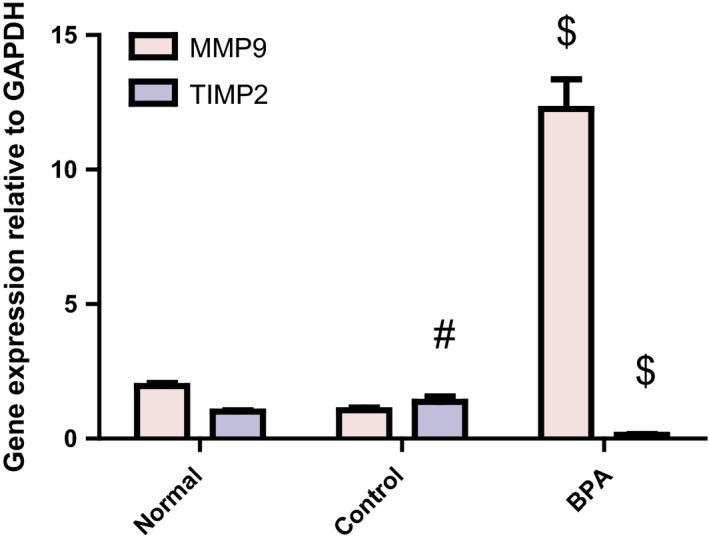
Hepatic MMP9 and TIMP2 mRNA expressions relative to GAPDH in male rats receiving corn oil and/or bisphenol A (50 mg/kg body weight) for 8 weeks. Values were expressed as mean ± SD (n = 6/group). #Significantly different from normal group at P < 0.001, $Significantly different from control group at P < 0.001 using anova followed by Tukey–Kramer as a post hoc test. BPA, bisphenol A; MMP‐9, matrix metallopeptidase‐9; TIMP‐2, tissue inhibitor of matrix metalloproteinase‐2.
Cytokines
Corn oil alone significantly reduced serum pro‐inflammatory cytokine (IL‐1β) and increased serum anti‐fibrotic cytokine (IL‐10) levels as matched to the normal group (P < 0.01). However, BPA‐intoxicated group showed a significant increase in serum IL‐1β level and a significant reduction in serum IL‐10 level in comparison with the control group (P < 0.001). Moreover, BPA induced a significant increase in the ratio between IL‐1β and IL‐10 relative to the control group (P < 0.001), suggesting a shift of cytokine balance towards the pro‐inflammatory cytokine (IL‐1β) (Table 2).
Hepatic oxidative stress
Corn oil possesses antioxidant properties as it significantly lowered hepatic lipid peroxidation and increased both enzymatic and non‐enzymatic antioxidants as compared to the normal group. BPA induced oxidative stress in rat liver as manifested by the significant increase in MDA as well as the significant decrease in reduced GSH contents and CAT activity with respect to control group (P < 0.001) (Table 2).
Liver apoptotic markers
In vehicle‐treated control group, the level of hepatic caspase‐3 protein was significantly lower than the normal value (P < 0.001), whereas the number of immunopositive BCL2‐stained hepatocytes was higher than the normal value (P < 0.05). The BCL2 immune staining of liver sections of normal and vehicle receiving rats revealed strong positive cytoplasmic immunoreactions for BCL2 while those receiving BPA for 8 weeks showed weak positive cytoplasmic immunoreactions for BCL2. Administration of BPA caused also a significant increase in hepatic pro‐apoptotic protein caspase‐3 level and a significant decrease in the number of immunopositive BCL2‐stained hepatocytes compared to the control group (P < 0.001) (Figures 4 and 5A–D).
Figure 4.
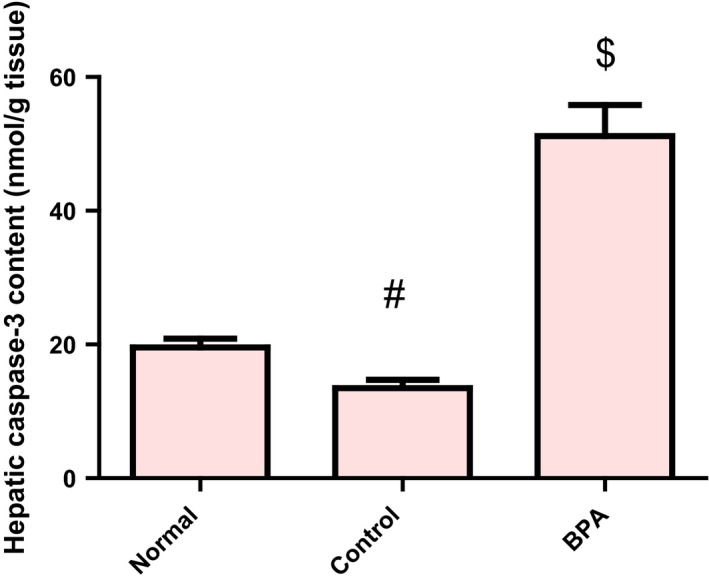
Hepatic caspase‐3 protein level in male rats receiving corn oil and/or bisphenol A (50 mg/kg body weight) for 8 weeks. Values were expressed as mean ± SD (n = 6/group). #Significantly different from normal group at P < 0.001, $Significantly different from control group at P < 0.001 using anova followed by Tukey–Kramer as a post hoc test. BPA, bisphenol A.
Figure 5.
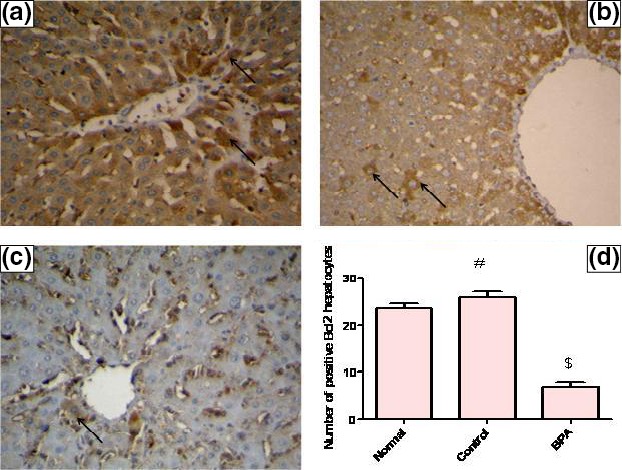
Representative photomicrographs of liver sections illustrating expression of BCL2 antigen by immunohistochemical staining (avidin–biotin–peroxidase stain with H counter stain, magnification ×400). (A) A liver section of an adult male albino rat of normal group, showing strong positive cytoplasmic immunoreactions for BCL2 (arrows). (B) A liver section taken from an adult male albino rat of control group receiving vehicle (corn oil), showing strong positive cytoplasmic immunoreactions for BCL2 (arrows). (C) A liver section obtained from a BPA‐intoxicated adult male albino rat, showing a weak positive cytoplasmic immunoreaction for BCL2 (arrow). (D) Number of BCL2‐positive hepatocytes/μm2 surface area of liver tissue in the studied groups of adult male albino rats. Values were expressed as mean ± SD (n = 6/group). #Significantly different from normal group at P < 0.05, $significantly different from control group at P < 0.001 using anova followed by Tukey–Kramer as a post hoc test. BPA, bisphenol A; BCL2, B‐cell lymphoma 2.
Correlations
Positive correlations between serum ALT, IL‐1β, IL‐1β/IL‐10, hepatic MDA, caspase‐3 and fibrosis markers, MMP‐9 gene expression as well as the ECM turnover marker were observed. On the other hand, there were negative correlations between hepatic GSH, CAT, BCL2 and fibrotic markers (Table 3).
Table 3.
Correlation between liver fibrosis markers and each of liver function index, serum inflammatory markers, hepatic oxidative stress and apoptosis among studied groups
| Fibrosis markers | Liver function | Inflammatory markers | Oxidative stress markers | Apoptotic markers | ||||
|---|---|---|---|---|---|---|---|---|
| ALT | IL‐1β | IL‐1β/IL‐10 | MDA | GSH | CAT | Caspase‐3 | BCL2 | |
| Hyp | r = 0.99a | r = 0.97a | r = 0.95a | r = 0.98a | r = −0.97a | r = −0.99a | r = 0.98a | r = −0.99a |
| Collagen area% | r = 0.99a | r = 0.98a | r = 0.97a | r = 0.98a | r = −0.95a | r = −0.99a | r = 0.98a | r = −0.99a |
| MMP‐9 | r = 0.99a | r = 0.99a | r = 0.99a | r = 0.98a | r = −0.96a | r = −0.99a | r = 0.98a | r = −0.98a |
| TIMP‐2 | r = −0.95a | r = −0.95a | r = −0.91a | r = −0.97a | r = 0.96a | r = 0.95a | r = −0.96a | r = 0.96a |
| ECM turnover marker (MMP‐9/TIMP‐2mRNA) | r = 0.97a | r = 0.99a | r = 0.99a | r = 0.94a | r = −0.92a | r = −0.97a | r = 0.96a | r = −0.95a |
Significant at P < 0.0001.
Discussion
The major finding of the current study was that BPA (50 mg/kg body weight) administration for 8 weeks induced hepatic fibrosis in male rats. BPA mediated this deleterious effect through increasing the pro‐inflammatory cytokine (IL‐1β) and decreasing the anti‐inflammatory/anti‐fibrotic cytokine (IL‐10) as well as exaggerating oxidative stress through inhibition of the antioxidant enzyme activity of CAT and reduction in GSH content. All these effects enhanced hepatocyte apoptosis and hence fibrosis.
In this study, oral administration of BPA‐induced liver fibrosis represented by massive collagen deposition and the increase in Hyp content (fibrosis marker) in the liver tissues of rats. In addition, the expression of genes encoding MMP‐9 was enhanced and TIMP‐2 was decreased. These results are in agreement with the recent findings of Crespo et al. (2015) and Liang et al. (2015) where CCl4‐induced liver fibrosis in mice is associated with increased transcription of MMP‐9. The observed upregulation in MMP‐9 gene expression in liver fibrosis progression could be also attributed to activation of hepatic stellate cells generating MMP‐2, MMP‐9 and MMP‐3, which destroy the basement membrane, leading to recruitment of inflammatory cells to the site of injury (Kisseleva & Brenner 2008; Brenner 2009; Cohen‐Naftaly & Friedman 2011). It has been thought that MMP‐9 plays a primary role in liver fibrosis by breaking down the normal basolateral matrix. Thus, the subsequent decrease in ECM degradation by MMP‐9 can result in excessive ECM accumulation and hence liver fibrosis (Vempati et al. 2007). Increased hepatic MMP‐9 mRNA expression here might be also a compensatory mechanism for severe liver fibrosis in BPA‐intoxicated rats to degrade collagen fibers. Interestingly, similar to our results, a previous study revealed that TIMP‐2 activity was increased significantly only in CCl4 cirrhotic not fibrotic rat liver (Segovia‐Silvestre et al. 2011).
Increases in MMPs and TIMPs are frequent in fibrotic diseases, suggesting significant ECM remodelling, a requirement of fibrosis (Mandal et al. 2003). Normal ECM is crucial for preserving the homeostasis of all liver cells. Consequently, degradation of the ECM by MMP‐9 might alter cell matrix and cell–cell interactions promoting hepatocyte vulnerability to necrosis and/or apoptosis caused by prolonged BPA exposure (Davis et al. 2000; Ghatak et al. 2011). Current results indicated that BPA markedly increased the ratio between MMP‐9 and TIMP‐2 genes expressions (ECM turnover marker) in the liver. Likewise, an increase in the MMP to TIMP gene ratio is critical to depress ECM degradation enhancing further ECM deposition and exacerbation of liver fibrosis (Yoshida & Matsuzaki 2012).
Pro‐inflammatory cytokines and chemokines, including IL‐1β, IL‐6, TNF and monocyte chemoattractant protein‐1 (MCP‐1), are produced from hepatic macrophages in various murine models of liver injury (Tuñón et al. 2009; Zimmermann et al. 2012; Shen et al. 2015; Ferreira et al. 2016). Furthermore, the release of these cytokines can activate macrophages during infection, injury and inflammation (Kim et al. 2007; Li et al. 2012). IL‐1β also stimulates the production of other cytokines and recruits inflammatory cells (Tilg & Diehl 2000). The increased serum level of IL‐1β here in BPA‐intoxicated rats may be attributed to increased transcription of such cytokine during the inflammatory response. ROS and lipid peroxidation products could also activate nuclear factor‐kappa B (NF‐κB) stimulating the release of various cytokines, comprising TNF, IL‐6, transforming growth factor‐beta1 (TGF‐B1) and others upon BPA administration. Upregulation of pro‐inflammatory cytokines was able to induce hepatic inflammation by transporting mononuclear and polymorphonuclear leucocytes into inflamed tissues (Wei et al. 2014). The increase in IL‐1β level here coincided with a reduction in the serum level of IL‐10 following BPA administration. IL‐10 can downregulate various pro‐inflammatory macrophage functions. It is expressed during macrophage activation in liver injury. IL‐10 has a therapeutic role in downregulation of inflammation in vivo. Recombinant IL‐10 shows effectiveness against hepatic inflammation/fibrosis in acute alcoholic hepatitis (Thompson et al. 1998). This is consistent with positive correlations between hepatic Hyp content, fibrotic area, MMP‐9 mRNA expression and serum IL‐1β as well as IL‐1β/IL‐10 in the present study.
Oxidative stress is another harmful cellular effect of BPA on the liver (Bindhumol et al. 2003). Bisphenol A is decomposed to various metabolites such as bisphenol A radical produced via a reaction with radical oxygen (Sajiki 2001). In the liver, exposure to hepato‐toxicants triggers macrophage generation of ROS comprising superoxide anion and hydrogen peroxide (McCloskey et al. 1992). Disturbing the redox balance in cells could also contribute to BPA‐induced oxidative stress (Hasselberg et al. 2004). The cell possesses several defence mechanisms to combat oxidative stress, like free radicals scavenging antioxidant systems, including GSH, CAT and glutathione peroxidase (GPx). CAT catalyses dismutation of the superoxide anion into hydrogen peroxide, which is then converted to water by GPx, conferring protection against highly reactive hydroxyl radical, derived from hydrogen peroxide (Sayed‐Ahmed et al. 2010). This study revealed that BPA reduced hepatic GSH content and CAT activity. This is consistent with the previous findings that reported downregulation of gene expression of antioxidant enzymes (CAT, GPx, superoxide dismutase ‘SOD’) and reduction in their activities in the liver (Hassan et al. 2012), kidney (Kabuto et al. 2003) and sperm (Chitra et al. 2003) of male mice and rats exposed to BPA. Because GSH is an important cofactor for GPx activity (Huang et al. 2004), reduction in hepatic GSH content here may reduce GPx activity enhancing oxidative stress. Glutathione‐S‐transferase (GST) is another important enzyme that catalyses the conjugation of GSH to electrophilic centres on different of substrates and xenobiotics, including lipid peroxides enhancing their detoxification (Douglas 1987; Leaver & George 1998). Thus, exposure to BPA in the current study might increase the activity of GST to help in the detoxification of BPA and degradation of lipid peroxides at the expense of GSH content that participates in the catalytic activity of GST. It has been proposed that BPA exposure produces ROS by inhibiting the activities of antioxidant enzymes or that generation of ROS depletes enzymatic and non‐enzymatic antioxidants (Moon et al. 2012). In addition, both ALT and AST activities were increased significantly following BPA administration. This may be attributed to BPA‐induced oxidative damage to the liver releasing hepatic enzymes into the blood or to hyperactivity of the liver (Kourouma et al. 2015). In consistence, inflammatory response to BPA administration might be involved in BPA‐induced liver damage here because hepatic enzyme activities are strongly and positively correlated with the pro‐inflammatory cytokine (IL‐1β) level and IL‐1β/IL‐10 ratio.
In this study, oral administration of BPA induced an increase in the pro‐apoptotic protein caspase‐3 level and a reduction in the anti‐apoptotic protein (BCL2) immunoreactivity in the liver of male rats. It has been suggested that mitochondria are target organelles of BPA effects (Xia et al. 2014). They play an important role in apoptosis through releasing of intermembrane space proteins, including cytochrome c, a key mediator of apoptosis via activation of caspases in the cytosol (Kroemer et al. 2007; Vaux 2011). Therefore, the mechanism of BPA‐induced apoptosis is also likely to be due to an alteration in the ratio of expression of pro‐apoptotic to anti‐apoptotic proteins of the BCL‐2‐associated X (BAX) and BCL2 family on the outer mitochondrial membrane modulating the release of apoptogenic factors (Kluck et al. 1997; Xu et al. 2002). Oxidative stress plays also an important role in apoptosis. In line with this, BPA was found to enhance oxidative stress and provoke cellular apoptosis in hepatocytes in vitro (Asahi et al. 2010). It was proposed that BPA‐induced hepatic mitochondria dysfunction was likely to be due to oxidative stress. Damaged mitochondria can produce more ROS (Moon et al. 2012). Mitochondria are susceptible to ROS due to deterioration of the antioxidant defence and DNA repair enzyme systems (Turrens 1997). Furthermore, accumulation of ROS in the mitochondria induces mitochondrial dysfunction, DNA depletion and cellular apoptosis (Ott et al. 2007). A previous study showed that hepatocyte apoptosis seem to be a key stone in hepatic inflammation and fibrosis (Malhi et al. 2010).
Interestingly, we observed that liver function indices, ECM matrix turnover markers, as well as inflammatory markers, hepatic oxidative stress and apoptotic markers, all were significantly improved in the corn oil group when compared with the normal group. Supporting this notion it has been reported that mice fed with corn oil‐based diet for 8 weeks displayed hepatoprotection against ethanol and iron‐induced liver injury. This could be attributed to attenuation of hepatic oxidative stress and steatosis, preservation of the enzymatic antioxidant defence system (CAT, GPx), modulation of the inflammatory cascade and fibrogenic pathways, protecting against liver fibrosis (Tan et al. 2013). However, we did not observe such improvement in liver functions and structure in the BPA group although it was dissolved in the same dose of corn oil as in the vehicle control group. This might be attributed to the use of corn oil concurrently with BPA as a protective agent rather than its use as a prophylactic therapy for the prevention of BPA hepatotoxic effects. Another explanation could be the fact that BPA‐induced liver damage was so severe that corn oil co‐administration failed to reduce such damage to approach the control group. In concordance, previous studies indicated BPA induced deleterious effects on the liver and other organs even it was dissolved in corn oil (El Ghazzawy et al. 2011; Yıldız & Barlas 2013; Kourouma et al. 2015; Kazemi et al. 2016).
In conclusion, the current study established that inflammation, oxidative stress and apoptosis are implicated in BPA‐induced liver injury and fibrosis (Figure 6). These results strongly suggest that BPA exposure is harmful to the liver. So, its exposure should be avoided and workers in plastic factories should undergo periodical monitoring of liver functions. Corn oil seems to keep and improve liver function and structure in the normal state. However, these beneficial effects did not occur in the group received BPA dissolved in corn oil. Therefore, clinical and experimental studies should be conducted in the future to determine prophylactic strategies for people chronically exposed to BPA. In addition, further studies should be performed to compare between the hepatotoxic effect of BPA dissolved in corn oil and in another aqueous vehicle.
Figure 6.
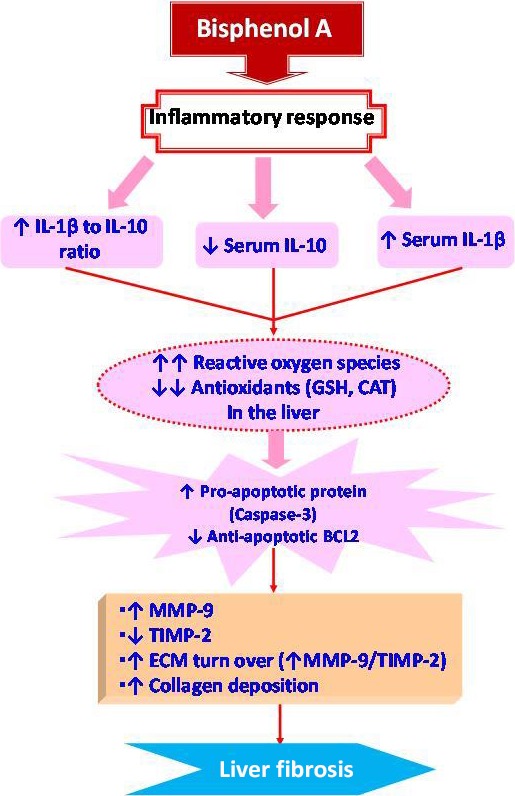
Proposed pathways by which liver fibrosis is programmed in BPA‐exposed male rats.
Conflict of interests
The authors declare that there are no conflicts of interest to disclose.
Acknowledgements
This work was not supported or funded by any Scientific Research Foundation.
References
- Aebi H. (1984) Catalase in vitro . Methods Enzymol. 105, 121–126. [DOI] [PubMed] [Google Scholar]
- Asahi J., Kamo H., Baba R. et al (2010) Bisphenol A induces endoplasmic reticulum stress‐associated apoptosis in mouse non‐parenchymal hepatocytes. Life Sci. 87, 431–438. [DOI] [PubMed] [Google Scholar]
- Beutler E., Duron O. & Kelly M.B. (1963) Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 61, 882–888. [PubMed] [Google Scholar]
- Biedermann S., Tschudin P. & Grob K. (2010) Transfer of bisphenol A from thermal printer paper to the skin. Anal. Bioanal. Chem. 398, 571–576. [DOI] [PubMed] [Google Scholar]
- Bindhumol V., Chitra K.C. & Mathur P.P. (2003) Bisphenol A induces reactive oxygen species generation in the liver of male rats. Toxicology 188, 117–124. [DOI] [PubMed] [Google Scholar]
- Brenner D.A. (2009) Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc. 120, 361–368. [PMC free article] [PubMed] [Google Scholar]
- Chitra K.C., Latchoumycandane C. & Mathur P.P. (2003) Induction of oxidative stress by bisphenol A in the epididymal sperm of rats. Toxicology 185, 119–127. [DOI] [PubMed] [Google Scholar]
- Cohen‐Naftaly M. & Friedman S.L. (2011) Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap. Adv. Gastroenterol. 4, 391–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo I., San‐Miguel B., Fernández A., Ortiz de Urbina J., González‐Gallego J. & Tuñón M.J. (2015) Melatonin limits the expression of profibrogenic genes and ameliorates the progression of hepatic fibrosis in mice. Transl. Res. 165, 346–357. [DOI] [PubMed] [Google Scholar]
- Davis G., Bayless K.J., Davis M. & Meininger G.A. (2000) Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 156, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster S. & van Larebeke N. (2012) Endocrine‐disrupting chemicals: associated disorders and mechanisms of action. J. Environ. Public Health 2012, 713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti‐Kandarakis E., Bourguignon J.P., Giudice L. et al (2009) Endocrine‐disrupting chemicals: an Endocrine Society scientific statement. Endocr. Rev. 30, 293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas K.T. (1987) Mechanism of action of glutathione‐dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 59, 103–167. [DOI] [PubMed] [Google Scholar]
- Drury R.A. & Wallington E.A. (1980) Carleton's Histological Techniques. 5th ed Oxford, New York and Toronto: Oxford University Press; 144–145, 183–185. [Google Scholar]
- El Ghazzawy I.F., Meleis A.E., Farghaly E.F. & Solaiman A. (2011) Histological study of the possible protective effect of pomegranate juice on bisphenol‐A induced changes of the caput epididymal epithelium and sperms of adult albino rats. Alexandria J. Med. 47, 125–137. [Google Scholar]
- van Esterik J.C., Dolle M.E., Lamoree M.H. et al (2014) Programming of metabolic effects in C57BL/6JxFVB mice by exposure to bisphenol A during gestation and lactation. Toxicology 321, 40–52. [DOI] [PubMed] [Google Scholar]
- Ferreira D.W., Goedken M.J., Rommelaere S. et al (2016) Enhanced hepatotoxicity by acetaminophen in Vanin‐1 knockout mice is associated with deficient proliferative and immune responses. Biochim. Biophys. Acta 1862, 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S.L. (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134, 1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M., Shannon J.M., Morikawa O., Gauldie J., Hara N. & Mason R.J. (2003) Overexpression of tumor necrosis factor‐α diminishes pulmonary fibrosis induced by bleomycin or transforming growth factor‐β. Am. J. Respir. Cell Mol. Biol. 29, 669–676. [DOI] [PubMed] [Google Scholar]
- Ghatak S., Biswas A., Dhali G.K., Chowdhury A., Boyer J.L. & Santra A. (2011) Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol. Appl. Pharmacol. 251, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grijalva J. & Vakili K. (2013) Neonatal liver physiology. Semin. Pediatr. Surg. 22, 185–189. [DOI] [PubMed] [Google Scholar]
- Hanioka N., Jinno H., Nishimura T. & Ando M. (1998) Suppression of male‐specific cytochrome P450 isoforms by bisphenol A in rat liver. Arch. Toxicol. 72, 387–394. [DOI] [PubMed] [Google Scholar]
- Hassan Z.K., Elobeid M.A., Virk P. et al (2012) Bisphenol A induces hepatotoxicity through oxidative stress in rat model. Oxid. Med. Cell Longev. 2012, 194829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselberg L., Meier S. & Svardal A. (2004) Effects of alkylphenols on redox status in first spawning Atlantic cod (Gadus morhua). Aquat. Toxicol. 69, 95–105. [DOI] [PubMed] [Google Scholar]
- Huang J., Tan P.H., Tan B.K. & Bay B.H. (2004) GST‐pi expression correlates with oxidative stress and apoptosis in breast cancer. Oncol. Rep. 12, 921–925. [PubMed] [Google Scholar]
- Jiang Y., Xia W., Zhu Y. et al (2014) Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol. Lett. 228, 85–92. [DOI] [PubMed] [Google Scholar]
- Kabuto H., Hasuike S., Minagawa N. & Shishibori T. (2003) Effects of bisphenol A on the metabolisms of active oxygen species in mouse tissues. Environ. Res. 93, 31–35. [DOI] [PubMed] [Google Scholar]
- Kazemi S., Mousavi S.N., Aghapour F., Rezaee B., Sadeghi F. & Ali Moghadamnia Akbar A.A. (2016) Induction effect of bisphenol A on gene expression involving hepatic oxidative stress in rat. Oxid. Med. Cell. Longev. 2016, 6298515. doi:10.1155/2016/6298515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.B., Han A.R., Park E.Y. et al (2007) Inhibition of LPS‐induced iNOS, COX‐2 and cytokines expression by poncirin through the NF‐kappaB inactivation in RAW 264.7 macrophage cells. Biol. Pharm. Bull. 30, 2345–2351. [DOI] [PubMed] [Google Scholar]
- Kisseleva T. & Brenner D.A. (2008) Mechanisms of fibrogenesis. Exp. Biol. Med. (Maywood) 233, 109–122. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy‐Wetzel E., Green D.R. & Newmeyer D.D. (1997) The release of cytochrome c from mitochondria: a primary site for Bcl‐2 regulation of apoptosis. Science 275, 1132–1136. [DOI] [PubMed] [Google Scholar]
- Kourouma A., Quan C., Duan P. et al (2015) Bisphenol A induces apoptosis in liver cells through induction of ROS. Adv. Toxicol. 2015, 1–10. [Google Scholar]
- Kroemer G., Galluzzi L. & Brenner C. (2007) Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163. [DOI] [PubMed] [Google Scholar]
- Leaver M.J. & George S.G. (1998) A piscine glutathione S‐transferase which efficiently conjugates the end‐products of lipid peroxidation. Marine Environ. Res. 46, 71–74. [Google Scholar]
- Li Y.C., Kuan Y.H., Huang F.M. & Chang Y.C. (2012) The role of DNA damage and caspase activation in cytotoxicity and genotoxicity of macrophages induced by bisphenol‐A‐glycidyldimethacrylate. Int. Endod. J. 45, 499–507. [DOI] [PubMed] [Google Scholar]
- Liang B., Guo X., Jin J., Ma Y.‐C. & Zheng‐Quan Feng Z.‐Q. (2015) Glycyrrhizic acid inhibits apoptosis and fibrosis in carbon‐tetrachloride‐ induced rat liver injury. World J. Gastroenterol. 21, 5271–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Yu P., Qian W. et al (2013) Perinatal bisphenol A exposure and adult glucose homeostasis: identifying critical windows of exposure. PLoS ONE 8, e64143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H., Guicciardi M.E. & Gores G.J. (2010) Hepatocyte death: a clear and present danger. Physiol. Rev. 90, 1165–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M., Mandal A., Das S., Chakraborti T. & Sajal C. (2003) Clinical implications of matrix metalloproteinases. Mol. Cell. Biochem. 252, 305–329. [DOI] [PubMed] [Google Scholar]
- McCloskey T.W., Todaro J.A. & Laskin D.L. (1992) Lipopolysaccharide treatments of rats alters antigen expression and oxidative metabolism in hepatic macrophages and endothelial cells. Hepatology 16, 191–203. [DOI] [PubMed] [Google Scholar]
- Mohamad H.E., Askar M.E. & Hafez M.M. (2011) Management of cardiac fibrosis in diabetic rats; the role of peroxisome proliferator activated receptor gamma (PPAR‐gamma) and calcium channel blockers (CCBs). Diabetol. Metab. Syndr. 3, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon M.K., Kim M.J., Jung I.K. et al (2012) Bisphenol A impairs mitochondrial function in the liver at doses below the no observed adverse effect level. J. Korean Med. Sci. 27, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa H., Ohishi N. & Yagi K. (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 95, 351–358. [DOI] [PubMed] [Google Scholar]
- Ott M., Gogvadze V., Orrenius S. & Zhivotovsky B. (2007) Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922. [DOI] [PubMed] [Google Scholar]
- Pottenger L.H., Domoradzki J.Y., Markham D.A., Hansen S.C., Cagen S.Z. & Waechter J.M. Jr (2000) The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicol. Sci. 54, 3–18. [DOI] [PubMed] [Google Scholar]
- Reitman S. & Frankel S. (1957) A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Pathol. 28, 56–63. [DOI] [PubMed] [Google Scholar]
- Rochester J.R. (2013) Bisphenol A and human health: a review of the literature. Reprod. Toxicol. 42, 132–155. [DOI] [PubMed] [Google Scholar]
- Rönn M., Kullberg J., Karlsson H. et al (2013) Bisphenol A exposure increases liver fat in juvenile fructose‐fed Fischer 344 rats. Toxicology 303, 125–132. [DOI] [PubMed] [Google Scholar]
- Sajiki J. (2001) Decomposition of bisphenol A by radical oxygen. Environ. Int. 27, 315–320. [DOI] [PubMed] [Google Scholar]
- Salama S.M., Abdulla M.A., Alrashdi A.S., Ismail S., Alkiyumi S.S. & Golbabapour S. (2013) Hepatoprotective effect of ethanolic extract of Curcuma longa on thioacetamide induced liver cirrhosis in rats. BMC Complement. Altern. Med. 13, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed‐Ahmed M.M., Aleisa A.M., Al‐Rejaie S.S. et al (2010) Thymoquinone attenuates diethylnitrosamine induction of hepatic carcinogenesis through antioxidant signaling. Oxid. Med. Cell. Longev. 3, 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T.D. & Livak K.J. (2008) Analyzing real‐time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Segovia‐Silvestre T., Reichenbach V., Fernández‐Varo G. et al (2011) Circulating CO3‐610, a degradation product of collagen III, closely reflects liver collagen and portal pressure in rats with fibrosis. Fibrogenesis Tissue Repair. 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K., Feng X., Su R., Xie H., Zhou L. & Zheng S. (2015) Epigallocatechin 3‐gallate ameliorates bile duct ligation induced liver injury in mice by modulation of mitochondrial oxidative stress and inflammation. PLoS One 10, e0126278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Li C. & Cai L. (2004) Fluvastatin prevents nephropathy likely through suppression of connective tissue growth factor‐mediated extracellular matrix accumulation. Exp. Mol. Pathol. 76, 66–75. [DOI] [PubMed] [Google Scholar]
- Steib C.J., Gerbes A.L., Bystron M. et al (2007) Kupffer cell activation in normal and fibrotic livers increases portal pressure via thromboxane A2. J. Hepatol. 47, 228–238. [DOI] [PubMed] [Google Scholar]
- Sun Y., Irie M., Kishikawa N., Wada M., Kuroda N. & Nakashima K. (2004) Determination of bisphenol A in human breast milk by HPLC with column‐switching and fluorescence detection. Biomed. Chromatogr. 18, 501–507. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Sugihara A., Uchida K. et al (2002) Developmental effects of perinatal exposure to bisphenol‐A and diethylstilbestrol on reproductive organs in female mice. Reprod. Toxicol. 16, 107–116. [DOI] [PubMed] [Google Scholar]
- Tan T.C., Crawford D.H., Jaskowski L.A. et al (2013) A corn oil‐based diet protects against combined ethanol and iron‐induced liver injury in a mouse model of hemochromatosis. Alcohol. Clin. Exp. Res. 37, 1619–1631. [DOI] [PubMed] [Google Scholar]
- Thompson A.J. & Patel K. (2010) Antifibrotic therapies: will we ever get there? Curr. Gastroenterol. Rep. 12, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson K., Maltby J., Fallowfield J., McAulay M., Millward‐Sadler H. & Sheron N. (1998) Interleukin‐10 Expression and Function in Experimental Murine Liver Inflammation and Fibrosis. Hepatology 28, 1597–1606. [DOI] [PubMed] [Google Scholar]
- Tilg H. & Diehl A.M. (2000) Cytokines in alcoholic and nonalcoholic steatohepatitis. N. Engl. J. Med. 343, 1467–1476. [DOI] [PubMed] [Google Scholar]
- Tuñón M.J., Alvarez M., Culebras J.M. & González‐Gallego J. (2009) An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J. Gastroenterol. 15, 3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens J.F. (1997) Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 17, 3–8. [DOI] [PubMed] [Google Scholar]
- Vaux D.L. (2011) Apoptogenic factors released from mitochondria. Biochim. Biophys. Acta 1813, 546–550. [DOI] [PubMed] [Google Scholar]
- Vempati P., Karagiannis E.D. & Popel A.S. (2007) A biochemical model of matrix metalloproteinase 9 activation and inhibition. J. Biol. Chem. 282, 37585–37596. [DOI] [PubMed] [Google Scholar]
- Wan Y., Choi K., Kim S. et al (2010) Hydroxylated polybrominated diphenyl ethers and bisphenol A in pregnant women and their matching fetuses: placental transfer and potential risks. Environ. Sci. Technol. 44, 5233–5239. [DOI] [PubMed] [Google Scholar]
- Wei J., Lin Y., Li Y. et al (2011) Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high‐fat diet. Endocrinology 152, 3049–3061. [DOI] [PubMed] [Google Scholar]
- Wei J., Sun X., Chen Y. et al (2014) Perinatal exposure to bisphenol A exacerbates nonalcoholic steatohepatitis‐like phenotype in male rat offspring fed on a high‐fat diet. J. Endocrinol. 222, 313–325. [DOI] [PubMed] [Google Scholar]
- Weinhouse C., Anderson O.S., Bergin I.L. et al (2014) Dose‐dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ. Health Perspect. 122, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welshons W.V., Nagel S.C. & Vom Saal F.S. (2006) Large effects from small exposures: III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 147, S56–S69. [DOI] [PubMed] [Google Scholar]
- Xia W., Jiang Y., Li Y. et al (2014) Early‐life exposure to bisphenol a induces liver injury in rats involvement of mitochondria‐mediated apoptosis. PLoS ONE 9, e90443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiaohui Z., Yaling H., Hemei Y. & Suzuo L. (2007) Effects of SSTF on the expression of apoptosis associated gene Bcl2 and Bax by cardiomyocytes induced by H2O2 . Guangdong. Med. J. 28, 1590–1591. [Google Scholar]
- Xu J., Osuga Y., Yano T. et al (2002) Bisphenol A induces apoptosis and G2‐to‐M arrest of ovarian granulose cells. Biochem. Biophys. Res. Commun. 292, 456–462. [DOI] [PubMed] [Google Scholar]
- Yıldız N. & Barlas N. (2013) Hepatic and renal functions in growing male rats after bisphenol A and octylphenol exposure. Hum. Exp. Toxicol. 32, 675–686. [DOI] [PubMed] [Google Scholar]
- Yoshida K. & Matsuzaki K. (2012) Differential regulation of TGF‐b/Smad signaling in hepatic stellate cells between acute and chronic liver injuries. Front. Physiol. 3, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H.W., Trautwein C. & Tacke F. (2012) Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front. Physiol. 3, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]