

Abstract
Background & objectives:
Alport syndrome (AS) is an inherited disorder characterized by glomerulonephritis and end-stage renal disease (ESRD). The aim of this study was to identify the gene responsible for the glomerulopathy in a Chinese family with autosomal dominant AS using exome sequencing.
Methods:
A 4-generation, 30-member Chinese Han family was enrolled in this study. Exome sequencing was conducted in the proband of the family, and then direct sequencing was performed in family members of the pedigree and 100 normal controls.
Results:
A novel frameshift mutation, c.3213delA (p.Gly1072Glufs*69), in the collagen type IV alpha-4 gene (COL4A4) was found to be the genetic cause. Neither sensorineural hearing loss nor ocular abnormalities were present in the patients of this family. Other clinical features, such as age of onset, age of ESRD occurring and disease severity, varied among the patients of this family.
Interpretation & conclusions:
A novel frameshift mutation, c.3213delA (p.Gly1072Glufs*69) in the COL4A4 gene, was identified in the Chinese pedigree with autosomal dominant AS. Our findings may provide new insights into the cause and diagnosis of AS and also have implications for genetic counselling.
Keywords: Alport syndrome, collagen type IV alpha-4 gene, diagnosis, frameshift mutation, genetic counselling
Alport syndrome (AS), first described by Cecil A. Alport in 1927, is an inherited kidney disorder heralded with continuous microhaematuria, which rapidly progresses to proteinuria and chronic or end-stage renal disease (ESRD) by adolescence. It is often associated with high-tone sensorineural hearing loss and/or ocular abnormalities (dot-and-fleck retinopathy, anterior lenticonus and posterior polymorphous corneal dystrophy)1,2,3,4. The AS is considered to affect, <1/2000 individuals5, and it occurs in a variety of settings, exhibiting a widely variable clinical expression6. It is caused by defects in type IV collagen, a major structural component of basement membranes in the kidney, ear, eye, etc7. Although the majority of pedigrees are X-linked due to the collagen type IV alpha-5 gene (COL4A5) mutations at Xq22, autosomal recessive and dominant forms of this disorder are also recognized and have been shown to be caused by mutations in the COL4A3 and the COL4A4 (MIM 120131), head-to-head genes on chromosome 2q362,8. X-linked, autosomal recessive and autosomal dominant patterns of inheritance account for about 80, 15 and 5 per cent of patients with AS, respectively9. In general, the disease is more severe in males with X-linked AS and is equally severe in male and female homozygotes or compound heterozygotes in the autosomal AS4,5. Various mutations were reported to lead to a broad spectrum of disease phenotypes, ranging from mild renal insufficiency to ESRD4,10.
Mutation screening of these three large genes by Sanger sequencing is laborious, time consuming and expensive due to the broad size of these genes and lack of mutational hot spots11. The aim of this study was to identify the gene responsible for the glomerulopathy in a 4-generation Chinese Han pedigree by exome sequencing, using a fast, sensitive and cost-effective method12.
Material & Methods
This study was conducted in the Third Xiangya Hospital, Central South University, Changsha, PR China. A 4-generation, 30-member Chinese Han family was enrolled in this study (Fig. 1). Ten members of this family including seven males and three females had symptomatic glomerulopathy. Consanguineous marriage was denied by the family members. Blood samples (10 ml) were collected from 19 members of this family, including ten patients. Blood samples were also collected from 100 ethnically matched unrelated normal controls (male/female: 50/50, age 40.6 ± 8.4 yr) who were healthy volunteers, without diagnostic features of AS or family history of renal disease. The study was performed during the period of March 2012 and May 2013. The protocol of this study was approved by the Ethics Committee of the Third Xiangya Hospital, Central South University, and all participants signed informed consent.
Fig. 1.
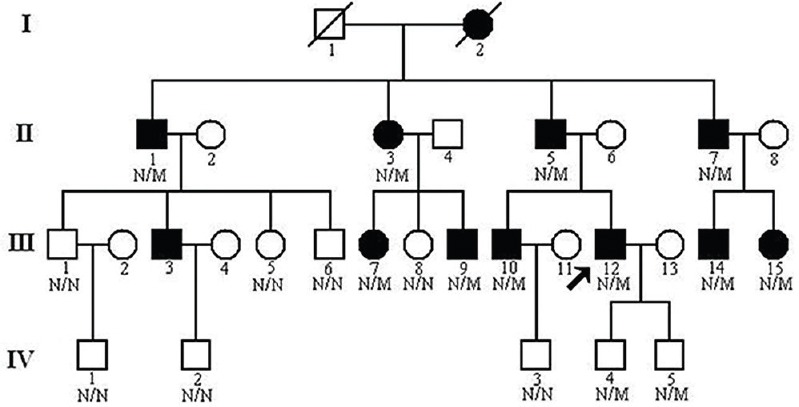
Pedigree of the family with autosomal dominant glomerulopathy. N, normal; M, collagen type IV alpha-4 c.3213delA mutation. Arrow indicates the proband.
Clinical data: Urinalysis and renal function evaluation were performed on all family members2. Members of this family were considered normal if urinalysis identified no more than trace amount of haematuria or proteinuria with normal renal ultrasound examination13. Kidney biopsy was performed for the proband, a 32 yr old male with normal renal function but continuous microhaematuria (III:12, Fig. 1). Light microscopy identified global and segmental sclerosis and mesangial expansion. Electron microscopy revealed that the glomerular basement membranes (GBMs) were irregularly thickened and splitting. No immunoglobulin A deposits were identified on immunofluorescence or electron microscopy. All patients underwent auditory and ophthalmological examinations. None of the family members showed any evidence of auditory, ophthalmological abnormality or leiomyomatosis.
Exome capture: Genomic DNA was isolated from venous blood using standard phenol-chloroform extraction method14,15. Three micrograms of genomic DNA was used to construct the exome library. Genomic DNA of the proband (III:12, Fig. 1) was sheared by sonication and hybridized to the Nimblegen SeqCap EZ Library (Roche, USA) for enrichment, following the manufacturer's protocol. The library enriched for target regions was sequenced on the HiSeq 2000 platform (Illumina, USA) to get paired-end reads with read length of 90 bp1. A mean exome coverage of 59.56× was obtained that provided sufficient depth to accurately call variants at 99.38 per cent of the targeted exome.
Read mapping and variant analysis: The human reference genome was obtained from the UCSC database (http://www.genome.ucsc.edu/), version hg19 (build 37.1). Sequence alignment was performed in the proband using the programme, SOAPaligner (http://soap.genomics.org.cn/soapaligner.html). Moreover, single nucleotide polymorphisms (SNPs) were called using SOAPsnp set with the default parameters after the duplicated reads (produced mainly in the polymerase chain reaction step) were deleted1,16. Short insertions or deletions (indels) affecting coding sequence or splicing sites were identified. The thresholds for calling SNPs and short indels included the number of unique mapped reads supporting an SNP ≥4, and the consensus quality score ≥20 (the quality score is a Phred score, generated by the program SOAPsnp1.03, quality score 20 represents 99 per cent accuracy of a base call), the estimated copy number not >2 and the distance between two SNPs larger than 5. All candidate mutations were filtered against the Single-nucleotide Polymorphism database (dbSNP137, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), 1000 genomes data (1000 genomes release_20100804), HapMap project (2010-08_phase II + III) and YanHuang1 (YH1) project1,12. SIFT prediction (http://sift.jcvi.org) was performed to predict whether an amino acid substitution affects the function of the protein17. Sanger sequencing was employed to validate the identified potential disease-causing variants with ABI3500 sequencer (Applied Biosystems, USA)18. Sequences of the primers were as follows: 5′-CAACTGGATCGTGTTGTGCA-3′ and 5′-GCAGTTTCTTTGATACACTTTGC-3′.
Results
Exome sequencing of the proband (III:12, Fig. 1) in the Chinese family with glomerulopathy was performed. A total of 5.16 billion bases of 90 bp paired-end read sequence were generated for the patient. Among the 5.16 billion bases, 5.02 billion (97.11 %) passed the quality assessment, 4.74 billion (94.45 %) aligned to the human reference sequence and 2.64 billion bases (52.59 %) mapped to the targeted bases with a mean coverage of 59.56-fold. Further, 96,772 genetic variants, including 13,800 non-synonymous variants, were identified in either the coding regions or the splice sites. A prioritization scheme was applied to identify the pathogenic mutation in the patient, similar to earlier studies1,19. Known variants identified in dbSNP137, 1000 genomes project, HapMap and YH1 were excluded. Applying the above strategy, the number of candidate genes was reduced by more than 89.76 per cent.
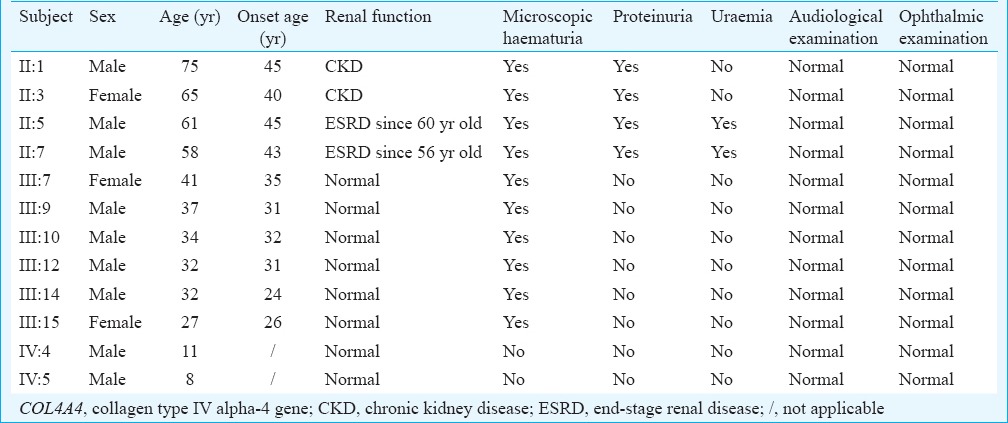
A novel frameshift variant, c.3213delA (p.Gly1072Glufs*69), in the exon 34 of the COL4A4 gene was identified in the proband. The frameshift mutation results in premature stop codon leading to a truncated protein. Such a variation was further confirmed by direct Sanger sequencing (Fig. 2). Mutations in other known kidney disease-causing genes were excluded. The p.Gly1072Glufs*69 variant cosegregated with patients and male carriers in the family, and none of the 100 ethnically matched unrelated controls carried the variant. It was also absent in an in-house database from BGI-Shenzhen with 2375 ethnically matched controls. Our data indicated that the variant, c.3213delA (p.Gly1072Glufs*69), in the COL4A4 gene was the disease-causing mutation in this family. Ten patients (II:1, II:3, II:5, II:7, III:7, III:9, III:10, III:12, III:14 and III:15) who carried this mutation had adult onset AS. Two (II:5 and II:7) of the 10 patients progressed to ESRD at 60 and 56 yr of age, respectively. There were two currently asymptomatic male members (IV:4 and IV:5) who carried the heterozygous p.Gly1072Glufs*69 mutation and they were 11 and eight years old, respectively. The main clinical manifestation was microscopic haematuria, which was present in all patients. Proteinuria was present in four of 10 patients, and chronic kidney disease (CKD) occurred in two patients (Table).
Fig. 2.
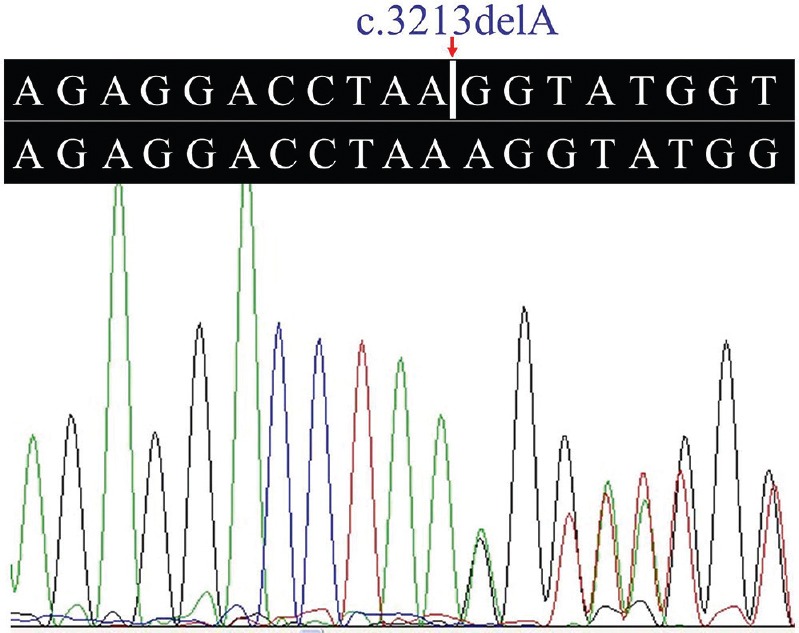
Heterozygous c.3213delA mutation in the collagen type IV alpha-4 gene.
Table.
Clinical data of COL4A4 c.3213delA mutation carriers (n = 12)
Discussion
AS is a clinically and genetically heterogeneous disorder characterized by persistent haematuria and development of ESRD at various ages7. It is a progressive hereditary nephropathy accounting for 1-2 per cent of all cases who need renal replacement therapy in Europe2,20. Alport patients have a thin, multilaminated basement membrane, which is thought to be more fragile and cause microscopic and gross haematuria. The GBM becomes disorganized and thickens with multiple interwoven layers with time6.
In our study a Chinese Han family was investigated that included 10 patients with the marked phenotypic heterogeneity of glomerulopathy. The pattern was most consistent with autosomal dominant inheritance due to male-to-male transmission. Male and female patients were equally, severely affected in this family. Exome sequencing revealed an A deletion at nucleotide 3213 (p.Gly1072Glufs*69) in the COL4A4 gene in the proband. Our data supported that the c.3213delA (p.Gly1072Glufs*69) variant was pathogenic and confirmed the autosomal dominant inheritance pattern in this family. The mutation potentially shortened the α4(IV) chain by 551 amino acids.
The COL4A4 gene is located at chromosome 2q36-q37. It is a large gene with 54 exons. The gene encodes the α4 chain (1690 amino acid residues) of type IV collagen, a major constituent of basement membranes. Although α1 and α2 chains are widely expressed in basement membranes, α3, α4 and α5 chains are specifically expressed in the glomerulus, inner ear and eye21. This family showed a milder phenotype compared to classic AS. The later onset of the development of ESRD, absence of deafness and eye signs have been described in families with autosomal dominant hereditary nephritis2,22.
Mutations in the COL4A4 gene produce abnormal α4(IV) chain which fails to incorporate properly into the triple helix of type IV collagen and leads to a destabilization of the molecular superstructure. Heterozygous mutations may lead to a less severe phenotype than that caused by homozygous mutations because there is still normal α4(IV) chain being produced2. In the family studied, two patients (II:5 and II:7, Fig. 1) progressed to ESRD and required dialysis since the age 60 and 56 yr, respectively.
Extrarenal manifestations were not observed in our family members, consistent with the report of carriers with heterozygous COL4A4 mutation having no hearing loss or the ocular manifestations23. The clinical manifestations of patients were consistent with those with heterozygous COL4A4 mutations, including late onset age of ESRD (an average onset age of ESRD of 58 yr), a lower incidence of hearing impairment and/or ocular changes2,3,20. In our study, all carriers were symptomatic except two obligate, non-penetrant boys (IV:4 and IV:5), which could be due to the young ages (11 and 8 yr). The symptomatic patients showed microhaematuria in the early decades of life and development of proteinuria, CKD and even ESRD after the age of 30 yr, but without deafness or any ocular abnormality. The phenotypic variability in this family may account for collaboration and compensation of the homologue genes, regulation of modifier genes and other acquired factors24.
Mutations in the COL4A4 gene have been reported to cause a spectrum of GBM disorders, ranging from autosomal recessive AS to autosomal dominant AS, benign familial haematuria (BFH) and thin basement membrane nephropathy (TBMN)2,25. Two patients in our study showed the late development of familial proteinuria, CKD and ESRD, consistent with the observation of familial microscopic haematuria, TBMN and late development of ESRD in a family with heterozygous COL4A4 c.3854delG24. Typical AS and BFH are distinct, with the former characterized by progressive nephritis with haematuria and proteinuria, sensorineural deafness and ocular abnormalities, while the latter characterized by prominent diffuse thinning of the GBM, life-long glomerular haematuria and normal renal function26. TBMN (MIM 141200) is more common than AS and affects at least one per cent of the population. It is usually diagnosed clinically when there is persistent glomerular haematuria, minimal proteinuria and normal renal function. Prognosis is usually excellent25. Given that mutation in an important domain of a gene may cause a monogenic disorder, whereas variant in a non-critical region may enhance susceptibility or cause a less severe phenotype of the disorder, COL4A4-related BFH and TBMN may be categorized as less severe forms of AS, consistent with the observations of familial microscopic haematuria, gross haematuria, TBMN and late development of proteinuria, CKD and ESRD in heterozygous carriers with COL4A4 mutation1,21,24,25. Further studies on the genetic and epigenetic factors modifying the expression and function of the COL4A4 gene may help understand the molecular basis of AS better.
In conclusion, this study identified not only the genetic cause of glomerulopathy in the family studied but also two asymptomatic family members harbouring the same COL4A4 c.3213delA mutation. This finding may provide new insights into the cause and diagnosis of AS and have implications for genetic counselling.
Acknowledgment
Authors thank the participants for their cooperation and their efforts in collecting the genetic information and DNA specimens. This work was supported by grants from the National Natural Science Foundation of China (81271921, 81101339, 81001476), the Sheng Hua Scholars Program of Central South University, PR China (H.D.), the Research Fund for the Doctoral Program of Higher Education of China (20110162110026), the Natural Science Foundation of Hunan Province, PR China (10JJ5029), the Construction Fund for Key Subjects of the Third Xiangya Hospital, Central South University and the Students Innovative Pilot Scheme of Central South University (YC12417), PR China.
Footnotes
Conflicts of Interest: None.
References
- 1.Guo Y, Yuan J, Liang H, Xiao J, Xu H, Yuan L, et al. Identification of a novel COL4A5 mutation in a Chinese family with X-linked Alport syndrome using exome sequencing. Mol Biol Rep. 2014;41:3631–5. doi: 10.1007/s11033-014-3227-1. [DOI] [PubMed] [Google Scholar]
- 2.Jefferson JA, Lemmink HH, Hughes AE, Hill CM, Smeets HJ, Doherty CC, et al. Autosomal dominant Alport syndrome linked to the type IV collage alpha 3 and alpha 4 genes (COL4A3 and COL4A4) Nephrol Dial Transplant. 1997;12:1595–9. doi: 10.1093/ndt/12.8.1595. [DOI] [PubMed] [Google Scholar]
- 3.Ciccarese M, Casu D, Ki Wong F, Faedda R, Arvidsson S, Tonolo G, et al. Identification of a new mutation in the alpha4(IV) collagen gene in a family with autosomal dominant Alport syndrome and hypercholesterolaemia. Nephrol Dial Transplant. 2001;16:2008–12. doi: 10.1093/ndt/16.10.2008. [DOI] [PubMed] [Google Scholar]
- 4.Kruegel J, Rubel D, Gross O. Alport syndrome – insights from basic and clinical research. Nat Rev Nephrol. 2013;9:170–8. doi: 10.1038/nrneph.2012.259. [DOI] [PubMed] [Google Scholar]
- 5.Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, et al. Alport syndrome in women and girls. Clin J Soc Nephrol. 2016;11:1713–20. doi: 10.2215/CJN.00580116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demosthenous P, Voskarides K, Stylianou K, Hadjigavriel M, Arsali M, Patsias C, et al. X-linked Alport syndrome in Hellenic families: phenotypic heterogeneity and mutations near interruptions of the collagen domain in COL4A5. Clin Genet. 2012;81:240–8. doi: 10.1111/j.1399-0004.2011.01647.x. [DOI] [PubMed] [Google Scholar]
- 7.Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010;21:876–83. doi: 10.1681/ASN.2009070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Momota R, Sugimoto M, Oohashi T, Kigasawa K, Yoshioka H, Ninomiya Y. Two genes, COL4A3 and COL4A4 coding for the human alpha3(IV) and alpha4(IV) collagen chains are arranged head-to-head on chromosome 2q36. FEBS Lett. 1998;424:11–6. doi: 10.1016/s0014-5793(98)00128-8. [DOI] [PubMed] [Google Scholar]
- 9.Kashtan CE. Alport syndrome and the X chromosome: implications of a diagnosis of Alport syndrome in females. Nephrol Dial Transplant. 2007;22:1499–505. doi: 10.1093/ndt/gfm024. [DOI] [PubMed] [Google Scholar]
- 10.Stokman MF, Renkema KY, Giles RH, Schaefer F, Knoers NV, van Eerde AM. The expanding phenotypic spectra of kidney diseases: insights from genetic studies. Nat Rev Nephrol. 2016;12:472–83. doi: 10.1038/nrneph.2016.87. [DOI] [PubMed] [Google Scholar]
- 11.Morinière V, Dahan K, Hilbert P, Lison M, Lebbah S, Topa A, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol. 2014;25:2740–51. doi: 10.1681/ASN.2013080912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiu X, Yuan J, Deng X, Xiao J, Xu H, Zeng Z, et al. A novel COL4A5 mutation identified in a Chinese Han family using exome sequencing. Biomed Res Int. 2014;2014:186048. doi: 10.1155/2014/186048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becknell B, Zender GA, Houston R, Baker PB, McBride KL, Luo W, et al. Novel X-linked glomerulopathy is associated with a COL4A5 missense mutation in a non-collagenous interruption. Kidney Int. 2011;79:120–7. doi: 10.1038/ki.2010.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan L, Song Z, Xu H, Gu S, Zhu A, Gong L, et al. EIF4G1 Ala502Val and Arg1205His variants in Chinese patients with Parkinson disease. Neurosci Lett. 2013;543:69–71. doi: 10.1016/j.neulet.2013.02.056. [DOI] [PubMed] [Google Scholar]
- 15.Di Pietro F, Ortenzi F, Tilio M, Concetti F, Napolioni V. Genomic DNA extraction from whole blood stored from 15- to 30-years at -20 °C by rapid phenol-chloroform protocol: a useful tool for genetic epidemiology studies. Mol Cell Probes. 2011;25:44–8. doi: 10.1016/j.mcp.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–4. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Li Y, Zhang D, Zhang H, Li Y, Lu F, et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 2011;7:e1002084. doi: 10.1371/journal.pgen.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Y, Yang H, Deng X, Song Z, Yang Z, Xiong W, et al. Genetic analysis of the S100B gene in Chinese patients with Parkinson disease. Neurosci Lett. 2013;555:134–6. doi: 10.1016/j.neulet.2013.09.037. [DOI] [PubMed] [Google Scholar]
- 19.Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–23. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marcocci E, Uliana V, Bruttini M, Artuso R, Silengo MC, Zerial M, et al. Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome. Nephrol Dial Transplant. 2009;24:1464–71. doi: 10.1093/ndt/gfn681. [DOI] [PubMed] [Google Scholar]
- 21.Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Novel COL4A4 splice defect and in-frame deletion in a large consanguine family as a genetic link between benign familial haematuria and autosomal Alport syndrome. Nephrol Dial Transplant. 2003;18:1122–7. doi: 10.1093/ndt/gfg157. [DOI] [PubMed] [Google Scholar]
- 22.Pochet JM, Bobrie G, Landais P, Goldfarb B, Grünfeld JP. Renal prognosis in Alport's and related syndromes: influence of the mode of inheritance. Nephrol Dial Transplant. 1989;4:1016–21. [PubMed] [Google Scholar]
- 23.Dagher H, Yan Wang Y, Fassett R, Savige J. Three novel COL4A4 mutations resulting in stop codons and their clinical effects in autosomal recessive Alport syndrome. Hum Mutat. 2002;20:321–2. doi: 10.1002/humu.9065. [DOI] [PubMed] [Google Scholar]
- 24.Pierides A, Voskarides K, Athanasiou Y, Ioannou K, Damianou L, Arsali M, et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24:2721–9. doi: 10.1093/ndt/gfp158. [DOI] [PubMed] [Google Scholar]
- 25.Rana K, Tonna S, Wang YY, Sin L, Lin T, Shaw E, et al. Nine novel COL4A3 and COL4A4 mutations and polymorphisms identified in inherited membrane diseases. Pediatr Nephrol. 2007;22:652–7. doi: 10.1007/s00467-006-0393-y. [DOI] [PubMed] [Google Scholar]
- 26.Kaneko K, Tanaka S, Hasui M, Nozu K, Krol RP, Iijima K, et al. A family with X-linked benign familial hematuria. Pediatr Nephrol. 2010;25:545–8. doi: 10.1007/s00467-009-1370-z. [DOI] [PubMed] [Google Scholar]