

Abstract
Background & objectives:
Clinically, nephrotic syndrome (NS) is a diverse group of symptoms; about 20 per cent of NS cases are resistant to steroid treatment, and within ten years they progress to end-stage renal disease. The present study was undertaken to identify the mutations of Wilms’ tumour 1 (WT1) gene in steroid-resistant NS (SRNS) children.
Methods:
A total of 173 children with SRNS and 100 children in the control group were enrolled in the study. DNA extraction was done, screened for WT1 (exons 8 and 9) gene amplified by polymerase chain reaction and direct sequencing. Karyotype analyses were done for WT1 mutation cases.
Results:
WT1 mutations were found in three of 173 SRNS cases (2 girls, 1 boy). All of them had intron 9 (IVS 9 + 4 C>T, 2; IVS + 5 G>A, 1) mutation. Of these three cases, one had familial and another two had sporadic history. Renal histology analysis showed two cases with focal segmental glomerulosclerosis (FSGS) and they had external female genitalia but 46, XY karyotype. Both of them had streak gonads. Of the three cases, one expired.
Interpretation & conclusions:
The findings of the present study indicate that all females with SRNS-FSGS should be screened for WT1 gene mutation to diagnose whether they have FS for possible gonadectomy.
Keywords: Denys-Drash syndrome, focal segmental glomerulosclerosis, Frasier syndrome, Indian children, steroid-resistant nephrotic syndrome, Wilms’ tumour 1
Nephrotic syndrome (NS) is the most frequent renal-related syndrome in childhood. According to the clinical definitions, it requires the presence of oedema, massive proteinuria, hypoalbuminaemia and high cholesterol1. As per the treatment response, NS was categorized as steroid sensitive/remission, steroid responders/dependent (further this group divided into frequent and infrequent relapse) and finally, steroid non-responder/steroid resistant2. According to renal histological findings, NS is classified by minimal change nephrotic (MCN), focal segmental glomerulosclerosis (FSGS) and diffuse mesangial sclerosis (DMS). Many genes (NPHS1, NPHS2, WT1, LAMB2, CD2AP, α-ACT4 and PLCE1) have been identified and well documented in familial and sporadic NS cases3,4. In India, the two available studies have focussed on NPHS2 gene only5,6.
Wilms’ tumour (WT1) gene is originally a tumour suppressor gene; with a span of about 50 kb and consists of 10 exons which encode mRNA transcript of about 3.2 kb. In the hot spot sites (exons 5, 7, 8 and 9), small amount of mutant effect is sufficient to disrupt urogenital function; its immensity expression is linked to podocyte differentiation during nephrogenesis and gonadal development7,8,9. Usually, 95 per cent of mutations within the WT1 gene causing steroid – resistant (SRNS) occur within the two exons 8 and 910. Frasier syndrome (FS) and Denys-Drash syndrome (DDS) are caused by the WT1 mutation. These disorders are characterised by NS, sex reversal, gonadoblastoma and WT. The WT, FS and DDS are successfully treated in majority (80%) of the cases7,11,12,13. There has been no study on WT1 mutation from south India, thus the molecular mechanisms of WT1 gene and its link to SRNS are poorly understood. We therefore, undertook this study in a tertiary care hospital in south India to analyse the WT1 (exon 8 and 9) gene mutations in children with SRNS.
Material & Methods
This study was conducted in the department of Paediatric Nephrology, Institute of Child Health and Hospital for Children (ICH & HC), Madras Medical College between 2008 and 2012. Failure to respond to oral prednisolone of 2 mg/kg/day for four weeks followed by three doses of pulse methyl prednisolone was considered as steroid resistance. The children with SRNS were followed up for a minimum of six months before including in the study. All initial steroid-sensitive NS (SSNS) patients who subsequently turned to be SRNS (frequent relapse/late non-responders), chronic kidney disease (CKD) cases with a history of SRNS (aged 1-12 yr) were included in the study. Children with SSNS and history of positive HIV and HbsAg were excluded from the study. Children with minor illness (such as fever and diarrhoea) were taken as control, all of them aged between 3-12 years. Both cases and controls were selected randomly from the outpatient department. Ethical approval was obtained from the institutional ethical committee and written informed consents were obtained from the patient's parents. Blood sample (5 ml) was collected; genomic DNA was isolated from blood leucocytes according to a standard salting out method14. The primer sets cover the WT1 exons (gene expressed region) and introns based on a previously published report15, genomic DNA was amplified by polymerase chain reaction (PCR) using flanking specific sequences for confirmation of the genotype primer sets - forward 5’-CCT TTA ATG AGA TCC CCT TTT CC-3’and reverse 5’-GGG GAA ATG TGG GGT GTT TCC-3’, forward 5’-CCT CAC TGT GCC CAC ATT GT-3’ and reverse 5’-GCA CTA TTC CTT CTC TCA ACT GAG-3’. Amplification with the primer pairs resulted in 391 and 349 bp products. PCR reaction was performed in 25 μl reactions (0.5 μg genomic DNA, 200 pmol of each primer, 0.5 mM dNTPs, 3 mM MgCl2, 1 unit of Taq DNA polymerase (Genet Bio, South Korea) and ×10 PCR buffer (Genet Bio) with initial denaturation at 94°C for 10 min followed by 30 cycles of one min at 94°C, one min at 58°C (annealing) and two mins at 72°C (extension) in a thermal cycler (Eppendorf AG, Germany). After amplification, the PCR products were electrophoresed in 1.5 per cent agarose gels containing 0.5 µg/ml ethidium bromide. The gels were run in ×0.5 TBE (Tris-borate-EDTA) buffer for 40 min at 100 volt and visualized under ultraviolet transilluminator and documented in a gel documentation unit (Vilber – Lourmat, France). The PCR product was purified in PureLink™ kit (Thermo Fisher Scientific, USA) and products were loaded in the sequencer (Genetic analyzer, 3500 from Applied Biosystems, USA).
Results & Discussion
A total of 173 children with SRNS were studied, including 107 boys (mean age 7.4 ± 3.76 yr) and 66 girls (6.7 ± 3.38 yr). Renal biopsy was done for 71 SRNS cases [MCN, 46 (31 boys, 15 girls); FSGS, 18 (11 boys, 7 girls); DMS, 7 (2 boys, 5 girls)]. One hundred (63 boys, 37 girls, age 6.19 ± 1.85 and 5.8 ± 1.77 yr, respectively) controls were also screened. Molecular genetic analysis confirmed that three patients (2 girls and 1 boy) had intron 9 mutation (Table, Figure) and no mutation was identified in control group. In mutation cases, the first girl had a history of familial NS with two previous (sisters) deaths; the other two had sporadic. Karyotype analysis was carried out for all the three cases. Two of them were phenotypically females, but genetically they were males (sex reversal). Their renal histology showed FSGS. Abdominal scans of these two patients showed normal uterus with streak gonads and considerable risk of gonadoblastoma. Gonadectomy was done for both cases, and tissue biopsy was reported as dysgerminoma stage I. The first patient underwent a live donor (mother) renal transplant; and recovered with a stable graft function. The second was diagnosed as end-stage renal disease (ESRD) and underwent haemodialysis and finally died. The third case was a seven year old nephrotic boy; histopathology report was MCNs. He had normal genotype, and was on steroid treatment (infrequent relapses) with regular follow up.
Table.
Summary of Wilms’ tumour 1 mutation in steroid-resistant nephrotic syndrome cases
Figure.
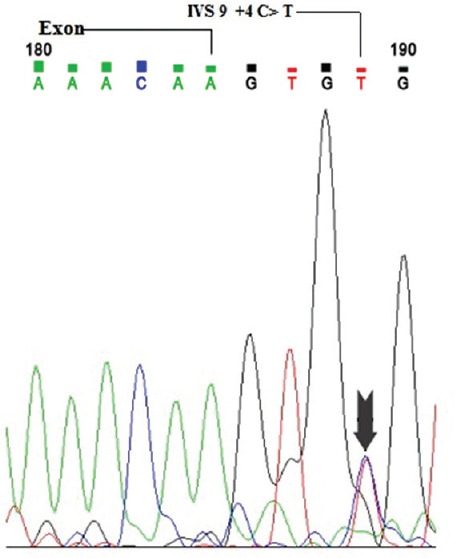
The chromatogram indicates heterozygous mutation in 9 exon/intron junction (arrow indicates Cytosine (C) to Thymine (T) conversion at position +4 intron region) was identified in the steroid-resistant nephrotic syndrome case.
Frasier et al16 initially reported a pair of 46, XY monozygotic twins with pure gonadal dysgenesis, gonadoblastoma and streak gonads. Most of the FS cases have proteinuria in childhood and progressively with age they develop ESRD. FS is caused by mutations in the intronic region of 8 and 9; it leads to loss of lysine, threonine and serine (KTS) isoforms. Drash et al17 described a child with male pseudohermaphroditism, WT and nephropathy in association with sex chromosomal abnormality. Most of the DDS cases have germ line, missense and heterozygous form mutations, and these occur in exons 7, 8 and 918. The classical presentation of DDS is a child with ambiguous genitalia, in exceptional cases they may be normal male or female19. DDS usually has an early onset (first 3 years of life), but FS progresses slowly20 (puberty period).
Usually five nucleotides are altered in 9 intronic region these are +2T>C, +4 C>T, +5 G>A, +5 G>T, +6 T>A, most frequent being +4 C>T position7,10. In our study two patients had +4 C>T nucleotide mutation. The onset was at five and six years, respectively. On the contrary, one Chinese study reported 4 C>T mutation in a female child with streak gonad, who had ESRD occurring at 8 months21, Fujita et al22 reported a three year old girl with SRNS and FS. Interestingly, one unusual FS case was reported with an IVS 9 + 4 C>T mutation, genotypically the case was a male and presented with diaphragmatic hernia, hypospadias and unilateral cryptorchidism23. According to the reports, WT1 mutation mostly occurred in SRNS girls7,10,11,12,13,24. Early (pre-pubertal period) diagnosis may help in the management of FS and to offer a reliable prognosis11,12,19,22. Sinha et al13 reported SRNS-FSGS girls with 9.5 per cent mutation, it is higher than our study. In our study, only one female child had familial history, her first sibling had physical development delay, unable to sit, stand and walk without support; she died at the age of three years. The second sibling had NS with irregular follow up, she died at the age of 12. In this study, it was confirmed that the third girl had intronic mutation. Denamur et al25 reported a mother of a girl with NS having proteinuria since the age six, she had normal phenotype and normal genital development with pregnancy. Her child had DDS with 46, XY genotype, WT1 intronic mutation identified for both. In another report, siblings of two female patients had glomerular disease and developed CKD; both of them had intronic mutation26. WT1 mutations occur rarely in boys, in our study, one boy had WT1 mutation; the same type also reported in an earlier study20.
Genotype and phenotype correlations are not always possible in WT1 mutation cases27,28. Previous reports29,30 and the present study indicate that WT1 gene alteration, and phenotype correlation between boys and girls are difficult to interpret. Early diagnosis of FS in girls helps trace the streak gonads from early gonadectomy.
In conclusion, in our study, two of 66 girls and one of 107 boys with SRNS had WT1 mutation; the boy had no other morphological abnormality. The pathogenesis of WT1 mutations in the early-onset age in female NS children becomes imperative to be examined. In future, studies need to be performed on a large sample to confirm the findings.
Acknowledgment
Authors thank the Director, (ICH&HC, Egmore, Chennai) for the approval of the study, and staffs and social workers of the Deparment of Paediatric Nephrology, ICH & HC for their assistance and data collection. Authors acknowledge Prof. P.S. Rahumathulla, former Head, Department of Animal Genetics and Breeding, Madras Veterinary College (MVC), Chennai, for providing laboratory facilities.
References
- 1.Bagga A, Mantan M. Nephrotic syndrome in children. Indian J Med Res. 2005;122:13–28. [PubMed] [Google Scholar]
- 2.Hogg RJ, Portman RJ, Milliner D, Lemley KV, Eddy A, Ingelfinger J. Evaluation and management of proteinuria and nephrotic syndrome in children: recommendations from a pediatric nephrology panel established at the National Kidney Foundation conference on proteinuria, albuminuria, risk, assessment, detection, and elimination (PARADE) Pediatrics. 2000;105:1242–9. doi: 10.1542/peds.105.6.1242. [DOI] [PubMed] [Google Scholar]
- 3.Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119:e907–19. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 4.Mbarek IB, Abroug S, Omezzine A, Pawtowski A, Gubler MC, Bouslama A, et al. Novel mutations in steroid-resistant nephrotic syndrome diagnosed in Tunisian children. Pediatr Nephrol. 2011;26:241–9. doi: 10.1007/s00467-010-1694-8. [DOI] [PubMed] [Google Scholar]
- 5.Choudhry S, Bagga A, Hari P, Sharma S, Kalaivani M, Dinda A. Efficacy and safety of tacrolimus versus cyclosporine in children with steroid-resistant nephrotic syndrome: a randomized controlled trial. Am J Kidney Dis. 2009;53:760–9. doi: 10.1053/j.ajkd.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 6.Vasudevan A, Siji A, Raghavendra A, Sridhar TS, Phadke KD. NPHS2 mutations in Indian children with sporadic early steroid resistant nephrotic syndrome. Indian Pediatr. 2012;49:231–3. doi: 10.1007/s13312-012-0057-x. [DOI] [PubMed] [Google Scholar]
- 7.Scholz H, Kirschner KM. A role for the Wilms’ tumor protein WT1 in organ development. Physiology (Bethesda) 2005;20:54–9. doi: 10.1152/physiol.00048.2004. [DOI] [PubMed] [Google Scholar]
- 8.Klattig J, Sierig R, Kruspe D, Makki MS, Englert C. WT1-mediated gene regulation in early urogenital ridge development. Sex Dev. 2007;1:238–54. doi: 10.1159/000104774. [DOI] [PubMed] [Google Scholar]
- 9.Kreidberg JA. WT1 and kidney progenitor cells. Organogenesis. 2010;6:61–70. doi: 10.4161/org.6.2.11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mucha B, Ozaltin F, Hinkes BG, Hasselbacher K, Ruf RG, Schultheiss M, et al. Mutations in the Wilms’ tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr Res. 2006;59:325–31. doi: 10.1203/01.pdr.0000196717.94518.f0. [DOI] [PubMed] [Google Scholar]
- 11.Aucella F, Bisceglia L, De Bonis P, Gigante M, Caridi G, Barbano G, et al. WT1 mutations in nephrotic syndrome revisited. High prevalence in young girls, associations and renal phenotypes. Pediatr Nephrol. 2006;21:1393–8. doi: 10.1007/s00467-006-0225-0. [DOI] [PubMed] [Google Scholar]
- 12.Auber F, Lortat-Jacob S, Sarnacki S, Jaubert F, Salomon R, Thibaud E, et al. Surgical management and genotype/phenotype correlations in WT1 gene-related diseases (Drash, Frasier syndromes) J Pediatr Surg. 2003;38:124–9. doi: 10.1053/jpsu.2003.50025. [DOI] [PubMed] [Google Scholar]
- 13.Sinha A, Sharma S, Gulati A, Sharma A, Agarwala S, Hari P, et al. Frasier syndrome: early gonadoblastoma and cyclosporine responsiveness. Pediatr Nephrol. 2010;25:2171–4. doi: 10.1007/s00467-010-1518-x. [DOI] [PubMed] [Google Scholar]
- 14.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruf RG, Schultheiss M, Lichtenberger A, Karle SM, Zalewski I, Mucha B, et al. Prevalence of WT1 mutations in a large cohort of patients with steroid-resistant and steroid-sensitive nephrotic syndrome. Kidney Int. 2004;66:564–70. doi: 10.1111/j.1523-1755.2004.00775.x. [DOI] [PubMed] [Google Scholar]
- 16.Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr. 1964;64:740–5. doi: 10.1016/s0022-3476(64)80622-3. [DOI] [PubMed] [Google Scholar]
- 17.Drash A, Sherman F, Hartmann WH, Blizzard RM. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr. 1970;76:585–93. doi: 10.1016/s0022-3476(70)80409-7. [DOI] [PubMed] [Google Scholar]
- 18.da Silva TE, Nishi MY, Costa EM, Martin RM, Carvalho FM, Mendonca BB, et al. A novel WT1 heterozygous nonsense mutation (p.K248X) causing a mild and slightly progressive nephropathy in a 46, XY patient with Denys-Drash syndrome. Pediatr Nephrol. 2011;26:1311–5. doi: 10.1007/s00467-011-1847-4. [DOI] [PubMed] [Google Scholar]
- 19.Megremis S, Mitsioni A, Fylaktou I, Tzeli SK, Komianou F, Stefanidis CJ, et al. Broad and unexpected phenotypic expression in Greek children with steroid-resistant nephrotic syndrome due to mutations in the Wilms’ tumor 1 (WT1) gene. Eur J Pediatr. 2011;170:1529–34. doi: 10.1007/s00431-011-1450-5. [DOI] [PubMed] [Google Scholar]
- 20.Klamt B, Koziell A, Poulat F, Wieacker P, Scambler P, Berta P, et al. Syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/-KTS splice isoforms. Hum Mol Genet. 1998;7:709–14. doi: 10.1093/hmg/7.4.709. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Ding J, Zhao D, Yu Z, Fan Q, Chen Y, et al. WT1 gene mutations in Chinese children with early onset nephrotic syndrome. Pediatr Res. 2010;68:155–8. doi: 10.1203/PDR.0b013e3181e4c9e3. [DOI] [PubMed] [Google Scholar]
- 22.Fujita S, Sugimoto K, Miyazawa T, Yanagida H, Tabata N, Okada M, et al. A female infant with Frasier syndrome showing splice site mutation in Wilms’ tumor gene (WT1) intron 9. Clin Nephrol. 2010;73:487–91. doi: 10.5414/cnp73487. [DOI] [PubMed] [Google Scholar]
- 23.Melo KF, Martin RM, Costa EM, Carvalho FM, Jorge AA, Arnhold IJ, et al. An unusual phenotype of Frasier syndrome due to IVS9+ 4C>T mutation in the WT1 gene: predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J Clin Endocrinol Metab. 2002;87:2500–5. doi: 10.1210/jcem.87.6.8521. [DOI] [PubMed] [Google Scholar]
- 24.Yang YH, Zhao F, Feng DN, Wang JJ, Wang CF, Huang J, et al. Wilms’ tumor suppressor gene mutations in girls with sporadic isolated steroid-resistant nephrotic syndrome. Genet Mol Res. 2013;12:6184–91. doi: 10.4238/2013.December.4.5. [DOI] [PubMed] [Google Scholar]
- 25.Denamur E, Bocquet N, Mougenot B, Da Silva F, Martinat L, Loirat C, et al. Mother-to-child transmitted WT1 splice-site mutation is responsible for distinct glomerular diseases. J Am Soc Nephrol. 1999;10:2219–23. doi: 10.1681/ASN.V10102219. [DOI] [PubMed] [Google Scholar]
- 26.Demmer L, Primack W, Loik V, Brown R, Therville N, McElreavey K. Frasier syndrome: a cause of focal segmental glomerulosclerosis in a 46, XX female. J Am Soc Nephrol. 1999;10:2215–8. doi: 10.1681/ASN.V10102215. [DOI] [PubMed] [Google Scholar]
- 27.Guaragna MS, Lutaif AC, Bittencourt VB, Piveta CS, Soardi FC, Castro LC, et al. Frasier syndrome: four new cases with unusual presentations. Arq Bras Endocrinol Metabol. 2012;56:525–32. doi: 10.1590/s0004-27302012000800011. [DOI] [PubMed] [Google Scholar]
- 28.McTaggart SJ, Algar E, Chow CW, Powell HR, Jones CL. Clinical spectrum of Denys-Drash and Frasier syndrome. Pediatr Nephrol. 2001;16:335–9. doi: 10.1007/s004670000541. [DOI] [PubMed] [Google Scholar]
- 29.Lipska BS, Ranchin B, Iatropoulos P, Gellermann J, Melk A, Ozaltin F, et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. 2014;85:1169–78. doi: 10.1038/ki.2013.519. [DOI] [PubMed] [Google Scholar]
- 30.Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. 2001;106:319–29. doi: 10.1016/s0092-8674(01)00453-6. [DOI] [PubMed] [Google Scholar]