

Abstract
To identify genetic variants influencing plasma lipid concentrations, we first used genotype imputation and meta-analysis to combine three genome-wide scans totaling 8,816 individuals and comprising 6,068 individuals specific to our study (1,874 individuals from the FUSION study of type 2 diabetes and 4,184 individuals from the SardiNIA study of aging-associated variables) and 2,758 individuals from the Diabetes Genetics Initiative, reported in a companion study in this issue. We subsequently examined promising signals in 11,569 additional individuals. Overall, we identify strongly associated variants in eleven loci previously implicated in lipid metabolism (ABCA1, the APOA5-APOA4-APOC3-APOA1 and APOE-APOC clusters, APOB, CETP, GCKR, LDLR, LPL, LIPC, LIPG and PCSK9) and also in several newly identified loci (near MVK-MMAB and GALNT2, with variants primarily associated with high-density lipoprotein (HDL) cholesterol; near SORT1, with variants primarily associated with low-density lipoprotein (LDL) cholesterol; near TRIB1, MLXIPL and ANGPTL3, with variants primarily associated with triglycerides; and a locus encompassing several genes near NCAN, with variants strongly associated with both triglycerides and LDL cholesterol). Notably, the 11 independent variants associated with increased LDL cholesterol concentrations in our study also showed increased frequency in a sample of coronary artery disease cases versus controls.
Coronary artery disease (CAD) and stroke are the leading causes of morbidity, mortality and disability in industrialized countries, and the prevalence of these diseases is increasing rapidly in developing countries1. A main underlying pathology is atherosclerosis, a process of cumulative deposition of LDL cholesterol in the arteries supplying blood to the heart and brain that eventually leads to impaired or absent blood supply and myocardial infarction or stroke1. Consistent and compelling evidence has demonstrated association between lipoprotein-associated lipid concentrations and cardiovascular disease incidence worldwide2–4. Whereas high concentrations of LDL cholesterol are associated with increased risk of CAD, high concentrations of HDL cholesterol are associated with decreased risk of CAD. Specifically, it has been estimated that each 1% decrease in LDL cholesterol concentrations reduces the risk of coronary heart disease by ~1% (ref. 5), and each 1% increase in HDL cholesterol concentrations reduces the risk of coronary heart disease by ~2% (ref. 6). A recent meta-analysis of data on 150,000 individuals, including 3,000 with CAD-related deaths, shows that the two factors are independently associated with CAD risk7. There is evidence that a high concentration of triglycerides is an additional, independent risk factor for cardiovascular disease8,9, although whether this association is causal is still under debate.
Smoking, diet and physical activity all have a role in determining individual lipid profiles. Still, family studies suggest that in many populations, about half of the variation in these traits is genetically determined10,11, and it is clear that LDL cholesterol, HDL cholesterol and triglyceride concentrations are strongly influenced by the genetic constitution of each individual. Furthermore, genetic variants that increase LDL cholesterol concentrations—such as rare variants in the LDL receptor (LDLR) and apolipoprotein B (APOB) genes and common variants in the apolipoprotein E (APOE) gene—have also been associated with increased susceptibility to coronary heart disease12. Thus, the available evidence demonstrates not only that genetic variants account for a substantial fraction of individual variation in lipid concentrations, but also that lipid concentrations are associated with the risk of CAD.
Although several genes and genetic variants have been found that associate with individual variation in lipid concentrations, additional variants influencing these traits remain to be identified. As with other complex traits, identification of genes influencing lipid concentrations is likely to be much enhanced by large sample sizes. Thus, we decided to combine genome-wide association scan data from two of our studies, including 1,874 individuals from the FUSION study of type 2 diabetes13 and 4,184 individuals from the SardiNIA study of aging-associated variables10,14, with data on 2,758 individuals from the Diabetes Genetics Initiative15,16. Here, we describe results of a combined analysis of the three genome-wide association scans involving a total of 8,816 individuals and our follow-up assessments of up to 11,569 individuals, which were done in order to verify common genetic variants associated with plasma concentrations of LDL cholesterol, HDL cholesterol and triglycerides. Our results identify >25 independent common variants associated with individual variation in lipid concentrations (each with P < 5 × 10−8). Some are located in previously implicated loci, indicating that our approach was valid, and others are found in loci where genetic variants have not been previously implicated in lipid metabolism. Our results also provide promising, albeit not definitive, evidence of association between several other common variants and lipid concentrations. In a companion manuscript, Kathiresan and colleagues from the Diabetes Genetics Initiative report results of their own follow-up genotyping of SNPs selected on the basis of our combined analysis of the three scans, their original analyses, and previously published reports. Their independent follow-up samples and genotyping further support the newly identified loci reported here.
RESULTS
Genome-wide association scans
To survey the genome for common variants associated with plasma concentrations of HDL cholesterol, LDL cholesterol and triglyceride concentrations, we conducted genome-wide association scans on two different populations. In one scan, after we excluded markers on the basis of quality-control filters (see Methods), we examined 304,581 SNPs with minor allele frequency (MAF) >1% from the Illumina HumanHap300 BeadChip and a GoldenGate panel designed to improve coverage around type 2 diabetes (T2D) candidate genes in 1,874 Finnish individuals from the Finland–United States Investigation of NIDDM Genetics (FUSION) study13. In a second scan, after quality-control filtering, we examined 356,539 SNPs (MAF > 5%) from the Affymetrix 500K Mapping Array Set in 4,184 individuals from the SardiNIA Study of Aging10,14. The Sardinian sample is organized into a number of small-to medium-sized pedigrees. We took advantage of this relatedness to reduce genotyping costs: we genotyped 1,412 individuals with the Affymetrix 500K Mapping Array Set (organized into groups of 2–3 individuals per nuclear family) and then propagated their genotypes to the remaining individuals, who were genotyped using only the Affymetrix 10K Mapping Array14,17,18 (see Methods). To increase statistical power, we also contacted the authors of a previously published study15 to obtain results for 347,010 SNPs (MAF > 5%) genotyped in 2,758 Finnish and Swedish individuals from the Diabetes Genetics Initiative (DGI) using the Affymetrix 500K Mapping Array Set. Further details of the DGI study and independent follow-up analyses are provided in a companion manuscript16. All three initial scans excluded individuals taking lipid lowering therapies, for a total of 8,816 phenotyped individuals (Table 1). Informed consent was obtained from all study participants and ethics approval was obtained from the participating institutions.
Table 1.
Characteristics of samples used in genome-wide and follow-up analyses
| Samples | Phenotyped individualsa (% female) | Demographics
|
Median trait concentrations (quartile ranges)
|
||||
|---|---|---|---|---|---|---|---|
| Geographic origin | Median age (quartile range) | Median BMI (quartile range) | HDL-C (mg/dl) | LDL-C (mg/dl) | Triglycerides (mg/dl) | ||
| Genome-wide analyses (n = 8,816) | |||||||
| FUSION | |||||||
| Type 2 diabetics | 773 (41%) | Finland | 63.0 (11.1) | 29.8 (6.1) | 44.9 (15.9) | 135.6 (44.5) | 150.6 (106.3) |
| Controls | 1,101 (48%) | Finland | 62.0 (14.5) | 26.6 (5.0) | 54.6 (21.7) | 141.1 (44.9) | 103.7 (60.2) |
| SardiNIA | 4,184 (57%) | Sardinia (in Italy) | 42.4 (28.0) | 24.9 (6.4) | 62.7 (18.6) | 124.6 (47.6) | 70.0 (54.0) |
| DGI | 2,758 (51%) | Finland, Sweden | 62.8 (15.5) | 27.3 (5.4) | 46.2 (15.9) | 148.3 (51.8) | 121.7 (81.9) |
| Follow-up samples (n = 11,569) | |||||||
| FUSION | |||||||
| Type 2 diabetics | 970 (41%) | Finland | 60.0 (11.0) | 30.2 (6.5) | 49.1 (17.0) | 123.5 (51.6) | 139.1 (90.4) |
| Controls | 1,249 (39%) | Finland | 59.0 (10.5) | 26.4 (4.9) | 56.1 (21.3) | 138.4 (46.1) | 103.2 (55.8) |
| ISIS | |||||||
| Myocardial infarction survivors | 1,254 (28%) | United Kingdom | 52.0 (14.0) | 26.0 (6.0) | 40.6 (12.4) | 144.0 (48.4) | n/a |
| Controls | 1,252 (35%) | United Kingdom | 48.0 (14.0) | 24.0 (5.0) | 49.9 (16.3) | 124.2 (41.4) | 132.0 (102.8) |
| HAPI | 861 (46%) | United States | 43.0 (22.0) | 25.9 (5.9) | 55.8 (18.0) | 139.1 (56.0) | 68.5 (38.0) |
| SUVIMAX | 1,551 (62%) | France | 50.0 (9.0) | 23.3 (4.1) | 61.9 (21.9) | 135.8 (41.4) | 80.0 (41.6) |
| BWHHS | 3,358 (100%) | United Kingdom | 69.0 (9.0) | 26.9 (6.1) | 61.9 (23.2) | 158.3 (54.2) | 141.8 (90.4) |
| Caerphilly | 1,074 (0%) | United Kingdom | 57.0 (8.0) | 26.1 (4.1) | 51.5 (17.0) | 142.3 (54.3) | 132.9 (102.8) |
Individuals known to be on lipid lowering therapies were excluded; see Methods.
Because the three studies used different marker sets with an overlap of only 44,998 SNPs across studies, we used information on patterns of haplotype variation in the HapMap CEU samples (release 21)19 to infer missing genotypes in silico and to facilitate comparison between the studies13. Imputation analyses were carried out with Markov Chain Haplotyping software (MaCH; see URLs section in Methods). For our analyses, we only considered SNPs that were either genotyped or could be imputed with relatively high confidence; that is, SNPs for which patterns of haplotype sharing between sampled individuals and those genotyped by the HapMap consistently indicated a specific allele. Comparison of imputed and experimentally derived genotypes in our samples yielded estimated error rates of 1.46% (for imputation based on Illumina genotypes) to 2.14% (imputation based on Affymetrix genotypes) per allele, consistent with expectations from HapMap data. For additional details of quality-control and imputation procedures, see Methods and Supplementary Table 1 online.
We then conducted a series of association analyses to relate the ~2,261,000 genotyped and/or imputed SNPs with plasma concentrations of HDL cholesterol, LDL cholesterol and triglyceride concentrations. For each SNP, lipid concentrations were regressed onto allele counts in a regression model that also included gender, age and age2 as covariates. For the FUSION sample, we analyzed T2D cases and controls separately, and added additional covariates accounting for birth province and study subset. For the DGI sample, we analyzed cases and controls together using an additional covariate to indicate diabetes status. For SNPs genotyped in the lab, allele counts were discrete (0, 1 or 2), whereas for SNPs genotyped in silico, allele counts were fractional (between 0.0 and 2.0, depending on the imputed number of copies of the allele for each individual; see Methods). To allow for relatedness in the FUSION and SardiNIA samples, we estimated regression coefficients in the context of a variance component model that modeled background polygenic effects17. As usual20,21, modeling polygenic effects is important in the context of an association study such as this one, because ignoring relatedness among sampled individuals can lead to misleading P values and inflated false-positive rates.
Figure 1 shows the results of a meta-analysis of the initial scans from all three studies, comprising a total of 8,816 participants. The genomic control22 parameters for this meta-analysis were 1.04, 1.02 and 1.01 (for HDL cholesterol, LDL cholesterol and triglycerides, respectively), suggesting that population stratification and unmodeled relatedness had negligible impact on our association results. Stage 1 results indicate strong association with lipids for 18 loci where at least one SNP exceeds the arbitrary threshold of P < 5 × 10−7 (Table 2). Several loci previously implicated in lipid metabolism show strong evidence for association, including regions near CETP (strongest association at rs3764261, P < 10−18, HDL cholesterol concentration increase of 2.42 mg/dl per A allele), LPL (rs12678919, P < 10−10, 2.44 mg/dl increase per G allele), LIPC (rs10468017, P < 10−10, 1.76 mg/dl increase per T allele), ABCA1 (rs4149274, P ~7.4 × 10−8, 1.51 mg/dl increase per G allele) and LIPG (rs4939883, P ~1.4 × 10−7, 1.87 mg/dl increase per C allele) associated with HDL cholesterol concentrations; the APOE-APOC1-APOC4-APOC2 cluster (rs4420638, P < 10−20, 8.02 mg/dl increase per G allele), APOB (rs515135, P < 10−13, 6.08 mg/dl increase per C allele) and LDLR (rs6511720, P < 10−9, 8.03 mg/dl increase per C allele) associated with LDL cholesterol concentrations; and near the APOA5-APOA4-APOC3-APOA1 cluster (rs964184, P < 10−15, 18.12 mg/dl increase per G allele), GCKR (rs1260326, P < 10−14, 10.25 mg/dl increase per T allele) and LPL (rs6993414, P < 10−12, 14.20 mg/dl increase per A allele) associated with triglyceride concentrations. At several of these loci, the SNP showing strongest association was in linkage disequilibrium (LD) with previously identified variants (r2 > 0.80) or had itself been previously reported to show association. However, at other loci—in particular, the regions near LIPC, LIPG, LDLR and APOB—strongly associated SNPs were in only weak LD with previously identified variants (r2 < 0.30) and thus were likely to represent new signals (Supplementary Table 2 online). At the GCKR locus, the strongest observed association was with a coding SNP, consistent with the results of a recent detailed analysis of the region (S. Kathiresan and M. Orho-Melander, personal communication). In addition to SNPs in these known loci, several other SNPs showed strong association in our initial genome-wide analysis. For example, SNPs near the GRIN3A, GALNT2, CELSR2-PSRC1-SORT1, NCAN-SF4 and TRIB1 genes all had P values <5 × 10−7 for at least one of the three lipid traits in our initial analysis (Table 2). We observed association with distinct gene sets for each of the three traits, consistent with the modest degree of correlation between the traits (the correlation between HDL and LDL cholesterol was essentially zero in our samples, the correlation between HDL cholesterol and triglycerides was approximately −0.4 and the correlation between LDL cholesterol and triglycerides was 0.3 in the SardiNIA sample and 0.1 in FUSION).
Figure 1.
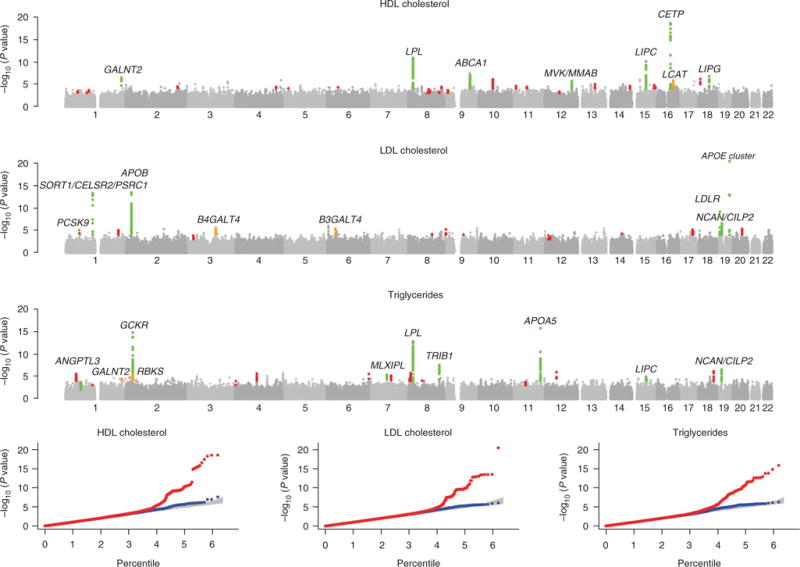
Summary of genome-wide association scans. The figure summarizes combined genome-wide association scan results in the top 3 panels (plotted as −log10 P value for HDL cholesterol, LDL cholesterol and triglycerides). Loci that were not followed up are in gray. Loci that were followed-up are in green (combined dataset yielded convincing evidence of association, P < 5 × 10−8), orange (combined dataset yielded promising evidence of association, P < 10−5), or red (combined dataset did not suggest association, P > 10−5). The three panels in the bottom row display quantile-quantile plots for test statistics. The red line corresponds to all test statistics, the blue line corresponds to results after excluding statistics at replicated loci (in green, top panel), and the gray area corresponds to the 90% confidence region from a null distribution of P values (generated from 100 simulations).
Table 2.
Summary of GWAS meta-analysis stage 1 results (includes all signals with P < 5 × 10−7)
| Locus
|
Association signal
|
Corroborating signals (P < 10−6)
|
Nearby genes
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Position (Mb) | Allele(+/−) | Freq (+) | Effect (mg/dl) | P value | SNPs | LD groups (r2 < 0.2) | (Relative position) (−upstream, +downstream) |
| HDL cholesterol (n = 8,656) | |||||||||
| rs3764261 | 16 | 55.6 | A/C | 0.29 | 2.42 | 2.8 × 10−19 | 14 | 2 | CETP (−2.4 kb) |
| rs12678919 | 8 | 19.9 | G/A | 0.12 | 2.44 | 1.3 × 10−11 | 84 | 2 | LPL (+19.5 kb) |
| rs10468017 | 15 | 56.5 | T/C | 0.32 | 1.76 | 8.6 × 10−11 | 18 | 2 | LIPC (−45.7 kb) |
| rs1323432 | 9 | 101.4 | A/G | 0.87 | 1.93 | 2.5 × 10−8 | 4 | 1 | GRIN3A (Intron 6); PPP3R2 (−5.7 kb) |
| rs4149274 | 9 | 104.7 | G/A | 0.69 | 1.51 | 7.4 × 10−8 | 20 | 1 | ABCA1 (Intron 5) |
| rs4939883 | 18 | 45.4 | C/T | 0.86 | 1.87 | 1.4 × 10−7 | 2 | 1 | LIPG (+47.9 kb) |
| rs4846914 | 1 | 226.6 | A/G | 0.62 | 1.15 | 2.9 × 10−7 | 4 | 1 | GALNT2 (Intron 1) |
| LDL cholesterol (n = 8,589) | |||||||||
| rs4420638 | 19 | 50.1 | G/A | 0.16 | 8.02 | 3.2 × 10−21 | 2 | 1 | APOE/APOC cluster |
| rs515135 | 2 | 21.2 | C/T | 0.83 | 6.08 | 3.1 × 10−14 | 116 | 3 | APOB (−19.1kb) |
| rs602633 | 1 | 109.5 | G/T | 0.80 | 6.09 | 4.8 × 10−14 | 8 | 1 | CELSR2 (+3.1kb); PSRC1 (+668 bp); SORT1 (−30 kb) |
| rs6511720 | 19 | 11.1 | C/A | 0.91 | 8.03 | 6.8 × 10−10 | 1 | 1 | LDLR (Intron 1) |
| rs2228603 | 19 | 19.2 | C/T | 0.93 | 6.46 | 1.8 × 10−7 | 3 | 1 | NCAN (Pro92Ser) |
| Triglycerides (n = 8,684) | |||||||||
| rs964184 | 11 | 116.2 | G/C | 0.12 | 18.12 | 1.5 × 10−16 | 29 | 2 | APOA5 (+11.2 kb) |
| rs1260326 | 2 | 27.6 | T/C | 0.40 | 10.25 | 1.5 × 10−15 | 52 | 2 | GCKR (Leu446Pro) |
| rs6993414 | 8 | 19.9 | A/G | 0.46 | 14.20 | 1.4 × 10−13 | 85 | 2 | LPL (+78.1 kb) |
| rs2954029 | 8 | 126.6 | A/T | 0.56 | 6.42 | 2.8 × 10−8 | 15 | 1 | TRIB1 (+40.3 kb) |
| rs10401969 | 19 | 19.3 | T/C | 0.92 | 12.28 | 2.3 × 10−7 | 5 | 1 | NCAN (+44.7 kb); SF4 (Intron 8) |
The table summarizes association signals observed in the analysis of lipid concentrations in three GWAS scans. Chromosome assignments, position and gene annotations all refer to the March 2006 Genome Build (UCSC). Alleles are ordered such that the first allele (+) is associated with increased lipid levels. Effect sizes are measured as additive effects, which correspond to the average change in phenotype when one (−) allele is replaced with one (+) allele. Corroborating signals refer to the number of additional SNPs within 1 Mb with P < 10−6. The number of LD groups (r2 < 0.2) among these corroborating signals was calculated using LD information from the HapMap CEU sample. P values in bold exceed a threshold of 5 × 10−8, which corresponds to a false-positive rate of 0.05 after adjustment for 1 million independent tests, comparable to the number of independent common SNPs in the Phase II CEU HapMap. For each locus, the most strongly associated SNP is indicated together with its position relative to nearby genes, with a focus on genes previously implicated in lipid metabolism. In the nearby gene column, positions are relative to the transcription start for the nearest gene.
Follow-up of initial findings
To further evaluate these and other promising findings from our initial scan, we examined a subset of SNPs in six additional cohorts of European ancestry, totaling 11,569 individuals (Table 1). These follow-up analyses were conducted in several stages. In a first round of follow-up analysis, SNPs included on the Affymetrix arrays (genotyped in SardiNIA and DGI) and imputed or genotyped in FUSION were selected for follow-up on the basis of a preliminary meta-analysis. We selected a total of 100 SNPs in this manner for examination in the ISIS23,24, HAPI25,26 and SUVIMAX27,28 samples, and 67 SNPs for examination in FUSION stage 2 samples. Once imputation of HapMap SNPs was completed for SardiNIA and DGI samples and an additional meta-analysis carried out, we examined nine additional SNPs in loci not selected for initial follow-up in the FUSION stage 2 and SUVIMAX samples. Finally, we genotyped a single SNP in each of the 21 loci showing promising evidence for replication in the initial stage 2 samples in the Caerphilly29,30 and BWHHS31 samples (Supplementary Fig. 1 and Supplementary Methods online).
Table 3 provides a summary of the stage 2 results and a combined analysis of the data from both stage 1 and stage 2. The table includes the SNP with the strongest association signal at each locus and a selection of additional SNPs that also show strong association but only weak LD with the most strongly associated SNP (r2 < 0.30). All loci with a P value <5 × 10−7 in our initial analysis were confirmed except for the association signal near GRIN3A. Supplementary Table 3 online provides stage 2 results for all SNPs, and Supplementary Table 4 online provides more detailed results for the SNPs highlighted in Table 3.
Table 3.
Summary of most significant stage 1 and stage 2 results
| SNP | Chr | Pos(Mb) | Alleles (+/−) | Freq (+) | Effect (mg/dl) | Association P values
|
Sample sizes
|
Nearby genes | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage 1 (two-sided) | Stage 2 (one-sided) | Combined (two-sided) | Stage 1 | Stage 2 | |||||||
| SNPs associated with HDL cholesterol | |||||||||||
| rs3764261 | 16 | 55.6 | A/C | 0.69 | 3.47 | 2.8 × 10−19 | 6.4 × 10−43 | 2.3 × 10−57 | 8,656 | 8,072 | CETP |
| rs1864163 | 16 | 55.6 | G/A | 0.80 | 4.12 | 3.0 × 10−17 | 4.3 × 10−28 | 6.9 × 10−39 | 8,656 | 3,684 | CETP |
| rs9989419 | 16 | 55.5 | G/A | 0.65 | 1.72 | 8.0 × 10−16 | 1.8 × 10−17 | 3.2 × 10−31 | 8,656 | 6,981 | CETP |
| rs12596776 | 16 | 55.5 | G/C | 0.13 | 1.26 | 3.7 × 10−5 | 1.0 × 10−4 | 2.8 × 10−8 | 8,656 | 7,030 | CETP |
| rs1566439 | 16 | 55.6 | C/T | 0.45 | 0.96 | 2.0 × 10−5 | 2.1 × 10−4 | 3.3 × 10−8 | 8,656 | 4,881 | CETP |
| rs4775041 | 15 | 56.5 | C/G | 0.67 | 1.38 | 2.8 × 10−9 | 9.6 × 10−13 | 3.2 × 10−20 | 8,656 | 11,426 | LIPC |
| rs261332 | 15 | 56.5 | A/G | 0.19 | 1.41 | 1.7 × 10−9 | 1.3 × 10−7 | 2.3 × 10−15 | 8,656 | 6,956 | LIPC |
| rs10503669 | 8 | 19.9 | A/C | 0.10 | 2.09 | 3.2 × 10−10 | 9.4 × 10−11 | 4.1 × 10−19 | 8,656 | 11,431 | LPL |
| rs2197089 | 8 | 19.9 | A/G | 0.42 | 1.38 | 3.4 × 10−8 | 3.2 × 10−5 | 1.0 × 10−11 | 8,656 | 3,644 | LPL |
| rs6586891 | 8 | 20 | A/C | 0.34 | 1.00 | 3.5 × 10−5 | 9.7 × 10−6 | 2.9 × 10−9 | 8,656 | 7,017 | LPL |
| rs2144300 | 1 | 226.6 | T/C | 0.40 | 1.11 | 6.6 × 10−7 | 4.0 × 10−9 | 2.6 × 10−14 | 8,656 | 11,406 | GALNT2 |
| rs2156552 | 18 | 45.4 | T/A | 0.84 | 1.20 | 8.4 × 10−7 | 7.1 × 10−7 | 6.4 × 10−12 | 8,656 | 11,437 | LIPG |
| rs4149268 | 9 | 104.7 | C/T | 0.355 | 0.82 | 3.3 × 10−7 | 2.2 × 10−5 | 1.2 × 10−10 | 8,656 | 11,327 | ABCA1 |
| rs2338104 | 12 | 108.4 | G/C | 0.45 | 0.48 | 1.9 × 10−6 | 7.6 × 10−4 | 3.4 × 10−8 | 8,656 | 11,399 | MVK/MMAB |
| rs255052 | 16 | 66.6 | A/G | 0.17 | 0.74 | 1.5 × 10−6 | 0.0087 | 1.2 × 10−7 | 8,656 | 4,534 | LCAT |
| rs1323432 | 9 | 101.4 | A/G | 0.88 | −0.03 | 2.5 × 10−8 | 0.82 | 7.7 × 10−4 | 8,656 | 8,176 | GRIN3A |
| SNPs associated with LDL cholesterol | |||||||||||
| rs4420638 | 19 | 50.1 | G/A | 0.82 | 6.61 | 3.2 × 10−21 | 4.9 × 10−24 | 3.0 × 10−43 | 8,589 | 10,806 | APOE/C1/C4 |
| rs10402271 | 19 | 50 | G/T | 0.67 | 2.62 | 9.8 × 10−6 | 1.5 × 10−5 | 1.2 × 10−9 | 8,589 | 6,519 | APOE/C1/C4 |
| rs599839 | 1 | 109.5 | A/G | 0.77 | 5.48 | 1.2 × 10−13 | 2.7 × 10−21 | 6.1 × 10−33 | 8,589 | 10,783 | CELSR2/PSRC1/SORT1 |
| rs6511720 | 19 | 11.1 | G/T | 0.90 | 9.17 | 6.8 × 10−10 | 3.3 × 10−19 | 4.2 × 10−26 | 8,589 | 7,442 | LDLR |
| rs562338 | 2 | 21.2 | G/A | 0.18 | 4.89 | 1.2 × 10−11 | 3.6 × 10−12 | 5.6 × 10−22 | 8,589 | 10,849 | APOB |
| rs754523 | 2 | 21.2 | G/A | 0.28 | 2.78 | 7.0 × 10−7 | 1.3 × 10−6 | 8.3 × 10−12 | 8,589 | 6,542 | APOB |
| rs693 | 2 | 21.1 | A/G | 0.42 | 2.44 | 1.2 × 10−7 | 0.0034 | 3.1 × 10−9 | 8,589 | 3,222 | APOB |
| rs11206510 | 1 | 55.2 | T/C | 0.81 | 3.04 | 7.5 × 10−6 | 5.4 × 10−7 | 3.5 × 10−11 | 8,589 | 10,805 | PCSK9 |
| rs16996148 | 19 | 19.5 | G/T | 0.89 | 3.32 | 2.4 × 10−6 | 8.3 × 10−5 | 2.7 × 10−9 | 8,589 | 10,841 | NCAN/CILP2 |
| rs2254287 | 6 | 33.3 | G/C | 0.38 | 1.91 | 2.9 × 10−6 | 0.0015 | 5.1 × 10−8 | 8,589 | 7,440 | B3GALT4 |
| rs12695382 | 3 | 120.4 | A/G | 0.90 | 2.23 | 4.9 × 10−6 | 0.0067 | 1.0 × 10−6 | 8,589 | 10,802 | B4GALT4 |
| SNPs associated with triglycerides | |||||||||||
| rs780094 | 2 | 27.7 | T/C | 0.39 | 8.59 | 1.7 × 10−14 | 2.0 × 10−19 | 6.1 × 10−32 | 8,684 | 9,723 | GCKR |
| rs11127129 | 2 | 28.0 | C/G | 0.79 | 3.77 | 2.0 × 10−4 | 3.2 × 10−4 | 4.7 × 10−7 | 8,684 | 9,700 | RBKS/GCKR |
| rs12286037 | 11 | 116.2 | T/C | 0.94 | 25.82 | 1.1 × 10−7 | 1.6 × 10−22 | 1.0 × 10−26 | 8,684 | 9,738 | APOA5/A4/C3/A1 |
| rs662799 | 11 | 116.2 | G/A | 0.05 | 16.88 | 4.3 × 10−8 | 2.7 × 10−10 | 2.4 × 10−15 | 8,684 | 3,248 | APOA5/A4/C3/A1 |
| rs2000571 | 11 | 116.1 | A/G | 0.17 | 6.93 | 4.7 × 10−5 | 8.7 × 10−5 | 5.7 × 10−8 | 8,684 | 3,209 | APOA5/A4/C3/A1 |
| rs486394 | 11 | 116.0 | C/A | 0.28 | 1.50 | 1.7 × 10−4 | 0.0073 | 7.4 × 10−6 | 8,684 | 3,597 | APOA5/A4/C3/A1 |
| rs10503669 | 8 | 19.9 | C/A | 0.895 | 11.57 | 1.4 × 10−9 | 1.6 × 10−14 | 3.9 × 10−22 | 8,684 | 9,711 | LPL |
| rs2197089 | 8 | 19.9 | G/A | 0.58 | 3.38 | 3.1 × 10−11 | 0.0029 | 1.1 × 10−12 | 8,684 | 3,202 | LPL |
| rs6586891 | 8 | 20.0 | C/A | 0.66 | 4.60 | 2.4 × 10−4 | 5.0 × 10−4 | 1.1 × 10−6 | 8,684 | 3,622 | LPL |
| rs17321515 | 8 | 126.6 | A/G | 0.56 | 6.42 | 6.8 × 10−8 | 1.0 × 10−6 | 7.0 × 10−13 | 8,684 | 5,312 | TRIB1 |
| rs17145738 | 7 | 72.4 | C/T | 0.84 | 8.21 | 4.1 × 10−6 | 5.0 × 10−8 | 2.0 × 10−12 | 8,684 | 9,741 | MLXIPL |
| rs1748195 | 1 | 62.8 | C/G | 0.70 | 7.12 | 2.3 × 10−4 | 5.4 × 10−8 | 1.7 × 10−10 | 8,684 | 9,559 | ANGPTL3 |
| rs16996148 | 19 | 19.5 | G/T | 0.92 | 6.10 | 6.3 × 10−7 | 2.4 × 10−4 | 2.5 × 10−9 | 8,684 | 9,707 | NCAN/CILP2 |
| rs4775041 | 15 | 56.5 | C/G | 0.67 | 3.62 | 7.3 × 10−5 | 2.9 × 10−5 | 1.6 × 10−8 | 8,684 | 8,462 | LIPC |
| rs2144300 | 1 | 226.6 | C/T | 0.60 | 4.25 | 4.9 × 10−4 | 2.4 × 10−4 | 7.9 × 10−7 | 8,684 | 8,473 | GALNT2 |
The table summarizes association signals after follow-up of the promising SNPs in stage 2 samples. Column 1 headings are as described for Table 2, with the addition of one-sided P values for the stage 2 samples, in which we tested for the same direction of effect as Stage 1—consistent with current best practice for replication of GWAS findings. The effect sizes shown were estimated from stage 2 samples only. SNPs with a combined (stage 1 + 2) P value <10−5 were included, although we also show GRIN3A for completeness because it was significant in the initial scan. Rows corresponding to SNPs with a combined P value < 5 × 10−8 are in boldface. SNPs in this table may not match those in Table 2, which only displays the strongest signal in each locus. The discrepancy also reflects our bias towards genotyped Affymetrix 500K SNPs in the Stage 2 follow-up. Association P values for each of the six stage 2 samples are shown in Supplementary Table 4.
Overall, we observed the strongest evidence for association (P < 10−20) between HDL cholesterol and SNPs in CETP (rs3764261, rs1864163 and rs9989419; the three are in weak LD with each other), LIPC (rs4775041) and LPL (rs10503669); between LDL cholesterol and SNPs in the APOE-APOC cluster (rs4420638), near the CELSR2-PSRC1-SORT1 (rs599839), LDLR (rs6511720) and APOB (rs562338) genes; and between triglycerides and SNPs near the GCKR (rs780094), APOA5-APOA4-APOC3-APOA1 (rs12286037) and LPL (rs10503669) genes (P values and effect sizes are shown in Table 3). In each case, we observed strong evidence for association in both stages of genotyping (P < 5 × 10−7). The association of LDL cholesterol concentrations with the CELSR2-PSRC1-SORT1 locus is particularly notable, because variants in the region have not been previously implicated in lipid metabolism (Supplementary Fig. 2c online). There is no obvious connection between the genes closest to the association signal, CELSR2 and PSRC1, and lipid metabolism. One possibility is that rs599839 or an associated variant influences expression of SORT1, a nearby gene that mediates endocytosis and degradation of lipoprotein lipase32. In our sample, allele A at rs599839 was associated with an increase of 5.48 mg/dl in LDL cholesterol concentrations. Notably, the same rs599839 allele has recently been associated with an increased risk of CAD in an independent study33, suggesting that the association to CAD risk might be mediated by the effect on LDL cholesterol concentrations.
Another tier of loci also remains significant after adjustment for 1,000,000 independent tests. This tier includes additional SNPs for loci within the previous tier and also SNPs near ABCA1, LIPC, LIPG and PCSK9 (Table 3). Of note, although polymorphisms in all of these genes have a well-established role in lipid metabolism, some of the signals we identified do not overlap with established associations and likely point to new risk alleles (Supplementary Table 2). For example, in PCSK9, variants previously associated with LDL cholesterol concentrations have r2 < 0.10 with the variants identified here (Supplementary Table 2). Other examples of newly identified risk alleles include LIPG (rs2156552), LIPC (rs4775041) and LDLR (rs6511720).
This tier also includes six loci where genetic variants have not previously been implicated in lipid metabolism. We found association between HDL cholesterol and SNPs near GALNT2 and near MVK and MMAB (Supplementary Fig. 2a,b); between LDL cholesterol and triglycerides and SNPs in an extended region near NCAN and CILP2 (Supplementary Fig. 2d,h); and between triglycerides and SNPs near TRIB1, MLXIPL and ANGPTL3 (Supplementary Fig. 2e–g). Among genes in these six regions, we observed the clearest connections to cholesterol and lipoprotein metabolism for MLXIPL, which encodes a protein that binds and activates specific motifs in the promoters of triglyceride synthesis genes, and for ANGPTL3, whose protein homolog is a major regulator of lipid metabolism in mice34. Rare variants in a related gene, ANGPTL4, have been associated with HDL and triglyceride concentrations in humans35. A connection to lipid metabolism has also been observed for MVK and MMAB, two neighboring genes that are regulated by SREBP2 and that share a common promoter36. MVK encodes mevalonate kinase, which catalyzes an early step in cholesterol biosynthesis, and MMAB encodes a protein that participates in a metabolic pathway that degrades cholesterol.
In the other three loci, we did not find any established connections to cholesterol metabolism. The signals near GALNT2 and TRIB1 each overlap a single gene. GALNT2 encodes a widely expressed glycosyltransferase that could potentially modify a lipoprotein or receptor. TRIB1 encodes a G-protein–coupled receptor-induced protein involved in the regulation of mitogen-activated protein kinases37 and may regulate lipid metabolism through this pathway. In contrast, the association signal near NCAN extends for over 500 kb and encompasses 20 genes. In our combined data, rs16996148 (an Affymetrix array SNP near CILP2) was selected for follow-up and showed strong association with both LDL cholesterol (P ~ 2.7 × 10−9) and triglycerides (P ~ 2.5 × 10−9). The allele that is associated with increased LDL cholesterol concentrations is also associated with increased triglyceride concentrations, consistent with the modest positive correlation between the two traits but in contrast to other SNPs associated with both LDL cholesterol and triglycerides that showed association with only one of the traits in our sample. Notably, in the analysis of our three genome-wide association scans and imputed HapMap SNPs, a nonsynonymous coding SNP in the NCAN gene (rs2228603, Pro92Ser) showed the strongest evidence for association (P ~ 1.8 ×10−7). This SNP was not included in our initial follow-up analysis, which considered only SNPs on the Affymetrix arrays, but it was in strong LD with rs16996148 (r2 = 0.89). NCAN is a nervous system-specific proteoglycan involved in neuronal pattern formation, remodeling of neuronal networks and regulation of synaptic plasticity38, with no obvious relation to LDL cholesterol or triglyceride concentrations.
A final tier of genes has one or more SNPs with a P value <10−5 when stage 1 and stage 2 data are considered together (Table 3). Among these genes is LCAT, which encodes a protein with a well-established role in lipid metabolism, and for which well-characterized, but rare, genetic variants have been shown to considerably affect lipid concentrations39. Our signal supports a single unconfirmed report of a common variant influencing HDL concentrations40. Two other association signals of note are located near the B3GALT4 and B4GALT4 genes. Similarly to GALNT2, these genes encode glycosyltransferases, and thus our results may implicate glycosyltransferases as having a previously unrecognized influence on variation in lipid concentrations: it is possible that they affect lipid concentrations by modifying lipoprotein receptors41.
A summary of evidence for association between HDL cholesterol, LDL cholesterol and triglycerides and all markers genotyped or imputed in our initial survey of the genome is available online (see URLs section in Methods). This should enable other investigators to combine our results with their own data or to select SNPs for followup in other samples. As an example of the utility of this resource, in a companion report, Kathiresan and colleagues16 used the DGI data and the meta-analysis resource to select a set of SNPs for examination in a sample of >18,000 individuals. They report convincing statistical evidence for six newly identified loci at P < 5 × 10−8, all of which overlap with those in our study.
Association with coronary artery disease
In view of the well-established associations between lipid concentrations and CAD, we examined whether the alleles associated with lipid concentrations in the present study were also associated with CAD in the Wellcome Trust Case Control Consortium (WTCCC) sample of ~2,000 CAD cases and an expanded reference panel of ~13,000 British individuals42 (including ~3,000 random controls and ~2,000 cases for each of five common diseases). Given the relatively modest changes in LDL cholesterol concentrations associated with the alleles we identified (changes of ~2–9 mg/dl per allele), we expected that a subset of SNPs might also be associated with a small increase in susceptibility to CAD. Notably, the results show that all of the alleles that were associated with increased LDL cholesterol concentrations in our sample were more common among CAD cases than in the expanded reference panel (Table 4). Among eleven independent alleles (r2 < 0.30 between nearby alleles) associated with increased LDL cholesterol concentrations in our sample (all with P < 10−6 in our sample), all eleven showed increased frequency among CAD cases (P = 2−11 = 0.0005). The increase was significant (P < 0.05) for eight of the SNPs, and nearly so (P < 0.06) for another two (Table 4, penultimate column). Although the associated risk estimates are small (relative risk increases of 1.04–1.29 per allele, see Table 4), it is extremely unlikely (P < 10−11) that 10 of the 11 SNPs would show suggestive association with CAD at P < 0.06 by chance, making the connection between LDL and associated SNPs and CAD particularly worthy of note. Overall, although we observed a correlation between the strength of the observed association with CAD and the impact of each allele on LDL cholesterol concentrations (Spearman correlation coefficient r = 0.71, P = 0.015), we also found some alleles that had a strong association with LDL cholesterol but no significant association with CAD (for example, rs562338 in the APOB locus). We did not find a similar pattern of association for alleles associated with the other lipid traits (Supplementary Table 5 online), although alleles associated with increased triglyceride concentrations near TRIB1 (for example, at rs17321515) were also associated with increased risk of CAD (P = 0.0008). Although the data suggest that nearly all alleles associated with increased LDL cholesterol concentrations will be associated with increased risk of CAD (given a large sample size), the converse is not true, as expected. Alleles at the chromosome 9 locus that show strong association with CAD, coronary heart disease and myocardial infarction33,42–44 do not seem to influence lipid concentrations in our sample (P = 0.31 for association between LDL cholesterol and the SNP most strongly associated with CAD, rs1333049, in our stage 1, and P > 0.50 for HDL cholesterol and triglycerides). Additional studies will show whether these variants are also associated with longevity45, stroke46 and the other health outcomes associated with LDL cholesterol concentrations.
Table 4.
Association between coronary artery disease and LDL cholesterol–associated SNPs
| Association with coronary artery disease (WTCCC)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus
|
LDL-C association (current study)
|
Expanded reference set
|
CAD cases
|
||||||||
| SNP | Chr | Position (Mb) | Alleles (+/−) | P value (two-sided) | n | Frequency of LDL+ allele | n | Frequency of LDL+ allele | P value (one sided) | OR (95% CI) | Nearby genes |
| rs4420638 | 19 | 50.1 | G/A | 3.0 × 10−43 | 12,281 | 0.184 | 1,926 | 0.209 | 1.0 × 10−4 | 1.17 (1.08–1.28) | APOE/C1/C4 |
| rs10402271 | 19 | 50.0 | G/T | 1.2 × 10−9 | 12,256 | 0.319 | 1,921 | 0.339 | 0.0068 | 1.10 (1.02–1.18) | APOE/C1/C4 |
| rs599839 | 1 | 109.5 | A/G | 6.1 × 10−33 | 12,292 | 0.778 | 1,923 | 0.808 | 1.3 × 10−5 | 1.20 (1.10–1.31) | PSRC1/SORT1 |
| rs6511720a | 19 | 11.1 | G/T | 4.2 × 10−26 | 12,301 | 0.890 | 1,926 | 0.902 | 6.7 × 10−4 | 1.29 (1.10–1.52) | LDLR |
| rs562338 | 2 | 21.2 | G/A | 5.6 × 10−22 | 12,288 | 0.824 | 1,924 | 0.830 | 0.18 | 1.04 (0.95–1.14) | APOB |
| rs754523 | 2 | 21.2 | G/A | 8.3 × 10−12 | 12,292 | 0.332 | 1,926 | 0.353 | 0.0042 | 1.10 (1.03–1.18) | APOB |
| rs693 | 2 | 21.1 | A/G | 3.1 × 10−9 | 12,292 | 0.520 | 1,924 | 0.536 | 0.028 | 1.07 (1.00–1.14) | APOB |
| rs11206510 | 1 | 55.2 | T/C | 3.5 × 10−11 | 12,284 | 0.807 | 1,925 | 0.825 | 0.0042 | 1.13 (1.03–1.23) | PCSK9 |
| rs16996148 | 19 | 19.5 | G/T | 2.7 × 10−9 | 12,182 | 0.915 | 1,921 | 0.922 | 0.055 | 1.11 (0.98–1.26) | NCAN/CILP2 |
| rs2254287a | 6 | 33.3 | G/C | 5.1 × 10−8 | 12,301 | 0.385 | 1,926 | 0.399 | 0.039 | 1.07 (0.99–1.14) | B3GALT4 |
| rs12695382 | 3 | 120.4 | A/G | 1.0 × 10−6 | 12,292 | 0.865 | 1,924 | 0.874 | 0.051 | 1.09 (0.98–1.20) | B4GALT4 |
The table summarizes association between coronary artery disease and the alleles associated with LDL-C concentrations in our study. Evidence for association was evaluated in the Wellcome Trust Case Control Consortium panel and was not adjusted for additional covariates, because these are not available for the bulk of study participants. Rows corresponding to SNPs that show association with LDL cholesterol with P < 5 × 10−8 in our sample are in boldface.
Expected genotype counts for rs6511720 and rs2254287 were imputed in the WTCCC samples, averaged over cases and controls to estimate allele frequencies and then analyzed using logistic regression to estimate odds ratios. The approach results in unbiased estimates of the odds ratio but can result in estimates of case and control frequencies that are ‘shrunk’ towards the null.
DISCUSSION
Genes at the loci implicated in our study affect the entire cycle of formation, activity and turnover of lipoproteins and triglycerides. Thus, they encode many of the apolipoproteins themselves (APOE, APOB and APOA5), but they also encode a transcription factor activating triglyceride synthesis (MLXIPL), an enzyme involved in cholesterol biosynthesis (MVK), transporters of cholesterol (ABCA1) and cholesterol ester (CETP), a lipoprotein receptor (LDLR), potential receptor-modifying glycosyltransferases (B4GALT4, B3GALT4 and GALNT2), lipases (LPL, LIPC and LIPG) and a protein involved in cholesterol degradation (MMAB), an inhibitor of lipase (ANGPTL3) and a possible endocytic receptor for LPL (SORT1). Notably, some of the loci we identify (near TRIB1 and in the large region surrounding NCAN, for example) include no obvious functional candidates, and further studies to pinpoint the genes and mechanisms involved could lead to important new insights about lipid metabolism.
In multiple regression models, the variants identified here together accounted for only about 5–8% of the variation in the three lipid traits in the populations studied, leaving much of the heritability of these traits unexplained. The missing genetic factors might be accounted for by a much longer list of loci with common variants of small effect, by rare variants of large effect that have been missed by the association approach, or by interactions between these and other genetic and environmental factors. To clarify the overall role of the loci reported here, it will be critical to resequence the exons and conserved regions in a large number of individuals, in order to identify and evaluate all potential variants within each gene or cluster of genes. This resequencing effort will help identify the functional variants involved in each region. In addition, resequencing may identify nonsense, nonsynonymous or other changes that are associated with variability in lipid concentrations, clarifying the identity of the genes involved in regions with multiple candidates. Resequencing of certain candidate genes has shown that such rare variants can sometimes be identified in individuals at the extremes of lipid concentration distributions47; thus, focused studies of the regions identified here in individuals with dyslipidaemia could be particularly informative.
Several of the loci newly identified in this report are potentially attractive drug targets. Furthermore, the ability to stratify individuals on the basis of specific genetic profiles may provide future benefits for optimization of therapy, given that lipid lowering drugs are already widely prescribed to help manage individual lipid profiles and reduce the risk of cardiovascular events2. For monogenic forms of diseases that lead to dysregulation of HDL cholesterol, LDL cholesterol or triglyceride concentrations, it is clear that individuals with different mutations require different therapeutic regimens48,49. Thus, it is our hope that common variants at the loci identified here will lead to development of novel therapeutics and influence optimal treatment profiles for each individual, resulting in improved management of blood lipid concentrations and reduction of cardiovascular disease risk.
METHODS
Genome-wide association scans
We used standard protocols to genotype the Illumina 317K HumanHap 300 BeadChip and Affymetrix 500K and 10K Mapping Array Sets in the FUSION and SardiNIA samples, respectively. We collaborated with the authors of a previously published study15 to integrate their results into our analysis. To facilitate comparison of results among the three studies, and to better assess the effects of unmeasured variants, we first identified stretches of haplotype shared between individuals in our sample and those in the HapMap CEU sample and then used these shared stretches to impute missing genotypes. This resulted in a total of ~2,261,000 SNPs that were either genotyped or imputed with high confidence in all three samples.
Association analysis
We first analyzed each study independently. For each marker, we identified a reference allele and calculated statistics summarizing its evidence for association with HDL cholesterol, LDL cholesterol and triglycerides. Association models include gender, age and age2 as covariates, and additional covariates appropriate to each study. These statistics were then combined across studies taking into account both the number of phenotyped individuals in each study and the direction and magnitude of the estimated effect.
Follow-up
SNPs from the loci showing the strongest evidence for association in the genome-wide scans were selected for analysis in follow-up samples. In our initial round of follow-up, we favored SNPs that were successfully genotyped in both the DGI and SardiNIA studies. As in the analysis of the initial scans, we first conducted analyses within each sample separately and then combined the resulting summary statistics by meta-analysis.
Coronary artery disease analysis
Individual genotype data for this analysis were obtained from the WTCCC website. We first imputed all relevant untyped SNPs using the HapMap CEU as a reference population and carried out tests for association with a likelihood-ratio test.
Supplementary Material
Acknowledgments
We are indebted to the many volunteers who generously participated in these studies. We thank our colleagues from the DGI for sharing prepublication data; M. Erdos, P. Chines, P. Deodhar, K. Kubalanza, A. Sprau and M. Tong of FUSION and E. Pugh, K. Doheny and Center for Inherited Disease Research (CIDR) investigators for expert technical work; N. Rosenberg for helpful discussions about population genetics; the SardiNIA Research Clinic staff; and the Amish Research Clinic staff. This study makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data are available from the WTCCC website. Funding for the WTCCC project was provided by the Wellcome Trust under award 076113. The Caerphilly study was funded by the Medical Research Council (UK). The Caerphilly study was undertaken by the former MRC Epidemiology Unit (South Wales) and was funded by the Medical Research Council of the United Kingdom. The data archive is maintained by the Department of Social Medicine, University of Bristol. This work was supported in part by the Intramural Research Program of the National Institute on Aging (NIA), by extramural grants from National Human Genome Research Institute (NHGRI), the National Diabetes and Digestive and Kidney Diseases (NIDDK) and the National Heart Lung and Blood Institute (NHLBI), by the American Diabetes Association, the Department of Veterans Affairs, the British Heart Foundation, the Medical Research Council of the United Kingdom and the French Ministry of Higher Education and Research. FUSION genome-wide genotyping was carried out by the Johns Hopkins University Genetic Resources Core Facility (GRCF) SNP Center at CIDR with support from CIDR NIH (contract N01-HG-65403) and the GRCF SNP Center. Additional support for the SardiNIA study was provided by the mayors, administration and residents of Lanusei, Ilbono, Arzana and Elini and the head of Public Health Unit ASL4 in Sardinia. C.J.W. is the recipient of a postdoctoral fellowship from the American Diabetes Association. G.R.A. and K.L.M. are Pew Scholars for the Biomedical Sciences.
Footnotes
URLs. Association data, http://www.sph.umich.edu/csg/abecasis/public/lipids/; Markov Chain Haplotyping Package, http://www.sph.umich.edu/csg/abecasis/MaCH.
Note: Supplementary information is available on the Nature Genetics website.
References
- 1.Mackay J, Mensah GA. The Atlas of Heart Disease and Stroke. World Health Organization; Geneva: 2004. [Google Scholar]
- 2.Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. Br Med J. 2003;326:1423. doi: 10.1136/bmj.326.7404.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuulasmaa K, et al. Estimation of contribution of changes in classic risk factors to trends in coronary-event rates across the WHO MONICA Project populations. Lancet. 2000;355:675–687. doi: 10.1016/s0140-6736(99)11180-2. [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, et al. Cholesterol fractions and apolipoproteins as risk factors for heart disease mortality in older men. Arch Intern Med. 2007;167:1373–1378. doi: 10.1001/archinte.167.13.1373. [DOI] [PubMed] [Google Scholar]
- 5.Grundy SM, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 6.Gotto AM, Jr, Brinton EA. Assessing low levels of high-density lipoprotein cholesterol as a risk factor in coronary heart disease: a working group report and update. J Am Coll Cardiol. 2004;43:717–724. doi: 10.1016/j.jacc.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 7.Prospective Studies Collaboration. Blood cholesterol and vascular mortality by age, sex and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 8.Bansal S, et al. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. J Am Med Assoc. 2007;298:309–316. doi: 10.1001/jama.298.3.309. [DOI] [PubMed] [Google Scholar]
- 9.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. J Am Med Assoc. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 10.Pilia G, et al. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet. 2006;2:e132. doi: 10.1371/journal.pgen.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollin TI, et al. A genome-wide scan of serum lipid levels in the Old Order Amish. Atherosclerosis. 2004;173:89–96. doi: 10.1016/j.atherosclerosis.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Breslow JL. Genetics of lipoprotein abnormalities associated with coronary artery disease susceptibility. Annu Rev Genet. 2000;34:233–254. doi: 10.1146/annurev.genet.34.1.233. [DOI] [PubMed] [Google Scholar]
- 13.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scuteri A, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saxena R, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 16.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. doi: 10.1038/ng.75. advance online publication 13 January 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen WM, Abecasis GR. Family-based association tests for genome-wide association scans. Am J Hum Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burdick JT, Chen WM, Abecasis GR, Cheung VG. In silico method for inferring genotypes in pedigrees. Nat Genet. 2006;38:1002–1004. doi: 10.1038/ng1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The International HapMap Consortium. The International HapMap Project. Nature. 2005;437:1299–1320. [Google Scholar]
- 20.George VT, Elston RC. Testing of association between polymorphic markers and quantitative traits in pedigrees. Genet Epidemiol. 1987;4:193–201. doi: 10.1002/gepi.1370040304. [DOI] [PubMed] [Google Scholar]
- 21.Abecasis GR, Cardon LR, Cookson WOC. A general test of association for quantitative traits in nuclear families. Am J Hum Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 23.ISIS-3 Collaborative Group. ISIS-3: a randomised comparison of streptokinase vs tissue plasminogen activator vs anistreplase and of aspirin plus heparin vs aspirin alone among 41,299 cases of suspected acute myocardial infarction. ISIS-3 (Third International Study of Infarct Survival) Collaborative Group. Lancet. 1992;339:753–770. [PubMed] [Google Scholar]
- 24.Keavney B, et al. Lipid-related genes and myocardial infarction in 4685 cases and 3460 controls: discrepancies between genotype, blood lipid concentrations, and coronary disease risk. Int J Epidemiol. 2004;33:1002–1013. doi: 10.1093/ije/dyh275. [DOI] [PubMed] [Google Scholar]
- 25.Post W, et al. Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum Hered. 2007;64:214–219. doi: 10.1159/000103630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Post W, et al. Determinants of coronary artery and aortic calcification in the Old Order Amish. Circulation. 2007;115:717–724. doi: 10.1161/CIRCULATIONAHA.106.637512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hercberg S, et al. The SU.VI.MAX Study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch Intern Med. 2004;164:2335–2342. doi: 10.1001/archinte.164.21.2335. [DOI] [PubMed] [Google Scholar]
- 28.Hercberg S, et al. A primary prevention trial using nutritional doses of antioxidant vitamins and minerals in cardiovascular diseases and cancers in a general population: the SU.VI.MAX study–design, methods, and participant characteristics. SUpplementation en VItamines et Mineraux AntioXydants. Control Clin Trials. 1998;19:336–351. doi: 10.1016/s0197-2456(98)00015-4. [DOI] [PubMed] [Google Scholar]
- 29.The Caerphilly and Speedwell Collaborative Group. Caerphilly and Speedwell collaborative heart disease studies. J Epidemiol Community Health. 1984;38:259–262. doi: 10.1136/jech.38.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bainton D, et al. Plasma triglyceride and high density lipoprotein cholesterol as predictors of ischaemic heart disease in British men. The Caerphilly and Speedwell Collaborative Heart Disease Studies. Br Heart J. 1992;68:60–66. doi: 10.1136/hrt.68.7.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawlor DA, Bedford C, Taylor M, Ebrahim S. Geographical variation in cardiovascular disease, risk factors, and their control in older women: British Women’s Heart and Health Study. J Epidemiol Community Health. 2003;57:134–140. doi: 10.1136/jech.57.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen MS, Jacobsen C, Olivecrona G, Gliemann J, Petersen CM. Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem. 1999;274:8832–8836. doi: 10.1074/jbc.274.13.8832. [DOI] [PubMed] [Google Scholar]
- 33.Samani NJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koishi R, et al. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30:151–157. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- 35.Romeo S, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy C, Murray AM, Meaney S, Gafvels M. Regulation by SREBP-2 defines a potential link between isoprenoid and adenosylcobalamin metabolism. Biochem Biophys Res Commun. 2007;355:359–364. doi: 10.1016/j.bbrc.2007.01.155. [DOI] [PubMed] [Google Scholar]
- 37.Kiss-Toth E, et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem. 2004;279:42703–42708. doi: 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- 38.Rauch U, Feng K, Zhou XH. Neurocan: a brain chondroitin sulfate proteoglycan. Cell Mol Life Sci. 2001;58:1842–1856. doi: 10.1007/PL00000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuivenhoven JA, et al. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38:191–205. [PubMed] [Google Scholar]
- 40.Pare G, et al. Genetic analysis of 103 candidate genes for coronary artery disease and associated phenotypes in a founder population reveals a new association between endothelin-1 and high-density lipoprotein cholesterol. Am J Hum Genet. 2007;80:673–682. doi: 10.1086/513286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magrane J, Casaroli-Marano RP, Reina M, Gafvels M, Vilaro S. The role of O-linked sugars in determining the very low density lipoprotein receptor stability or release from the cell. FEBS Lett. 1999;451:56–62. doi: 10.1016/s0014-5793(99)00494-9. [DOI] [PubMed] [Google Scholar]
- 42.The Welcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helgadottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 44.McPherson R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barzilai N, et al. Unique lipoprotein phenotype and genotype associated with exceptional longevity. J Am Med Assoc. 2003;290:2030–2040. doi: 10.1001/jama.290.15.2030. [DOI] [PubMed] [Google Scholar]
- 46.Baigent C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 47.Cohen JC, et al. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 48.Naoumova RP, et al. Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: long-term follow-up and treatment response. Arterioscler Thromb Vasc Biol. 2005;25:2654–2660. doi: 10.1161/01.ATV.0000190668.94752.ab. [DOI] [PubMed] [Google Scholar]
- 49.Lind S, et al. Autosomal recessive hypercholesterolaemia: normalization of plasma LDL cholesterol by ezetimibe in combination with statin treatment. J Intern Med. 2004;256:406–412. doi: 10.1111/j.1365-2796.2004.01401.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.