

Abstract
Objective:
To identify genetic risk factors associated with susceptibility to age-related cognitive decline in African Americans (AAs).
Methods:
We performed a genome-wide association study (GWAS) and an admixture-mapping scan in 3,964 older AAs from 5 longitudinal cohorts; for each participant, we calculated a slope of an individual's global cognitive change from neuropsychological evaluations. We also performed a pathway-based analysis of the age-related cognitive decline GWAS.
Results:
We found no evidence to support the existence of a genomic region which has a strongly different contribution to age-related cognitive decline in African and European genomes. Known Alzheimer disease (AD) susceptibility variants in the ABCA7 and MS4A loci do influence this trait in AAs. Of interest, our pathway-based analyses returned statistically significant results highlighting a shared risk from lipid/metabolism and protein tyrosine signaling pathways between cognitive decline and AD, but the role of inflammatory pathways is polarized, being limited to AD susceptibility.
Conclusions:
The genetic architecture of aging-related cognitive in AA individuals is largely similar to that of individuals of European descent. In both populations, we note a surprising lack of enrichment for immune pathways in the genetic risk for cognitive decline, despite strong enrichment of these pathways among genetic risk factors for AD.
Aging-related cognitive decline is a multifactorial process and is likely to be promoted by accumulated brain injury due to chronic conditions of aging, including Alzheimer disease (AD),1 cardiovascular risk factors,2 type II diabetes mellitus,3,4 cerebrovascular diseases,2 and inflammatory diseases.5
When trajectories of cognitive decline are compared between individuals of African American (AA) ancestry and individuals of non-Hispanic European ancestry (EA), there is no appreciable difference at the average rate of decline in these 2 populations.6 However, differences in the prevalence and effect size of different risk factors for cognitive decline—such as AD, small and large vessel neurovascular disease, and lipid profiles—have been noted previously.7,8 These differences suggest that it is important to explore the role of such risk factors in distinct human populations so that interventions to mitigate cognitive decline can be more effectively targeted.
To identify genetic loci influencing age-related cognitive decline in AAs, we first used admixture mapping, an approach that scans the genome for disease-related genetic variants that differ in frequency across populations.9 In addition, we performed a genome-wide association study (GWAS) using the same 3,964 AA participants and outcome measure to identify genetic variants that are associated with the individual's trajectory of cognitive decline. We evaluated the mechanism of the association by examining pathologic phenotypes, gene expression, and biological pathway data. Together, these analyses shed light on the role of genetic ancestry and on genes and pathways that contribute to the pathogenesis of age-related cognitive decline in AAs.
METHODS
Additional information is included in the e-Methods at Neurology.org/ng.
Study participants and cohorts.
Samples are from participants in 5 longitudinal studies of cognition, the Religious Orders Study (ROS), the Rush Memory and Aging Project (MAP), the Chicago Health and Aging Project (CHAP), the Minority Aging Research Study (MARS), and the Indianapolis-Ibadan Dementia Project (IIDP). A description of the original cohorts contributing samples is provided in the e-Methods.
Clinical and cognitive evaluation.
Table e-1 summarizes the specific longitudinal cognitive measures that are collected in each cohort. A detailed description of the clinical and cognitive evaluation and statistical modeling is provided in the e-Methods.
Genotyping, quality control, and genotype imputation.
DNA from participants was extracted from whole blood, lymphocytes, or frozen postmortem brain tissue. The CHAP and IIDP participants were genotyped on the Illumina Human Omni1Quad. The ROS, MAP, and MARS participants were genotyped on the Illumina Omni Express. We used BEAGLE software to impute the ungenotyped markers using reference haplotype panels from the 1000 Genomes Project (Phase I Version 3).10 EIGENSTRAT software11 was used to calculate eigenvectors that explained genetic differences in ancestry among participants in the study (figure e-1). A description of the genotype imputation and quality control is provided in the e-Methods.
Genome-wide association studies.
We tested the association of each imputed single nucleotide polymorphism (SNP) (allelic dosages) and residual cognitive decline slope extracted from the mixed-effects models. The association analysis was performed using linear regression (with the first 2 ancestry principal components as a covariate) using the PLINK toolkit.12 The meta-analysis was performed using METAL software.13
Admixture analysis.
To detect chromosomal segments of distinct ancestry in AAs, we used the HAPMIX algorithm.9 The quantitative and case-control admixture scan was performed using MIXSCORE (version 1.3) software.14 A description of the admixture analysis is provided in the e-Methods.
Pathway and protein-protein interaction analysis.
We performed a pathway enrichment analysis using MAGENTA software15 and adopting Ingenuity and BIOCARTA databases as pathway information resources.
A protein-protein interaction network was reconstructed with the online tool DAPPLE16 with 1,000 permutations and 2 interacting binding degrees as a cutoff.
Gene expression quantitative trait locus analysis.
The gene expression data from primary CD14+CD16− monocytes and CD4+ T cells from 461 healthy humans are part of the Immunological Variation Project.17 A description of the expression analysis is provided in the e-Methods.
Standard protocol approvals, registrations, and patient consents.
All blood draws and data analyses were performed in compliance with protocols approved by the institutional review boards of each institution. Written informed consent was obtained from all participants.
RESULTS
Demographic and phenotypic characteristics of participants.
After stringent data quality control, 3,964 AA individuals from 5 study cohorts (CHAP, IIDP, ROS, MAP, and MARS) were available for our analysis (table e-2). All of these cohorts recruit participants who are older than 65 years and do not have dementia at the time of enrollment. The participants are then followed longitudinally using a battery of cognitive tests. A subject's performance on each test was standardized, and an average aggregate measure of global cognitive performance was computed for each evaluation.18 Each individual had 2 or more neuropsychological evaluations, and a linear mixed-effects model was used to calculate a residual that captures the slope of an individual's global cognitive change (figure e-2; e-Methods). This residual was then used as a quantitative trait in our analyses, as we have done in a prior GWAS.19
Genome-wide admixture scan for age-related cognitive decline.
To examine whether genetic ancestry influences age-related cognitive decline in AA participants, we conducted a genome-wide admixture study. This technique estimates the proportion of African or EA that is present at every point in the genome of each individual. The estimated average percentage of EA in the AA participants used in this analysis was 21.2 ± 12.3%, consistent with other studies of AA participants9,20 (figure e-3).
In our primary analysis, we performed a quantitative admixture evaluation,14 which associates local ancestry to the continuous phenotype—in this case, the slope of cognitive decline. The quantitative admixture scan did not identify any single region of the genome that meets the stringent threshold of genome-wide significance (p < 1 × 10−5) (figure 1A). We also conducted a different admixture scan using an “extreme of outcome” strategy. Specifically, we defined the top 20% of individuals (n = 792) in the distribution of cognitive decline trajectories as “cases” and the 20% with lowest cognitive decline trajectories as “controls.” In this analysis, we observed no admixture peak of excess European or African ancestry in the cases relative to the control participants, consistent with the results of the quantitative admixture scan (figure 1B). However, while no single chromosomal region shows a statistically significant difference in ancestry, we find that cases have a higher proportion of African ancestry across their genome compared to controls (80.0% ± 11.4% African ancestry in cases and 78.4% ± 13.3% in controls; t test p < 0.0001). This result suggests that there may be several different regions of the genome that contain risk alleles that are more frequent in AA populations but that each of them may have only a modest influence on the risk of cognitive decline.
Figure 1. Genome-wide quantitative admixture scans in 3,964 African Americans.

(A) Locus-specific quantitative admixture association scan. The plot shows −log10-transformed p values for all genotyped single nucleotide polymorphisms. The dotted gray line indicates genome-wide significant threshold for admixture association. (B) Case-control admixture analysis using an extreme of outcome strategy.
GWAS for age-related cognitive decline.
While genetic ancestry at any single locus does not appear to have a strong role in age-related cognitive decline, genetic variants do influence this trait.19,21–24 We therefore proceeded with a GWAS meta-analysis across the 5 AA cohorts (n = 3,964 participants) to identify common genetic variants that contribute to age-related cognitive decline. Quantile-quantile plots of the distribution of test statistics showed little deviation from the expected null distribution over most of the genome (figure 2A), and the genomic inflation factor (λGC) was 0.997, indicating no substantial population stratification in the study.
Figure 2. Genome-wide association study meta-analysis of 3,964 African Americans.

(A) Quantile-quantile (QQ) plot of the genome-wide association study (GWAS) meta-analysis. Expected −log10 p values are those expected under the null hypothesis. Observed are the GWAS association results derived by linear regression of all imputed and genotyped single nucleotide polymorphisms (SNPs). (B) Manhattan plot of the cognitive decline GWAS meta-analysis. The plot shows −log10-transformed p values for all 13.9M SNPs tested. AA = African American.
The top results of the GWAS are listed in table 1. As expected, common variants such as rs429358 that partially reflect the effect of the APOE locus7,25 showed association at a genome-wide level of significance, consistent with the previously reported role of the APOE ε4 haplotype in cognitive decline.19 In addition to APOE, we discovered 20 loci with suggestive (p < 10−6) and 4 loci (nearest genes are TRPS1, TEK, GABRA4, and ZNF737) with highly suggestive (p < 10−7) levels of association to the rate of cognitive decline in our study (table 1; figure 2B).
Table 1.
Top results of genome-wide scan for the rate of age-related cognitive decline in African Americans
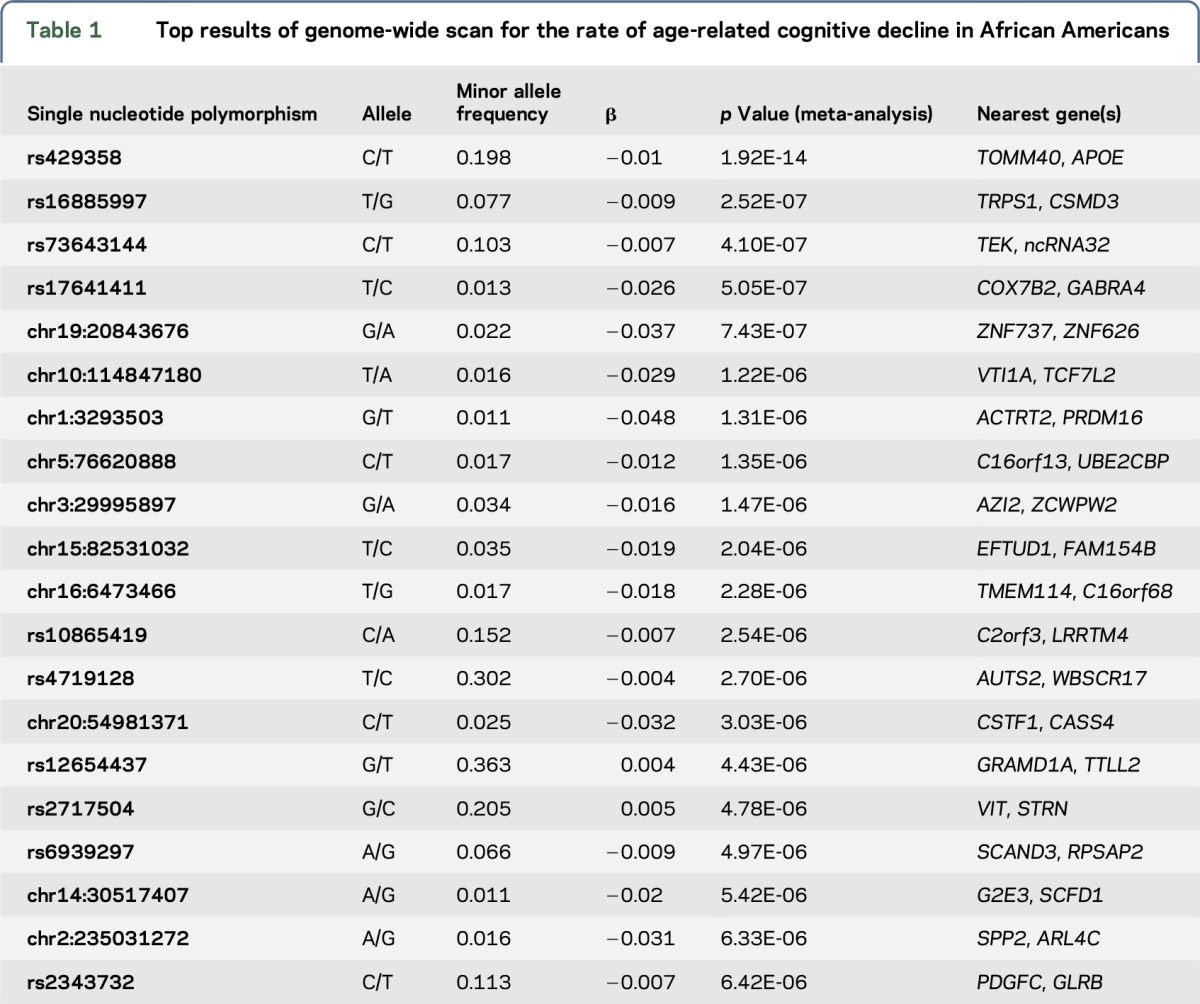
The highly suggestive variant rs16885997 is located 911 kb from TRPS1, a gene encoding a zinc finger transcription factor that represses GATA-regulated genes and binds to a dynein light chain protein. Of interest, the TRPS1 haplotype associated with a higher rate of cognitive decline has previously been associated with greater total (p = 2 × 10−8) and high-density lipoprotein (p = 6 × 10−11) cholesterol levels,26 suggesting a possible link with neurovascular disease.
A complementary strategy to evaluate the role of the 4 highly suggested loci involves their evaluation in network analyses: specifically, we assessed whether the genes found within these 4 loci connect to an existing AD susceptibility network. Of interest, in a protein-protein interaction analysis using DAPPLE, the TEK gene on chr 9p21 (table 1) interacts with validated AD susceptibility genes identified in a recent GWAS25 including CD2AP, PTK2B, BIN1, and INPP5D (figure e-4). This network analysis is significant (permuted p = 0.021), suggesting that the TEK locus may have a role in cognitive decline through an effect on known AD susceptibility networks.
Since AD susceptibility alleles have been reported to influence the trajectory of cognitive decline,19,21 we also evaluated the role of the known AD risk alleles discovered in EA participants in our AA results. As outlined in table e-3, we find evidence that the index SNPs in the ABCA7 and MS4A loci are associated with age-related cognitive decline in AAs using a gene-based multiple hypothesis correction.27
Immune system–mediated pathways are enriched in the AD GWAS but not in cognitive decline.
To systematically identify biological connections between putative susceptibility loci in our analysis, we conducted a pathway enrichment analysis using MAGENTA software15 and highly curated databases as pathway information resources. We found evidence of enrichment of associations involved in lipid/cholesterol metabolism and protein tyrosine kinase signaling pathways in relation to cognitive decline in AA participants (figure 3). We replicated the role of these 2 pathways in an analysis of the same trait in the participants of EA from the ROS and MAP cohorts (table e-4).
Figure 3. Pathway enrichment analysis of African American age-related cognitive decline and Alzheimer disease genome-wide association studies.
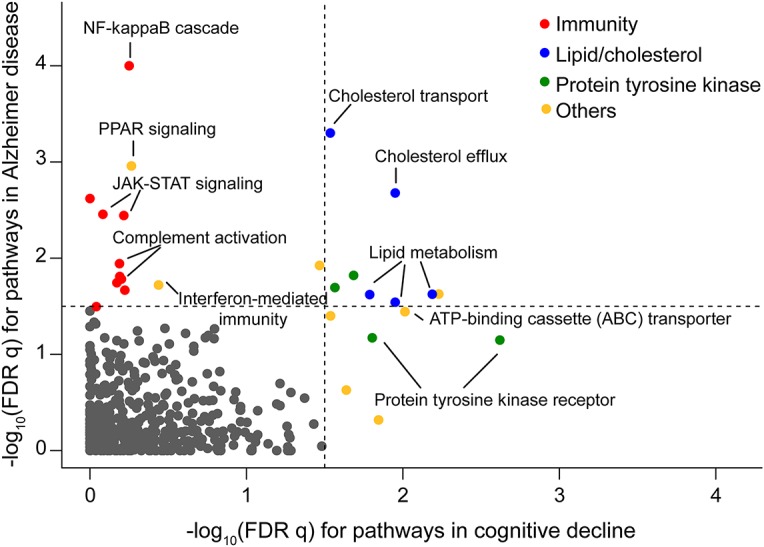
The figure plots the −log10 (false discovery rate [FDR] q value) derived using MAGENTA for pathways enriched in association for Alzheimer disease susceptibility on the y axis, and the results of the same analysis for the genetic architecture of aging-related cognitive decline that we define in our participants on the x axis. The dotted lines denote the threshold of significance (FDR < 0.05) in each analysis. Each point represents one pathway; enriched pathways are color coded with the color key included at the top right aspect of the figure. Selected pathways are annotated to illustrate the types of pathways that we find to be enriched.
We then compared our pathway enrichment results for cognitive decline with results for AD susceptibility using data from a recent large AD GWAS.25 In the AD GWAS pathway analysis, we found that cholesterol/lipid metabolism and immune processes were enriched (p < 10−3; false discovery rate 0.1). When comparing our AA cognitive decline results with the results of a similar analysis in relation to AD in AA participants using a recently published study,7 we find that “lipid/cholesterol metabolism” and “protein kinase signaling processes” are enriched in both analyses, but we observed striking polarization of immunity-related pathways in the AD GWAS (figure 3; table e-3). The pathways including “complement activation,” “interferon-mediated immunity,” and “NF-kappaB cascade” are enriched for AD GWAS results but not for loci associated with cognitive decline (figure 3). These results suggest that, proportionally, the involvement of innate immunity may be much more important in AD than that in aging-related cognitive decline.
DISCUSSION
Our large study represents an important step forward in understanding the role of genetic risk factors in aging-related cognitive decline in the AA population. We were well powered to find susceptibility loci of an even modest effect in our admixture analysis, but we failed to find any single locus with a significant association in this analysis that relies on differences in the genetic risk between the AA and EA populations. Thus, we have not found evidence to support the existence of genetic risk factors that are unique to AA individuals and have a strong effect on aging-related cognitive decline at the population level.
Nonetheless, the greater average proportion of African ancestry in AA individuals with more rapid cognitive decline suggests that there are likely to be a number of genetic risk factors that, while shared between African and European populations, may have a more detrimental role in AA participants. Such a variant may have a more detrimental role for the AA population because of an increase in frequency, a larger effect size, or an interaction with an environmental variable that is enriched in AA individuals.
Our study holds many other intriguing results that warrant further investigation such as the low frequency variants that are strongly associated with cognitive decline in AA individuals and are not found in EA individuals. This prevents us from evaluating these variants in existing data from EA participants, and thus, replication in large cohorts of AA participants will be necessary to validate these observations. Similarly, the other suggestive association of the TEK locus, which connects to a pathway of validated AD susceptibility genes, will require further investigation in AA participants given the very low frequency of the risk allele in EA populations. Finally, we also examined the role of the most suggestively associated variants in the published results of the AD GWAS in AA participants, and we saw no association with this different trait, although this study is not powered to rule out a modest effect in AD for the variants that we report in this study.
Leveraging several large recent studies of AD and cognitive decline in AA and EA participants in addition to our new AA study, we report an intriguing perspective on broader patterns in the genetic architecture of AD and cognitive decline. The fact that certain pathways are significantly enriched for associations with both traits is not surprising since AD is an important driver of cognitive decline in older age. However, the degree of polarization in the enrichment of immune pathways is striking and was unexpected. While there are certainly some susceptibility variants for cognitive decline with presumed effects on immune function (such as MS4A genes and perhaps TRPS1), the relative role of immune pathways is clearly different in terms of susceptibility to these 2 conditions. This is important in prioritizing target validation and drug discovery efforts in shared pathways as well as framing the deployment of compounds that may be discovered in the future: patient stratification may be required to develop an optimal therapy for an individual, based on the relative role of different pathophysiologic processes that contribute to that individual's cognitive decline.
Overall, this study illustrates the successful application of a study design that does not target cognitive performance in older age—which is an amalgam of the effects of developmental factors, life experience, and degenerative processes—but rather targets cognitive decline. This trait allows us to specifically interrogate the genetic architecture of factors that contribute to cognitive loss with age, which occurs in all participants, regardless of their maximal cognitive attainment. Our focus on cohorts that represent samples of the older population makes our results relevant to the experience of our aging population, particularly the AA population. Our pathway analyses are consistent with the narrative that neurovascular risk factors, as well as AD risk factors, are important for cognitive well-being in older age in AA individuals and individuals of other ancestries. The admixture analyses highlight the fact that there are probably no genetic risk factors for cognitive decline that are common and unique to populations with African ancestry: such genetic risk factors appear to be present in both the European and African component of an AA individual's genome, but the allele frequency and effect size of a susceptibility variant can vary between populations, making an allele easier to discover in one or another population.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the study participants in the CHAP, IIDP, ROS, MAP, and MARS cohorts for their contributions. They are grateful to Bogdan Pasaniuc and Paul McLaren for assistance with running the MIXSCORE and HAPMIX tools.
GLOSSARY
- AA
African American
- AD
Alzheimer disease
- CHAP
Chicago Health and Aging Project
- EA
European ancestry
- GWAS
genome-wide association study
- IIDP
Indianapolis-Ibadan Dementia Project
- MAP
Rush Memory and Aging Project
- MARS
Minority Aging Research Study
- ROS
Religious Orders Study
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Towfique Raj and Lori B. Chibnik: analysis. Cristin McCabe: data generation. Andus Wong: interpretation of data. Joseph M. Replogle and Lei Yu: analysis. Sujuan Gao, Frederick W. Unverzagt, and Barbara Stranger: interpretation of data. Jill Murrell, Lisa Barnes, Hugh C. Hendrie, and Tatiana Foroud: provided samples. Anna Krichevsky: interpretation of data. David A. Bennett: provided samples. Kathleen S. Hall: provided samples and study design. Denis A. Evans: provided samples. Philip L. De Jager: study design, data generation, analysis, and interpretation of data.
STUDY FUNDING
This research is supported by NIH Grants R01 AG030146 and R01 AG09956. T.R. is supported by grants from the NIH (F32 AG043267) and the Alzheimer's Association.
DISCLOSURE
Towfique Raj reports no disclosures. Lori B. Chibnik has received research support from NIH NIA (K25 AG41906). Cristin McCabe reports no disclosures. Andus Wong is employed by Merck Research Laboratories Boston. Joseph M. Replogle has received research support from the National Institute of General Medical Sciences (NIGMS) University of California San Francisco (UCSF) Medical Scientist Training Program. Lei Yu has received research support from NIA (RF1AG015819, R01AG017917, R01AG036042, R01AG033678, and R01AG038651), NIH/NIA (U01AG046152), NIH (R01DK099269 and R01AG048056), and National Institute of Neurological Disorders and Stroke (U18NS082140). Sujuan Gao has received research support from NIH (RO1 AG09956 and P30 AG010133-16). Frederick W. Unverzagt has served on the editorial boards of Neuropsychology, Journal of the International Neuropsychological Society, and Current Alzheimer Research; has consulted for UCB Pharma and Piramal Life Science; has received research support from Posit Science, Inc., R01 AG045157, R01 AG019181, U01 NS041588, P30 AG10133, UL1 TR001108, and R01 AG045350; has consulted for Eli Lilly, Inc.; and has served as a consultant and expert witness testimony in medicolegal tort cases. Barbara Stranger has served on the editorial board of PLoS Genetics. Jill Murrell holds a patent for Method for screening for Alzheimer's disease (Patent number: 5,879,883, issued, March 9, 1999) and Transgenic mouse expressing APP770-V717F (Patent number: 6,184,435, issued, February 6, 2001); has received research support from NIA/NIH (1RC2AG036650-01, PHS P30 AG10133, R01 AG09956, PHS 3 U24 AG21886, and R01 AG025688) and NINR/NIH (U01 NR004508); and receives royalty payments for Transgenic mouse expressing APP770-V717F. Lisa Barnes has served on the editorial boards of Journal of Aging and Health and has received research support from NIA (P30-AG10161, RF1-AG22018, P30AG17917, RF1AG51641, U2CAAG54397, R01AG51635, R01AG52583, and RF1AG54057) and NIMHD (P20-MD6886). Hugh C. Hendrie reports no disclosures. Tatiana Foroud has served on scientific advisory boards of the National Advisory Council on Alcohol Abuse and Alcoholism and Washington University Alzheimer's Disease Research Center; has received funding for travel and/or speaker honoraria from Michael J Fox Foundation for Parkinson's Research, NIH, University of Pittsburgh, and University of Chicago; and has received research support from NIH, Columbia University, San Diego State University, University of California, San Diego, University of Mass, University of Pennsylvania, and the Michael J Fox Foundation for Parkinson's Research. Anna Krichevsky reports no disclosures. David A. Bennett has served on the scientific advisory boards of Vigorous Minds, Takeda Pharm, and AbbVie; has served on the editorial boards of Neurology, Current Alzheimer Research, and Neuroepidemiology; and has received research support from NIH (P30AG10161, RF1AG15819, R01AG17917, R01AG36042, U01AG46152, U01AG46161, R01AG33678, and R01AG34374). Kathleen S. Hall has received research support from the National Institute on Aging (5RO1AG009956-18). Denis A. Evans has served on scientific advisory boards of McGill University, and has received research support from NIH (AG11101, AG030146, AG041843, AG019363, and ES024700) and Alzheimer's Association (NIRG 302587). Philip L. De Jager has served on scientific advisory boards of TEVA Neuroscience and Genzyme/Sanofi; has received funding for travel and/or speaker honoraria from Biogen IDEC, Source Healthcare Analytics, Pfizer Inc., and TEVA; has served on the editorial boards of Journal of Neuroimmunology, Neuroepigenetics, and Multiple Sclerosis; and has received research support from Biogen IDEC, GSK, Vertex, Genzyme/Sanofi, and National MS Society. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol 2011;7:137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desmond DW, Moroney JT, Paik MC, et al. Frequency and clinical determinants of dementia after ischemic stroke. Neurology 2000;54:1124–1131. [DOI] [PubMed] [Google Scholar]
- 3.Croxson SC, Jagger C. Diabetes and cognitive impairment: a community-based study of elderly subjects. Age Ageing 1995;24:421–424. [DOI] [PubMed] [Google Scholar]
- 4.Reijmer YD, van den Berg E, Ruis C, Kappelle LJ, Biessels GJ. Cognitive dysfunction in patients with type 2 diabetes. Diabetes Metab Res Rev 2010;26:507–519. [DOI] [PubMed] [Google Scholar]
- 5.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 2009;64:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes LL, Wilson RS, Li Y, Gilley DW, Bennett DA, Evans DA. Change in cognitive function in Alzheimer's disease in African-American and white persons. Neuroepidemiology 2006;26:16–22. [DOI] [PubMed] [Google Scholar]
- 7.Reitz C, Jun G, Naj A, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ε4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 2013;309:1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deo RC, Reich D, Tandon A, et al. Genetic differences between the determinants of lipid profile phenotypes in African and European Americans: the Jackson Heart Study. PLoS Genet 2009;5:e1000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price AL, Tandon A, Patterson N, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet 2009;5:e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1000 Genomes Project Consortium, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasaniuc B, Zaitlen N, Lettre G, et al. Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a Breast Cancer Consortium. PLoS Genet 2011;7:e1001371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segre AV, Consortium D, investigators M, et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 2010;6:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossin EJ, Lage K, Raychaudhuri S, et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet 2011;7:e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raj T, Rothamel K, Mostafavi S, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 2014;344:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson RS, Mendes De Leon CF, Barnes LL, et al. Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA 2002;287:742–748. [DOI] [PubMed] [Google Scholar]
- 19.De Jager PL, Shulman JM, Chibnik LB, et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging 2012;33:1017.e1–1017.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng CY, Kao WH, Patterson N, et al. Admixture mapping of 15,280 African Americans identifies obesity susceptibility loci on chromosomes 5 and X. PLoS Genet 2009;5:e1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chibnik LB, Shulman JM, Leurgans SE, et al. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol 2011;69:560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glahn DC, Kent JW Jr, Sprooten E, et al. Genetic basis of neurocognitive decline and reduced white-matter integrity in normal human brain aging. Proc Natl Acad Sci USA 2013;110:19006–19011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C, Pierce BL. Genetic susceptibility to accelerated cognitive decline in the US Health and Retirement Study. Neurobiol Aging 2014;35:1512.e11–1512.e18. [DOI] [PubMed] [Google Scholar]
- 24.Bressler J, Fornage M, Demerath EW, et al. Fat mass and obesity gene and cognitive decline: the Atherosclerosis Risk in Communities Study. Neurology 2013;80:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu JZ, McRae AF, Nyholt DR, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet 2010;87:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.