

Abstract
Like many Gram-negative pathogens, Shigella rely on a complex type III secretion system (T3SS) to inject effector proteins into host cells, take over host functions, and ultimately establish infection. Despite these critical roles, the energetics and regulatory mechanisms controlling the T3SS and pathogen virulence remain largely unclear. In this study, we present a series of high resolution crystal structures of Spa47 and use the structures to model an activated Spa47 oligomer, finding that ATP hydrolysis may be supported by specific side chain contributions from adjacent protomers within the complex. Follow-up mutagenesis experiments targeting the predicted active site residues validate the oligomeric model and determined that each of the tested residues are essential for Spa47 ATPase activity, although they are not directly responsible for stable oligomer formation. Although N-terminal domain truncation was necessary for crystal formation, it resulted in strictly monomeric Spa47 that is unable to hydrolyze ATP, despite maintaining the canonical ATPase core structure and active site residues. Coupled with studies of ATPase inactive full-length Spa47 point mutants, we find that Spa47 oligomerization and ATP hydrolysis are needed for complete T3SS apparatus formation, a proper translocator secretion profile, and Shigella virulence. This work represents the first structure-function characterization of Spa47, uniquely complementing the multitude of included Shigella T3SS phenotype assays and providing a more complete understanding of T3SS ATPase-mediated pathogen virulence. Additionally, these findings provide a strong platform for follow-up studies evaluating regulation of Spa47 oligomerization in vivo as a much needed means of treating and perhaps preventing shigellosis.
Keywords: arginine finger, ATPase, crystal structure, oligomerization, type III secretion system (T3SS), secretion regulation, Shigella, Spa47, type III secretion apparatus (T3SA)
Introduction
Many Gram-negative pathogens rely on highly conserved type III secretion systems (T3SSs)3 to cause infection by supporting direct injection of effector proteins into the targeted host cell cytoplasm (1–4). One example from this class includes Shigella spp., which is the causative agent of bacillary dysentery in humans and a significant contributor to diarrheal disease. Diarrheal disease is a worldwide threat responsible for 1 in 9 child deaths and is the second leading post neonatal cause of death in children under the age of 5 (5). Many enteric pathogens contribute to these alarming statistics, but Shigella alone are responsible for an estimated 90 million infections and greater than 100,000 deaths annually (6). The majority of Shigella infections occur in developing regions where access to antibiotics are limited and even when they are available, the recent emergence of multidrug-resistant strains has drastically reduced the number of viable treatment options for shigellosis (7). Together, these concerns underscore the need to better understand the mechanisms of Shigella virulence and specifically the means by which Shigella invade human host cells.
Shigella rely entirely on the actions of a complex T3SS to inject effector proteins into the cytoplasm of host cells, initiating cellular invasion of the colonic epithelium and onset of infection (8–10). The Shigella T3SS consists of ∼54 genes that reside on a 220-kb virulence plasmid (11). The “entry region” of the virulence plasmid contains the mxi, spa, and ipa operons that code for the type III secretion apparatus (T3SA) itself (12). The T3SA resembles a nano-needle and syringe, which consists of a basal body that spans the inner and outer Shigella membranes, a ∼2.5-nm inner diameter needle that extends from the bacterial surface to provide a passageway for protein translocation and a tip complex that regulates secretion and interacts with the host cell membrane (13, 14). The mechanisms supporting protein secretion through the Shigella T3SA remain largely unclear; however, we recently characterized Spa47 as an active T3SS ATPase, suggesting that it may provide the energy necessary to power secretion (15).
T3SS ATPases have been identified in several T3SSs (15–22) and share significant sequence identity with one another and with the catalytic β subunit of F1 ATP synthase, which forms an activated heterohexamer capable of driving ATP synthesis or hydrolysis when part of the complete F1F0 ATP synthase complex (23). Similarly, several T3SS ATPases also form activated oligomers in vitro, although the stoichiometries appear to range from trimers to dodecamers and are specific to each ATPase (15, 19, 20, 24–26). The precise role(s) that the ATPases and ATPase stoichiometry plays in T3SS activity remains controversial, although T3SS ATPase deletion mutants and loss of function mutations have resulted in attenuated virulence phenotypes (26–30). This implicates the importance of T3SS ATPases in proper T3SS function and as a potential player in virulence regulation. Some of the most highly studied virulence-associated T3SS ATPases are those from Escherichia coli and Salmonella, which have been shown to recognize T3SS chaperones (29, 30) and unfold the bound effector to facilitate secretion (31). Although these and related works have contributed significantly to the understanding of T3SS ATPases in general, the mechanism(s) driving enzyme activation and the specific role(s) that T3SS ATPases play in many pathogens remain unstudied and unclear.
Here, we report the first crystal structure of the Shigella T3SS ATPase Spa47 and use it to guide biochemical studies mapping key residues/regions involved in ATP hydrolysis and Spa47 oligomer formation. We additionally identify an “arginine finger” that is responsible for Spa47 activation upon oligomer formation and show that either mutation of the conserved arginine or removal of the identified N-terminal oligomerization domain completely inactivates Spa47. Finally, each of the diverse ATPase-inactive Spa47 mutants were expressed in Shigella and extensively tested for phenotype, providing insight into the specific roles of Spa47 in Shigella T3SS activity and virulence. The findings presented here show that Shigella virulence is entirely reliant on ATPase active Spa47 and that its activity is driven by oligomerization, contributing new insight into Shigella virulence mechanisms and providing a promising target for much needed anti-infective agents. In addition, this work provides one of the most comprehensive characterizations of a T3SS ATPase in a single organism to date, highlighting potentially critical similarities and differences to the currently limited number of studied T3SS homologs from other bacterial pathogens, and supports a novel model describing an active population of subhexameric T3SS ATPases.
Results
Crystal Structure of Spa47
Expression and purification of full-length Spa47 produces ATPase active recombinant protein that can be isolated as discrete monomeric and oligomeric species (15). The oligomeric form of the enzyme exhibits significantly higher ATPase activity than the monomer, suggesting that Spa47 is activated through oligomer formation. We have attempted to crystallize both the isolated monomer and oligomeric fractions but have been unable to generate diffracting crystals of either form of the protein. Secondary structure prediction profiles and sequence alignment with several T3SS ATPases (Fig. 1) identify significant levels of sequence identity (e.g. 41% to EscN and 37% to FliI) and directed the design of a Spa47Δ1–79 construct lacking the predicted N-terminal oligomerization domain. The Spa47Δ1–79 construct provided milligram quantities of highly soluble and stable monomeric protein, which readily crystallized overnight to produce rod-shaped crystals. The resulting 2.4 Å Spa47 structure consists of two copies of Spa47 in the asymmetric unit that do not appear to represent a physiological dimer. The Spa47 structure (Fig. 2A) is reminiscent of known AAA ATPases, containing a canonical α/β Rossmann fold (32) consisting of a mostly parallel 9-stranded twisted β-sheet (β3, β2, β7, β6, β8, β10, β5, β9, and β11) closely associated with four α-helices on one side (α5–α8) and two helices on the other (α3 and α4). The C-terminal domain of the Spa47 structure consists of a well defined 3-helix bundle (α11-α13) resembling that of the Salmonella flagellar ATPase, FliI (Fig. 2B). This contrasts with the 5-helix bundle observed in the C terminus of the E. coli T3SS ATPase EscN (Fig. 2C). Structural differences in this region are consistent with suggestions that the C-terminal domain of T3SS ATPases is not involved in ATP hydrolysis but rather recognition and presentation of species-specific T3SS substrates to the secretion machinery (31).
FIGURE 1.

Multiple sequence alignment of Spa47 and related ATPase homologs. Protein sequence alignment of Spa47 (T3SS ATPase, S. flexneri), F1β subunit (ATP synthase, Bos taurus), FliI (Flagellar ATPase, Salmonella typhimurium), EscN (T3SS ATPase, E. coli), InvC (T3SS ATPase, Salmonella gallinrium), and SsaN (T3SS ATPase, S. typhimurium) was performed using the Uniprot multiple sequence alignment tool, Clustal Omega, with single fully conserved residues (*), conservation between groups with strongly similar properties (:), and weakly similar properties (.) identified. Helical and β-sheet regions, as predicted by the PSIPRED structure prediction server, are color-coded red and blue, respectively. Vertical lines separate the predicted N-terminal oligomerization, ATPase, and C-terminal domains. Predicted Walker A (P-loop) and Walker B regions and the fully conserved residues targeted for alanine screening in this study are also identified in the alignment. Uniprot accession numbers used for sequence alignment were Q6XVW8, P00829, P26465, Q7DB71, B5RDL8, P74857 for Spa47, F1 ATPase B subunit, FliI, EscN, InvC, and SsaN, respectively.
FIGURE 2.

2.4 Å X-ray crystal structure of the Shigella T3SS ATPase Spa47. A, crystal structure of Spa47Δ1–79 with coloration corresponding to the predicted domains highlighted in the bar diagram of the Spa47 sequence, where the truncated N-terminal oligomerization domain is represented by a thick dashed line and the black-shaded region corresponds to the N-terminal residues of the construct that are not included in the structure. The gray portion of the Spa47 structure corresponds to the catalytic ATPase domain, the cyan region represents the C-terminal domain, and the Walker A (P-loop) and B motifs are shown in red and blue, respectively. The secondary structure elements are identified, and the dashed lines within the Spa47 structure represent disordered regions with unassigned structure (four N-terminal residues and those connecting α6-β7 and α8-β10). β9 is located directly behind β10 and not visible in this orientation. B and C, alignment of Spa47 (gray) with the catalytic ATPase and C-terminal domains of FliI (green, B) and EscN (blue, C) illustrates the high degree of conservation within the catalytic core of the related ATPases, especially within the nucleotide-binding region (black boxes). D, a closer view of the nucleotide binding site within the aligned structures identifies the conserved active site Lys165 and Glu188 residues positioned near the FliI-bound ADP shown with a yellow carbon skeleton. The PDB codes for the Spa47, FliI, and EscN structures are 5SWJ, 2DPY, and 2OBM, respectively. The crystal structures were rendered using PyMOL.
Key Catalytic Residues Are Spatially Conserved in Spa47
Structural alignment of Spa47Δ1–79 and the nucleotide (ADP)-bound homologs FliI and EscN clearly illustrates the striking structural homology within the catalytic cores of these T3SS ATPases (Fig. 2, B and C). The structural similarities are reflected by the average pairwise RMSDs for Cα atoms between Spa47 and FliI (1.43 Å over 310 atoms) and between Spa47 and EscN (1.41 Å over 306 atoms). Structure and sequence comparisons identify a conserved P-loop (Walker A domain) in Spa47 located between β4 and α3, which is likely involved in substrate binding and coordination (32). Further comparison of the conserved residues and structural elements within the active site of Spa47 identifies individual side chains likely involved in Spa47-catalyzed ATP hydrolysis (Fig. 2D). The first identified residue is a lysine at position 165 that we previously hypothesized to be involved in Spa47 activity (15) based on studies finding that the equivalent P-loop lysine in FliI and EscN stabilizes ATP binding through electrostatic interaction with its β-phosphate (16, 29). Additionally, strong similarities in spatial orientation of the conserved glutamate 188 side chain in Spa47 and the equivalent glutamate residues in FliI and EscN were also observed (Fig. 2D), placing Glu188 in an appropriate position to coordinate a catalytic water as shown for both EscN and the catalytic β-subunit of F1 ATP synthase (29, 33).
Generating an Activated Hexameric Spa47 Model
Biochemical studies together with recent cryo-electron tomography studies have begun to uncover the importance of oligomerization in activating and regulating ATPase activity within the context of T3SSs. Unfortunately, a lack of atomic resolution data for T3SS ATPase oligomers has been a major hurdle in studying the mechanism driving ATPase activation and effector secretion. Here, we generated an in silico model of hexameric Spa47 based on recent high resolution cryo-electron tomography data identifying Spa47 as a homohexamer in the context of the intact T3SA (34) and the propensity of AAA and AAA+ ATPases to form activated hexamers (Fig. 3A) (35). The model was generated by aligning our 2.4 Å Spa47 structure onto the 2.8 Å heterohexameric F1 α3β3 ATP synthase structure (PDB code 1BMF) (36). The hexameric Spa47 model results in good alignment to F1 ATP synthase with an overall backbone RMSD of 2.06 Å over 1846 Cα atoms and no major gaps or overlapping regions within the model (Fig. 3B). Structurally, the Spa47 hexamer results in a ∼100 Å diameter ring-like complex with a central channel measuring ∼20 Å in diameter, perhaps providing a passageway for secreted T3SS proteins. Looking closely at the active site containing the AMP-PNP substrate modeled from the F1 ATP synthase structure highlights the spatial relationship between the nucleotide substrate and the Lys165 and Glu188 active site residues identified earlier (Figs. 2D and 3B). Viewing the active site in the context of a Spa47 oligomer additionally identifies an arginine (Arg350) that extends into the ATP binding site located on an adjacent protomer within the complex (Fig. 3B), suggesting that it may also be important for ATP hydrolysis. Similar examples of “arginine fingers” are found in many oligomeric AAA+ ATPases including F1 ATP synthase, where an arginine side chain from a regulatory α-subunit contributes to the active site of the neighboring catalytic β-subunit, completing the active site and perhaps stabilizing the binding of the largely negative ATP (37, 38). Interestingly, in our model, the arginine in the Spa47 hexamer is also presented to the active site by a neighboring molecule in the oligomer and would be unlikely to be involved in catalysis in a monomeric form of the enzyme, making Spa47 Arg350 a key residue in the biochemical studies presented in the following sections.
FIGURE 3.

Hexameric model of activated Spa47. A, Spa47 modeled as an activated homohexamer, based on the F1 ATP synthase structure. Each of the Spa47 monomer subunits are colored independently. B, surface representation of the hexameric model shown in top view and at a 90° rotation. Each of the six nucleotide-binding regions within the hexamer are located at a separate protomer interface with one example outlined. A closer view of the Spa47 active site looking from within the hexamer pore shows the conserved active site Lys165 and Glu188 side chains from one protomer (gray) and an arginine from the adjacent protomer (blue) in relation to the bound ATP homolog (AMPPNP) modeled from its location in the F1 ATP synthase structure.
Structural Influences of Engineered Spa47 Point Mutants
The predicted active site residues Lys165 and Glu188, as well as Arg350, were each mutated to alanine to directly investigate their roles in Spa47-catalyzed ATP hydrolysis and Shigella virulence. Each of the mutations were engineered in the full-length Spa47 backbone for biophysical characterization and Shigella phenotype analysis, as well as in the Spa47Δ1–79 construct to allow structural characterization of the targeted mutations. Three mutant crystal structures were determined, ranging from 2.7 to 1.8 Å resolution (Table 1). Comparing each of the mutant structures to wild type confirms that the mutations are present and that they have little effect on the global structure of the proteins (Fig. 4).
TABLE 1.
Data collection, phasing, and refinement statistics
One crystal was used for each data set.
| Spa47Δ1–79 | Spa47Δ1–79 K165A | Spa47Δ1–79 E188A | Spa47Δ1–79 R350A | |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | SSRL 7–1 | SSRL 14–1 | SSRL 14–1 | SSRL 14–1 |
| Wavelength (Å) | 1.1271 | 0.9795 | 0.9795 | 0.9795 |
| Space group | P21 | P21 | P21 | P21 |
| Cell dimensions | ||||
| a, b, c (Å) | 43.96, 153.92, 54.63 | 44.40, 154.06, 54.94 | 45.09, 152.60, 54.58 | 44.16, 153.66, 54.88 |
| α, β, γ (°) | 90.0, 109.83, 90.0 | 90.0, 110.29, 90.0 | 90.0, 110.58, 90.0 | 90.0, 110.14, 90.0 |
| Resolution (Å)a | 35–2.40 (2.49–2.40) | 40–2.15 (2.23–2.15) | 35–2.70 (2.80–2.70) | 40–1.80 (1.86–1.80) |
| No. of reflections | 26490 | 37547 | 18848 | 63430 |
| CC½ | 0.988 (0.400) | 0.997 (0.474) | 0.990 (0.601) | 0.999 (0.511) |
| I/σI | 8.2 (1.2) | 15.1 (1.6) | 9.6 (1.6) | 20.9 (1.3) |
| Completeness (%) | 99.5 (99.7) | 99.9 (100.0) | 99.6 (100.0) | 99.8 (100.0) |
| Redundancy | 7.0 (6.7) | 5.0 (4.3) | 4.3 (4.3) | 3.4 (3.3) |
| Refinement | ||||
| Resolution (Å) | 35–2.40 (2.49–2.40) | 40–2.15 (2.23–2.15) | 35–2.70 (2.80–2.70) | 40–1.80 (1.86–1.80) |
| Rwork/Rfree | 0.196/0.237 | 0.194/0.232 | 0.200/0.259 | 0.178/0.216 |
| No. of atoms | ||||
| Protein | 5274 | 5319 | 5275 | 5265 |
| Ligand/ion | 10 | 10 | 10 | 10 |
| Water | 209 | 374 | 106 | 449 |
| B-factors | ||||
| Protein | 49.8 | 42.6 | 49.1 | 40.2 |
| Ligand/ion | 89.3 | 73.0 | 97.3 | 85.3 |
| RMSDs | ||||
| Bond lengths (Å) | 0.004 | 0.003 | 0.004 | 0.007 |
| Bond angles (°) | 0.887 | 0.710 | 0.940 | 1.140 |
| Ramachandran | ||||
| Preferred (%) | 98.5 | 97.8 | 97.1 | 97.9 |
| Outliers (%) | 0 | 0 | 0 | 0 |
| PDB code | 5SWJ | 5SYP | 5SWL | 5SYR |
a Values in parentheses are for the highest resolution shell.
FIGURE 4.

Structure alignments between wild-type Spa47Δ1–79 and the ATPase inactive alanine point mutants used in this study. The wild-type Spa47Δ1–79 structure is shown in gray and is aligned to the Spa47Δ1–79 K165A (purple), Spa47Δ1–79 E188A (red), and Spa47Δ1–79 R350 (fuchsia) structures shown in A, B, and C, respectively. The bars located below each structure provides a visual representation of the regions included (white) and missing (shaded) in each structure as well as the relative location of each engineered point mutant within the Spa47 sequence and structure (red asterisk). The PDB codes for Spa47Δ1–79, Spa47Δ1–79 K165A, Spa47Δ1–79 E188A, and Spa47Δ1–79 R350A used in the figure are 5SWJ, 5SYP, 5SWL, and 5SYR, respectively.
Influence of Engineered Mutations on Spa47 Oligomer Formation
Full-length Spa47 purifies as discrete monomeric and oligomeric species with the oligomer exhibiting significantly higher levels of ATP hydrolysis activity than the monomer (15). Size exclusion chromatography (SEC) and sedimentation velocity analytical ultracentrifugation (SV-AUC) suggest the activated oligomer is a novel homotrimer that is thus far unique to Spa47, although the driving force for oligomerization and the implications of an activated Spa47 trimer in vivo remain unclear. To explore this further, we examined the influence of the engineered K165A, E188A, and R350A active site point mutations on Spa47 oligomer formation and distribution. SEC analysis of these Spa47 mutants found that they all purify as primarily monomeric species with dominant peaks in the SEC chromatogram at ∼79 ml with a minor oligomeric population eluting at ∼52 ml (Fig. 5A). This distribution has been consistently observed for wild-type Spa47, suggesting that the targeted active site residues are not involved in the formation of the activated oligomeric species, although they are required for ATPase activity (see activity results in Fig. 6). This finding is especially interesting because all other reported active T3SS oligomers exhibit a minimum of hexameric stoichiometry, and oligomerization is frequently dependent on nucleotide binding and/or hydrolysis (39–41).
FIGURE 5.
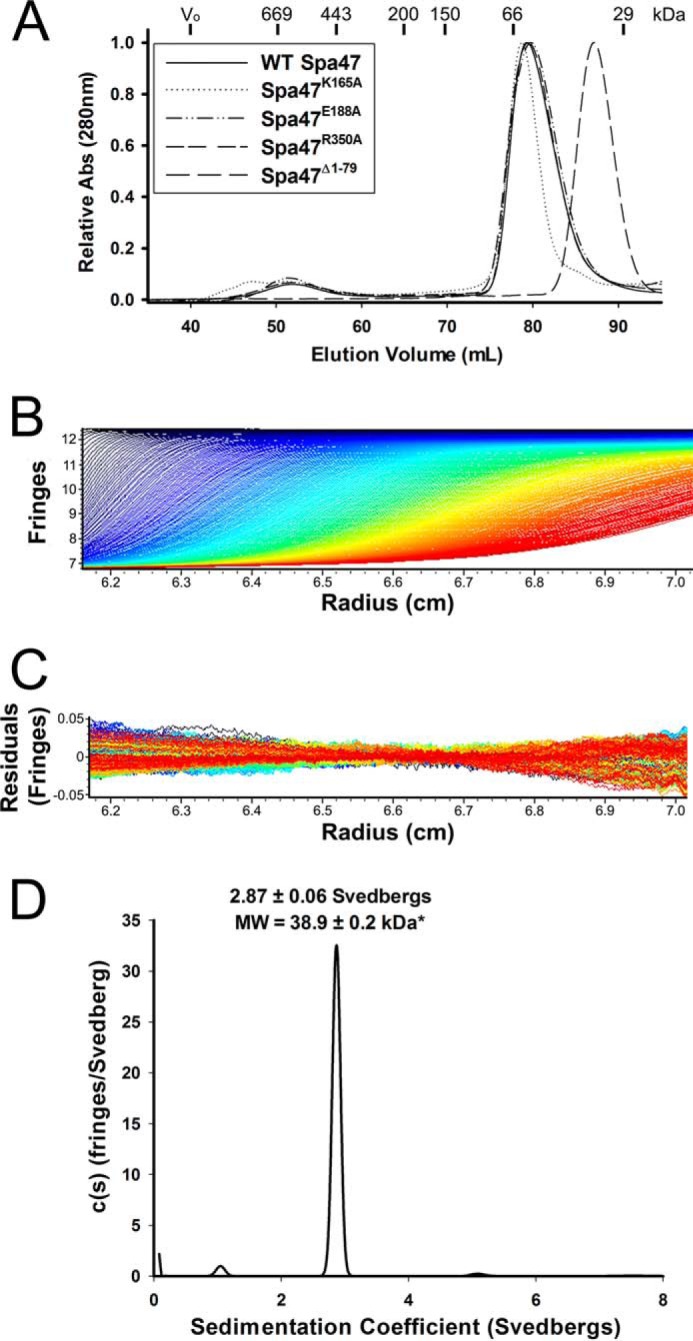
Analysis of Spa47 oligomeric distribution. A, size exclusion chromatography elution profiles of wild-type Spa47, Spa47K165A, Spa47E188A, and Spa47R350A are all consistent with the formation of monomeric and single oligomeric species. The weak signal observed prior to the early eluting oligomer for the K165A mutant is attributed to nucleic acids and does not result from a change in oligomerization profile. The Spa47Δ1–79 SEC elution profile exhibits a single shifted elution peak, consistent with the reduced molecular mass of the Spa47 N-terminal truncation construct and an essentially exclusive monomeric protein distribution. The void volume and the elution volumes corresponding to commercial protein molecular mass standards are included at the top of the chromatogram. B, representative interference scans of purified Spa47Δ1–79 monitored during SV-AUC. C, representative residuals from fitting the data to a continuous c(s) distribution model as described under “Experimental Procedures.” D, representative sedimentation coefficient distribution (c(s) versus S) showing that Spa47Δ1–79 sediments as an essentially homogeneous species with a sedimentation coefficient of 2.87 ± 0.06 S, corresponding to a calculated molecular mass of 38.9 ± 0.2 kDa and a Spa47Δ1–79 monomer. Reported sedimentation coefficients represent means ± S.D. from three independent measurements.
FIGURE 6.

ATPase activity of engineered Spa47 constructs. Kinetic analysis of ATP hydrolysis by each of the engineered Spa47 constructs used in this study. Wild-type Spa47 monomeric and oligomeric species are ATPase active with an increase in hydrolysis efficiency for the Spa47 oligomer. Isolated monomeric and oligomeric forms of Spa47K165A, Spa47E188A, and Spa47R350A, as well as the monomeric form of the oligomerization-deficient Spa47Δ1–79 construct were all inactive, resulting in background levels of ATP hydrolysis. Each data point represents the means ± S.D. of three independent measurements from two separate protein preparations.
In addition to the Spa47 alanine point mutants, SEC analysis was performed on the Spa47Δ1–79 construct used to solve the X-ray crystal structure. Removal of the N-terminal domain resulted in a single late eluting peak (87 ml) that is consistent with the reduced monomeric molecular mass of the truncated construct, suggesting that removal of the N-terminal domain prevents Spa47 oligomerization (Fig. 5A). Although SEC provides a quick and relatively simple method for the analysis of protein samples, the strong dependence of SEC on protein shape, in addition to molecular mass, makes accurate stoichiometry determination difficult. This is exemplified by the drastic overestimation of the molecular mass of the early eluting Spa47 oligomers by SEC (Fig. 5A) compared with the more reliable molecular mass predictions recently provided by SV-AUC (15). SV-AUC directly measures the sedimentation coefficient for each species in a solution, providing an accurate and robust means of identifying multiple species in a solution and an ideal technique to evaluate the Spa47Δ1–79 oligomerization profile in this study. SV-AUC analysis was performed on Spa47Δ1–79, as was done previously for the full-length construct (15), finding that Spa47Δ1–79 exists nearly exclusively as a single species with a sedimentation coefficient of 2.87 ± 0.06 S, corresponding to a calculated molecular mass of 38.9 ± 0.2 kDa (Fig. 5, B–D). This finding is in good agreement with the known Spa47Δ1–79 monomeric molecular mass of 38.964 kDa and confirms that the Spa47Δ1–79 construct does not oligomerize under the tested conditions. The new structure data provided by the Spa47 N-terminal deletion construct together with its inability to form higher order oligomers makes Spa47Δ1–79 a valuable tool for uncovering the critical link between Spa47 oligomer formation and activation.
Predicted Spa47 Active Site Residues and Oligomerization Are Required for ATPase Activity
Several Spa47 residues were predicted to be involved in enzyme activity based on their conservation among homologous proteins and their location within the Spa47 active site (Figs. 2D and 3B). Each of the full-length Spa47 constructs (wild-type Spa47, Spa47K165A, Spa47E188A, and Spa47R350A), as well as the N-terminal truncation construct Spa47Δ1–79, were expressed and purified as described under “Experimental Procedures.” The stable oligomeric species of each construct were isolated using SEC. As seen in Fig. 5A, each of the individual point mutants produced discrete monomeric and oligomeric populations; however, the Spa47Δ1–79 construct resulted in the purification of only monomeric species. The effect of each construct and oligomer state on ATPase activity was determined using a radioactivity assay that directly quantifies ATP to ADP conversion as a function of time (Fig. 6). The activity results show that the full-length wild-type Spa47 behaves as previously reported with activity observed for both the isolated monomeric and oligomeric species, again finding that the preformed oligomeric Spa47 complex hydrolyzes ATP at a much greater rate than the isolated full-length monomer (15). The Spa47K165A construct was unable to hydrolyze ATP as either an isolated monomeric or oligomeric species, which is consistent with previous activity data for both Spa47 and an equivalent mutation in the Salmonella SPI-1 ATPase InvC (15, 26). Similar results were found for the newly developed Spa47E188A and Spa47R350A mutants, which also lack ATPase activity under both oligomeric and monomeric conditions, confirming that each of the Spa47 residues tested in this study are in fact critical for ATP hydrolysis and providing valuable new constructs that helped to uncover the mechanism behind Spa47-catalyzed ATP hydrolysis.
We additionally examined the impact of oligomerization on Spa47 ATPase activity using the stable monomeric Spa47Δ1–79 construct. Interestingly, though Spa47Δ1–79 contains the native sequence and all of the regions proposed to be involved in ATP hydrolysis, it is catalytically inactive, as seen for the predicted active site mutants, including R350A. Together, these results support the idea that Spa47 oligomerization presents arginine 350 to the active site of an adjacent protomer as suggested in our model (Fig. 3).
Active Spa47 Is Required for Proper T3SS Translocon Formation and Shigella flexneri Host Cell Invasion
Each of the engineered Spa47 constructs were expressed in a S. flexneri spa47 null strain to better understand the role that Spa47 plays in the Shigella T3SS and ultimately its virulence. A gentamicin protection assay quantified the ability of the complemented strains to invade cultured eukaryotic host cells, an event that relies entirely on a functional Shigella T3SS (4). We showed previously that complementing a spa47 null S. flexneri strain with the inactive Spa47K165A mutant resulted in the same lack of invasiveness seen for the null strain, suggesting that ATPase active Spa47 is required for proper T3SS function (15). This study expands these findings to include the Spa47E188A and Spa47R350A constructs and the oligomerization-deficient Spa47Δ1–79 mutant. When compared with the wild-type S. flexneri clinical isolate 2457T, the spa47 null strain was essentially non-invasive, exhibiting 0.8 ± 1.1% invasiveness, whereas complementation with the wild-type Spa47 construct resulted in nearly complete phenotype reinstatement at 81.4 ± 10.8% invasiveness (Table 2). Complementation with the catalytically inactive Spa47K165A, Spa47E188A, Spa47R350A, or Spa47Δ1–79 constructs failed to restore virulence with 0.5 ± 0.2, 0.2 ± 0.2, 0.1 ± 0.2, and 0.2 ± 0.1% invasion, respectively. Together with previous studies linking Spa47 to T3SS-based cellular invasion (15, 28), these findings strongly implicate Spa47 ATPase activity as an essential factor in Shigella virulence. It is perhaps even more interesting to consider the role of the identified “arginine finger” in Spa47 activation, in which Arg350 is clearly required for ATP hydrolysis (Fig. 6), although it is located quite distantly from the active site in the context of a Spa47 monomer. This again supports the hypothesis that a multisubunit active site is critical for activity both in vitro and in vivo.
TABLE 2.
The effect of directed Spa47 mutations on Shigella hemolysis and invasion
| S. flexneri strain | Complementation | Relative invasion (% ± S.D.)a | Relative hemolysis (% ± S.D.)b |
|---|---|---|---|
| 2457T (WT) | None | 100 | 100 |
| spa47 null | None | 0.8 ± 1.1 | 0.9 ± 2.0 |
| spa47 null | spa47/pWPsf4 | 81.7 ± 10.8 | 81.4 ± 11.6 |
| spa47 null | spa47 K165A/pWPsf4 | 0.5 ± 0.2 | 1.1 ± 1.9 |
| spa47 null | spa47 E188A/pWPsf4 | 0.2 ± 0.2 | 0.9 ± 2.0 |
| spa47 null | spa47 R350A/pWPsf4 | 0.1 ± 0.2 | 1.2 ± 2.5 |
| spa47 null | spa47Δ1–79/pWPsf4 | 0.2 ± 0.1 | 0.9 ± 2.2 |
a The ability of the Shigella mutants to invade cultured host cells was measured by a standard gentamicin protection assay (see “Experimental Procedures”). Invasion results are presented as the percentage of invasion by the wild-type S. flexneri strain 2457T.
b The ability of each Spa47 Shigella mutant to lyse red blood cells was tested using a hemolysis assay that quantifies released hemoglobin following incubation of erythrocytes with the described Shigella mutants. Hemolysis results are presented as the percentages of hemoglobin released by the wild-type S. flexneri strain (2457T). The experiments were all repeated in triplicate, and results represent the means ± standard deviation.
Although functional Spa47 is clearly required for the critical series of events supporting Shigella invasion, it remains unclear exactly which steps Spa47 contributes to and which roles the engineered mutants are unable to perform. Based on the link between T3SS ATPases and protein secretion, we first turned to a hemolysis assay that quantifies the ability of Shigella to insert the T3SS translocon proteins into red blood cell membranes, lysing the infected host cells. A wild-type hemolysis phenotype requires proper formation of the T3SA and secretion of the translocator proteins IpaB and IpaC but is independent of effector secretion into the host cytoplasm and effector activity. This makes the hemolysis assay a powerful complementary tool when used with the invasion assay. Here, each of the Shigella strains described earlier for invasion were tested for hemolytic activity and compared with wild-type S. flexneri (Table 2). As expected, the spa47 null strain was non-hemolytic exhibiting only 0.9 ± 2.0% hemolysis, confirming that a complete lack of Spa47 expression prevents formation and/or insertion of a functional T3SA translocon. Complementation of the spa47 null strain with wild-type Spa47 results in a recovery of hemolysis phenotype (81.4 ± 11.6%). Complementation with Spa47K165A, Spa47E188A, Spa47R350A, or Spa47Δ1–79 constructs failed to restore hemolytic ability with 1.1 ± 1.9, 0.9 ± 2.0, 1.2 ± 2.5, and 0.9 ± 2.2% hemolysis, respectively. The inability of the catalytically inactive Spa47 mutants to restore hemolytic capabilities together with the lack of invasion phenotype (Table 2) suggests that the invasion defect results at least in part from an inability to form a complete and properly functioning T3SA and in particular an inability to insert an IpaB/IpaC translocon pore into the host cell membrane.
Shigella T3SS Translocator Secretion Requires T3SS ATPase Activity
The T3SS translocator proteins IpaB and IpaC reside at the tip of the mature Shigella T3SA where they interact with the host cell membrane (42). It is the insertion of IpaB and IpaC into the membrane that forms the translocon pore and ultimately allows secretion of effectors into the targeted host cell cytoplasm (43). The hemolysis results presented in this study suggest that Shigella strains lacking the gene for Spa47 or strains complemented with ATPase inactive Spa47 mutants are not able to properly insert the translocon into the host membrane (Table 2). We hypothesize that this is a direct result of an inability of the mutant strains to secrete IpaB and IpaC and properly deliver them to the host cell membrane. To test this hypothesis, the role of Spa47 in both uninduced and induced translocator secretion profiles was examined for S. flexneri. We first looked at the uninduced secretion profiles of each of the Shigella strains examined above. In this assay, the levels of the translocator proteins secreted into the supernatant of overnight Shigella cultures are quantified as a function of the expressed Spa47 mutations. A properly formed T3SA displays low levels of translocator secretion (“leakage”) into the overnight culture supernatant, indicating that the T3SA is properly formed, and the bacteria are poised for active secretion of effectors into the host cell (43). Here, the assay control is a Shigella strain expressing wild-type Spa47, which serves as the benchmark for secretion comparison. Significantly reduced levels of IpaB and IpaC secretion are observed for the spa47 null strain with only 25.4 ± 7.0 and 19.1 ± 17.7% IpaB and IpaC secreted, respectively. Complementation with Spa47K165A, Spa47E188A, Spa47R350A, or Spa47Δ1–79 constructs result in a similar attenuation in both IpaB and IpaC secretion levels as shown in Fig. 7 (A and B). The cytoplasmic levels of IpaB and IpaC were also quantified to ensure that the decrease in translocator secretion from the mutant strains is not a result of attenuated expression levels or lack of available IpaB and IpaC. The results clearly show that IpaB and IpaC levels are consistent within the Shigella cytoplasm of each strain (Fig. 7C). Additionally, a bacterial cell lysis control was performed by blotting and probing for cytoplasmic and supernatant levels of the T3SS chaperone protein IpgC, which is natively expressed but not secreted by Shigella. As expected, IpgC was detected in the Shigella whole cell extract samples but was not observed in the supernatant of the uninduced or actively secreting Shigella cultures, confirming that lysis did not significantly contribute to the detected levels of secreted IpaB and IpaC (data not shown).
FIGURE 7.
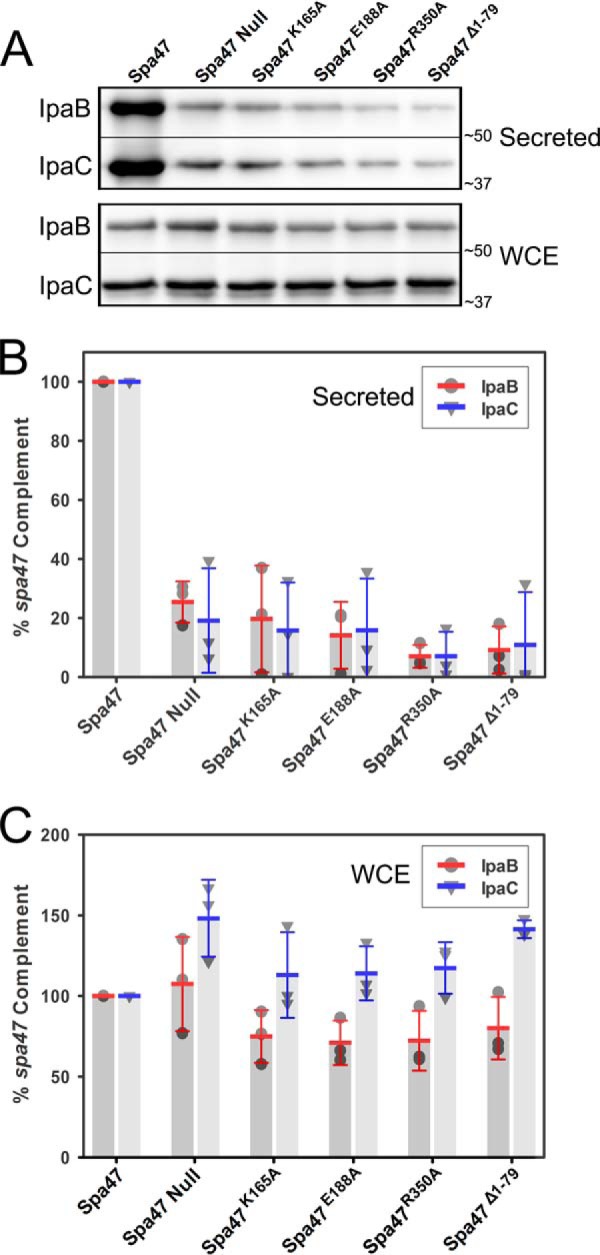
Immunoblot analysis of uninduced translocator secretion profiles of engineered Spa47 mutant Shigella strains. A, the levels of IpaB and IpaC secreted (“leaked”) into overnight Shigella culture media were compared for Shigella strains expressing engineered ATPase inactive Spa47 mutants, as well as positive and negative controls expressing wild-type Spa47 and a spa47 null strain, respectively. The secreted proteins were detected by Western blot and compared with one another using densitometry analysis. B, the protein levels are reported relative to a S. flexneri strain expressing wild-type Spa47 (normalized to 100%) and are reported for each strain. C, the levels of IpaB and IpaC isolated from the bacterial whole cell extract (WCE) were also visualized and are shown relative to the protein levels isolated from the control strain expressing wild-type Spa47. The reported values represent the means ± S.D. from three independent analyses.
In addition to quantifying the uninduced secretion profiles shown in Fig. 7, the small molecule Congo red was used to mimic in vivo activation of the T3SS and the ability to rapidly initiate T3SS protein secretion (44). Although the low levels of uninduced secretion require overnight growth and protein precipitation to observe secreted protein levels, Congo red-induced secretion is limited to 15 min, evaluating the ability of the T3SS to be activated as if it had come in contact with a host cell membrane (44). As expected, Congo red exposure to the S. flexneri strain expressing wild-type Spa47 results in efficient secretion of both IpaB and IpaC, whereas the spa47 null strain failed to actively secrete either IpaB or IpaC at wild-type levels (Fig. 8). The active site mutants Spa47K165A and Spa47E188A, the oligomerization-dependent active site mutant Spa47R350A, and the Spa47Δ1–79 construct lacking the N-terminal oligomerization domain all resulted in significantly attenuated active secretion profiles. The observed attenuation under both uninduced and induced conditions, together with the hemolysis and invasion results, suggests that the loss of Shigella virulence is due, at least in part, to reduced secretion efficiencies and incomplete formation of the T3SA.
FIGURE 8.
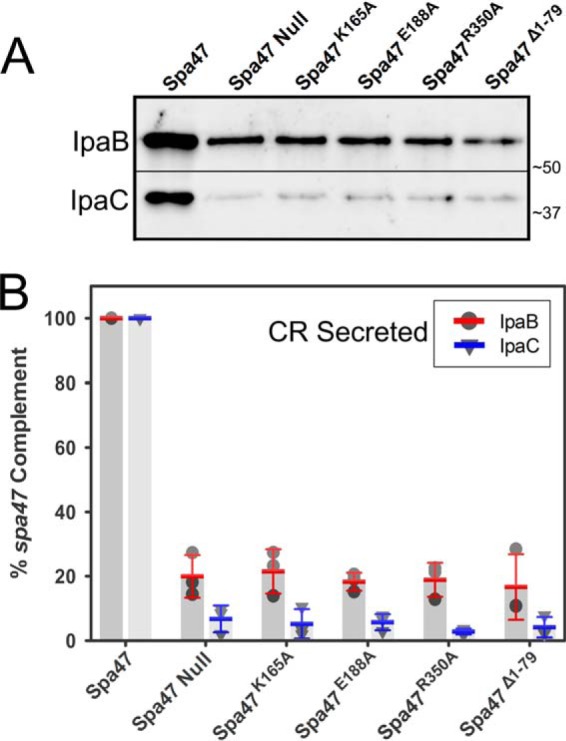
Immunoblot analysis of Congo red-induced T3SS secretion profiles for engineered Spa47 mutant Shigella strains. The levels of IpaB and IpaC actively secreted following Congo red induction were compared for Shigella strains expressing engineered ATPase inactive Spa47 mutants, as well as positive and negative controls expressing wild-type Spa47 and a spa47 null strain, respectively. A, actively secreted IpaB and IpaC were detected by Western blot and compared using densitometry analysis. B, the protein levels are reported relative to a S. flexneri strain expressing wild-type Spa47 (normalized to 100%) and are reported for each strain. The reported values represent the means ± S.D. from three independent analyses.
Spa47-catalyzed ATP Hydrolysis Supports Construction of the Shigella T3SA
One of the pioneering studies that began to unravel the role of Spa47 in the Shigella T3SS showed that a spa47 knock-out Shigella strain was only able to construct a partial T3SA, consisting of portions of the basal body and lacking an external needle apparatus (28). The phenotype results presented in this study are consistent with these findings, additionally suggesting that the presence of Spa47 alone is not enough to restore phenotype, but that Spa47-catalyzed ATP hydrolysis is essential for the proper secretion of T3SS proteins, including those that make up the external needle and tip complex. Here, we have extended these studies to directly examine the effect of each of the ATPase inactive Spa47 mutations on the ability of Shigella to assemble a complete T3SA. Fluorescent antibodies were used to probe T3SA needle and tip formation in Shigella strains including the wild-type S. flexneri clinical isolate 2457T, a spa47 null strain, and complemented strains expressing wild-type Spa47, Spa47K165A, Spa47E188A, Spa47R350A, and Spa47Δ1–79. The level of fluorescently immunolabeled T3SS needle protein MxiH was quantified for each strain using flow cytometry. The wild-type Shigella strain and the Spa47 complemented strain both exhibited a positive shift in MxiH-labeled fluorescence intensity histograms compared with those of the spa47 null strain and mxiH null strain control (Fig. 9A). Each of the engineered Spa47 mutants resulted in fluorescence intensity histograms that overlay almost perfectly with both the spa47 null and mxiH null strains, suggesting that they are unable to form external MxiH needles. A similar experiment was conducted using polyclonal antibodies against the constitutively expressed T3SS tip protein, IpaD. Multiple copies of IpaD are known to reside at the tip of the T3SA (45, 46), located far enough from the bacterial membranes and lipopolysaccharide layer to be efficiently detected by immunolabeling (47). The cytometry results for IpaD are essentially identical to those of the MxiH labeling experiment, with significant labeling for strains expressing wild-type Spa47, again consistent with a properly formed apparatus and accessible IpaD at the needle tip (Fig. 9B). The MxiH null and spa47 null strains report background levels of fluorescence intensity resulting from a lack of IpaD on the bacterial surface. Each of the strains expressing the engineered Spa47 mutants were also unable to mobilize IpaD to the bacterial surface and resulted in fluorescence intensity histograms that overlay with the mxiH null and spa47 null strain histograms (Fig. 9B). Taken together with the secretion assays presented here, these findings support the hypothesis that ATPase active Spa47 is required for proper protein secretion and T3SA formation and that the resulting lack of an external needle complex is responsible for the avirulent phenotype observed for the engineered strains expressing ATPase inactive Spa47 mutants. The rapidly increasing resolution offered by cryo-electron tomography and the capabilities of single-molecule super-resolution fluorescence techniques will undoubtedly help to uncover more specifics related to the dependence of T3SA formation on Spa47 activity, but the work presented here provides a foundational understanding of the role(s) that Spa47 plays in Shigella T3SS activation and virulence.
FIGURE 9.
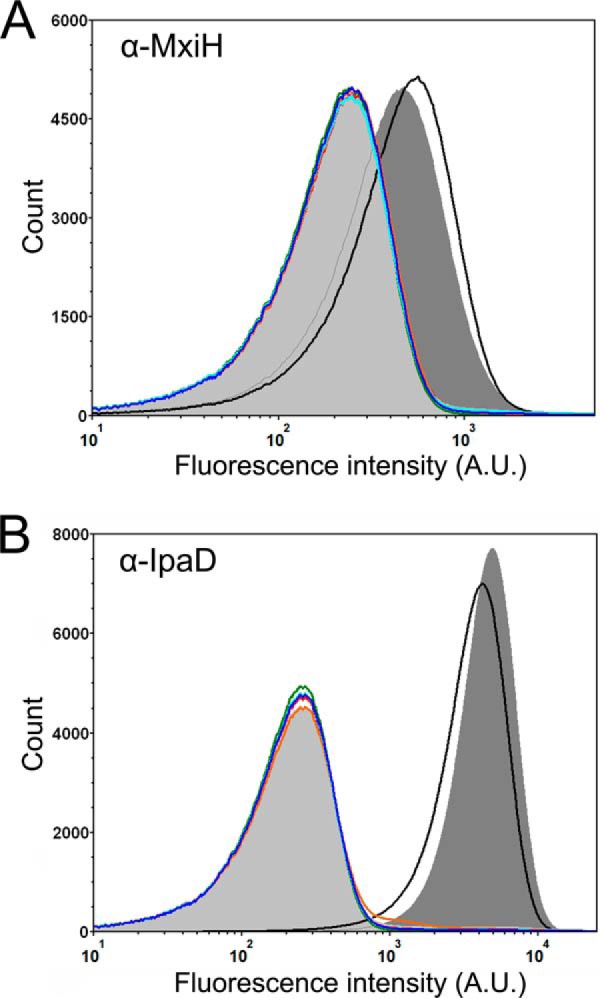
Flow cytometry detection of surface localized Shigella T3SS components. A, fluorescence intensity histograms depicting the levels of surface localization of MxiH for the S. flexneri 2457T strain (black), mxiH null strain (orange), spa47 null S. flexneri strain (light gray shading), and the spa47 null strain complemented with wild-type Spa47 (dark gray shading), Spa47K165A (red), Spa47E188A (blue), Spa47R350A (cyan), and Spa47Δ1–79 (green). B, fluorescence intensity histograms depicting the levels of surface localization of the T3SS tip protein IpaD for the same strains shown in A. The histograms include 500,000 individual intensity measurements per condition and are representative of independent triplicate analysis. The decreased levels of the MxiH and IpaD for all of the ATPase inactive Spa47 mutants are consistent with the in vitro experiments, suggesting that Spa47-catalyzed ATP hydrolysis drives/supports T3SA needle formation and Shigella virulence.
Discussion
Like many Gram-negative pathogens, Shigella rely on a T3SS as their primary virulence factor (3, 48). Studies aimed to better understand the structure and function of the Shigella T3SS are essential, because Shigella not only represents a worldwide health concern but serves as a valuable in situ/in vivo model for T3SS studies. As a model T3SS expressing-pathogen, Shigella are non-motile, can be studied under BSL-2 conditions, encode their T3SS on a virulence plasmid, express only one T3SS, rely entirely on their T3SS for virulence, and are currently the only pathogen for which a complete stepwise maturation of the secretion apparatus has been observed and described (43, 49–51). Despite these advantages, many of the details surrounding the activation of its T3SS and ultimately the ability of Shigella to regulate infection remain unclear and a high priority for understanding and controlling T3SS-based infections.
Sequence similarity to known ATPases including the catalytic β-subunit of F1 ATP synthase and the Salmonella flagellar ATPase FliI suggested that the Shigella T3SS protein Spa47 is a T3SS-associated ATPase and that it could in turn provide the energy required to assemble and activate the T3SA. We recently showed that Spa47 is in fact an ATPase, that it exhibits oligomerization-dependent ATPase activity, and that active Spa47 appears to be required for T3SS-mediated cellular invasion by Shigella (15). These findings are consistent with pioneering T3SS ATPase studies showing that Salmonella motility is controlled through tight regulation of the flagellar ATPase, FliI, by what appears to be control over ATPase oligomerization (52, 53). Additionally, work with the SPI-1 ATPase in Salmonella showed that InvC recognizes T3SS substrate and facilitates chaperone release and unfolding of secreted proteins (31). Building on these studies and elucidating the roles of Spa47 in Shigella will fill a significant gap in the current understanding of the otherwise generally well characterized Shigella T3SS and provide a model to help describe T3SS activation and regulation.
The implication of ATPases in T3SS energetics and regulation makes them a high impact target for structural investigations; however, the same stability challenges and heterogeneous oligomer distributions that have plagued biochemical characterization have limited structural studies as well. In fact, the three T3SS ATPases that have previously been crystallized all required stabilization and control over oligomerization by either removing the N-terminal oligomerization domain (29, 30, 54) or cocrystallization of the ATPase with a chaperone as seen in the recently reported FliI/FliH heterotrimer (55). In turn, we engineered a Spa47 N-terminal deletion construct (Spa47Δ1–79) by removing the Spa47 oligomerization domain and allowing crystallization. The highly soluble product resulted in several high resolution Spa47 crystal structures of the catalytic core and C-terminal domain of Spa47 (Figs. 2 and 4 and Table 1), adding to the short list of available T3SS ATPase structures and guiding studies to help uncover the relationships between Spa47 oligomerization, ATPase activity, T3SS function, and overall Shigella virulence.
Although full-length Spa47 can be purified, removal of the 79 N-terminal Spa47 residues prevents oligomerization and results in a homogeneous monomeric distribution (Fig. 5). Furthermore, the monomeric Spa47Δ1–79 construct is catalytically inactive, although the crystal structure identifies a conserved Rossmann fold, Walker domains, and predicted nucleotide binding site that align well with other ATPases, including EscN and FliI (Fig. 2). Together, this suggests that the Spa47Δ1–79 construct contains the essential components for ATP hydrolysis, whereas its inability to oligomerize is responsible for preventing activation, providing a likely means of regulating Shigella T3SS ATPase activity in vivo. Comparing oligomerization characteristics for each of the four T3SS ATPases with solved structures reveals several striking differences among the sequentially and structurally well conserved proteins, suggesting that T3SS associated secretion mechanisms may diverge at the level of ATPase function and apparatus activation. Specifically, although varying degrees of N-terminal truncations within FliI, EscN, SsaN, and now Spa47 provide predominantly monomeric stoichiometries and led to successful protein crystallization (29, 30, 54), the monomeric truncated FliIΔ7 and SsaNΔ1–89 constructs maintain low level ATPase activity (30, 41). In contrast, the monomeric EscNΔ1–102 and Spa47Δ1–79 constructs are completely ATPase inactive with no evidence for ability to form higher order homo-oligomers (29). The implications of these findings are not completely clear but suggest that E. coli and Shigella T3SS activation relies entirely on N-terminal interactions that drive ATPase oligomerization and that other family members including FliI and SsaN are capable of monomer-derived ATPase activity. Alternatively, monomeric FliI and SsaN N-terminal truncation constructs may undergo transient oligomer formation, which supports the low level ATPase activity, as we believe is the case for isolated full-length Spa47 monomer.
To better understand the mechanism driving oligomerization-based activation of Spa47, we generated an in silico Spa47 hexameric model. Because no atomic homo-oligomeric structures exist for T3SS ATPases, we generated the model by aligning six copies of Spa47 with the F1 α3β3 ATP synthase structure solved by Walker and co-workers (36) (Fig. 3). Comparison of the hexameric Spa47 model and the F1 ATP synthase structure finds striking similarities within the catalytic cores of the individual protomers and at the active site interfaces between adjacent protomers, with an overall RMSD of 2.06 Å (data not shown). Additionally, the resulting ring-like Spa47 hexamer is consistent with recent cryo-electron tomography results identifying Spa47 as a homohexamer in the context of the Shigella T3SA basal body (34). Looking specifically at the six active site regions within the Spa47 hexamer model identifies an arginine side chain (Arg350) that extends into the nucleotide binding site of an adjacent Spa47 protomer (Fig. 3). The location of this “arginine finger” in Spa47 is consistent with an equivalent active site arginine found in F1 ATP synthase that requires F1 oligomer formation for an α-subunit protomer to contribute its arginine to the active site of an adjacent catalytic β subunit, supporting ATP binding and hydrolysis (56). Sequence alignment of several known T3SS ATPases shows that the residues positioned equivalently to arginine 350 in Spa47 are highly conserved (Fig. 1), suggesting that they have a key role in protein function, although it has only been directly investigated in EscN (29) and now Spa47.
We generated Spa47 alanine point mutants to test the influence of the predicted catalytic active site residues and found that Spa47K165A, Spa47E188A, and Spa47R350A all purify as discrete oligomeric species as seen for wild-type Spa47 (Fig. 5). The activity profiles, however, show that the isolated monomeric and oligomeric species of each of these mutants are catalytically “dead” (Fig. 6). The oligomerization-deficient Spa47Δ1–79 construct used for crystallization was also completely inactive, although it maintains an intact catalytic core as evident in the crystal structure (Fig. 2). These findings not only show that Spa47 oligomerization and the conserved “arginine finger” are essential to Spa47 activation but also show that Spa47 oligomerization capabilities are independent of Spa47 activity, suggesting that catalysis is not required to bring monomeric Spa47 together. Rather, it seems likely that the 6-fold symmetry of the Shigella T3SA sorting platform and interaction with one or several of the neighboring T3SS proteins identified by cryo-electron tomography (34) facilitates the assembly of Spa47 oligomers in vivo. The appeal of this model is that the monomeric forms of Spa47 that exist in the Shigella cytoplasm would not be catalytically active and would not wastefully hydrolyze cellular stores of ATP. Once incorporated into the T3SA as an activated oligomeric complex, the Spa47 would immediately begin to hydrolyze ATP, activate the T3SA, and support Shigella virulence. It is also important to consider that Spa47 is currently the only T3SS ATPase for which an active subhexameric oligomer has been identified, and although it is admittedly unclear what role (if any) this putative trimeric complex plays in vivo, it has provided an invaluable tool for discerning the mechanistic role of Spa47 oligomerization in enzyme activation and T3SS function. The observed increase in activity levels by the oligomer and the AUC assignment of the Spa47 oligomer geometry as consistent with a triangular trimer (15) suggest that the active site protomer interfaces within the trimeric complex likely mimic those of the hexamer observed by electron microscopy (34, 57) and are modeled based on our recent Spa47 structures (Figs. 2 and 3). Additionally, it is tempting to speculate that the stable, active Spa47 trimer may be involved in dynamic processes within the basal body where the observed hexamer is actually comprised of two stable triangular trimers, allowing rapid and efficient formation and dissociation with the T3SA sorting platform and ultimately regulation of T3SS activity.
Complex and dedicated single-molecule super-resolution fluorescence studies will likely be required to fully uncover the dynamics of Spa47 within the context of the sorting platform; however, now armed with a better structural and biochemical understanding of Spa47-catalyzed ATP hydrolysis, we were able to test our hypothesis that Spa47 activation could serve as a potent regulator of Shigella T3SS activity and virulence. Previous works have shown that eliminating the T3SS ATPases altogether from pathogens such as Salmonella, Yersinia, E. coli, and Shigella results in reduction in T3SS protein secretion and have been shown in some cases to reduce virulence (20, 22, 28, 30, 58, 59). Additionally, loss of function InvC mutants in Salmonella lack external T3SS needles, suggesting that ATPase activity is required for their formation (60), although the full range of effects on the T3SS remains largely unclear. We took advantage of the numerous T3SS phenotype assays developed for Shigella and tested our ATPase inactive Spa47 mutants for their ability to complement proper T3SS activity and virulence phenotype in a spa47 null Shigella strain. The results for all of the tests are surprisingly clear. Each of the T3SS ATPase inactive Shigella mutants exhibited drastically reduced secretion phenotypes that are coupled with incomplete T3SA formation and a complete loss of Shigella virulence.
The work reported in this study provides the only structure function analysis of the Shigella T3SS ATPase Spa47 that we are aware of and begins to unravel the influence of Spa47 activation on the Shigella T3SS and pathogen virulence. In addition, the advantages of Shigella as a model T3SS organism and the ability to purify stable recombinant activated Spa47 oligomers provides exciting opportunities to answer questions concerning T3SS ATPase activation and the role of Spa47 oligomerization with respect to T3SS protein recognition and secretion. There is clearly still a lot of work to be done in this area, but with this foundational work characterizing Spa47, we can now add Shigella to the short list of pathogens with a characterized T3SS ATPase and expand the mechanistic understanding of T3SS function in general. It is our hope that these findings will help support the development of much needed non-antibiotic-based anti-infective agents against Shigella and other pathogens reliant on T3SSs for infection.
Experimental Procedures
Materials
WT S. flexneri corresponds to the serotype 2a 2457T strain originally isolated in 1954 (61). The S. flexneri spa47 null strain was engineered by Abdelmounaaïm Allaoui as described by Jouihri et al. (28). The S. flexneri mxiH null strain was engineered by Ariel Blocker as described by Blocker et al. (62). Rabbit polyclonal antibodies against IpaB, IpaC, IpaD, IpgC, and MxiH were generous gifts from William Picking and Wendy Picking (University of Kansas). The anti-IpaB, IpaC, IpaD, and IpgC antibodies were all provided as antisera and diluted 1:500 prior to use in the described experiments. The anti-MxiH antibodies were provided as protein G-purified IgG at a concentration of 9.8 mg/ml and diluted 1:500 prior to use. Selectivity and sensitivity of each antibody was evaluated by performing SDS-PAGE followed by Western blot analyses against 80 ng of the appropriate recombinant, purified target protein, as well as against a total protein extract obtained from wild-type Shigella. E. coli strains and 2× ligation mix were from Novagen (Madison, WI). Restriction enzymes, the pTYB21 protein expression plasmid, PCR buffer, Phusion high fidelity polymerase, and chitin resin were purchased from New England Biolabs (Ipswich, MA). Oligonucleotide primers and the synthesized Spa47 gene were from Integrated DNA Technologies (Coralville, IA). The Superdex 16/600 size exclusion, 5-ml Q FF, and 5-ml Heparin HP columns were purchased from General Electric (Pittsburgh, PA). ATP was from Sigma-Aldrich, and [α-32P]ATP was from PerkinElmer Life Sciences. DTT and ampicillin were from Gold Biotechnology (St. Louis, MO). Defibrinated sheep red blood cells were from Colorado Serum Company (Denver, CO). All other solutions and chemicals were of reagent grade.
Methods: Cloning
The Spa47 gene was purchased as a double-stranded gBlock product from Integrated DNA Technologies with modifications for cloning into the expression plasmid pTYB21 as described previously (15). Briefly, 5′ SapI and 3′ PstI restriction sites flanked the purchased Spa47 gene with a silent mutation made within the Spa47 gene to remove a native internal SapI restriction site. The SapI/PstI-digested gene was ligated into the expression plasmid pTYB21, which encodes an N-terminal chitin-binding domain and intein linker. The ligated product was transformed into E. coli Nova Blue cells by heat shock and screened for the presence of spa47 via PCR. Sequences were verified by Sanger sequencing (Genewiz, Inc., South Plainfield, NJ). spa47 was cloned into the plasmid pWPsf4 for expression in Shigella by introducing NdeI and BamHI restriction sites at the 5′ and 3′ ends, respectively, and ligating into the digested vector backbone. Sequences were again verified by Sanger sequencing prior to transformation into electro-competent S. flexneri strains via electroporation. The Spa47 point mutations and Spa47Δ1–79 N-terminal truncation were generated in both pTYB21 and pWPsf4 using inverse PCR followed by sequence verification and transformation into E. coli and S. flexneri, respectively.
Protein Expression and Purification
Spa47 and each of the Spa47 mutants encoded in pTYB21 were transformed into E. coli Tuner(DE3) cells and were expressed and purified as previously described (15). Briefly, the expression strains were grown to mid log phase in Terrific Broth medium containing 0.1 mg/ml ampicillin at 37 °C, 200 rpm. The culture was then cooled to 17 °C before induction with 1 mm isopropyl β-d-1-thiogalactopyranoside for ∼20 h (17 °C, 200 rpm). All subsequent steps were carried out at 4 °C unless otherwise stated. The cells were pelleted by centrifugation, resuspended in binding buffer (20 mm Tris, 500 mm NaCl, pH 7.9), and lysed by sonication. The soluble protein was released from the cells by sonication, and the supernatant was run over a chitin affinity column to capture the chitin-binding domain-intein-Spa47 fusion complex followed by intein cleavage in binding buffer containing 50 mm DTT. Column elutions were collected over the course of several days until no further elution of Spa47 was observed by SDS-PAGE (∼4 days). The elution fractions were diluted, resulting in a final buffer concentration of 20 mm Tris, 100 mm NaCl, 5 mm DTT, pH 7.9, prior to purification by negative selection over tandem 5-ml Q Sepharose FF anion exchange columns and a 5-ml Heparin HP column. The purified Spa47 in the flow through was concentrated using an Amicon Ultra centrifugal filter unit with a 30-kDa molecular mass cutoff and further purified/characterized using a Superdex 200 16/600 size exclusion column equilibrated with 20 mm Tris, 100 mm NaCl, 5 mm DTT, pH 7.9. Because of a complete lack of tryptophan in Spa47, all Spa47 concentrations were determined using a Bradford protein assay as done and validated previously (15). All concentrations are reported in monomer concentration units for consistency and clarity.
Spa47 Crystallization
The Spa47 N-terminal oligomerization domain was identified by sequence alignment to related ATPase homologs (Fig. 1) using the PSIPRED structure prediction server (63) and the UniProt multiple sequence alignment tool Clustal Omega (64). Crystallization of Spa47Δ1–79 and point mutation variants was performed using standard vapor diffusion methods. Initial crystallization hits were identified from the commercial MCSG4 crystallization screen condition F10 (Microlytic) and subsequently optimized to 0.1 m Tris, pH 8.5, 0.2 m ammonium acetate, 0.2 m lithium sulfate, 20–26% PEG 4000, and 4.5–9.5% (±)-2-methyl-2,4-pentanediol. Crystals were grown at room temperature (22 °C) using 10 mg/ml protein at 3:1 protein:well drop ratios. For all constructs, crystallization was also successful in the presence of 1.5–3 molar excess of nucleotide (ADP, ATP, ADP-AlF3, and ATPγS), although nucleotide was not observed in the electron density for any of the structures.
Data Collection and Structure Determination
Crystallographic data were collected to 2.4 Å (Spa47Δ1–79), 2.15 Å (Spa47Δ1–79 K165A), 2.70 Å (Spa47Δ1–79 E188A), and 1.80 Å (Spa47Δ1–79 R350A), on Beamlines 7-1 and 14-1 at the Stanford Synchrotron Radiation Lightsource (Table 1). The data were processed using HKL2000 (65). The crystals belong to space group P 21 and contain two molecules in the asymmetric unit (Matthews coefficient = 2.3, 45% solvent). The Spa47Δ1–79 structure was solved by molecular replacement using the homologous FliI structure (PDB code 2DPY) as a search model. The refined Spa47Δ1–79 structure was used as a search model for each of the point mutant variants. Phenix.refine (66), as implemented in the PHENIX software package (67), was used to perform individual b-factor, positional, and translation–libration–screw refinement. Model building was performed using Coot (68). Structure validation was performed using Molprobity (69). Final refinement statistics are shown for each crystal in Table 1. The PyMOL Molecular Graphics System (version 1.8; Schrödinger) was used to align structures, generate the activated Spa47 model, and create figures. PDBeFold (71) was used to calculate RMSD values.
Sedimentation Velocity Analytical Ultracentrifugation
SV-AUC experiments were conducted using an Optima XL-I (Beckman Coulter, Fullerton, CA) analytical ultracentrifuge equipped with an interference optical detection system. 1.0 mg/ml Spa47Δ1–79 and a buffer reference were loaded into Beckman charcoal-epon two sector cells with 12-mm path lengths and quartz windows. The samples were analyzed at 20 °C and 40,000 rpm using interference detection and scanning until complete sedimentation was achieved. The data were regularized with a confidence interval of 0.95 and analyzed using the software program Sedfit (72) with a continuous c(s) distribution and 500 scans as done previously for wild-type Spa47 (15). The Spa47 partial specific volume, buffer density, and viscosity used in the analysis (0.741 ml/g, 1.0031 g/ml, and 0.01018 Poise, respectively) were calculated using Sednterp (73).
ATP Hydrolysis Activity Assay
A multiple time point activity assay was used to determine reaction rates for all of the Spa47 constructs in each available oligomeric state. The assay was carried out under the conditions previously found to provide the most robust results (15). Briefly, the reactions were initiated by combining protein samples with a prepared ATP solution resulting in a final concentration of 1 mm ATP, 10 mm MgCl2, and 0.5 μCi (∼300 nm) [α-32P]ATP. Samples were then removed from the reaction every 30 s and rapidly quenched with a final concentration of 250 mm EDTA. The level of ATP hydrolysis was quantified by first separating the remaining [α-32P]ATP substrate from the [α-32P]ADP product using thin layer chromatography. The separated species were detected with a Storm PhosphorImager (Molecular Dynamics) and quantified using the associated ImageQuant software (Molecular Dynamics). The final concentration of Spa47 in all reaction conditions was held constant at 1.35 μm.
S. flexneri Invasion of Epithelial Cells
S. flexneri invasion of cultured HeLa cells was monitored by a gentamicin protection assay as previously described (74). Sterile 24-well plates were seeded with passaged HeLa cells and grown overnight in DMEM supplemented with 10% fetal calf serum, penicillin, and streptomycin at 100% relative humidity, 37 °C, and 5% CO2. Tested S. flexneri strains were streaked onto tryptic soy agar plates containing 0.025% Congo red and grown overnight at 37 °C. Small cultures containing appropriate antibiotics to maintain the transformed plasmid were inoculated from the agar plates and grown to and A600 of ∼0.4 at 37 °C and 200 rpm. Equivalent bacterial loads were introduced to the cultured HeLa cells, and the plates were centrifuged at 1000 × g to synchronize contact between the bacteria and HeLa cells. The inoculated cells were incubated at 37 °C for 30 min, rinsed to remove the extracellular bacteria, and treated with 50 μg/ml gentamicin to selectively kill the Shigella that had not successfully invaded the HeLa cells. The internalized bacteria were visualized and quantified by lysing the host cells with 1% agarose in water and overlaying with a 2× lysogeny broth agar solution. Overnight incubation at 37 °C resulted in colony formation that is quantified and used to provide relative levels of invasiveness between the tested S. flexneri strains.
Shigella Induced Erythrocyte Hemolysis
The effects of the engineered Spa47 mutations on T3SS-mediated hemolysis of red blood cells were determined using a slightly modified version of a previously described method (75). Briefly, S. flexneri expressing wild-type Spa47 or the Spa47 mutants described in this study were grown overnight on TSA-Congo red plates, and a small number of isolated colonies were used to inoculate 10 ml of tryptic soy broth containing appropriate antibiotics. The cultures were grown to an A600 of 1.0, collected by centrifugation, and gently resuspended in 200 μl of PBS. 50 μl of each bacterial mix was then combined with ∼5 × 108 red blood cells in a 96-well microtiter plate and centrifuged at 2300 × g and 30 °C for 15 min to initiate contact between the bacteria and the red blood cells. The plate was then incubated at 37 °C for 1 h and resuspended following the addition of 100 μl of cold PBS. The resuspended mix was centrifuged at 2300 × g and 10 °C for 15 min to separate cellular components and the hemoglobin that was released following red blood cell lysis. The levels of released hemoglobin in the supernatant were quantified by measuring absorbance at 545 nm and compared with the levels resulting from S. flexneri expressing wild-type Spa47.
Quantitation of S. flexneri T3SS Translocator Secretion
The effects of the engineered Spa47 mutations on uninduced effector secretion (“leakage”) was determined using a previously described method (76). Briefly, S. flexneri expressing wild-type Spa47 or one of the engineered Spa47 mutants were grown overnight on TSA-Congo red plates, and a small number of isolated colonies were used to inoculate 10 ml of tryptic soy broth containing appropriate antibiotics. Cultures were grown at 37 °C overnight (16 h). The saturated overnight cultures were then placed on ice for 5 min prior to isolation of the culture supernatant from the bacteria via centrifugation at 3750 × g for 10 min at 4 °C. The isolated supernatants were treated with TCA to a final concentration of 10% and allowed to sit on ice for 30 min to precipitate the soluble protein, including the secreted translocator proteins. The precipitated proteins were collected by centrifugation at 12,000 × g for 15 min at 4 °C, gently rinsed with 5% TCA followed by ice cold acetone, and finally resuspended in SDS-PAGE sample buffer. The isolated bacterial cell pellets were also rinsed thoroughly and resuspended in in SDS-PAGE sample buffer. Both the secreted protein samples and whole cell extracts from each strain were separated using SDS-PAGE, transferred to PVDF membranes by Western blot, and probed using anti-IpaB and anti-IpaC rabbit polyclonal antibodies and an Alexa 647 goat anti-rabbit secondary antibody. IpaB and IpaC levels were detected and compared using a Bio-Rad ChemiDoc imaging system and the associated Image Lab analysis software.
The small diazo dye Congo red effectively induces active secretion of translocator proteins through the Shigella T3SS by mimicking the natural trigger resulting from host cell membrane interaction (44). Thus, Congo red exposure serves as a valuable tool that allows for concerted activation of T3SSs from Shigella strains expressing various protein mutants such as the engineered Spa47 mutants used in this study. The protocol has been described in detail elsewhere (70). Briefly, S. flexneri strains lacking the gene for Spa47, strains expressing wild-type Spa47, and strains expressing the engineered Spa47 mutants were grown overnight on TSA-Congo red plates, and a small number of isolated colonies were used to inoculate 10 ml of tryptic soy broth containing appropriate antibiotics. Cultures were grown at 37 °C to an A600 of 1.0 before they were cooled on ice to temporarily slow protein expression and secretion. The cultures were centrifuged and rinsed to separate the bacteria from the culture supernatant and any proteins that had been secreted up to this point. The cells were then resuspended in sodium phosphate buffer containing 0.28 mg/ml Congo red and were incubated at 37 °C for 15 min to promote active T3SS secretion. Cultures were then chilled on ice for 5 min to limit further secretion and the bacteria were separated from the protein-containing supernatant by centrifugation at 13,000 × g for 15 min at 4 °C. The secreted proteins within the supernatant from each strain were separated using SDS-PAGE, transferred to PVDF membranes by Western blot, and probed using anti-IpaB and anti-IpaC rabbit polyclonal antibodies and an Alexa 647 goat anti-rabbit secondary antibody. Secreted IpaB and IpaC levels were detected and compared using a Bio-Rad ChemiDoc imaging system and the associated Image Lab analysis software.
The cytoplasmic Shigella T3SS chaperone, IpgC, is natively expressed at relatively high levels and is not secreted from the cytoplasm at any point, making it an ideal lysis control for each of the secretion assays described above. Briefly, Western blot analysis was used to probe for IpgC in the whole cell extract of isolated Shigella and in the supernatant of overnight and Congo red-induced cultures. The results found that although IpgC was present in pelleted Shigella whole cell extracts, it was not detected in the culture supernatants with IpaB and IpaC, confirming that cell lysis does not contribute significantly to the levels of observed protein secretion (data not shown).
Spa47-dependent Formation of the T3SA
The ability of S. flexneri strains expressing ATPase inactive Spa47 mutants to assemble extracellular portions of the T3SA was investigated using flow cytometry coupled with fluorescence detection. Each of the tested S. flexneri strains were streaked onto TSA-Congo red plates, and a small number of isolated colonies were used to inoculate 15 ml of tryptic soy broth containing appropriate antibiotics. The cultures were grown to an A600 of 0.8 at 37 °C and 200 rpm, collected by centrifugation at 4 °C, and gently rinsed with cold PBS. The cells were then chemically fixed for 15 min in 4% formaldehyde in PBS at room temperature (20–22 °C). The fixed cells were rinsed and labeled with rabbit polyclonal antibodies against either MxiH or IpaD. The bacteria were then treated with an Alexa 647 goat anti-rabbit secondary antibody to fluorescently label the targeted proteins for detection by flow cytometry. The cells were rinsed again and analyzed using a BD Accuri C6 flow cytometer collecting 500,000 instances/run. The resulting data sets were analyzed with De Novo FCS Express 5 flow cytometry software to determine whether the target proteins were present on the bacterial surface for each strain.
Author Contributions
N. E. D., J. L. B., and R. A. B. designed the studies. N. E. D., J. L. B., R. A. B., and J. M. B. performed the Shigella phenotype studies, expressed and purified the Spa47 constructs, and performed the biophysical characterizations. Y. M. set up the crystallization experiments. Y. M. and S. J. J. collected the X-ray diffraction data sets and solved the Spa47 structures. N. E. D. wrote the manuscript with assistance from all of the authors. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Abby Benninghoff for providing access to the flow cytometer and Abram Bernard for critical reading of the manuscript. We also acknowledge the RapiData2016 workshop and the SSRL staff for assistance in providing crystallographic training to Yalemi Morales.
This work was supported in part by National Institutes of Health Grants 1K22AI099086-01A1, National Science Foundation MRI Grant 1530862, and R. Gaurth Hansen endowment funds (to N. E. D.) and by individual Utah State University Undergraduate Research and Creative Opportunity Grants (to J. L. B. and J. M. B.). The crystallographic data were collected at the Stanford Synchrotron Radiation Lightsource (Stanford University), which is supported by U.S. Dept. of Energy Contract DE-AC02-76SF00515 and National Institutes of Health NIGMS Grant P41GM103393. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5SWJ, 5SYP, 5SWL, and 5SYR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- T3SS
- type III secretion system
- T3SA
- type III secretion apparatus
- SEC
- size exclusion chromatography
- SV-AUC
- sedimentation velocity analytical ultracentrifugation
- TSA
- tryptic soy agar
- RMSD
- root mean square deviation
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- PDB
- Protein Data Bank
- AMPPNP
- adenosine 5′-(β,γ-imino)triphosphate.
References
- 1. Schroeder G. N., and Hilbi H. (2008) Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Portaliou A. G., Tsolis K. C., Loos M. S., Zorzini V., and Economou A. (2016) Type III secretion: building and operating a remarkable nanomachine. Trends Biochem. Sci. 41, 175–189 [DOI] [PubMed] [Google Scholar]
- 3. Galán J. E., Lara-Tejero M., Marlovits T. C., and Wagner S. (2014) Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 68, 415–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cossart P., and Sansonetti P. J. (2004) Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248 [DOI] [PubMed] [Google Scholar]
- 5. World Health Organization (Updated April 2013) Diarrhoeal Disease, Fact Sheet no. 330, http://www.who.int/mediacentre/factsheets/fs330/en/
- 6. Kotloff K. L., Nataro J. P., Blackwelder W. C., Nasrin D., Farag T. H., Panchalingam S., Wu Y., Sow S. O., Sur D., Breiman R. F., Faruque A. S., Zaidi A. K., Saha D., Alonso P. L., Tamboura B., et al. (2013) Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209–222 [DOI] [PubMed] [Google Scholar]
- 7. Dutta S., Ghosh A., Ghosh K., Dutta D., Bhattacharya S. K., Nair G. B., and Yoshida S. (2003) Newly emerged multiple-antibiotic-resistant Shigella dysenteriae type 1 strains in and around Kolkata, India, are clonal. J. Clin. Microbiol. 41, 5833–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carayol N., and Tran Van Nhieu G. (2013) The inside story of Shigella invasion of intestinal epithelial cells. Cold Spring Harb. Perspect. Med. 3, a016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Enninga J., Mounier J., Sansonetti P., and Tran Van Nhieu G. (2005) Secretion of type III effectors into host cells in real time. Nat. Methods 2, 959–965 [DOI] [PubMed] [Google Scholar]
- 10. Ashida H., Mimuro H., and Sasakawa C. (2015) Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front. Immunol. 6, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Venkatesan M. M., Goldberg M. B., Rose D. J., Grotbeck E. J., Burland V., and Blattner F. R. (2001) Complete DNA sequence and analysis of the large virulence plasmid of Shigella flexneri. Infect. Immun. 69, 3271–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buchrieser C., Glaser P., Rusniok C., Nedjari H., D'Hauteville H., Kunst F., Sansonetti P., and Parsot C. (2000) The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38, 760–771 [DOI] [PubMed] [Google Scholar]
- 13. Schroeder G. N., Jann N. J., and Hilbi H. (2007) Intracellular type III secretion by cytoplasmic Shigella flexneri promotes caspase-1-dependent macrophage cell death. Microbiology 153, 2862–2876 [DOI] [PubMed] [Google Scholar]
- 14. Chatterjee S., Chaudhury S., McShan A. C., Kaur K., and De Guzman R. N. (2013) Structure and biophysics of type III secretion in bacteria. Biochemistry 52, 2508–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burgess J. L., Jones H. B., Kumar P., Toth R. T. 4th, Middaugh C. R., Antony E., and Dickenson N. E. (2016) Spa47 is an oligomerization-activated type three secretion system (T3SS) ATPase from Shigella flexneri. Protein Sci. 25, 1037–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan F., and Macnab R. M. (1996) Enzymatic characterization of FliI: an ATPase involved in flagellar assembly in Salmonella typhimurium. J. Biol. Chem. 271, 31981–31988 [DOI] [PubMed] [Google Scholar]
- 17. Minamino T., and Namba K. (2008) Distinct roles of the FliI ATPase and proton motive force in bacterial flagellar protein export. Nature 451, 485–488 [DOI] [PubMed] [Google Scholar]
- 18. Blaylock B., Riordan K. E., Missiakas D. M., and Schneewind O. (2006) Characterization of the Yersinia enterocolitica type III secretion ATPase YscN and its regulator, YscL. J. Bacteriol. 188, 3525–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stone C. B., Johnson D. L., Bulir D. C., Gilchrist J. D., and Mahony J. B. (2008) Characterization of the putative type III secretion ATPase CdsN (Cpn0707) of Chlamydophila pneumoniae. J. Bacteriol. 190, 6580–6588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrade A., Pardo J. P., Espinosa N., Pérez-Hernández G., and González-Pedrajo B. (2007) Enzymatic characterization of the enteropathogenic Escherichia coli type III secretion ATPase EscN. Arch. Biochem. Biophys. 468, 121–127 [DOI] [PubMed] [Google Scholar]
- 21. Yoshida Y., Miki T., Ono S., Haneda T., Ito M., and Okada N. (2014) Functional characterization of the type III secretion ATPase SsaN encoded by Salmonella pathogenicity island 2. PLoS One 9, e94347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eichelberg K., Ginocchio C. C., and Galán J. E. (1994) Molecular and functional characterization of the Salmonella typhimurium invasion genes invB and invC: homology of InvC to the F0F1 ATPase family of proteins. J. Bacteriol. 176, 4501–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walker J. E. (2013) The ATP synthase: the understood, the uncertain and the unknown. Biochem. Soc. Trans. 41, 1–16 [DOI] [PubMed] [Google Scholar]
- 24. Müller S. A., Pozidis C., Stone R., Meesters C., Chami M., Engel A., Economou A., and Stahlberg H. (2006) Double hexameric ring assembly of the type III protein translocase ATPase HrcN. Mol. Microbiol. 61, 119–125 [DOI] [PubMed] [Google Scholar]
- 25. Claret L., Calder S. R., Higgins M., and Hughes C. (2003) Oligomerization and activation of the FliI ATPase central to bacterial flagellum assembly. Mol. Microbiol. 48, 1349–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akeda Y., and Galán J. E. (2004) Genetic analysis of the Salmonella enterica type III secretion-associated ATPase InvC defines discrete functional domains. J. Bacteriol. 186, 2402–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Woestyn S., Allaoui A., Wattiau P., and Cornelis G. R. (1994) YscN, the putative energizer of the Yersinia Yop secretion machinery. J. Bacteriol. 176, 1561–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jouihri N., Sory M. P., Page A. L., Gounon P., Parsot C., and Allaoui A. (2003) MxiK and MxiN interact with the Spa47 ATPase and are required for transit of the needle components MxiH and MxiI, but not of Ipa proteins, through the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 49, 755–767 [DOI] [PubMed] [Google Scholar]
- 29. Zarivach R., Vuckovic M., Deng W., Finlay B. B., and Strynadka N. C. (2007) Structural analysis of a prototypical ATPase from the type III secretion system. Nat. Struct. Mol. Biol. 14, 131–137 [DOI] [PubMed] [Google Scholar]
- 30. Allison S. E., Tuinema B. R., Everson E. S., Sugiman-Marangos S., Zhang K., Junop M. S., and Coombes B. K. (2014) Identification of the docking site between a type III secretion system ATPase and a chaperone for effector cargo. J. Biol. Chem. 289, 23734–23744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akeda Y., and Galán J. E. (2005) Chaperone release and unfolding of substrates in type III secretion. Nature 437, 911–915 [DOI] [PubMed] [Google Scholar]
- 32. Walker J. E., Saraste M., Runswick M. J., and Gay N. J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimabukuro K., Yasuda R., Muneyuki E., Hara K. Y., Kinosita K. Jr., and Yoshida M. (2003) Catalysis and rotation of F1 motor: cleavage of ATP at the catalytic site occurs in 1 ms before 40 degree substep rotation. Proc. Natl. Acad. Sci. U.S.A. 100, 14731–14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu B., Morado D. R., Margolin W., Rohde J. R., Arizmendi O., Picking W. L., Picking W. D., and Liu J. (2015) Visualization of the type III secretion sorting platform of Shigella flexneri. Proc. Natl. Acad. Sci. U.S.A. 112, 1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ogura T., and Wilkinson A. J. (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6, 575–597 [DOI] [PubMed] [Google Scholar]
- 36. Abrahams J. P., Leslie A. G., Lutter R., and Walker J. E. (1994) Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 [DOI] [PubMed] [Google Scholar]
- 37. Senior A. E., Weber J., and Nadanaciva S. (2000) The catalytic transition state in ATP synthase. J. Bioenerg. Biomembr. 32, 523–529 [DOI] [PubMed] [Google Scholar]
- 38. Hanson P. I., and Whiteheart S. W. (2005) AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 39. Kazetani K., Minamino T., Miyata T., Kato T., and Namba K. (2009) ATP-induced FliI hexamerization facilitates bacterial flagellar protein export. Biochem. Biophys. Res. Commun. 388, 323–327 [DOI] [PubMed] [Google Scholar]
- 40. Shiue S. J., Kao K. M., Leu W. M., Chen L. Y., Chan N. L., and Hu N. T. (2006) XpsE oligomerization triggered by ATP binding, not hydrolysis, leads to its association with XpsL. EMBO J. 25, 1426–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Minamino T., Kazetani K., Tahara A., Suzuki H., Furukawa Y., Kihara M., and Namba K. (2006) Oligomerization of the bacterial flagellar ATPase FliI is controlled by its extreme N-terminal region. J. Mol. Biol. 360, 510–519 [DOI] [PubMed] [Google Scholar]
- 42. Blocker A., Gounon P., Larquet E., Niebuhr K., Cabiaux V., Parsot C., and Sansonetti P. (1999) The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Epler C. R., Dickenson N. E., Olive A. J., Picking W. L., and Picking W. D. (2009) Liposomes recruit IpaC to the Shigella flexneri type III secretion apparatus needle as a final step in secretion induction. Infect. Immun. 77, 2754–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parsot C., Menard R., Gounon P., and Sansonetti P. J. (1995) Enhanced secretion through the Shigella flexneri Mxi-Spa translocon leads to assembly of extracellular proteins into macromolecular structures. Mol. Microbiol. 16, 291–300 [DOI] [PubMed] [Google Scholar]
- 45. Epler C. R., Dickenson N. E., Bullitt E., and Picking W. L. (2012) Ultrastructural analysis of IpaD at the tip of the nascent MxiH type III secretion apparatus of Shigella flexneri. J. Mol. Biol. 420, 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheung M., Shen D. K., Makino F., Kato T., Roehrich A. D., Martinez-Argudo I., Walker M. L., Murillo I., Liu X., Pain M., Brown J., Frazer G., Mantell J., Mina P., Todd T., et al. (2015) Three-dimensional electron microscopy reconstruction and cysteine-mediated crosslinking provide a model of the type III secretion system needle tip complex. Mol. Microbiol. 95, 31–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Espina M., Olive A. J., Kenjale R., Moore D. S., Ausar S. F., Kaminski R. W., Oaks E. V., Middaugh C. R., Picking W. D., and Picking W. L. (2006) IpaD localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect Immun. 74, 4391–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diepold A., and Armitage J. P. (2015) Type III secretion systems: the bacterial flagellum and the injectisome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dickenson N. E., and Picking W. D. (2012) Forster resonance energy transfer (FRET) as a tool for dissecting the molecular mechanisms for maturation of the Shigella type III secretion needle tip complex. Int. J. Mol. Sci. 13, 15137–15161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dickenson N. E., Zhang L., Epler C. R., Adam P. R., Picking W. L., and Picking W. D. (2011) Conformational changes in IpaD from Shigella flexneri upon binding bile salts provide insight into the second step of type III secretion. Biochemistry 50, 172–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stensrud K. F., Adam P. R., La Mar C. D., Olive A. J., Lushington G. H., Sudharsan R., Shelton N. L., Givens R. S., Picking W. L., and Picking W. D. (2008) Deoxycholate interacts with IpaD of Shigella flexneri in inducing the recruitment of IpaB to the type III secretion apparatus needle tip. J. Biol. Chem. 283, 18646–18654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. González-Pedrajo B., Fraser G. M., Minamino T., and Macnab R. M. (2002) Molecular dissection of Salmonella FliH, a regulator of the ATPase FliI and the type III flagellar protein export pathway. Mol. Microbiol. 45, 967–982 [DOI] [PubMed] [Google Scholar]
- 53. Minamino T., and MacNab R. M. (2000) FliH, a soluble component of the type III flagellar export apparatus of Salmonella, forms a complex with FliI and inhibits its ATPase activity. Mol. Microbiol. 37, 1494–1503 [DOI] [PubMed] [Google Scholar]
- 54. Imada K., Minamino T., Tahara A., and Namba K. (2007) Structural similarity between the flagellar type III ATPase FliI and F1-ATPase subunits. Proc. Natl. Acad. Sci. U.S.A. 104, 485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Minamino T., Kinoshita M., Inoue Y., Morimoto Y. V., Ihara K., Koya S., Hara N., Nishioka N., Kojima S., Homma M., and Namba K. (2016) FliH and FliI ensure efficient energy coupling of flagellar type III protein export in Salmonella. Microbiologyopen 5, 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Komoriya Y., Ariga T., Iino R., Imamura H., Okuno D., and Noji H. (2012) Principal role of the arginine finger in rotary catalysis of F1-ATPase. J. Biol. Chem. 287, 15134–15142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Makino F., Shen D., Kajimura N., Kawamoto A., Pissaridou P., Oswin H., Pain M., Murillo I., Namba K., and Blocker A. J. (2016) The architecture of the cytoplasmic region of type III secretion systems. Sci. Rep. 6, 33341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bozue J., Cote C. K., Webster W., Bassett A., Tobery S., Little S., and Swietnicki W. (2012) A Yersinia pestis YscN ATPase mutant functions as a live attenuated vaccine against bubonic plague in mice. FEMS Microbiol. Lett. 332, 113–121 [DOI] [PubMed] [Google Scholar]
- 59. Kato J., Lefebre M., and Galán J. E. (2015) Structural features reminiscent of ATP-driven protein translocases are essential for the function of a type III secretion-associated ATPase. J. Bacteriol. 197, 3007–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sukhan A., Kubori T., Wilson J., and Galán J. E. (2001) Genetic analysis of assembly of the Salmonella enterica serovar typhimurium type III secretion-associated needle complex. J. Bacteriol. 183, 1159–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Formal S. B., Dammin G. J., Labrec E. H., and Schneider H. (1958) Experimental Shigella infections: characteristics of a fatal infection produced in guinea pigs. J. Bacteriol. 75, 604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Blocker A., Jouihri N., Larquet E., Gounon P., Ebel F., Parsot C., Sansonetti P., and Allaoui A. (2001) Structure and composition of the Shigella flexneri “needle complex,” a part of its type III secreton. Mol. Microbiol. 39, 652–663 [DOI] [PubMed] [Google Scholar]
- 63. McGuffin L. J., Bryson K., and Jones D. T. (2000) The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405 [DOI] [PubMed] [Google Scholar]
- 64. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 66. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kenjale R., Wilson J., Zenk S. F., Saurya S., Picking W. L., Picking W. D., and Blocker A. (2005) The needle component of the type III secreton of Shigella regulates the activity of the secretion apparatus. J. Biol. Chem. 280, 42929–42937 [DOI] [PubMed] [Google Scholar]
- 71. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 72. Lebowitz J., Lewis M. S., and Schuck P. (2002) Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Laue T., Shah B., Ridgeway T., and Pelletier S. (1992) Analytical ultracentrifugation in biochemistry and polymer science. In Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S., Rowe A., and Horton J. C., eds) Royal Society of Chemistry, London [Google Scholar]
- 74. Niesel D. W., Chambers C. E., and Stockman S. L. (1985) Quantitation of HeLa cell monolayer invasion by Shigella and Salmonella species. J. Clin. Microbiol. 22, 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sansonetti P. J., Ryter A., Clerc P., Maurelli A. T., and Mounier J. (1986) Multiplication of Shigella flexneri within HeLa cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect. Immun. 51, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ménard R., Sansonetti P. J., and Parsot C. (1993) Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 175, 5899–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]