

Abstract
The ryanodine receptor (RyR) channel pore is formed by four S6 inner helices, with its intracellular gate located at the S6 helix bundle crossing region. The cytoplasmic region of the extended S6 helix is held by the U motif of the central domain and is thought to control the opening and closing of the S6 helix bundle. However, the functional significance of the S6 cytoplasmic region in channel gating is unknown. Here we assessed the role of the S6 cytoplasmic region in the function of cardiac RyR (RyR2) via structure-guided site-directed mutagenesis. We mutated each residue in the S6 cytoplasmic region of the mouse RyR2 (4876QQEQVKEDM4884) and characterized their functional impact. We found that mutations Q4876A, V4880A, K4881A, and M4884A, located mainly on one side of the S6 helix that faces the U motif, enhanced basal channel activity and the sensitivity to Ca2+ or caffeine activation, whereas mutations Q4877A, E4878A, Q4879A, and D4883A, located largely on the opposite side of S6, suppressed channel activity. Furthermore, V4880A, a cardiac arrhythmia-associated mutation, markedly enhanced the frequency of spontaneous openings and the sensitivity to cytosolic and luminal Ca2+ activation of single RyR2 channels. V4880A also increased the propensity and reduced the threshold for arrhythmogenic spontaneous Ca2+ release in HEK293 cells. Collectively, our data suggest that interactions between the cytoplasmic region of S6 and the U motif of RyR2 are important for stabilizing the closed state of the channel. Mutations in the S6/U motif domain interface likely destabilize the closed state of RyR2, resulting in enhanced basal channel activity and sensitivity to activation and increased propensity for spontaneous Ca2+ release and cardiac arrhythmias.
Keywords: calcium, calcium channel, calcium imaging, calcium intracellular release, calcium-binding protein, ryanodine receptor, sarcoplasmic reticulum (SR)
Introduction
The ryanodine receptor type 2 (RyR2)5 is an intracellular Ca2+ channel located in the sarcoplasmic reticulum (SR) membrane of cardiac muscle cells and mediates the release of SR Ca2+, which triggers muscle contraction (1–3). RyR2-mediated SR Ca2+ release can also influence membrane excitability and cardiac rhythm. Naturally occurring mutations in RyR2 have been linked to various forms of cardiac arrhythmias and cardiomyopathies. Most of the disease-associated RyR2 mutations characterized enhance RyR2 activity by sensitizing the channel to activation by luminal and/or cytosolic Ca2+ (4, 5). However, the exact molecular mechanisms underlying the action of most disease-causing RyR2 mutations are unknown. The crystal structures of the N-terminal and other domains of RyR have provided precise and detailed insights into the structural basis of action of disease-causing mutations located in these domains (6–13). For instance, most of the N-terminal disease mutations are located in domain-domain interfaces. It is believed that domain-domain interactions in the N-terminal region are important for stabilizing the closed state of the RyR channel. Mutations in these domain interfaces may weaken the domain-domain interactions and thus destabilize the closed state of the channel, rendering the channel to open spontaneously and more sensitive to activation (6, 8, 12). Hence, identifying domain-domain interactions that are important for stabilizing the closed state of the channel is important for understanding the molecular mechanisms of action of disease-associated RyR mutations. Unfortunately, atomic resolution structures are not available for most part of the RyR channel. Our understanding of the molecular mechanisms of disease-associated mutations located in regions without detailed structural information has been challenging.
Recently, a near-atomic resolution (3.8 Å) 3D structure of the skeletal muscle RyR (RyR1) has been solved by cryo-electron microscopy (14). Although, at this resolution, the structure may not provide the exact locations of amino acid side chains, it does provide a useful and important structural framework and guidance for structure-function relationship studies of RyR2, especially of the channel domain, where a better resolution was observed. The RyR2 channel domain is one of the disease-causing mutation hot spots, but the mechanisms of disease mutations located in this region are largely undefined (4, 5). Based on the 3D structure of RyR, the channel conduction pathway is formed by the membrane regions of the four S6 inner helices. The constriction point of the pathway or the intracellular gate is located in the S6 helix bundle crossing region. The cytoplasmic region of the extended S6 helix interacts with the U motif of the central domain (14). Given its direct connection to the gate, it is believed that conformational changes in the cytoplasmic region of S6 control the opening and closure of the gate located in the bundle crossing region (14, 15). Hence, interactions between the S6 cytoplasmic region and U motif may be involved in stabilizing the closed state of the channel. To test this hypothesis, in this study, we investigate the functional significance of the cytoplasmic region of S6. We mutated each residue in this region and determined their impact on channel activation. We found that mutating residues on the side of the S6 helix that faces the U motif enhanced channel activity. Interestingly, a disease-causing V4880A mutation (16) located in this region markedly increased the basal activity of the channel in the near absence of activating Ca2+ and sensitized the channel to activation by cytosolic and luminal Ca2+ activation. The V4880A mutation also enhanced arrhythmogenic spontaneous Ca2+ release in HEK293 cells. Our data support the notion that the cytoplasmic region of S6 is important for stabilizing the closed state of the channel. Mutations in this region may disrupt the S6/U motif interaction and destabilize the closed state, thus increasing the basal activity and sensitivity of the channel to stimuli and the propensity for spontaneous Ca2+ release and cardiac arrhythmias.
Results
Role of the Cytoplasmic Region of the S6 Inner Helix in RyR2 Activation
Structural studies suggest that the cytoplasmic region of the S6 inner helix interacts with the U motif of the central domain (14, 15), but the functional significance of this S6/U motif interaction is unknown. To assess the role of the S6 cytoplasmic region in RyR2 function, we mutated each residue in the S6 cytoplasmic region (4876QQEQVKEDM4884) of the mouse RyR2 (Fig. 1) and determined its impact on the response of RyR2 to caffeine activation. As shown in Fig. 2, the amplitude of Ca2+ release in HEK293 cells expressing the RyR2 WT increased progressively with each repeated addition of caffeine (from 0.05 to 0.5 mm) and then decreased with further additions of caffeine (from 1.0 to 5 mm). This decrease likely resulted from the depletion of intracellular Ca2+ stores by the prior additions of caffeine (0.025 to 0.5 mm) (Fig. 2, A and F). The caffeine response of HEK293 cells expressing the V4880A and K4881A mutants was shifted to the left (Fig. 2, H, I, and L), whereas the caffeine response of HEK293 cells expressing the Q4877A, E4878A, Q4879A, and D4883A mutants was shifted to the right (Fig. 2, B–F). The caffeine responses of the Q4876A, E4882A, and M4884A mutants are similar to that of the WT (Fig. 2, G and J–L). Thus, the V4880A and K4881A mutations enhance the response of RyR2 to caffeine activation, whereas the Q4877A, E4878A, Q4879A, and D4883A mutations inhibit it.
FIGURE 1.
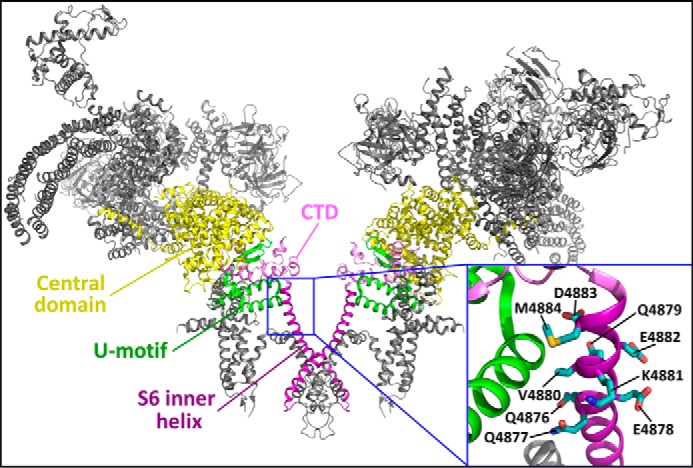
Location of the S6 cytoplasmic region in the three-dimensional structure of RyR2. The 3D structure of two RyR2 monomers (30) is shown. The central domain, U motif, CTD, and S6 inner helix are highlighted. The inset is a close-up view showing the cytoplasmic region of the S6 inner helix encompassing residues 4876QQEQVKEDM4884.
FIGURE 2.

Effect of mutations in the cytoplasmic region of S6 on caffeine activation of RyR2. A–E and G–K, HEK293 cells were transfected with RyR2 WT (A), Q4877A (B), E4878A (C), Q4879A (D), E4883A (E), Q4876A (G), V4880A (H), K4881A (I), E4882A (J), or M4884A (K). The fluorescence intensity of the Fluo-3-loaded transfected cells before and after repeated additions of increasing concentrations of caffeine (0.025–5 mm) was monitored continuously. F and L, Ca2+ release-cumulative caffeine concentration relationships in HEK293 cells transfected with RyR2 WT and mutants that shift the caffeine response curve to the right (F) or mutants that have little effect on caffeine response or shift the caffeine response curve to the left (L). The amplitude of each caffeine peak was normalized to that of the maximum peak for each experiment. Data shown are mean ± S.E. (n = 3–4; *, p < 0.05 versus WT.
Mutations in the Cytoplasmic Region of S6 Alter the Basal Activity and Ca2+-dependent Activation of [3H]ryanodine Binding
To gain further insights into the role of the S6 cytoplasmic region in RyR2 function, we used [3H]ryanodine binding assays to assess the impact of mutations in the S6 cytoplasmic region on the basal activity of the RyR2 channel and the sensitivity of the channel to Ca2+-dependent activation. Because ryanodine only binds to the open state of the RyR channel, [3H]ryanodine binding has been widely used as an index for RyR channel activity. As shown in Fig. 3, in the near absence of activating Ca2+ (<16 nm), [3H]ryanodine binding to RyR2 WT shows a basal activity of ∼3% of the maximum binding. On the other hand, the level of [3H]ryanodine binding to the Q4876A, V4880A, K4881A, and M4884A mutants in the near absence of activating Ca2+ (13%, 31%, 21%, and 10%, respectively) was significantly higher than that to the WT (p < 0.01) (Fig. 3, B and C). In contrast, in the near absence of activating Ca2+, the Q4877A, E4878A, Q4879A, E4882A, and D4883A mutants displayed a reduced basal level of [3H]ryanodine binding (1%, 0.1%, 0.2%, 0.7%, and 0.1%, respectively) (Fig. 3, A–C), which is significantly lower than that to the WT (p < 0.01). These observations indicate that the cytoplasmic region of S6 is an important determinant of the basal activity of RyR2. Furthermore, the Q4876A, V4880A, K4881A, and M4884A mutations reduced the EC50 of Ca2+ dependent activation of [3H]ryanodine binding compared with the WT, whereas the Q4877A, E4878A, Q4879A, and D4883A mutations increased it (Fig. 3D). These data indicate that the S6 cytoplasmic region also plays an important role in the Ca2+-dependent activation of RyR2. The expression level of RyR2 WT and mutants is shown in Fig. 4. Although all mutants in the S6 cytoplasmic region can be expressed in HEK293 cells (Fig. 4A), the level of expression of most of the mutants was significantly reduced (Fig. 4B).
FIGURE 3.

Effect of the S6 inner helix mutations on [3H]ryanodine binding to RyR2. A and B, [3H]Ryanodine binding to cell lysate prepared from HEK293 cells expressing RyR2 WT and the S6 mutants Q4877A, E4878A, Q4879A, E4882A, and D4883A (A) or the S6 mutants Q4876A, V4880A, K4881A, and M4884A (B) was carried out at various Ca2+ concentrations (∼0.2 nm to 0.1 mm), 800 mm KCl, and 5 nm [3H]ryanodine. The amounts of [3H]ryanodine binding at various Ca2+ concentrations were normalized to the maximal binding (100%). C, mutating the cytoplasmic region of the S6 inner helix of RyR2 affects the basal level of [3H]ryanodine binding to RyR2 in the near absence of activating Ca2+ (<20 nm). D, the EC50 values of Ca2+ activation of [3H]ryanodine binding to RyR2 WT and the S6 mutants. The data points shown are mean ± S.E. from three to five separate experiments (*p < 0.05 versus WT).
FIGURE 4.

Expression of S6 mutants in HEK293 cells. A, HEK293 cells were transfected with RyR2 WT or the S6 inner helix mutants. The same amount of transfected HEK293 cell lysate protein was used for Western blotting (WB) using the anti-RyR antibody (34c) and anti-β-actin antibody. B, besides the use of the same amount of WT and mutant cell lysate protein for loading, the expression levels of the WT and S6 mutants were further normalized to that of β-actin. Data shown are mean ± S.E. from five separate experiments (*, p < 0.05 versus WT; NS, not significant).
The V4880A Mutation Increases the Frequency of Openings of Single RyR2 Channels in the Near Absence of Activating Ca2+
Among the mutations in the S6 cytoplasmic region that enhance RyR2 activity, V4880A displays the most dramatic impact on [3H]ryanodine binding (Fig. 3). Importantly, V4880A is a naturally occurring RyR2 mutation associated with catecholaminergic polymorphic ventricular tachycardia (16). To further understand the mechanism of action of this disease-causing RyR2 mutation, we assessed the impact of V4880A on RyR2 gating using single channel recordings in planar lipid bilayers. As shown in Fig. 5, a single RyR2 WT channel exhibited very few opening events at 45 nm cytosolic Ca2+ (Fig. 5, A and C). However, at the same cytosolic Ca2+ concentration, single V4880A mutant channels showed a markedly increased frequency of openings with an event frequency of 4.68 ± 1.00 events/s (Fig. 5, B and C), which is significantly higher than that of single RyR2 WT channels (1.01 ± 0.56 events/s) (p < 0.01). The V4880A mutation also increased the open probability (Po) (Fig. 5D) and decreased the mean closed time (Tc) (Fig. 5F), but had no effect on mean open time (To) (Fig. 5E) of single RyR2 channels. These data suggest that the V4880A mutation destabilizes the closed state of the RyR2 channel, leading to enhanced channel openings in the near absence of activating Ca2+, which is consistent with the increased basal level of [3H]ryanodine binding (Fig. 3).
FIGURE 5.

Effect of the disease-causing RyR2 mutation V4880A on the gating of single RyR2 channels. A and B, single-channel activities of the RyR2 WT (A) and the V4880A mutant (B) were recorded in a symmetrical recording solution containing 250 mm KCl and 25 mm Hepes (pH 7.4). EGTA was added to either the cis or trans chamber to determine the orientation of the incorporated channel. The side of the channel to which an addition of EGTA inhibited the activity of the incorporated channel presumably corresponds to the cytosolic (cyto) face. The Ca2+ concentration on both the cytosolic and the luminal face of the channel was adjusted to ∼45 nm. Recording potentials were −20 mV. Openings are downward, and baselines are indicated (short bars). C–F, the event frequency (C), Po (D), To (E), and Tc (F) of single RyR2 WT and V4880A (VA) mutant channels at 45 nm cytosolic Ca2+ are shown. Data shown are mean ± S.E. from nine RyR2 WT and 11 V4880A single channels (**, p < 0.01; *, p < 0.05 versus WT; NS, not significant).
The V4880A Mutation Increases the Sensitivity of Single RyR2 Channels to Cytosolic Ca2+ Activation
RyR2 can be activated by cytosolic or luminal Ca2+. To determine whether the V4880A mutation affects the activation of RyR2 by cytosolic Ca2+, we carried out a single-channel analysis of the RyR2 WT and V4880A mutant in the presence of various cytosolic Ca2+ concentrations and in the near absence of luminal Ca2+ (∼45 nm). As shown in Fig. 6, single RyR2 WT channels were activated by cytosolic Ca2+ with an EC50 of ∼0.25 μm. On the other hand, single V4880A mutant channels were activated by cytosolic Ca2+ with an EC50 of ∼0.15 μm. Thus, the V4880A mutation enhances the cytosolic Ca2+ activation of RyR2.
FIGURE 6.

Effect of V4880A on the cytosolic Ca2+ activation of single RyR2 channels. A and B, single-channel activities of RyR2 WT (A) and the V4880A (B) mutant were recorded in a symmetrical recording solution containing 250 mm KCl and 25 mm Hepes (pH 7. 4). The Ca2+ concentration on the cytoplasmic (cyto) and luminal face of the channel was first adjusted to ∼45 nm. The cytosolic Ca2+ concentration was then increased from 45 nm to various levels by addition of aliquots of CaCl2 solution. C, the relationships between open probability and pCa of single RyR2 WT (solid circles) and the V4880A mutant (open circles). The data points shown are mean ± S.E. from four RyR2-WT and 11 V4880A single channels.
The V4880A Mutation Increases the Luminal Ca2+ Sensitivity of Single RyR2 Channels
We next assessed whether the V4880A mutation also affects the sensitivity of RyR2 to activation by luminal Ca2+. Single RyR2 WT and V4880A mutant channels were first inactivated by reducing the cytosolic Ca2+ concentration to ∼45 nm using EGTA. The luminal Ca2+ concentration was then increased stepwise from 45 nm to 40 mm. As shown in Fig. 7, single RyR2 WT channels were barely active at luminal Ca2+ concentrations of less than 1 mm in the near absence of cytosolic Ca2+ (45 nm). However, under the same conditions, single V4880A mutant channels were activated by luminal Ca2+ at 2.5 μm (Fig. 7C). Therefore, the V4880A mutation also increases the luminal Ca2+ activation of RyR2.
FIGURE 7.

Effect of V4880A on the luminal Ca2+ activation of single RyR2 channels. A and B, single-channel activities of RyR2 WT (A) and V4880A mutant (B) were recorded in a symmetrical recording solution containing 250 mm KCl and 25 mm Hepes (pH 7.4). The Ca2+ concentration on the cytoplasmic (cyto) and the luminal face of the channel was first adjusted to ∼45 nm. The luminal Ca2+ concentration was then increased to various levels by addition of aliquots of CaCl2 solution. C, the relationships between open probability and luminal pCa of single RyR2 WT (solid circles) and the V4880A mutant (open circles). The data points shown are mean ± S.E. from five RyR2-WT and four V4880A single channels.
The V4880A Mutation Enhances the Propensity for Store Overload-induced Ca2+ Release in HEK293 Cells
We have shown previously that enhanced luminal and/or cytosolic Ca2+ activation of RyR2 increases the propensity for spontaneous Ca2+ release during store Ca2+ overload, a phenomenon we referred to as store overload-induced Ca2+ release (SOICR) (17, 18). Thus, it is possible that the V4880A mutation that enhances both cytosolic and luminal Ca2+ activation of RyR2 may also increase the propensity for SOICR. To test this possibility, we monitored SOICR in HEK293 cells expressing the RyR2 WT or the V4880A mutant using single-cell Ca2+ imaging. SOICR was induced in these cells by elevating extracellular Ca2+ concentrations (0–2.0 mm) as described previously (17, 18). As shown in Fig. 8, the fraction of cells that displayed Ca2+ oscillations was significantly higher in HEK293 cells expressing the V4880A mutant at 0.2–2.0 mm extracellular Ca2+ compared with that in cells expressing the RyR2 WT. Thus, the V4880A mutation indeed enhances the propensity for SOICR.
FIGURE 8.

Effect of the V4880A mutation on the propensity for SOICR. A and B, stable, inducible HEK293 cells expressing RyR2 WT (A) and V4880A (B) were loaded with Fura-2/AM. The cells were then perfused continuously with KRH buffer containing increasing levels of extracellular Ca2+ (0–2 mm) to induce SOICR. Fura-2 ratios were recorded using epifluorescence single-cell Ca2+ imaging. Caff, caffeine. C, percentages of RyR2 WT (220 cells) and V4880A (171 cells) cells that display Ca2+ oscillations at various extracellular Ca2+ concentrations. Data shown are mean ± S.E. (n = 3; **, p < 0.01 versus WT).
The V4880A Mutation Reduces the Threshold for SOICR
SOICR is thought to occur when the store Ca2+ load exceeds a threshold level (SOICR threshold) (17–23). To assess whether the V4880A mutation affects the SOICR threshold, we measured the endoplasmic reticulum (ER) luminal Ca2+ dynamics using the ER luminal Ca2+ sensing protein D1ER (22, 24). As shown in Fig. 9, HEK293 cells expressing RyR2 WT displayed spontaneous ER Ca2+ oscillations upon elevating extracellular Ca2+ from 0 to 2 mm. We determined the SOICR activation threshold (FSOICR) at which SOICR occurred and the SOICR termination threshold (Ftermi) at which SOICR terminated. Consistent with its increased propensity for SOICR, the V4880A mutation reduced the SOICR activation threshold (79% versus 93% in the WT) (p < 0.05) (Fig. 9C). The V4880A mutation also reduced the SOICR termination threshold (53% versus 62% in the WT) (p < 0.05) (Fig. 9D). The fractional Ca2+ release during SOICR (activation threshold − termination threshold) in V4880A mutant cells (26%) was also reduced compared with that in RyR2 WT cells (32%) (p < 0.05) (Fig. 9E). There was no significant difference in the store capacity (Fmax − Fmin) between RyR2 WT and the V4880A mutant cells (Fig. 9F). It should be noted that SOICR did not occur in control HEK293 cells expressing no RyR2 and that SOICR was not affected by the IP3R inhibitor xestospongin C (23), indicating that SOICR is mediated by RyR2. Together, these data indicate that the V4880A mutation enhances the propensity for SOICR by reducing the SOICR threshold.
FIGURE 9.

Effect of V4880A on SOICR activation and termination thresholds. A and B, stable, inducible HEK293 cell lines expressing RyR2 WT (A) and V4880A (B) were transfected with the FRET-based ER luminal Ca2+-sensing protein D1ER 48 h before single-cell FRET imaging. Expression of the RyR2 WT and mutant was induced 24 h before imaging. The cells were perfused with KRH buffer containing increasing levels of extracellular Ca2+ (0–2 mm) to induce SOICR. This was followed by the addition of 1.0 mm tetracaine to inhibit SOICR and then 20 mm caffeine to deplete the ER Ca2+ stores. FRET recordings from representative RyR2 WT (total 57 cells, A) and mutant V4880A cells (total 53 cells, B) are shown. C and D, to minimize the influence of CFP/YFP cross-talk, we used relative FRET measurements to calculate the activation threshold (C) and termination threshold (D) using the equations shown in A. FSOICR indicates the FRET level at which SOICR occurs, whereas Ftermi represents the FRET level at which SOICR terminates. E, the fractional Ca2+ release was calculated by subtracting the termination threshold from the activation threshold. The maximum FRET signal Fmax is defined as the FRET level after tetracaine treatment. The minimum FRET signal Fmin is defined as the FRET level after caffeine treatment. F, the store capacity was calculated by subtracting Fmin from Fmax. Data shown are mean ± S.E. (n = 4; *, p < 0.05 versus WT; NS, not significant).
Discussion
RyRs are the largest known ion channels, important for a number of cellular processes ranging from muscle contraction to neuronal function (1–3). Despite their pivotal significance, the structure-function relationships of RyRs are poorly understood, in part because of their gigantic size and the lack of atomic resolution structures. Yan et al. (14) have recently made a major advance in our understanding of the structure-function relationships of especially the ion-conduction pathway of RyR by solving the 3D structure of RyR at a near-atomic resolution (3.8 Å). Based on this structure, the ion-conducting pathway of RyR is formed by four elongated S6 inner helices. The N-terminal region of the S6 helix is embedded in the membrane, where it forms the central pore cavity, whereas the middle region of the S6 helix, corresponding to the helix bundle crossing region located at the border zone between the membrane and cytoplasm, encompasses the constriction point or the ion gate of the channel (Fig. 1). These structural features are consistent with our previous functional studies demonstrating the critical role of the S6 helix bundle crossing region in the regulation and gating of RyR2 (25–29). However, the functional significance of the C-terminal (cytoplasmic) region of the S6 inner helix is unknown.
The C-terminal region of S6 is located in the cytoplasm, where it interacts with the U motif of the central domain of RyR. It has recently been proposed that the central domain of RyR acts as a transducer that converts the conformational changes in the large cytosolic assembly to the channel pore domain (14, 15, 30). Because the intracellular gate of RyR is formed by the S6 helices, movements of the cytoplasmic region of S6 would directly affect the opening and closure of the gate located in the helix bundle crossing region of S6. Hence, interaction between the cytoplasmic region of S6 and U motif would be expected to be critical for controlling the gating of the channel, but functional evidence is lacking. To assess the functional significance of this S6/U motif interaction, we mutated each residue in the S6 cytoplasmic region (4876QQEQVKEDM4884) (Fig. 1). We found that the Q4876A, V4880A, K4881A, and M4884A mutations increase the activity of RyR2, whereas the Q4877A, E4878A, Q4879A, and D4883A mutations suppress it. Thus, the cytoplasmic region of S6 is an important determinant of channel activation. The EF-hand motif located in the central domain has also been proposed to be a critical determinant of Ca2+-dependent activation of the RyR channel (31, 32). However, deletion of the entire EF-hand motif does not affect the sensitivity of RyR2 to cytosolic Ca2+ activation (33). This indicates that the EF-hand motif is unlikely to be critical for cytosolic Ca2+ activation of RyR. Thus, it is necessary and important to perform detailed functional studies to confirm the significance of specific domains/regions implicated by structural studies.
Interestingly, mutations that increase RyR2 activity are mainly located on one side of the S6 helix that faces the U motif, whereas mutations that decrease the RyR2 activity are largely located on the opposite side of the S6 helix (Fig. 3). How mutations on different sides of the S6 helix affect channel activity in an opposite manner is unclear. Mutations located in the S6/U motif domain interface are expected to disrupt S6/U motif interactions. Such interactions may be involved in the stabilization of the closed state of the channel. Hence, mutations in the S6/U motif interface would destabilize the closed state of RyR2. In support of this view, we indeed found that the Q4876A, V4880A, K4881A, and M4884A mutations located in the S6/U motif interface enhanced basal activity and increased the sensitivity of RyR2 to activation. Further, the V4880A mutation increased the opening event frequency of single RyR2 channels in the near absence of activating Ca2+ and reduced the mean closed time but had no effect on the mean open time of single RyR2s. These data are consistent with the notion that the V4880A mutation destabilizes the closed state of the channel.
We have shown previously that the S6 helix bundle crossing region is critical for channel activation by luminal and cytosolic Ca2+ (29). Mutations in the helix bundle crossing region reduced the open duration and open probability of single RyR2 channels. Hence, it is possible that the Q4877A, E4878A, Q4879A, and D4883A mutations located in the cytoplasmic region of S6 may affect Ca2+ activation or destabilize the open state of RyR2. Further studies are required to test these possibilities.
Recently, des Georges et al. (34) identified a Ca2+ binding site located in the interface between the central domain and the carboxyl-terminal domain (CTD) of RyR1. Based on this structural information, one can speculate that binding of Ca2+ at the interface between the central domain and CTD may cause an outward movement of the S6 helices, leading to dilation of the S6 helix bundle and, thus, opening of the gate. The V4880A mutation, which destabilizes the closed state of the channel, may reduce the free energy that is required to open the gate and, thus, makes the channel more sensitive to activation by Ca2+. Des Georges et al. (34) also identified an ATP binding site located in the interface between the S6-CTD junction and the U motif. Interestingly, the interface between the cytoplasmic region of S6 and the U motif constitutes part of the binding pocket for ATP. Based on our data that S6/U-motif interaction is important for stabilizing the closed state of the channel, ATP, a physiological agonist of RyRs, may enhance RyR2 channel activity by in part destabilizing the closed state of the channel. Hence, the S6/U motif interface represents an important regulatory element of RyR channel gating.
The channel domain is one of the hot spots of disease-causing RyR2 mutations (4, 5). The mechanisms underlying the action of most of the disease mutations located in the channel domain are largely unknown. The V4880A mutation located in the cytoplasmic region of S6 is a disease-causing mutation (16). We found that the V4880A mutation markedly increases the frequency of spontaneous channel openings in the absence of activating Ca2+, indicating that this mutation destabilizes the closed state of the channel. The V4880A mutation also sensitizes the RyR2 channel to activation by cytosolic or luminal Ca2+ and enhances the propensity for spontaneous Ca2+ release in HEK293 cells. Interestingly, the Val4880 residue is located in the S6/U motif interface, where its hydrophobic side chain may interact with the hydrophobic side chains of residues Ile4173, Phe4174, and Val4177 of the U motif (Fig. 10). Thus, like the disease-causing N-terminal RyR mutations that weaken domain interactions in the N-terminal region of RyR (6, 8, 12), the disease-causing V4880A mutation may destabilize the closed state of the channel by weakening the hydrophobic interactions between the S6 helix and the U motif of the Central domain.
FIGURE 10.

Residues in the cytoplasmic region of S6 that are potentially involved in S6/U-motif interactions. Images show the 3D structures of the U motif, the CTD, and the S6 inner helix of RyR2 (30). A, mutations that enhance the activity of RyR2, including Q4876A, V4880A, K4881A, and M4884A, are located on one side of the S6 helix that faces the U motif. B, mutations that suppress RyR2 activity are located on the opposite side of the S6 helix that faces the conduction pore. C, potential hydrophobic interactions among residues Val4880 in the S6 helix and Ile4173, Phe4174, and Val4177 in the U motif of the central domain (14, 30).
In summary, our data demonstrate that the cytoplasmic region of the S6 inner helix of RyR2 is an important determinant of channel stability and activation. Disease-causing RyR2 mutations in this region enhance the propensity for spontaneous Ca2+ release and the susceptibility to cardiac arrhythmias by weakening the interaction between S6 and the U motif and destabilizing the closed state of the channel. Stabilizing the S6/U motif interaction may present a therapeutic approach for suppressing enhanced channel activity.
Experimental Procedures
Construction of RyR2 Mutations Located in the Cytoplasmic Region of the S6 Inner Helix
The S6 RyR2 mutations (Q4876A, Q4877A, E4878A, Q4879A, V4880A, E4882A, D4883A, and M4884A) were generated by the overlap extension method using PCR (25, 35). Briefly, the NruI-NotI (in the vector) fragment containing the Q4876A, Q4877A, E4878A, Q4879A, V4880A, E4882A, D4883A, or M4884A mutation was obtained by overlapping PCR and used to replace the corresponding WT fragment in the full-length mouse RyR2 cDNA in pcDNA5. All mutations were confirmed by DNA sequencing.
Generation of Stable, Inducible HEK293 Cell Lines Expressing RyR2 WT and the V4880A Mutant
Stable, inducible HEK293 cell lines expressing RyR2 WT and V4880A were generated using the Flp-In T-REx Core Kit from Invitrogen. Briefly, Flp-In T-REx HEK293 cells were co-transfected with the inducible expression vector pcDNA5/FRT/TO containing the WT or mutant RyR2 cDNA and the pOG44 vector encoding the Flp recombinase in 1:5 ratios using the Ca2+ phosphate precipitation method. The transfected cells were washed with PBS 24 h after transfection, followed by a change into fresh medium for 24 h. The cells were then washed again with PBS, harvested, and plated onto new dishes. After the cells had attached (∼4 h), the growth medium was replaced with a selection medium containing 200 μg/ml hygromycin (Invitrogen). The selection medium was changed every 3–4 days until the desired number of cells were grown. The hygromycin-resistant cells were pooled, aliquoted (1 ml), and stored at −80 °C. These positive cells are believed to be isogenic because the integration of RyR2 cDNA is mediated by the Flp recombinase at a single FRT site.
Caffeine-induced Ca2+ Release in HEK293 Cells
The free cytosolic Ca2+ concentration in transfected HEK293 cells was measured using the fluorescence Ca2+ indicator dye Fluo-3 AM (Molecular Probes). HEK293 cells grown on 100-mm tissue culture dishes for 18–20 h after subculture were transfected with 12–16 μg of RyR2 WT or the S6 inner helix mutant cDNAs. Cells grown for 18–20 h after transfection were washed four times with PBS and incubated in Krebs-Ringer-Hepes (KRH) (125 mm NaCl, 5 mm KCl, 1.2 mm KH2PO4, 6 mm glucose, 1.2 mm MgCl2, 2 mm CaCl2, and 25 mm HEPES (pH 7.4)) buffer without MgCl2 and CaCl2 at room temperature for 40 min and at 37 °C for 40 min. After being detached from culture dishes by pipetting, cells were collected by centrifugation at 1000 rpm for 2 min in a Beckman TH-4 rotor. Cell pellets were loaded with 10 μm Fluo-3 AM in high glucose Dulbecco's modified Eagle medium at room temperature for 60 min, followed by washing with KRH buffer plus 2 mm CaCl2 and 1.2 mm MgCl2 (KRH+ buffer) three times and resuspended in 150 μl of KRH+ buffer plus 0.1 mg/ml BSA and 250 μm sulfinpyrazone. The Fluo-3 AM-loaded cells were added to 2 ml (final volume) KRH+ buffer in a cuvette. The fluorescence intensity of Fluo-3 AM at 530 nm was measured before and after repeated additions of various concentrations of caffeine (0.025–5 mm) in an SLM-Aminco series 2 luminescence spectrometer with 480 nm excitation at 25 °C (SLM Instruments). The peak levels of each caffeine-induced Ca2+ release were determined and normalized to the highest level (100%) of caffeine-induced Ca2+ release for each experiment.
[3H]Ryanodine Binding
HEK293 cells were grown to 95% confluence in a 75-cm2 flask, dissociated with PBS, and plated in 100-mm tissue culture dishes at ∼10% confluence 18∼20 h before transfection with RyR2 WT and the S6 inner helix mutant cDNAs. After transfection for 24 h, the cells were harvested and lysed in lysis buffer containing 25 mm Tris, 50 mm HEPES (pH 7.4), 137 mm NaCl, 1% CHAPS, 0.5% egg phosphatidylcholine, 2.5 mm DTT, and a protease inhibitor mixture (1 mm benzamidine, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, 2 μg/ml aprotinin, and 0.5 mm PMSF) on ice for 60 min. Cell lysate was obtained after removing the unsolubilized materials by centrifugation twice in a microcentrifuge at 4 °C for 30 min each. Equilibrium [3H]ryanodine binding to cell lysates was performed as described previously (36) with some modifications. [3H]Ryanodine binding was carried out in a total volume of 300 μl of binding solution containing 30 μl of cell lysate, 800 mm KCl, 25 mm Tris, 50 mm Hepes (pH 7.4), 5 nm [3H]ryanodine, and CaCl2 to set free [Ca2+] from pCa 9.89 to pCa 4 and a protease inhibitor mixture at 37 °C for 20 min. Free Ca2+ concentrations were calculated using the computer program of Fabiato and Fabiato (37). The binding mixture was diluted with 5 ml of ice-cold washing buffer containing 25 mm Tris (pH 8.0) and 250 mm KCl and immediately filtered through Whatman GF/B filters presoaked with 1% polyethylenimine. The filters were washed three times, and the radioactivity associated with the filters was determined by liquid scintillation counting. Nonspecific binding was determined by measuring [3H]ryanodine binding in the presence of 50 μm unlabeled ryanodine. All binding assays were done in duplicate.
Western Blotting
HEK293 cells grown for 24 h after transfection with RyR2 WT and the S6 inner helix mutant cDNAs were washed with PBS plus 2.5 mm EDTA and harvested in the same solution by centrifugation for 8 min at 700 × g in an IEC Centra-CL2 centrifuge. The cells were then washed with PBS without EDTA and centrifuged again at 700 × g for 8 min. The PBS-washed cells were solubilized in a lysis buffer containing 25 mm Tris, 50 mm Hepes (pH 7.4), 137 mm NaCl, 1% CHAPS, 0.5% soy bean phosphatidylcholine, 2.5 mm DTT, and a protease inhibitor mixture (1 mm benzamidine, 2 μg/ml leupeptin, 2 μg/ml pepstatin A, 2 μg/ml aprotinin, and 0.5 mm PMSF). This mixture was incubated on ice for 1 h. Cell lysate was obtained by centrifuging twice at 16,000 × g in a microcentrifuge at 4 °C for 30 min to remove unsolubilized materials. The RyR2 WT and mutant proteins were subjected to SDS-PAGE (5% gel) (38) and transferred onto nitrocellulose membranes at 100 V for 2 h at 4 °C in the presence of 0.01% SDS (39). The nitrocellulose membranes containing the transferred proteins were blocked for 60 min with 1% casein blocker (Bio-Rad). The blocked membrane was incubated with anti-RyR antibody (34C) (Thermo Scientific, MA3-925, lot no. PG200294, 1:1000 dilution) and then incubated with secondary anti-mouse IgG (heavy and light) antibodies conjugated to horseradish peroxidase (1:20,000 dilution). After washing for 10 min three times, the bound antibodies were detected using an enhanced chemiluminescence kit from Pierce. The intensity of each band was determined from its intensity profile obtained by ImageQuant LAS 4000 (GE Healthcare/Life Sciences), analyzed by ImageJ software, and normalized to that of β-actin (40).
Single-channel Recordings
Recombinant RyR2 WT and V4880A mutant channels were purified from cell lysate prepared from HEK293 cells transfected with the RyR2 WT or the V4880A mutant cDNA by sucrose density gradient centrifugation as described previously (18, 36). Heart phosphatidylethanolamine (50%) and brain phosphatidylserine (50%) (Avanti Polar Lipids), dissolved in chloroform, were combined and dried under nitrogen gas and resuspended in 30 μl of n-decane at a concentration of 12 mg of lipid/ml. Bilayers were formed across a 250-μm hole in a Delrin partition separating two chambers. The trans chamber (800 μl) was connected to the head stage input of an Axopatch 200A amplifier (Axon Instruments, Austin, TX). The cis chamber (1.2 ml) was held at virtual ground. A symmetrical solution containing 250 mm KCl and 25 mm Hepes (pH 7.4) was used for all recordings unless indicated otherwise. A 4-μl aliquot (≈1 μg of protein) of the sucrose density gradient-purified recombinant RyR2 WT or the V4880A mutant channels was added to the cis chamber. Spontaneous channel activity was always tested for sensitivity to EGTA and Ca2+. The chamber to which the addition of EGTA inhibited the activity of the incorporated channel presumably corresponds to the cytosolic side of the Ca2+ release channel. The direction of single-channel currents was always monitored from the luminal to the cytosolic side of the channel unless mentioned otherwise. Recordings were filtered at 2500 Hz. Data analyses were carried out using the pclamp 8.1 software package (Axon Instruments). Free Ca2+ concentrations were calculated using the computer program of Fabiato and Fabiato (37).
Single Cell Cytosolic Ca2+ Imaging
Cytosolic Ca2+ levels in stable, inducible HEK293 cells expressing RyR2 WT or the V4880A mutant were monitored using single-cell Ca2+ imaging and the fluorescent Ca2+ indicator dye Fura-2/AM as described previously (17, 18). Briefly, cells grown on glass coverslips for 8–18 h after induction (as indicated) by 1 μg/ml tetracycline (Sigma) were loaded with 5 μm Fura-2/AM in KRH buffer (125 mm NaCl, 5 mm KCl, 6 mm glucose, 1.2 mm MgCl2, and 25 mm Hepes (pH 7.4)) plus 0.02% pluronic F-127 and 0.1 mg/ml BSA for 20 min at room temperature (23 °C). The coverslips were then mounted in a perfusion chamber (Warner Instruments) on an inverted microscope (Nikon TE2000-S). The cells were perfused continuously with KRH buffer containing increasing extracellular Ca2+ concentrations (0, 0.1, 0.2, 0.3, 0.5, 1.0, and 2.0 mm). Caffeine (10 mm) was applied at the end of each experiment to confirm the expression of active RyR2 channels. Time-lapse images (0.25 frame/s) were captured and analyzed with Compix Simple PCI 6 software. Fluorescence intensities were measured from regions of interest centered on individual cells. Only cells that responded to caffeine were analyzed. The filters used for Fura-2 imaging were λexcitation = 340 ± 26 nm and 387 ± 11 nm and λemission = 510 ± 84 nm with a dichroic mirror (410 nm).
Single-cell Luminal Ca2+ Imaging
Luminal Ca2+ levels in HEK293 cells expressing RyR2 WT or the V4880A mutant were measured using single-cell Ca2+ imaging and the FRET-based ER luminal Ca2+-sensitive chameleon protein D1ER as described previously (22, 24). The cells were grown to 95% confluence in a 75-cm2 flask, passaged with PBS, and plated in 100-mm-diameter tissue culture dishes at ∼10% confluence 18–20 h before transfection with D1ER cDNA using the Ca2+ phosphate precipitation method. After transfection for 24 h, the growth medium was changed to an induction medium containing 1 μg/ml tetracycline. In intact cell studies, after induction for ∼22 h, the cells were perfused continuously with KRH buffer containing various concentrations of CaCl2 (0, 1, and 2 mm) and tetracaine (1 mm) for estimating the store capacity or caffeine (20 mm) for estimating the minimum store level by depleting the ER Ca2+ stores at room temperature (23 °C). Images were captured with Compix Simple PCI 6 software every 2 s using an inverted microscope (Nikon TE2000-S) equipped with an S-Fluor ×20/0.75 objective. The filters used for D1ER imaging were λexcitation = 436 ± 20 nm for CFP and λexcitation = 500 ± 20 nm for YFP, and λemission = 465 ± 30 nm for CFP and λemission = 535 ± 30 nm for YFP with a dichroic mirror (500 nm). The amount of FRET was determined from the ratio of the light emission at 535 and 465 nm.
Statistical Analysis
All values shown are mean ± S.E. unless indicated otherwise. To test for differences between groups, we used Student's t test (two-tailed) or one-way analysis of variance with post hoc test. p < 0.05 was considered to be statistically significant.
Author Contributions
B. S., W. G., X. T., J. Y., L. Z., R. W., and S. R. W. C. designed the research. B. S., W. G., X. T., J. Y., L. Z., and R. W. performed the research. B. S., W. G., X. T., J. Y., L. Z., R. W., and S. R. W. C. analyzed the data. B. S., W. G., R. W., and S. R. W. C. wrote the paper.
This work was supported by research grants from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, the Canada Foundation for Innovation, and the Heart and Stroke Foundation/Libin Cardiovascular Institute Professorship in Cardiovascular Research (to S. R. W. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- RyR
- ryanodine receptor
- SR
- sarcoplasmic reticulum
- Po
- open probability
- Tc
- mean closed time
- To
- mean open time
- SOICR
- store overload-induced Ca2+ release
- ER
- endoplasmic reticulum
- CTD
- carboxyl-terminal domain
- KRH
- Krebs-Ringer-Hepes
- CFP
- cyan fluorescent protein.
References
- 1. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 2. Fill M., and Copello J. A. (2002) Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922 [DOI] [PubMed] [Google Scholar]
- 3. Van and Petegem F. (2015) Ryanodine receptors: allosteric ion channel giants. J. Mol. Biol. 427, 31–53 [DOI] [PubMed] [Google Scholar]
- 4. Maclennan D. H., and Zvaritch E. (2011) Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim. Biophys. Acta 1813, 948–964 [DOI] [PubMed] [Google Scholar]
- 5. Priori S. G., and Chen S. R. (2011) Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 108, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amador F. J., Liu S., Ishiyama N., Plevin M. J., Wilson A., MacLennan D. H., and Ikura M. (2009) Crystal structure of type I ryanodine receptor amino-terminal β-trefoil domain reveals a disease-associated mutation “hot spot” loop. Proc. Natl. Acad. Sci. U.S.A. 106, 11040–11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lobo P. A., and Van Petegem F. (2009) Crystal structures of the N-terminal domains of cardiac and skeletal muscle ryanodine receptors: insights into disease mutations. Structure 17, 1505–1514 [DOI] [PubMed] [Google Scholar]
- 8. Tung C. C., Lobo P. A., Kimlicka L., and Van Petegem F. (2010) The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 468, 585–588 [DOI] [PubMed] [Google Scholar]
- 9. Lobo P. A., Kimlicka L., Tung C. C., and Van Petegem F. (2011) The deletion of exon 3 in the cardiac ryanodine receptor is rescued by β strand switching. Structure 19, 790–798 [DOI] [PubMed] [Google Scholar]
- 10. Yuchi Z., Lau K., and Van Petegem F. (2012) Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure 20, 1201–1211 [DOI] [PubMed] [Google Scholar]
- 11. Amador F. J., Kimlicka L., Stathopulos P. B., Gasmi-Seabrook G. M., Maclennan D. H., Van Petegem F., and Ikura M. (2013) Type 2 ryanodine receptor domain A contains a unique and dynamic α-helix that transitions to a β-strand in a mutant linked with a heritable cardiomyopathy. J. Mol. Biol. [DOI] [PubMed] [Google Scholar]
- 12. Kimlicka L., Lau K., Tung C. C., and Van Petegem F. (2013) Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat. Commun. 4, 1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lau K., and Van Petegem F. (2014) Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nat. Commun. 5, 5397. [DOI] [PubMed] [Google Scholar]
- 14. Yan Z., Bai X. C., Yan C., Wu J., Li Z., Xie T., Peng W., Yin C. C., Li X., Scheres S. H., Shi Y., and Yan N. (2015) Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bai X. C., Yan Z., Wu J., Li Z., and Yan N. (2016) The central domain of RyR1 is the transducer for long-range allosteric gating of channel opening. Cell Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bagattin A., Veronese C., Bauce B., Wuyts W., Settimo L., Nava A., Rampazzo A., and Danieli G. A. (2004) Denaturing HPLC-based approach for detecting RYR2 mutations involved in malignant arrhythmias. Clin. Chem. 50, 1148–1155 [DOI] [PubMed] [Google Scholar]
- 17. Jiang D., Xiao B., Yang D., Wang R., Choi P., Zhang L., Cheng H., and Chen S. R. (2004) RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. U.S.A. 101, 13062–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang D., Wang R., Xiao B., Kong H., Hunt D. J., Choi P., Zhang L., and Chen S. R. (2005) Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 97, 1173–1181 [DOI] [PubMed] [Google Scholar]
- 19. Bassani J. W., Yuan W., and Bers D. M. (1995) Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268, C1313–9 [DOI] [PubMed] [Google Scholar]
- 20. Díaz M. E., Trafford A. W., O'Neill S. C., and Eisner D. A. (1997) Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J. Physiol. 501, 3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shannon T. R., Pogwizd S. M., and Bers D. M. (2003) Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 93, 592–594 [DOI] [PubMed] [Google Scholar]
- 22. Jones P. P., Jiang D., Bolstad J., Hunt D. J., Zhang L., Demaurex N., and Chen S. R. (2008) Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem. J. 412, 171–178 [DOI] [PubMed] [Google Scholar]
- 23. Tang Y., Tian X., Wang R., Fill M., and Chen S. R. (2012) Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ. Res. 110, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palmer A. E., Jin C., Reed J. C., and Tsien R. Y. (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U.S.A. 101, 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao M., Li P., Li X., Zhang L., Winkfein R. J., and Chen S. R. (1999) Molecular identification of the ryanodine receptor pore-forming segment. J. Biol. Chem. 274, 25971–25974 [DOI] [PubMed] [Google Scholar]
- 26. Chen S. R., Li P., Zhao M., Li X., and Zhang L. (2002) Role of the proposed pore-forming segment of the Ca2+ release channel (ryanodine receptor) in ryanodine interaction. Biophys. J. 82, 2436–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang R., Bolstad J., Brown C., Zhang L., and Chen S. R. (2003) Role of the TM10 transmembrane helix in cardiac ryanodine receptor function. Biophys. J. 84, 427a [Google Scholar]
- 28. Wang R., Bolstad J., Kong H., Zhang L., Brown C., and Chen S. R. W. (2004) The Predicted TM10 Transmembrane Sequence of the Cardiac Ca2+ Release Channel (Ryanodine Receptor) Is Crucial for Channel Activation and Gating. J. Biol. Chem. 279, 3635–3642 [DOI] [PubMed] [Google Scholar]
- 29. Chen W., Wang R., Chen B., Zhong X., Kong H., Bai Y., Zhou Q., Xie C., Zhang J., Guo A., Tian X., Jones P. P., O'Mara M. L., Liu Y., Mi T., et al. (2014) The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med. 20, 184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peng W., Shen H., Wu J., Guo W., Pan X., Wang R., Chen S. R., and Yan N. (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science [DOI] [PubMed] [Google Scholar]
- 31. Zalk R., Clarke O. B., des Georges A., Grassucci R. A., Reiken S., Mancia F., Hendrickson W. A., Frank J., and Marks A. R. (2015) Structure of a mammalian ryanodine receptor. Nature 517, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Efremov R. G., Leitner A., Aebersold R., and Raunser S. (2015) Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517, 39–43 [DOI] [PubMed] [Google Scholar]
- 33. Guo W., Sun B., Xiao Z., Liu Y., Wang Y., Zhang L., Wang R., and Chen S. R. (2016) The EF-hand Ca2+ Binding domain is not required for cytosolic Ca2+ activation of the cardiac ryanodine receptor. J. Biol. Chem. 291, 2150–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. des Georges A., Clarke O. B., Zalk R., Yuan Q., Condon K. J., Grassucci R. A., Hendrickson W. A., Marks A. R., and Frank J. (2016) Structural basis for gating and activation of RyR1. Cell 167, 145–157.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction [see comments]. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 36. Li P., and Chen S. R. (2001) Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J. Gen. Physiol. 118, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fabiato A., and Fabiato F. (1979) Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. 75, 463–505 [PubMed] [Google Scholar]
- 38. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–665 [DOI] [PubMed] [Google Scholar]
- 39. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu Y., Sun B., Xiao Z., Wang R., Guo W., Zhang J. Z., Mi T., Wang Y., Jones P. P., Van Petegem F., and Chen S. R. (2015) Roles of the NH2-terminal domains of cardiac ryanodine receptor in Ca2+ release activation and termination. J. Biol. Chem. 290, 7736–7746 [DOI] [PMC free article] [PubMed] [Google Scholar]