

Abstract
Hemophilia A is a bleeding disorder caused by a deficiency in coagulation factor VIII (fVIII) that affects 1 in 5,000 males. Current prophylactic replacement therapy, although effective, is difficult to maintain due to the cost and frequency of injections. Hepatic clearance of fVIII is mediated by the LDL receptor-related protein 1 (LRP1), a member of the LDL receptor family. Although it is well established that fVIII binds LRP1, the molecular details of this interaction are unclear as most of the studies have been performed using fragments of fVIII and LRP1. In the current investigation, we examine the binding of intact fVIII to full-length LRP1 to gain insight into the molecular interaction. Chemical modification studies confirm the requirement for lysine residues in the interaction of fVIII with LRP1. Examination of the ionic strength dependence of the interaction of fVIII with LRP1 resulted in a Debye-Hückel plot with a slope of 1.8 ± 0.5, suggesting the involvement of two critical charged residues in the interaction of fVIII with LRP1. Kinetic studies utilizing surface plasmon resonance techniques reveal that the high affinity of fVIII for LRP1 results from avidity effects mediated by the interactions of two sites in fVIII with complementary sites on LRP1 to form a bivalent fVIII·LRP1 complex. Furthermore, although fVIII bound avidly to soluble forms of clusters II and IV from LRP1, only soluble cluster IV competed with the binding of fVIII to full-length LRP1, revealing that cluster IV represents the major fVIII binding site in LRP1.
Keywords: endocytosis, factor VIII (FVIII), lipoprotein receptor, lipoprotein receptor-related protein (LPR), surface plasmon resonance (SPR), LRP, blood coagulation, ligand, lysine
Introduction
Factor VIII (fVIII)3 is an essential blood coagulation cofactor whose deficiency leads to hemophilia A (1), an inherited bleeding disorder that affects 1 in 5,000 males. The majority of hemophilia A patients have <1% of normal fVIII levels and must be injected with fVIII to arrest bleeding episodes. Prophylactic treatment, in which low levels of fVIII are maintained in the circulation, is highly effective in preventing bleeding episodes. However, the short half-life of fVIII (10–14 h) (2) requires that it be administered every other day or 3 times a week to be effective (3). The demanding injection schedule often leads to a lack of adherence to a prophylactic treatment protocol, and thus the development of a fVIII molecule with a longer half-life in the circulation would be of tremendous benefit to these patients.
Once secreted from liver endothelial cells (4, 5), fVIII circulates as a two-chain protein non-covalently linked by a metal cation. The fVIII heavy chain consists of the A1 and A2 domains along with a variable length B domain (85–190 kDa), whereas the light chain comprises the A3, C1, and C2 domains (80 kDa). In the circulation, fVIII binds tightly to von Willebrand factor (6), which prevents its interaction with hepatic receptors responsible for the rapid clearance of fVIII from the circulation (7, 8). Upon vasculature injury, fVIII is activated by thrombin, resulting in its dissociation from von Willebrand factor. Active fVIII (fVIIIa) serves as a cofactor for the serine protease factor IXa, which activates factor X, an enzyme that forms a complex with factor V to catalyze the conversion of prothrombin to thrombin. Thrombin is the final activator in the pathway and converts fibrinogen to fibrin to form the fibrin clot. The activity of fVIIIa is short lived as the A2 domain rapidly dissociates from fVIIIa with a half-life of 2 min (9, 10).
The levels of circulating fVIII are not only regulated by its synthesis but also by its clearance in the liver. In vitro binding studies (7, 11–15) and genetic studies (16, 17) reveal that the low density lipoprotein receptor-related protein 1 (LRP1) is a major hepatic receptor responsible for fVIII removal. The identification of LRP1 as a receptor involved in fVIII catabolism was originally reported by Saenko et al. (11) and Lenting et al. (7) when they noted a direct interaction between fVIII and LRP1 and observed that LRP1-expressing cells, but not LRP1-deficient cells, were able to mediate the internalization of 125I-labeled fVIII (11). These studies also established that LRP1 binds fVIII in a process inhibited by receptor-associated protein (RAP). RAP binds tightly to LRP1 and functions as a molecular chaperone in the endoplasmic reticulum by preventing ligands from associating with newly synthesized LRP1 (18–24) and is widely used to antagonize ligand binding to this receptor. Genetic studies confirmed the importance of LRP1 in mediating the clearance of fVIII by demonstrating that hepatic deletion of the Lrp1 gene in mice resulted in a 2-fold increase in plasma levels of fVIII (16, 17) and significantly delayed the clearance of intravenously injected fVIII (16).
LRP1, a member of the LDL receptor family, is an endocytic receptor that is abundantly expressed in the liver in hepatocytes and resident macrophages (25, 26). LRP1 contains complement-type repeats (CRs), EGF repeats, β-propeller domains, a transmembrane domain, and a cytoplasmic domain. The CR modules are organized into four clusters (clusters I–IV), which are highly conserved regions where most LRP1 ligands bind. Ligand binding by this family of receptors appears to involve the docking of two or more lysine residues into acidic pockets located within the CR modules of the receptor (27). This has been clearly demonstrated for RAP, which contains two binding sites for LRP1. The first binding site is located within domains 1 and 2 of RAP (D1D2), and recent studies reveal that lysine 60 in D1 and lysine 191 in D2 interact with distinct sites on LRP1 to form a bivalent D1D2·LRP1 complex (28). A second LRP1 binding site is located in domain 3 of RAP, and here lysines 256 and 270 are key residues that are necessary for high affinity binding of this domain to LRP1 (29, 30).
Considerable interest exists in manipulating fVIII to increase its half-life in the circulation, and one strategy to achieve this would be to reduce its binding to LRP1. Indeed, numerous studies have characterized the binding of fVIII to LRP1, although most studies (12–15) have used fragments of these proteins, which do not give a clear picture of the molecular interactions involved. In the most comprehensive investigation, van den Biggelaar et al. (31) utilized hydrogen-deuterium exchange mass spectrometry and mutational analysis to conclude that the interaction between the fVIII light chain and cluster II from LRP1 occurs over an extended surface containing multiple lysine residues.
In the current investigation, we examine the binding of intact fVIII to full-length LRP1 to gain insight into the molecular interaction. Our studies imply an important role for at least two lysine residues in the interaction, and kinetic analysis reveal that fVIII forms a bivalent complex with LRP1. Finally, our data reveal that cluster IV on LRP1 represents the major fVIII binding site on this receptor.
Results
Chemical Modification of fVIII Lysine Residues Abolishes Its Binding to LRP1
Prior studies examined the binding of the fVIII light chain to cluster II of LRP1 and observed that chemical modification of lysine residues in the fVIII light chain abolished its binding to cluster II, suggesting that lysine residues are important for this binding interaction (31). To determine whether lysine residues are critical for the binding of the entire fVIII molecule to full-length LRP1, we performed a similar experiment by first coating B domain-deleted fVIII (BDD-fVIII) in microtiter wells. In one set of wells, bound BDD-fVIII was incubated with sulfo-NHS-biotin to specifically modify lysine residues. To confirm lysine modification of BDD-fVIII, the ability of streptavidin to bind to modified proteins in the microtiter wells was measured, and the results confirmed successful modification of lysine residues (Fig. 1A). Equal coating of BDD-fVIII and modified BDD-fVIII to the microtiter wells was confirmed by measuring the ability of the fVIII-specific antibody ESH8 to bind to the wells (Fig. 1B). To assess the impact of lysine modification on the ability of BDD-fVIII to bind LRP1, increasing concentrations of purified LRP1 were incubated with BDD-fVIII or modified BDD-fVIII, and following incubation and washing, the amount of LRP1 bound was quantified. The results (Fig. 1C) reveal that chemical modification of the BDD-fVIII lysine residues with sulfo-NHS-biotin substantially reduced its binding to LRP1, suggesting a critical role for these residues in the binding interaction of BDD-fVIII to full-length LRP1.
FIGURE 1.
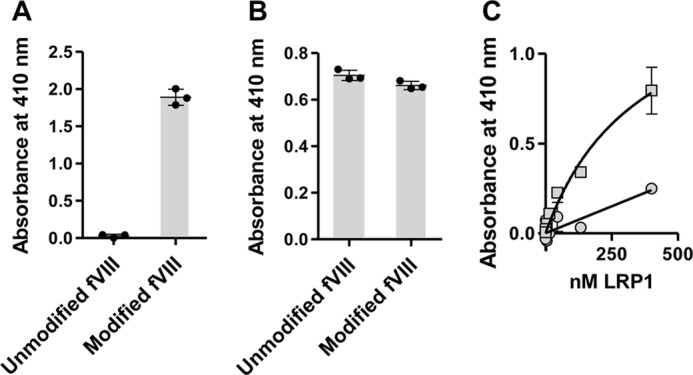
Modification of lysine residues on BDD-fVIII abolishes its binding to LRP1. A, microtiter wells were coated with BDD-fVIII (5 μg/ml) and then incubated with (Modified) or without (Unmodified) EZ-Link sulfo-NHS-biotin. Following incubation, the wells were incubated with streptavidin-alkaline phosphatase to confirm that BDD-fVIII was modified. B, following modification, wells were incubated with fVIII monoclonal antibody ESH8 to confirm even coating of fVIII. C, increasing concentrations of purified LRP1 were incubated with microtiter wells coated with BDD-fVIII (squares) or BDD-fVIII modified by sulfo-NHS-biotin (circles). Following washing, the amount of LRP1 bound was measured using anti-LRP1 monoclonal antibody 8G1. The experiments were repeated twice, and representative experiments are shown. Error bars represent S.E.
Ionic Strength Dependence of the Binding of BDD-fVIII to LRP1 Suggests the Involvement of Two Charged Residues
To determine whether fVIII binding to LRP1 is dependent upon ionic strength as would be predicted from the contribution of lysine residues to the binding, we examined the ionic strength dependence of BDD-fVIII binding to full-length LRP1 using surface plasmon resonance measurements. The results of these experiments are shown in Fig. 2 in the form of a Debye-Hückel plot where log10KD is plotted versus ionic strength. The data reveal that the binding of BDD-fVIII to LRP1 is highly dependent upon ionic strength. Importantly, these experiments can give insight into the number of ionic interactions involved in binding, which is derived from the slope of the graph. The data in Fig. 2 suggest the involvement of two charged residues in the binding of BDD-fVIII to full-length LRP1 (slope = 1.8 ± 0.5).
FIGURE 2.
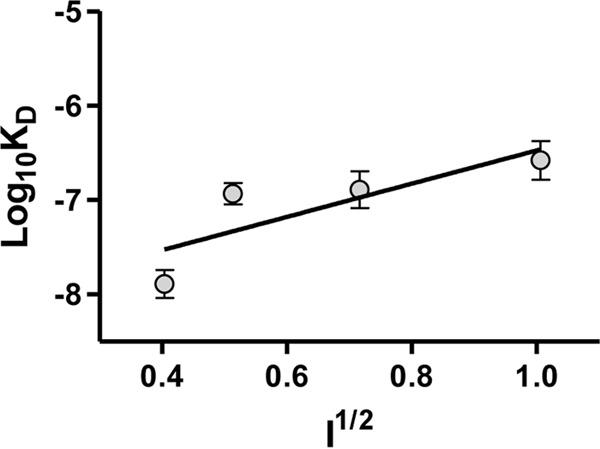
The binding of BDD-fVIII to LRP1 is ionic strength-dependent. A Debye-Hückel plot of BDD-fVIII binding to LRP1 is shown. The KD value at each ionic strength (150, 250, 500, and 1,000 mm NaCl) was measured by equilibrium SPR measurements. The values represent the mean of three independent experiments. Error bars represent S.E. A slope of 1.8 ± 0.5 was determined by linear regression analysis (r2 = 0.7).
fVIII Binds to LRP1 via Bivalent Interactions
The data from Fig. 2 reveal an important contribution of at least two charged residues in the interaction of fVIII with LRP1, suggesting that the high affinity of fVIII for LRP1 results from avidity effects mediated by the interaction of lysine residues located in two distinct regions of the fVIII molecule with complementary sites located on LRP1 to form a bivalent fVIII·LRP1 complex (Fig. 3A). To test this model, we performed kinetic measurements investigating the binding of full-length fVIII (FL-fVIII) as well as BDD-fVIII to LRP1 immobilized on an SPR chip. The results of these experiments are shown in Fig. 3, B and C, and reveal that the experimental data are well described by this model with the fit parameters summarized in Table 1. The kinetic data reveal KD values of 31 ± 4 and 38 ± 21 nm for BDD-fVIII and FL-fVIII, respectively. We also attempted to fit the SPR data to a model in which fVIII binds to a single class of sites on LPR1. The fit to this model was poor. Finally, additional models were evaluated, but none of them fit as well as the bivalent model shown in Fig. 3A.
FIGURE 3.

Binding of FL-fVIII and BDD-fVIII to LRP1 fits a bivalent binding model. A, bivalent binding model for the interaction of two distinct regions on fVIII with complementary sites on LRP1. B and C, increasing concentrations of FL-fVIII (2.5, 7.4, 22.2, 66.6, and 200 nm) (B) or BBD-fVIII (12.5, 25, 50, 100, and 200 nm) (C) were injected over the LRP1-coupled chip. The experimental data are shown in black, whereas fits to the bivalent binding model are shown in blue. Three independent experiments were performed, and representative experiments are shown. RU, resonance units.
TABLE 1.
Kinetic and equilibrium constants for the binding of BDD- and FL-fVIII LRP1
Values are means ± S.D. from three different experiments.
| Protein | ka1 | kd1 | ka2 | kd2 | KDa |
|---|---|---|---|---|---|
| m−1 s−1 | s−1 | s−1 | s−1 | nm | |
| BDD-fVIII | 5.3 ± 1.7 × 105 | 5.2 ± 1.2 × 10−2 | 4.2 ± 1.5 × 10−3 | 1.9 ± 0.5 × 10−3 | 31 ± 4 |
| FL-fVIII | 2.6 ± 1.0 × 105 | 5.2 ± 3.1 × 10−2 | 5.8 ± 1.6 × 10−3 | 1.4 ± 0.6 × 10−3 | 38 ± 21 |
a Determined from kinetic measurements using the following equation: KA1 = (ka1/kd1) × (1 + (ka2/kd2). KD1 was calculated as KD = 1/KA.
fVIII Preferentially Binds to Cluster IV in Full-length LRP1
Previous studies have shown that the fVIII light chain can bind to clusters II and IV of LRP1 (32, 33) and that fVIIIa binds to cluster III of LRP1 (34). However, a systematic investigation of the binding of intact fVIII with full-length LRP1 has not been investigated, and thus we conducted experiments to identify the sites on full-length LRP1 to which fVIII binds. LRP1 contains four clusters of CRs (Fig. 4A). Initial experiments measured the ability of soluble forms of LRP1 clusters II, III, and IV, which bind most LRP1 ligands, to interact with BDD-fVIII immobilized in microtiter wells. The results of these experiments revealed that BDD-fVIII bound to clusters II and clusters IV with KD values of 54 ± 14 and 20 ± 7 nm, respectively (Fig. 4B). Interestingly, no binding of BDD-fVIII to cluster III of LRP1 was observed (Fig. 4B). Control experiments confirmed that BDD-fVIII did not bind to microtiter wells coated with BSA (Fig. 4C). As an additional control experiment to confirm the integrity of the soluble clusters, we investigated the binding of RAP to these molecules. RAP bound to clusters II, III, and IV with expected KD values of 1.4 ± 0.2, 0.5 ± 0.1, and 0.8 ± 0.1 nm, respectively (Fig. 4D), confirming the integrity of the LRP1 clusters.
FIGURE 4.

BDD-fVIII binds to LRP1 clusters II and IV but not to cluster III. A, domain structure of LRP1. The extracellular chain contains four clusters of cysteine-rich ligand binding repeats (I–IV; red circles). An arrow shows the furin cleavage site. B, C, and D, increasing concentrations of clusters II (circles), III (squares), and IV (triangles) were incubated with microtiter wells coated with BDD-fVIII (B), BSA (C), or RAP (D). Following incubation and washing, the amount of clusters bound to each well was measured. The experiments were repeated twice, each in duplicate, and a representative experiment is shown in which replicate values are plotted.
We next sought to determine whether either cluster II, III, or IV was able to compete for the binding of BDD-fVIII to full-length LRP1. In these experiments, microtiter wells were first coated with full-length LRP1 and then incubated with 20 nm BDD-fVIII in the presence of increasing concentrations of soluble cluster II, III, or IV. After incubation and washing, the amount of fVIII bound to LRP1 was quantified. The results reveal that only cluster IV is able to compete for BDD-fVIII binding to full-length LRP1 with a KI of 27 ± 1 nm (Fig. 5A). Curiously, despite the fact that BDD-fVIII was able to bind to cluster II of LRP1, this molecule was unable to effectively compete for the binding of BDD-fVIII to LRP1.
FIGURE 5.
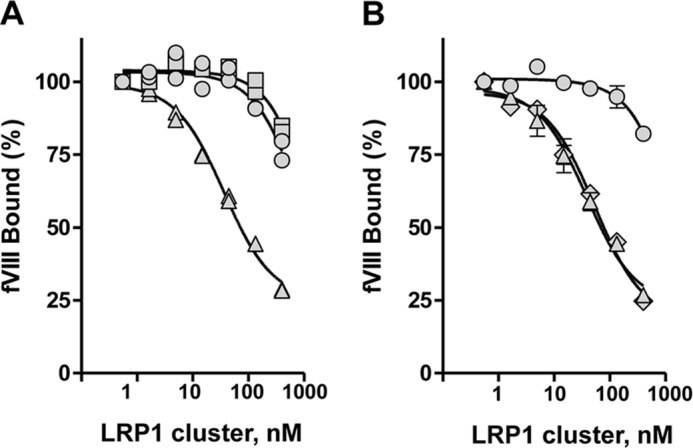
LRP1 cluster IV, but not cluster II, competes for the binding of BDD-fVIII to LRP1. A, BDD-fVIII (20 nm) was incubated in microtiter wells coated with LRP1 in the presence of increasing concentrations of LRP1 clusters II (circles), III (squares), and IV (triangles). Following incubation, bound BDD-fVIII was detected by a fVIII-specific antibody. This experiment was repeated twice with replicates. A representative experiment is shown with replicates plotted. B, BDD-fVIII (20 nm) was incubated in microtiter wells coated with LRP1 in the presence of increasing concentrations of LRP1 cluster II (circles), IV (triangles), or clusters II and IV combined (diamonds). Following incubation, bound BDD-fVIII was detected by a fVIII-specific antibody. This experiment was performed in triplicate, and the mean values are plotted. Error bars represent S.E.
If cluster IV represents the major binding site on LRP1 for fVIII, then combining cluster II with cluster IV would be expected to have little impact on the binding of fVIII to LRP1. To determine whether this is indeed the case, we incubated BDD-fVIII with LRP1-coated microtiter wells in the presence of increasing concentrations of cluster II, cluster IV, or combined clusters II and IV. The results reveal that combining cluster II and IV did not change the effectiveness of cluster IV to compete for the binding of BDD-fVIII with LRP1 (Fig. 5B), confirming that cluster IV represents the major LRP1 binding site for fVIII.
To gain additional mechanistic insight into the binding of BDD-fVIII to LRP1 clusters, we designed experiments to determine whether cluster IV could compete for the binding of BDD-fVIII to cluster II and whether cluster II could compete for the binding of BDD-fVIII to cluster IV. The results reveal that cluster IV is highly effective at competing for the binding of BDD-fVIII to cluster II (Fig. 6A). In contrast, cluster II was unable to compete for the binding of BDD-fVIII to cluster IV (Fig. 6B).
FIGURE 6.
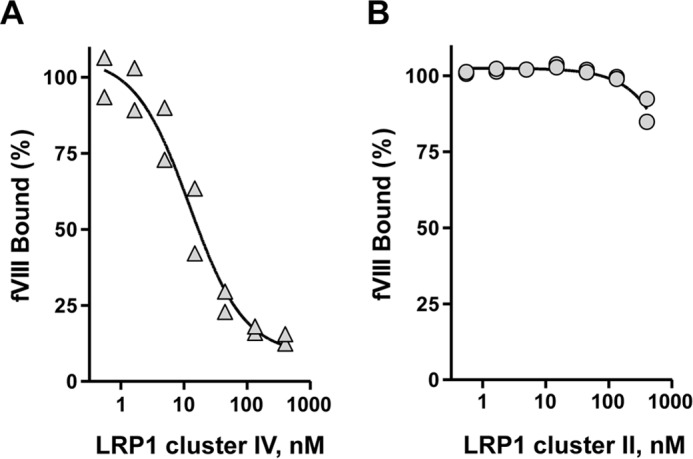
LRP1 cluster IV competes for the binding of BDD-fVIII to cluster II. A, 20 nm BDD-fVIII in the presence of increasing concentrations of cluster IV was incubated with microtiter wells coated with cluster II. Following incubation and washing, the amount of BDD-fVIII bound to cluster II was quantified. B, 20 nm BDD-fVIII in the presence of increasing concentrations of cluster II was incubated with microtiter wells coated with cluster IV. Following incubation and washing, the amount of BDD-fVIII bound to cluster IV was quantified. The data are represented as percent bound with 100% determined in the absence of competitor. These experiments were performed in duplicate, and replicates are plotted.
LRP1 Cluster IV, but Not Cluster II, Inhibits LRP1-mediated Cellular Uptake of 125I-labeled fVIII
We next examined the ability of clusters II and IV to block the LRP1-mediated uptake of 125I-BDD-fVIII by WI38 fibroblasts, a cell line that expresses large amounts of LRP1. The results indicate that an excess of cluster IV inhibits the LRP1-mediated uptake of 125I-labeled BDD-fVIII uptake by 60%, whereas an excess of cluster II had no significant effect on the uptake of 125I-labeled BDD-fVIII (Fig. 7A). In contrast, both cluster II and cluster IV inhibited the LRP1-mediated uptake of 125I-labeled tPA·PAI-1 complexes, an LRP1 ligand that is known to bind to both clusters II and IV (32) (Fig. 7B).
FIGURE 7.

Effect of clusters II and IV on LRP1-mediated internalization of BDD-fVIII (A) and tPA·PAI-1 (B) by WI-38 fibroblasts. WI-38 fibroblasts were seeded in 12-well tissue culture dishes (1 × 105 cells/well) and incubated with 20 nm 125I-BDD-fVIII (A) or 10 nm 125I-tPA·PAI-1 (B) for 4 h at 37 °C in the presence or absence of LRP1 cluster II or IV (500 nm). Following incubation, the amount of radiolabeled ligand internalized was determined, and the data were normalized to the amount of fVIII internalized in the absence of competitors. The results represent the mean of three independent experiments. Error bars represent S.E. (*, p ≤ 0.002, one-way analysis of variance with Tukey's multiple comparison post-test).
Discussion
In the current investigation, we conducted studies to characterize the binding of fVIII to full-length LRP1. First we demonstrated that modification of lysine residues in fVIII by reaction of the molecule with sulfo-NHS-biotin dramatically impacted its recognition by full-length LRP1. Although it is possible that, rather than the elimination of the lysine charges, the introduction of a biotin group results in a steric hindrance effect that causes reduced binding. This is not likely the case as our data reveal that the binding is highly dependent upon ionic strength. By examining the ionic strength dependence of the binding interaction between fVIII and LRP1, our studies suggest a critical role of two charged lysine residues within the fVIII molecule that are involved in its binding to LRP1. Furthermore, our kinetic data are consistent with a model in which the high affinity of fVIII for LRP1 results from avidity effects in which lysine residues located in distinct regions of fVIII interact with CR modules located in cluster IV of LRP1 to form a bivalent fVIII·LRP1 complex. Thus, the binding of fVIII to LRP1 conforms to the canonical model for ligand binding to LRP1 in which high affinity binding results from avidity effects in which multiple lysine residues located on the ligands dock into acidic pockets located within CRs of the receptor. This model was originally derived from investigation of the interaction of RAP D3 domain with LRP1 and the LDL receptor (27, 29). Together, these data confirmed a critical role for Lys-256 and Lys-270, which dock into acidic pockets located in CR modules of the receptor. RAP contains three domains, and another high affinity LRP1 binding site is located within the D1D2 domains (35, 36). Mutagenesis studies investigating the binding of D1D2 domains to LRP1 have revealed that D1D2 also forms a bivalent D1D2·LRP1 complex mediated by the interactions of lysine 60 in D1 and lysine 191 in D2 with sites on LRP1 (28). Interestingly, only when both lysine 60 and lysine 191 were mutated was binding of D1D2 to LRP1 ablated (28).
By using fragments of fVIII, previous studies have identified regions on three domains of fVIII that are capable of binding to LRP1. Initial studies discovered that a monoclonal antibody to a region within the A2 domain blocked binding of fVIII to LRP1 (11). Following these observations, Sarafanov et al. (14) discovered that the isolated A2 domain binds avidly to LRP1, and mutation of Lys-466 and Lys-499 to alanine reduced the affinity of the A2 domain for LRP1 by 4- and 3-fold, respectively. Interestingly, calculation of the accessible surface area (37) for Lys-466 and Lys-499 from the three-dimensional structures available for BDD-fVIII (38–40) reveals that these residues are buried in native fVIII and likely unavailable for binding to LRP1. This is consistent with the observation that this site may only be exposed upon thrombin cleavage of the heavy chain of fVIII (12). The major LRP1 binding sites for native forms of fVIII appear to be located within the light chain, specifically the A3 (13) and C1 domains (41). In elegant studies, van den Biggelaar (31) used hydrogen-deuterium exchange mass spectrometry and mutational analysis to identify lysine residues in the light chain that contribute to its interaction with cluster II from LRP1. These studies identified Lys-1693, Lys-1694, Lys-1813, Lys-1818, Lys-1827, and Lys-1967 as important residues within the A3 domain impacting the binding of fVIII light chain to cluster II. It should be pointed out, however, that calculation of the accessible surface area of Lys-1967 reveals that this residue is buried in native fVIII and therefore not available for binding to LRP1. In the C1 domain, Lys-2065 and Lys-2092 were identified to be important. Further studies are necessary to determine whether any of these lysine residues are indeed critical for intact fVIII binding to full-length LRP1. Although our ionic strength dependence data suggest the involvement of at least two lysine residues in the interaction, the study of van den Biggelaar et al. (31) did not identify any single pair of lysine residues that ablated binding when mutated, and the study concluded that fVIII interacts with LRP1 via an extended surface. This apparent discrepancy can be explained by the possibility that several lysine residues may compensate for one another. This also seems to be the case for the binding of D1D2 to LRP1 where we noted that although mutation of Lys-60 had a significant impact on binding of mutant D1D2 to LRP1 mutation of Lys-191 had a minimal impact on binding unless Lys-60 was also mutated (28).
The lysine residues potentially involved in the interaction of fVIII with LRP1 are shown in Fig. 8. The identification of two distinct regions located in fVIII that interact with LRP1 is in excellent agreement with the kinetic data derived from the current study. Interestingly, lysine residues within each domain of fVIII are spaced between 16 and 24 Å apart in excellent agreement with the 20–43-Å spacing of CR modules available from the limited structural information of various LDL receptor family members (27, 42, 43). Examination of Fig. 8 also reveals that certain lysine residues within the A3 domain are in close proximity and perhaps can compensate for binding as observed for the D1D2 domain of RAP (28).
FIGURE 8.

Structure of fVIII showing potential critical lysine residues for binding to LRP1. A, factor VIII domains are highlighted by different colors (A1 domain, yellow; A2 domain, green; A3 domain, blue; C1 domain, red; C2 domain, orange). Critical lysine residues potentially involved in LRP1 binding are colored in cyan. B, the distances between key lysine residues are shown. The figure was drawn with PyMOL from Protein Data Bank structure 3CDZ.
Our studies also identified cluster IV as the major LRP1 binding site for fVIII. Curiously, although fVIII bound to soluble forms of cluster II as well as cluster IV, only cluster IV was effective in competing for the binding of fVIII to full-length LRP1 and in preventing the LRP1-mediated cellular uptake of fVIII. In contrast, the LRP1-mediated cellular uptake of tPA·PAI-1 complexes, another LRP1 ligand, was inhibited by both cluster II and cluster IV. At this time, it is not clear why only cluster IV can block the binding and LRP1-mediated cellular uptake of fVIII, but we conclude from these results that the CRs in cluster II required for fVIII binding are not available in full-length LRP1. Possibly, this could result from an interaction of these repeats with a β-propeller domain also present on LRP1 that has been observed for the LDL receptor at low pH (42) or from the dimerization of LRP1, which is known to occur (44). These results stress the importance of examining the binding properties of full-length LRP1 and complementing these studies with cell-based experiments.
Although the role of LRP1 and other LDL receptor family members in the removal of fVIII from the circulation is well established (16, 17), it should be pointed out that numerous questions regarding the mechanisms of how this occurs remain that require further study. Interesting recent data have suggested the possibility that von Willebrand factor may also bind to LRP1 but only under conditions of shear stress (45, 46).
In summary, our studies suggest that it should be possible to ablate the binding of fVIII to LRP1 by selective mutation of a minimal number of lysine residues, although this may prove challenging due to the possibility of compensation from other residues. Our studies further reveal that cluster IV is the primary binding site on LRP1 that is responsible for the binding of fVIII. Combined, this information may lead to the development of an improved treatment for hemophilia A patients by developing an fVIII molecule with a longer half-life in the circulation. Further studies are necessary to determine the efficacy and feasibility of these strategies.
Experimental Procedures
Cell Lines, Proteins, Buffers, and Antibodies
Baby hamster kidney cells transfected with HSQ fVIII (47) as described previously (48) were generously provided by Pete Lollar (Emory University, Atlanta, GA) and used to express BDD-fVIII. The cells were maintained in DMEM/F-12 (Corning) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, and 100 μg/ml Geneticin. BDD-fVIII was purified as described (49) with the following modifications. Sulfopropyl Sepharose was equilibrated in 0.15 m NaCl, 20 mm HEPES, 5 mm CaCl2, 0.01% Tween 80, pH 7.4. After loading, the column was washed with the same buffer at 0.22 m NaCl. HSQ fVIII was eluted with a linear 0.22–0.65 m NaCl gradient in the same buffer. Fractions containing fVIII were pooled, added to a HiTrap Q HP column, and eluted with a linear 0.25–0.95 m NaCl gradient. WI38 cells, human lung fibroblast cells, were obtained from American Type Culture Collection (ATCC) and maintained in DMEM (Corning) supplemented with 10% FBS and penicillin/streptomycin. AIM V medium was purchased from Gibco. RAP was expressed and purified from bacteria as described previously (18). LRP1 was purified from placenta as described (50). Recombinant human LRP1 cluster II, III, and IV Fc chimera proteins were purchased from R&D Systems. Full-length factor VIII (Advate (antihemophilic factor (recombinant))) was generously provided by Dr. Andrey Sarafanov (United States Food and Drug Administration).
Lysine Modification of BDD-fVIII
BDD-fVIII was coated on 96-well plates at 5 μg/ml in 0.1 m NaHCO3, pH 9.6, overnight at 4 °C. Bound BDD-fVIII was modified by incubation with 10 mm EZ-Link sulfo-NHS-biotin (Thermo Fisher Scientific) for 2 h on ice. Successful modification was determined by incubation of BDD-fVIII-coated wells with sulfo-NHS-biotin followed by detection with streptavidin-alkaline phosphatase antibody diluted 1:2,000 (Gibco). Even coating of all wells was determined by incubation of coated wells with fVIII-specific antibody ESH8. All wells were blocked with assay buffer (20 mm HEPES, 0.15 m NaCl, 2 mm CaCl2, 0.1% Tween 80, and 1% BSA) for 1 h at 37 °C. Binding of LRP1 to BDD-fVIII or lysine-modified BDD-fVIII was performed by incubating different concentrations of purified LRP1 with the wells containing BDD-fVIII or lysine-modified BDD-fVIII overnight at 4 °C. After washing, LRP1 bound to BDD-fVIII was detected by 8G1 antibody followed by goat anti-mouse antibody conjugated to alkaline phosphatase at 1:3,000 dilution. Phosphatase substrate was diluted to 2 mg/ml in 0.1 m glycine, 1 mm MgCl2, and 1 mm ZnCl2, pH 10.4. The absorbance at 410 nm was measured, and the absorbance of blank wells containing only buffer was subtracted from the values.
Surface Plasmon Resonance
Binding of BDD-fVIII and FL-fVIII to LRP1 was measured using a BIAcore 3000 optical biosensor (GE Healthcare). LRP1 was amine-coupled to the chip to ∼9,000 resonance units, and ligand flowed over the surface at a rate of 20 μl/min in a buffer of 0.01 m HEPES, 150 mm NaCl, and 1 mm Ca2+ containing 0.005% Surfactant P. Ligand was dialyzed into the same buffer. Between sample runs, the chip was regenerated with 100 mm phosphoric acid. For ionic strength experiments, BDD-fVIII was dialyzed into buffer containing 10 mm HEPES, 1 mm CaCl2, 0.005% Surfactant P, and specific concentrations of NaCl.
Kinetic analysis of Surface Plasmon Resonance Data
The data were fit to Scheme 1 using numerical integration algorithms available in BIAevaluation software where A represents fVIII, B represents LRP1, AB represents complex I (Fig. 3A), and AB1 represents complex II (Fig. 3A). For numerical integration, the following equations were used.
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
| (Eq. 8) |
| (Eq. 9) |
To facilitate the fitting process, initial estimates for kd1 and kd2 were first obtained by fitting the dissociation data globally to a two-exponential decay model. The values obtained from these fits were then used to constrain kd1 and kd2 in the fit of the experimental data to Scheme 1. During this process, ka1 and ka2 were fit globally, whereas Rmax1 and Rmax2 were fit locally.
SCHEME 1.
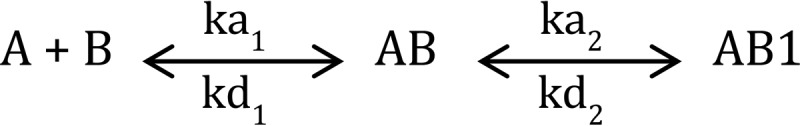
ELISA
96-well plates were coated with BDD-fVIII at 5 μg/ml in 0.1 m NaHCO3, pH 9.6, overnight at 4 °C. Even coating of wells was ensured by measuring the binding of ESH8 antibody to separate wells (data not shown). Wells were blocked with assay buffer (20 mm HEPES, 0.15 m NaCl, 2 mm CaCl2, 0.1% Tween 80, and 1% BSA) for 1 h at 37 °C. LRP1 cluster II, III, or IV diluted in assay buffer was added to the microtiter wells in duplicates at the indicated concentrations and incubated overnight at 4 °C. After washing, LRP1 clusters bound to BDD-fVIII were detected by human anti-Fc antibody conjugated to alkaline phosphatase at 1:1,000 dilution. Phosphatase substrate was diluted to 2 mg/ml in 0.1 m glycine, 1 mm MgCl2, and 1 mm ZnCl2, pH 10.4. Absorbance was measured at 410 nm. Binding to RAP controls was used to ensure quality of all LRP1 clusters. Binding of LRP1 clusters to BSA only-coated wells was also performed to detect background binding.
Competition Assays
Purified LRP1 or LRP1 cluster II/IV was coated between 3 and 5 μg/ml in TBS on 96-well plates overnight at 4 °C. Wells were blocked with assay buffer (20 mm HEPES, 0.15 m NaCl, 2 mm CaCl2, 0.1% Tween 80, and 1% BSA) for 1 h at 37 °C. Wells were washed three times with assay buffer. 100 μl of 20 nm BDD-fVIII in assay buffer was added to the wells in the absence or presence of competitor at the indicated concentrations. Binding of BDD-fVIII to LRP1 or LRP1 cluster II/IV was measured by 1:1,000 dilution of a mixture of anti-fVIII antibodies C4 and 413 and then detected by anti-mouse alkaline phosphatase-conjugated antibody. Phosphatase substrate was diluted to 2 mg/ml in 0.1 m glycine, 1 mm MgCl2, and 1 mm ZnCl2, pH 10.4, and readings were taken at 410 nm. The data are represented as percent bound with 100% determined by the amount of BDD-fVIII bound in the absence of competitor.
Cell-mediated Internalization Assays
BDD-fVIII was iodinated with iodogen as described (18). Cellular uptake assays were performed essentially as described (51). Briefly, WI38 cells were seeded into 12-well culture dishes (1 × 105 cells/well) and grown in DMEM supplemented with 10% FBS and 1× penicillin/streptomycin until cells reached ∼50% confluence. Cells were washed with PBS and incubated in serum-free DMEM containing 1 mm CaCl2, 20 mm HEPES, and 1.5% BSA for 1 h at 37 °C before the assay. Following washing, 500 μl of 125I-BDD-fVIII (20 nm) or 125I-tPA·PAI-1 (10 nm) was added to each well in the presence or absence of a competitor (cluster II or cluster IV). Following a 4-h incubation at 37 °C, cells were washed with PBS followed by 0.1 m glycine, pH 2.5, for 1 min. Cells were then washed with PBS and detached from the plate with trypsin with 50 mm EDTA and 50 μg/ml Proteinase K. Cells were spun down at 6,000 rpm for 3 min, and internalization was defined by radioactivity in the cell pellet. The cell numbers were counted in separate wells for normalization. The data are represented as percent internalized with 100% determined by the amount of 125I-BDD-fVIII internalized in the absence of a competitor.
Author Contributions
P. A. Y. and M. M. designed experiments, collected and analyzed data, and assisted in writing portions of the paper. D. K. S. designed experiments, assisted in analyzing data, and wrote the paper with P. A. Y. All authors approved the final version of the manuscript.
Acknowledgments
We thank Pete Lollar and Ernest Parker for providing baby hamster kidney cells transfected with HSQ fVIII and the purification protocols.
This work was supported in part by National Institutes of Health Grants HL114379 and HL120388 (to D. K. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- fVIII
- factor VIII
- LRP1
- low density lipoprotein receptor-related protein 1
- fVIIIa
- active fVIII
- RAP
- receptor-associated protein
- CR
- complement-type repeat
- D
- domain
- BDD
- B domain-deleted
- NHS
- N-hydroxysuccinimide
- FL
- full-length
- SPR
- surface plasmon resonance
- tPA
- tissue plasminogen activator
- PAI-1
- plasminogen activator inhibitor 1.
References
- 1. Saenko E. L., Ananyeva N. M., Tuddenham E. G., and Kemball-Cook G. (2002) Factor VIII—novel insights into form and function. Br. J. Haematol. 119, 323–331 [DOI] [PubMed] [Google Scholar]
- 2. Collins P. W., Blanchette V. S., Fischer K., Björkman S., Oh M., Fritsch S., Schroth P., Spotts G., Astermark J., Ewenstein B., and rAHF-PFM Study Group (2009) Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J. Thromb. Haemost. 7, 413–420 [DOI] [PubMed] [Google Scholar]
- 3. Fischer K., Pendu R., van Schooten C. J., van Dijk K., Denis C. V., van den Berg H. M., and Lenting P. J. (2009) Models for prediction of factor VIII half-life in severe haemophiliacs: distinct approaches for blood group O and non-O patients. PLoS One 4, e6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Everett L. A., Cleuren A. C., Khoriaty R. N., and Ginsburg D. (2014) Murine coagulation factor VIII is synthesized in endothelial cells. Blood 123, 3697–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fahs S. A., Hille M. T., Shi Q., Weiler H., and Montgomery R. R. (2014) A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood 123, 3706–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vlot A. J., Koppelman S. J., Meijers J. C., Dama C., van den Berg H. M., Bouma B. N., Sixma J. J., and Willems G. M. (1996) Kinetics of factor VIII-von Willebrand factor association. Blood 87, 1809–1816 [PubMed] [Google Scholar]
- 7. Lenting P. J., Neels J. G., van den Berg B. M., Clijsters P. P., Meijerman D. W., Pannekoek H., van Mourik J. A., Mertens K., and van Zonneveld A. J. (1999) The light chain of factor VIII comprises a binding site for low density lipoprotein receptor-related protein. J. Biol. Chem. 274, 23734–23739 [DOI] [PubMed] [Google Scholar]
- 8. Schwarz H. P., Lenting P. J., Binder B., Mihaly J., Denis C., Dorner F., and Turecek P. L. (2000) Involvement of low-density lipoprotein receptor-related protein (LRP) in the clearance of factor VIII in von Willebrand factor-deficient mice. Blood 95, 1703–1708 [PubMed] [Google Scholar]
- 9. Fay P. J., Haidaris P. J., and Smudzin T. M. (1991) Human factor VIIIa subunit structure. Reconstruction of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J. Biol. Chem. 266, 8957–8962 [PubMed] [Google Scholar]
- 10. Pipe S. W., Eickhorst A. N., McKinley S. H., Saenko E. L., and Kaufman R. J. (1999) Mild hemophilia A caused by increased rate of factor VIII A2 subunit dissociation: evidence for nonproteolytic inactivation of factor VIIIa in vivo. Blood 93, 176–183 [PubMed] [Google Scholar]
- 11. Saenko E. L., Yakhyaev A. V., Mikhailenko I., Strickland D. K., and Sarafanov A. G. (1999) Role of the low density lipoprotein-related protein receptor in mediation of factor VIII catabolism. J. Biol. Chem. 274, 37685–37692 [DOI] [PubMed] [Google Scholar]
- 12. Bovenschen N., van Stempvoort G., Voorberg J., Mertens K., and Meijer A. B. (2006) Proteolytic cleavage of factor VIII heavy chain is required to expose the binding-site for low-density lipoprotein receptor-related protein within the A2 domain. J. Thromb. Haemost. 4, 1487–1493 [DOI] [PubMed] [Google Scholar]
- 13. Bovenschen N., Boertjes R. C., van Stempvoort G., Voorberg J., Lenting P. J., Meijer A. B., and Mertens K. (2003) Low density lipoprotein receptor-related protein and factor IXa share structural requirements for binding to the A3 domain of coagulation factor VIII. J. Biol. Chem. 278, 9370–9377 [DOI] [PubMed] [Google Scholar]
- 14. Sarafanov A. G., Makogonenko E. M., Pechik I. V., Radtke K. P., Khrenov A. V., Ananyeva N. M., Strickland D. K., and Saenko E. L. (2006) Identification of coagulation factor VIII A2 domain residues forming the binding epitope for low-density lipoprotein receptor-related protein. Biochemistry 45, 1829–1840 [DOI] [PubMed] [Google Scholar]
- 15. Sarafanov A. G., Makogonenko E. M., Andersen O. M., Mikhailenko I. A., Ananyeva N. M., Khrenov A. V., Shima M., Strickland D. K., and Saenko E. L. (2007) Localization of the low-density lipoprotein receptor-related protein regions involved in binding to the A2 domain of coagulation factor VIII. Thromb. Haemost. 98, 1170–1181 [PubMed] [Google Scholar]
- 16. Bovenschen N., Mertens K., Hu L., Havekes L. M., and van Vlijmen B. J. (2005) LDL receptor cooperates with LDL receptor-related protein in regulating plasma levels of coagulation factor VIII in vivo. Blood 106, 906–912 [DOI] [PubMed] [Google Scholar]
- 17. Bovenschen N., Herz J., Grimbergen J. M., Lenting P. J., Havekes L. M., Mertens K., and van Vlijmen B. J. (2003) Elevated plasma factor VIII in a mouse model of low-density lipoprotein receptor-related protein deficiency. Blood 101, 3933–3939 [DOI] [PubMed] [Google Scholar]
- 18. Williams S. E., Ashcom J. D., Argraves W. S., and Strickland D. K. (1992) A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J. Biol. Chem. 267, 9035–9040 [PubMed] [Google Scholar]
- 19. Herz J., Goldstein J. L., Strickland D. K., Ho Y. K., and Brown M. S. (1991) 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J. Biol. Chem. 266, 21232–21238 [PubMed] [Google Scholar]
- 20. Strickland D. K., Ashcom J. D., Williams S., Battey F., Behre E., McTigue K., Battey J. F., and Argraves W. S. (1991) Primary structure of α2-macroglobulin receptor-associated protein. Human homologue of a Heymann nephritis antigen. J. Biol. Chem. 266, 13364–13369 [PubMed] [Google Scholar]
- 21. Willnow T. E., Rohlmann A., Horton J., Otani H., Braun J. R., Hammer R. E., and Herz J. (1996) RAP, a specialized chaperone, prevents ligand-induced ER retention and degradation of LDL receptor-related endocytic receptors. EMBO J. 15, 2632–2639 [PMC free article] [PubMed] [Google Scholar]
- 22. Willnow T. E., Armstrong S. A., Hammer R. E., and Herz J. (1995) Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 4537–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bu G., and Rennke S. (1996) Receptor-associated protein is a folding chaperone for low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 22218–22224 [DOI] [PubMed] [Google Scholar]
- 24. Bu G., Geuze H. J., Strous G. J., and Schwartz A. L. (1995) 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 14, 2269–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., and Strickland D. K. (2008) LDL Receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herz J., and Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Investig. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher C., Beglova N., and Blacklow S. C. (2006) Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol. Cell 22, 277–283 [DOI] [PubMed] [Google Scholar]
- 28. Prasad J. M., Young P. A., and Strickland D. K. (2016) High affinity binding of the receptor-associated protein D1D2 domains with the low density lipoprotein receptor-related protein (LRP1) involves bivalent complex formation: critical roles of lysines 60 and 191. J. Biol. Chem. 291, 18430–18439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Migliorini M. M., Behre E. H., Brew S., Ingham K. C., and Strickland D. K. (2003) Allosteric modulation of ligand binding to low density lipoprotein receptor-related protein by the receptor-associated protein requires critical lysine residues within its carboxyl-terminal domain. J. Biol. Chem. 278, 17986–17992 [DOI] [PubMed] [Google Scholar]
- 30. Dolmer K., Campos A., and Gettins P. G. (2013) Quantitative dissection of the binding contributions of ligand lysines of the receptor-associated protein (RAP) to the low density lipoprotein receptor-related protein (LRP1). J. Biol. Chem. 288, 24081–24090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van den Biggelaar M., Madsen J. J., Faber J. H., Zuurveld M. G., van der Zwaan C., Olsen O. H., Stennicke H. R., Mertens K., and Meijer A. B. (2015) Factor VIII interacts with the endocytic receptor low-density lipoprotein receptor-related protein 1 via an extended surface comprising “hot-spot” lysine residues. J. Biol. Chem. 290, 16463–16476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neels J. G., van Den Berg B. M., Lookene A., Olivecrona G., Pannekoek H., and van Zonneveld A. J. (1999) The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J. Biol. Chem. 274, 31305–31311 [DOI] [PubMed] [Google Scholar]
- 33. Meijer A. B., Rohlena J., van der Zwaan C., van Zonneveld A. J., Boertjes R. C., Lenting P. J., and Mertens K. (2007) Functional duplication of ligand-binding domains within low-density lipoprotein receptor-related protein for interaction with receptor associated protein, α2-macroglobulin, factor IXa and factor VIII. Biochim. Biophys. Acta 1774, 714–722 [DOI] [PubMed] [Google Scholar]
- 34. Kurasawa J. H., Shestopal S. A., Woodle S. A., Ovanesov M. V., Lee T. K., and Sarafanov A. G. (2015) Cluster III of low-density lipoprotein receptor-related protein 1 binds activated blood coagulation factor VIII. Biochemistry 54, 481–489 [DOI] [PubMed] [Google Scholar]
- 35. Lee D., Walsh J. D., Migliorini M., Yu P., Cai T., Schwieters C. D., Krueger S., Strickland D. K., and Wang Y.-X. (2007) The structure of receptor-associated protein (RAP). Protein Sci. 16, 1628–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jensen J. K., Dolmer K., Schar C., and Gettins P. G. (2009) Receptor-associated protein (RAP) has two high-affinity binding sites for the low-density lipoprotein receptor-related protein (LRP): consequences for the chaperone functions of RAP. Biochem. J. 421, 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willard L., Ranjan A., Zhang H., Monzavi H., Boyko R. F., Sykes B. D., and Wishart D. S. (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 31, 3316–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ngo J. C., Huang M., Roth D. A., Furie B. C., and Furie B. (2008) Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure 16, 597–606 [DOI] [PubMed] [Google Scholar]
- 39. Shen B. W., Spiegel P. C., Chang C. H., Huh J. W., Lee J. S., Kim J., Kim Y. H., and Stoddard B. L. (2008) The tertiary structure and domain organization of coagulation factor VIII. Blood 111, 1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svensson L. A., Thim L., Olsen O. H., and Nicolaisen E. M. (2013) Evaluation of the metal binding sites in a recombinant coagulation factor VIII identifies two sites with unique metal binding properties. Biol. Chem. 394, 761–765 [DOI] [PubMed] [Google Scholar]
- 41. Bloem E., van den Biggelaar M., Wroblewska A., Voorberg J., Faber J. H., Kjalke M., Stennicke H. R., Mertens K., and Meijer A. B. (2013) Factor VIII C1 domain spikes 2092-2093 and 2158-2159 comprise regions that modulate cofactor function and cellular uptake. J. Biol. Chem. 288, 29670–29679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rudenko G., Henry L., Henderson K., Ichtchenko K., Brown M. S., Goldstein J. L., and Deisenhofer J. (2002) Structure of the LDL receptor extracellular domain at endosomal pH. Science 298, 2353–2358 [DOI] [PubMed] [Google Scholar]
- 43. Jensen G. A., Andersen O. M., Bonvin A. M., Bjerrum-Bohr I., Etzerodt M., Thøgersen H. C., O'Shea C., Poulsen F. M., and Kragelund B. B. (2006) Binding site structure of one LRP-RAP complex: implications for a common ligand-receptor binding motif. J. Mol. Biol. 362, 700–716 [DOI] [PubMed] [Google Scholar]
- 44. Makarova A., Bercury K. K., Adams K. W., Joyner D., Deng M., Spoelgen R., Koker M., Strickland D. K., and Hyman B. T. (2008) The LDL receptor-related protein can form homo-dimers in neuronal cells. Neurosci. Lett. 442, 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rastegarlari G., Pegon J. N., Casari C., Odouard S., Navarrete A. M., Saint-Lu N., van Vlijmen B. J., Legendre P., Christophe O. D., Denis C. V., and Lenting P. J. (2012) Macrophage LRP1 contributes to the clearance of von Willebrand factor. Blood 119, 2126–2134 [DOI] [PubMed] [Google Scholar]
- 46. Wohner N., Legendre P., Casari C., Christophe O. D., Lenting P. J., and Denis C. V. (2015) Shear stress-independent binding of von Willebrand factor-type 2B mutants p.R1306Q & p.V1316M to LRP1 explains their increased clearance. J Thromb. Haemost. 13, 815–820 [DOI] [PubMed] [Google Scholar]
- 47. Lind P., Larsson K., Spira J., Sydow-Bäckman M., Almstedt A., Gray E., and Sandberg H. (1995) Novel forms of B-domain-deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur. J. Biochem. 232, 19–27 [DOI] [PubMed] [Google Scholar]
- 48. Healey J. F., Lubin I. M., Nakai H., Saenko E. L., Hoyer L. W., Scandella D., and Lollar P. (1995) Residues 484–508 contain a major determinant of the inhibitory epitope in the A2 domain of human factor VIII. J. Biol. Chem. 270, 14505–14509 [DOI] [PubMed] [Google Scholar]
- 49. Doering C. B., Healey J. F., Parker E. T., Barrow R. T., and Lollar P. (2002) High level expression of recombinant porcine coagulation factor VIII. J. Biol. Chem. 277, 38345–38349 [DOI] [PubMed] [Google Scholar]
- 50. Ashcom J. D., Tiller S. E., Dickerson K., Cravens J. L., Argraves W. S., and Strickland D. K. (1990) The human α2-macroglobulin receptor: identification of a 420-kD cell surface glycoprotein specific for the activated conformation of α2-macroglobulin. J. Cell Biol. 110, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prasad J. M., Migliorini M., Galisteo R., and Strickland D. K. (2015) Generation of a potent low density lipoprotein receptor-related protein 1 (LRP1) antagonist by engineering a stable form of the receptor-associated protein (RAP) D3 domain. J. Biol. Chem. 290, 17262–17268 [DOI] [PMC free article] [PubMed] [Google Scholar]