

Abstract
The cell wall of most Gram-positive bacteria contains equal amounts of peptidoglycan and the phosphate-rich glycopolymer wall teichoic acid (WTA). During phosphate-limited growth of the Gram-positive model organism Bacillus subtilis 168, WTA is lost from the cell wall in a response mediated by the PhoPR two-component system, which regulates genes involved in phosphate conservation and acquisition. It has been thought that WTA provides a phosphate source to sustain growth during starvation conditions; however, WTA degradative pathways have not been described for this or any condition of bacterial growth. Here, we uncover roles for the Bacillus subtilis PhoP regulon genes glpQ and phoD as encoding secreted phosphodiesterases that function in WTA metabolism during phosphate starvation. Unlike the parent 168 strain, ΔglpQ or ΔphoD mutants retained WTA and ceased growth upon phosphate limitation. Characterization of GlpQ and PhoD enzymatic activities, in addition to X-ray crystal structures of GlpQ, revealed distinct mechanisms of WTA depolymerization for the two enzymes; GlpQ catalyzes exolytic cleavage of individual monomer units, and PhoD catalyzes endo-hydrolysis at nonspecific sites throughout the polymer. The combination of these activities appears requisite for the utilization of WTA as a phosphate reserve. Phenotypic characterization of the ΔglpQ and ΔphoD mutants revealed altered cell morphologies and effects on autolytic activity and antibiotic susceptibilities that, unexpectedly, also occurred in phosphate-replete conditions. Our findings offer novel insight into the B. subtilis phosphate starvation response and implicate WTA hydrolase activity as a determinant of functional properties of the Gram-positive cell envelope.
Keywords: Bacillus, enzyme kinetics, enzyme mechanism, gram-positive bacteria, hydrolase, glpQ, phoD, phosphate starvation, wall teichoic acid
Introduction
A key distinguishing feature of the Gram-positive cell envelope is the presence of wall teichoic acids (WTAs)3 covalently attached to peptidoglycan (PG) in the cell wall. These anionic polymers typically feature repeating polyol-phosphate units and extend from PG to beyond the cell surface. WTAs are important for multiple cellular processes, including PG synthesis, morphogenesis, and the regulation of autolysins (1). WTA is also critical for the expression of virulence (2) and drug resistance in Staphylococcus aureus (3), spurring recent interest in the development of WTA biosynthesis inhibitors as lead antimicrobial compounds (3, 4).
Biosynthetic pathways for canonical poly(glycerol-phosphate) WTA in Bacillus subtilis 168 and poly(ribitol-phosphate) WTA in S. aureus have been intensively studied. In B. subtilis 168, synthesis is accomplished by sequential actions of the tag gene products (TagO, TagA, TagB, and TagF), producing WTA of 30–50 glycerol phosphate (GroP) units, that is then modified with α-glucose (TagE) and d-alanine and ultimately transferred (TagT, TagU, and TagV are implicated) to the 6′-hydroxyl on muramic acid in the glycan strands of PG (1). WTAs contribute roughly half the cell wall mass, including the entire wall phosphate content, yet are dispensable for viability in B. subtilis or S. aureus, although cells lacking WTA synthesis are phenotypically typified by aberrant growth and morphology (5, 6).
Throughout normal growth, bacteria turn over up to half the cell wall material per generation (7). Gram-negative organisms such as Escherichia coli use complex pathways to recover and recycle these turnover products, and although it is uncertain whether the same occurs in Gram-positive bacteria (8), it is perhaps implicit that WTA degradation occurs along with PG-lytic activities. Little, however, is known about this process. Early studies in B. subtilis and S. aureus found virtually identical turnover rates for WTA and PG (9), but it was likely that this WTA was in polymeric form still attached to PG fragments produced by autolysis. Direct evidence of WTA turnover was recorded over 3 decades ago with the discovery and partial characterization of so-called teichoicase activity in cell extracts from sporulating B. subtilis (10, 11), but the responsible enzymes and corresponding genes remain unidentified. More recently, B. subtilis impaired in the transfer of WTA to PG, contrary to S. aureus likewise impaired (12), was shown not to release WTA into the extracellular medium or accumulate WTA intermediates in the cell envelope (13), possible signs of WTA turnover and recovery. Nevertheless, the only enzymes shown to catalyze WTA degradation are of bacteriophage origin, for example the tail-spike protein GP12 from B. subtilis bacteriophage φ.29 (14, 15).
Although the relevance of WTA turnover during nutrient-sufficient bacterial growth is unknown, WTA turnover and metabolism are thought to be important aspects of the phosphate starvation response in B. subtilis (16). Following exhaustion of inorganic phosphate (Pi) from growth media, WTA is replaced with teichuronic acid, an alternative anionic polymer devoid of phosphate (17). The switch is transcriptionally regulated by the PhoPR two-component system, which responds to phosphate stress by repressing expression from WTA biosynthetic operons, while activating expression of the tuaABCEDFGH operon for teichuronic acid biosynthesis (18). Expression is also induced from genes annotated to function in phosphate scavenging, including genes encoding PhoA and PhoB that account for the majority of secreted alkaline phosphatase (APase) activity during phosphate limitation (19), the pst operon encoding a Pi transporter complex (20), and phosphodiesterase genes glpQ and phoD (21). That GlpQ and PhoD are also secreted proteins (22) has prompted notions of possible functions in WTA degradation, but this has not been examined to date.
The catalytic activity of PhoD has been studied to some degree (23, 24). In contrast, B. subtilis GlpQ has been poorly characterized; however, studies on homologous proteins such as E. coli GlpQ and UgpQ (25, 26) and protein D from Haemophilus influenzae (27) allude to possible catalytic activity and biological function. These enzymes demonstrate a strict requirement for glycerophosphodiester substrates, and there is evidence of roles for E. coli GlpQ in the utilization of GroP as a carbon source and UgpQ in phosphate acquisition, with deacylated glycerophospholipids as the presumed biological substrates in either case (26).
Here, we investigate roles for glpQ and phoD in WTA metabolism during phosphate starvation of B. subtilis 168. Deletion of either gene suppressed post-exponential growth after depletion of Pi from culture media, and the phosphate content in the cell wall of these mutants was significantly higher than in the parent strain. We confirmed teichoicase activity by GlpQ and PhoD in vitro using WTA isolated from B. subtilis 168, as well as synthetically derived analogs of WTA intermediates and WTA fragments. Both enzymes showed a preference for undecorated WTA, but catalyzed WTA depolymerization in distinct manners. Processivity studies, along with structural data, provide insight into the divergent catalytic activities. Finally, we present evidence of an involvement of glpQ and phoD, and thus WTA turnover, in cell envelope integrity. Our findings define functional roles for WTA degradation and recycling during the B. subtilis phosphate starvation response and offer novel insight on the relationship of WTA turnover to cell wall properties.
Results
glpQ and phoD Are Involved in WTA Metabolism during Phosphate Starvation of B. subtilis 168
Cultures of B. subtilis strains disrupted in the phoPR locus become static upon depletion of Pi from growth media (28). With glpQ and phoD among the most highly activated PhoP regulon genes (29), we sought to determine their involvement in the B. subtilis 168 phosphate starvation response, first by examining growth of single gene deletion mutants and a double deletion mutant, under phosphate-limited conditions. We were careful to ensure at the start of the experiment cells were not in a phosphate-limited state by using inocula from phosphate-replete cultures undergoing exponential growth, when phosphate is non-limiting. Unlike slow post-exponential growth of the wild-type strain following phosphate depletion, cultures of the mutant strains remained static (Fig. 1A), remarkably mirroring the effect from complete absence of PhoPR activity. Phosphate-limited growth of the deletion mutants was not stimulated by exogenous B. subtilis WTA, whereas that of wild type was (Fig. 1B), and mutant strains underwent quicker transition from vegetative growth to sporulation (Fig. 1C). Growth in phosphate-depleted cultures of the mutants resumed upon addition of KH2PO4 or GroP (Fig. 1D), indicating that phosphate starvation-induced APase activity and Pi transport are intact in these mutants. After addition of GroP, the amplitude of growth for the ΔphoD strain appeared higher than with wild type; however, the growth rates for these strains were largely similar. Notably, corresponding cultures for the ΔglpQ and double deletion mutants were markedly reduced both in the amplitude and rate of growth, indicating that recovery from phosphate starvation in the presence of GroP was attenuated in these strains compared with ΔphoD or wild-type cells. In addition, all strains exhibited similar phosphate-replete growth, verifying that the deletions did not hamper growth in the absence of phosphate stress (Fig. 1, B and D). Together, these findings support roles for glpQ and phoD in phosphate acquisition from WTA.
FIGURE 1.

glpQ and phoD are important for phosphate-limited growth of B. subtilis 168. A, phosphate-limited growth of B. subtilis strains. Plots show the mean absorbance at 600 nm, for 50-ml cultures, in phosphate-limited media. Data from three independent experiments are shown. B, growth kinetics in phosphate-replete media, phosphate-deplete media, and phosphate-deplete media supplemented with 0.5 mg/ml B. subtilis 168 WTA. Growth was carried out in 200 μl in a 96-well microplate at 37 °C. Post-exponential phosphate-starved growth in the wild-type strain is not visible under these conditions (middle panel) but is stimulated by exogenous WTA (right panel). Plots show the mean absorbance at 600 nm from two independent experiments. C, sporulation of B. subtilis strains. Sporulation of phosphate-starved cultures for each strain was induced in Schaffer's sporulation media according to established methods (65). Mean and S.E. from three independent experiments are shown. D, supplementation with KH2PO4 or GroP rescues phosphate-starved growth of B. subtilis strains. Growth was carried out in as in B until the exhaustion of Pi, and KH2PO4 or GroP was added to a final concentration of 2.5 mm. Plots show the mean absorbance at 600 nm from two independent experiments. A, B, and D, ●, WT; red square, ΔphoD; blue triangle, ΔglpQ; inverted pink triangle, ΔglpQΔphoD.
We hypothesized that an inability to turn over WTA during phosphate starvation would lead to an increase in the cell wall phosphate content, and measured cell wall phosphate in wild-type and mutant strains after phosphate-replete and phosphate-deplete growth. Consistent with previous reports (17, 30), wall phosphate decreased significantly (∼62%) in the wild-type strain after phosphate starvation (Fig. 2, A and B). Nevertheless, wall phosphate in the ΔphoD mutant and the double mutant was unchanged, whereas that of the ΔglpQ strain decreased by ∼ 36% (Fig. 2, A and B). Notably, the ΔglpQ mutant and the double mutant had higher wall phosphate than the ΔphoD mutant or the wild-type strain, after phosphate-replete growth (Fig. 2A). Overall, these results showed that deletion mutants retained significant amounts of WTA after phosphate starvation. Moreover, phosphate-starved deletion mutants contained comparable amounts of cell wall uronic acid as the wild-type strain (Fig. 2C) and exhibited similar secreted APase activity (Fig. 2D), both indicative of functional PhoPR activity. We conclude from these findings that glpQ and phoD play a major role in the loss of WTA in phosphate-starved B. subtilis that is separate from genetic repression of WTA synthesis.
FIGURE 2.

Loss of WTA during phosphate limitation of B. subtilis is dependent on glpQ and phoD. Wall phosphate content was normalized for absorbance after phosphate-replete (A) and phosphate-limited (B) growth. Means and S.E. from at least two independent measurements are shown. Differences between groups were assessed for significance by one-way analysis of variance. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, not significant. C, presence of uronic acid in the cell wall of phosphate-starved B. subtilis strains. Cell wall was isolated and digested as described under “Experimental Procedures.” Uronic acid was detected by the absorbance at 520 nm, after treatment of cell wall digest with concentrated H2SO4 and Na2B4O7 at 100 °C, followed by 3-phenylphenol as described previously (66). Data show the mean values, normalized for culture absorbance, from three independent experiments. Error bars are the S.E. D, APase activity in the supernatant of phosphate-limited cultures. Supernatant (25 ml) from phosphate-limited cultures of B. subtilis strains was filtered (0.22 μm) and then concentrated with a centrifugal filter (3-kDa molecular mass cutoff). The buffer was exchanged for 50 mm Tris-HCl (pH 8.0) containing 1 mm CaCl2 and 1 mm MgCl2, and the sample was further concentrated 10-fold. Total protein concentrations, as determined by the Bradford assay, were as follows: WT, 0.33 mg/ml; ΔphoD, 0.19 mg/ml; ΔglpQ, 0.08 mg/ml; ΔglpQΔphoD, 0.26 mg/ml. Assays were performed at room temperature, in a 96-well microplate, by mixing 10 μl of concentrated supernatant protein with 90 μl of a mixture comprising 50 mm Tris-HCl (pH 8.0), 1 mm MgCl2, 1 mm CaCl2, and 20 mm pNPP. Liberated pNP was detected continuously by monitoring absorbance at 410 nm. Data from three independent experiments are shown. Error bars, where visible, are the S.E.
GlpQ and PhoD Catalyze Degradation of WTA
The above results strongly suggested that glpQ and phoD encode WTA-active proteins. To test for teichoicase activity, we incubated purified recombinant GlpQ and PhoD with WTA isolated from B. subtilis 168 and examined reaction products by PAGE. Fig. 3A clearly shows PhoD was active on wild-type (glycosylated) WTA, while there appeared to be a slight decrease in apparent median polymer length after incubation with GlpQ, based on densitometric analysis. GlpQ and PhoD were next tested for activity on non-glycosylated WTA isolated from a ΔtagE mutant of B. subtilis 168 (31), and a substantial disappearance of polymeric material was observed in the presence of either enzyme (Fig. 3A), suggesting that modification by glucose reduces the susceptibility of WTA to degradation.
FIGURE 3.

GlpQ and PhoD degrade WTA. A, representative PAGE gels of WTA (0.4 mg/ml) after incubation with 1 μm GlpQ (lanes 2, 3, 7, and 8) or 0.25 μm PhoD (lanes 4, 5, 9, and 10). Reactions were quenched with EDTA after 60 min (lanes 2, 4, 7, and 9) or 120 min (lanes 3, 5, 8, and 10). Lanes 1 and 6, no enzyme controls. Densitometric scans plotting pixel density over distance migrated are shown below the gels; median polymer size of glycosylated WTA is indicated by the red vertical line. B, progress of GlpQ and PhoD reactions with WTA (∼ 220 μm Pi/0.2 mg/ml), based on Pi output. APase was added to GlpQ reactions 2 min prior to quenching. Pi generated in PhoD reactions was not affected by APase (data not shown), and data show the results of PhoD assays performed without added APase. Enzyme concentrations in reactions with glycosylated WTA are as follows: 0.031 μm (●); 0.063 μm (○); 0.13 μm (■); 0.25 μm (□); 0.5 μm (▴); and 1.0 μm (▵). Enzyme concentrations in reactions with non-glycosylated WTA are as follows: 0.0026 μm (●); 0.0052 μm (○); 0.010 μm (■); 0.021 μm (□); 0.042 μm (▴); and 0.083 μm (▵). Control reactions received buffer instead of enzyme, as well as APase in the case of GlpQ reactions. Plots show the mean from two independent experiments. Apparent turnover was estimated as the slope from linear regression of initial rate as a function of enzyme concentration. C, GlpQ and PhoD are active on WTA attached to PG. Cell wall from B. subtilis 168 and the ΔtagE mutant was isolated and purified as described under “Experimental Procedures” and then resuspended in ddH2O to an absorbance at 600 nm of ∼1.0. Reactions (50 μl) were performed at room temperature in 50 mm Tris-HCl (pH 8.0) containing 1 mm CaCl2, 1 mm MgCl2, 10 μl of cell wall (resulting in a final Pi concentration of ∼50 μm), and 1 μm GlpQ or PhoD. Reactions were quenched with EDTA at the time points indicated (GlpQ reactions received 0.5 units of APase 2 min prior to quenching), and the insoluble PG material was removed by centrifugation (21,000 × g) before Pi detection and quantification. Data from two independent experiments are shown. ●, PhoD; ○, GlpQ.
If GlpQ and PhoD catalyzed phosphodiester bond cleavage in WTA, Pi could be released from phosphomonoester products by the enzymes themselves or with exogenous APase. Thus, we assayed WTA degradation reactions for Pi production. Although Pi output from PhoD reactions increased proportionately over time and with increasing enzyme concentration (Fig. 3B), Pi was not detected above background levels in parallel reactions with GlpQ (data not shown). Indeed, dependence of Pi production on time and enzyme concentration in GlpQ reactions was evident only on addition of APase (Fig. 3B). Apparent turnover (phosphodiester bonds cleaved per unit of time) in these reaction conditions indicated a strong preference for non-glycosylated WTA by either enzyme, with faster kinetics for GlpQ (2.4 ± 0.1 and 1.9 ± 0.01 min−1 with glycosylated WTA; 110 ± 9.3 and 52.7 ± 0.9 min−1 with non-glycosylated WTA, for GlpQ and PhoD, respectively). The latter seemed to contradict PAGE findings, but it should be noted that polymeric species less than ∼20 units are not well detected by PAGE (32). Also, turnover was likely underestimated as it was not feasible to assess degradation kinetics with higher WTA concentrations due to increased background interference, particularly with non-glycosylated WTA. Nonetheless, we infer from these data that PhoD catalyzes phosphodiester cleavage of WTA and acts on the phosphomonoester products generated, whereas GlpQ strictly catalyzes phosphodiesterase activity. Pi was also released on incubating of GlpQ or PhoD with B. subtilis cell wall, demonstrating activity on WTA covalently linked to PG (Fig. 3C).
We endeavored to complement the phosphate starvation growth phenotype in mutant strains by introducing a copy of glpQ or phoD at the amyE locus. To do this, we used a xylose-inducible promoter (33), but these attempts were frustrated by leakiness in the induction system that seemed to mask the effect of complementing genes during phosphate-limited growth. More compelling results were achieved with experiments that showed exogenous WTA stimulated phosphate-starved growth in complemented strains (supplemental Fig. S1). Therefore, for further proof of the involvement of GlpQ and PhoD in WTA metabolism, we asked whether phosphate-starved growth in the mutants could be rescued by a partial hydrolysate produced by simultaneous treatment of WTA with both enzymes. As shown in Fig. 4, the extent of growth stimulation between mutants was comparable but was less than that in the parent strain, indicating that the mutants were unable to utilize polymeric WTA in the hydrolysate, and growth was stimulated only by the WTA degradation products generated in vitro. Collectively, these results suggest that the activities catalyzed by GlpQ and PhoD enable the utilization of WTA as a phosphate reserve under limiting conditions.
FIGURE 4.

Supplementation of phosphate-limited growth with teichoicase-treated WTA. B. subtilis 168 WTA was incubated with PhoD (0.5 μm) and GlpQ (1 μm) in 50 mm Tris-HCl (pH 8.0) containing 1 mm CaCl2, 1 mm MgCl2 for 2 h. Enzymes were removed using a centrifugal filter (10-kDa molecular mass cutoff). 10 μl of buffer (A) or collected filtrate (WTA partial hydrolysate) (B) were added to 190 μl of culture, prepared in phosphate-limited media, in individual wells of a 96-well microplate. Plots show the mean absorbance at 600 nm from three independent experiments. ●, WT; red square, ΔphoD; blue triangle, ΔglpQ; inverted pink triangle, ΔglpQΔphoD.
GlpQ and PhoD Catalyze Distinct Modes of WTA Degradation
Recognizing that the heterogeneity of WTA isolated from cells hindered enzyme kinetic studies and characterization of degradation products, chemically defined synthetic analogs of WTA intermediates were employed as substrates. We prepared tridecane-containing analogs of the TagF biosynthetic product (lipid φ.40 analog) with 14C incorporated into the GroP polymer, and monitored product formation after reactions with GlpQ or PhoD using high performance anion exchange chromatography (HPAEX) (15). GlpQ reaction products included GroP and species similar in apparent size to the starting substrate (Fig. 5A), consistent with exo-degradation that releases monomer units from the polymer terminus. The absence of Gro corroborated exclusive phosphodiesterase activity by this enzyme. Conversely, turnover of the lipid φ.40 analog by PhoD was accompanied by the generation of multiple products varying in size (Fig. 5B), suggesting endo-degradation. Weak signal intensity for individual product species was attributed to the dispersal of radioactivity originating in the starting substrate across numerous products, which were more clearly visualized using higher PhoD concentrations (supplemental Fig. S2). It was also evident that many PhoD-generated products would be undetectable by PAGE based on apparent sizes less than 20 polymer units (15).
FIGURE 5.

Degradation of lipid φ.40 analog by GlpQ and PhoD. HPAEX chromatograms of radioactive products from reactions of 14C-lipid φ.40 analog (30 μm, ∼1.2 mm Pi) with 0.04 μm GlpQ (A) or PhoD (B). Experiments were performed at least twice, with similar results. C, dependence of the initial rate of GroP production on GlpQ concentration. Initial rates were determined from GroP produced in 5 min. Turnover, estimated as the slope from linear regression, was 881 ± 25.6 min−1. D, dependence of the initial rate of 14C-lipid φ.40 analog degradation on PhoD concentration. Initial rates were determined from the amount of lipid φ.40 analog converted in 5 min. Estimated turnover was 62.4 ± 1.8 min−1. C and D, means and S.E. from three independent experiments are shown.
We took advantage of robust product characterization afforded from the use of the lipid φ.40 analog to investigate enzyme turnover using a 6-fold higher starting concentration of phosphodiesters than in reactions with B. subtilis WTA. Under these conditions GlpQ turnover was 8-fold faster (881 ± 25.6 min−1; Fig. 5C) than was apparent with the biological substrate, whereas PhoD turnover increased slightly (62.4 ± 1.8 min−1; Fig. 5D). It is worth noting that GlpQ turnover was based on GroP production, whereas PhoD turnover reflected the disappearance of starting material; thus, we have greater confidence in the rates for the former. In that context, the rate of GlpQ activity was linear with enzyme concentration over the range tested, although it was non-linear for PhoD. Notably, neither enzyme produced measurable activity with the glycosylated lipid φ.40 analog in the same reaction conditions (data not shown).
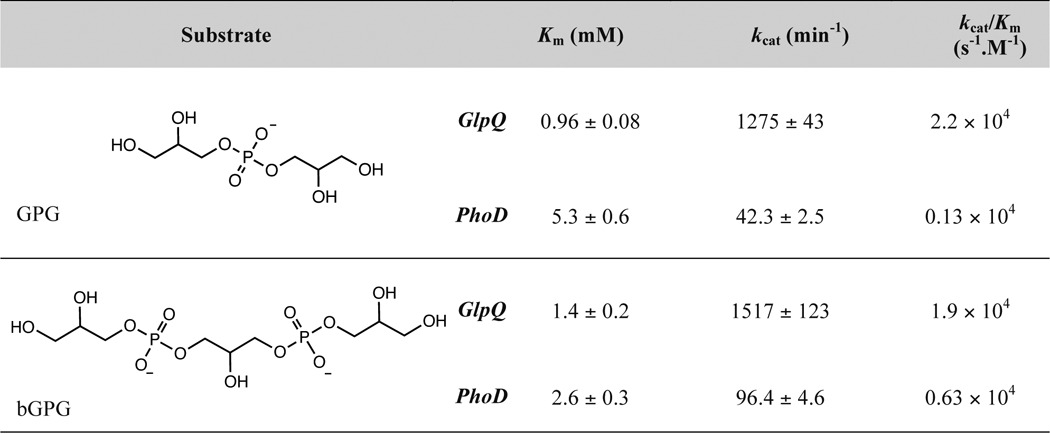
To directly compare catalytic activities of GlpQ and PhoD, we measured enzyme kinetic parameters on simple phosphate ester-containing substrates. As observed previously (24), PhoD was active on pNPP, bis-p-nitrophenyl phosphate, pNPC, and GroP (Table 1). GlpQ lacked measurable activity on these compounds, and thus catalytic activity was assessed on the WTA oligomer mimics glycerophosphoglycerol (GPG) and bis-glycerophosphoglycerol (bGPG). Compared with PhoD, GlpQ catalyzed faster turnover, with greater specificity for both substrates, with 30- and 16-fold greater kcat and kcat/Km values for GPG, and 16- and 3-fold greater kcat and kcat/Km values for bGPG (Table 2). Altogether, the findings portray PhoD as a promiscuous phosphodiesterase/phosphomonoesterase and GlpQ as highly selective for the poly(GroP) WTA backbone.
TABLE 1.
Kinetic parameters for hydrolysis of p-nitrophenyl phosphates and glycerol 3-phosphate by PhoD
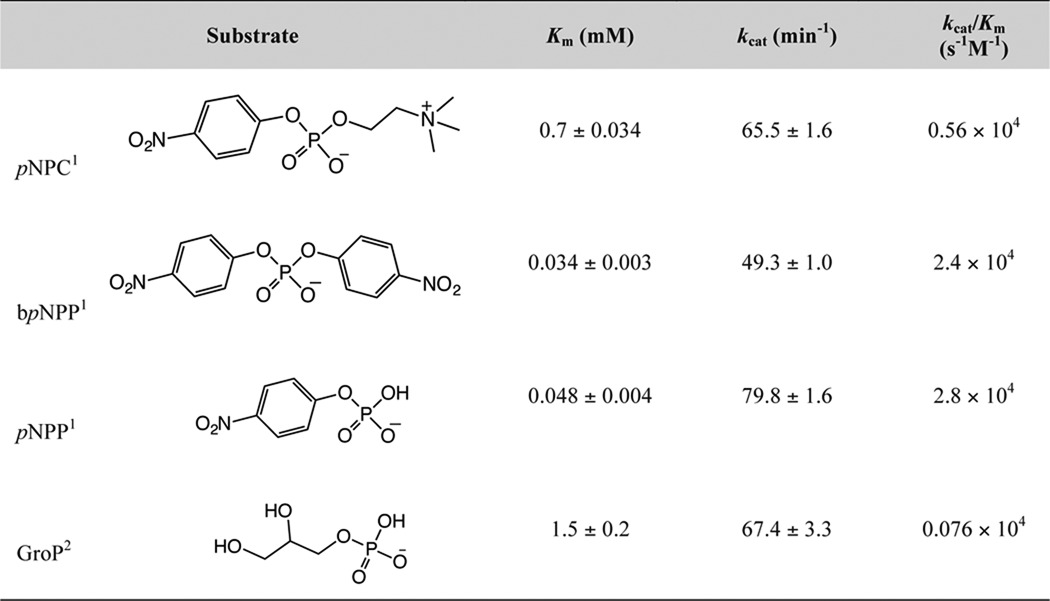
1 Assays were performed at room temperature in 50 mm Tris-HCl (pH 8.0) containing 1 mm MgCl2 and 1 mm CaCl2. Activity was measured by continuous detection of liberated pNP at 410 nm. pNP was quantified by interpolation to a standard curve. For determination of enzyme kinetic parameters, initial rates of pNP production were plotted as a function of substrate concentration and the data fit by non-linear regression (Prisim 6.0, GraphPad) to equation 1. Concentrations of pNPP, bpNPP, (bis-p-notrophenyl phosphate), and pNPC were between 0.0012 mm − 1.2 mm.
2 Reactions were performed as above, but activity assayed based on quantification of liberated Pi. GroP concentrations were 0.31 − 10 mm.
TABLE 2.
Kinetic parameters for hydrolysis of WTA oligomers by GlpQ and PhoD
Assays were performed as described in the “Experimental Procedures.” GPG and bGPG concentrations ranged from 0.039 − 2.5 mm for GlpQ reactions and 0.16 − 10 mm in PhoD reactions.
Processivity of Exolytic Degradation by GlpQ
Although the product distribution resulting from the action of GlpQ on lipid φ.40 analog was consistent with processive polymer degradation, a distributive mechanism could not be excluded. To resolve this ambiguity, we performed a substrate trapping experiment where degradation was initiated on the 14C-lipid φ.40 analog, and then an excess amount of unlabeled competing substrate was added to the reaction to trap dissociated enzyme. For the latter, we used a poly(GroP) polymer prepared with CDP-Gro as the acceptor, denoted CMP-poly(GroP) (15). As shown in Fig. 6A, turnover of the 14C-lipid φ.40 analog ceased upon addition of the trapping substrate, indicating that GlpQ has a distributive mechanism, dissociating from the polymer between catalytic events.
FIGURE 6.

Processivity and structure of GlpQ. A, reactions containing equimolar concentrations (2.5 μm) of GlpQ and 14C-lipid φ.40 analog were quenched after 5 min with urea. HPAEX chromatograms show radioactive products for the following: (i) no enzyme control; (ii) reaction in the absence of CMP-poly(GroP); (iii) reaction with 2.5 mm CMP-poly(GroP) added after 1 min; (iv) 2 min; and (v) reaction with buffer added after 2 min. B, crystal structure of GlpQ·Bicine. TIM barrel and GDPD-I domains are in gray and orange, respectively. Bicine is shown as yellow sticks and Ca2+ as a green sphere. Heteroatoms are colored by element (N, blue; O, red; P, orange). C, bound ligands in structures of GlpQ·Bicine, GlpQ·G3P-1 and GlpQ·G3P-2 are shown with the 2Fo − Fc electron density map contoured to 2σ in a gray mesh. Carbon atoms are in yellow, and heteroatoms are colored as in B. D, active sites of GlpQ·Bicine. GlpQ·G3P-1 (E) and GlpQ·G3P-2 (F) are shown with bound ligand, Ca2+, and selected residues. Metal-binding sites of complexes are shown adjacent, right. Carbon atoms are colored in cyan, and heteroatoms are colored by element. Ligands are shown as yellow sticks and Ca2+ as green spheres. GDPD-I and loop 11 are orange and magenta, respectively. G, electrostatic potential surface of GlpQ-GPG-2 and corresponding ribbon structure, colored as in D.
Structure of GlpQ
To gain molecular insight into GlpQ catalysis, we solved crystal structures of the enzyme in the presence and absence of GroP and refined the models to 1.48–1.62 Å resolution (Table 3). A Bicine molecule, supplied by the crystallization condition, bound GlpQ in the absence of GroP, and soaking of GlpQ·Bicine crystals with GPG yielded complexes GlpQ·G3P-1 from a 5-min soak and GlpQ·G3P-2 from a 1-h soak. Crystallized GlpQ hydrolyzed GPG, and thus our models represent the catalytically active enzyme.
TABLE 3.
X-ray data collection and refinement statistics
| GlpQ·Bicine (5T91) | GlpQ·G3P-1 (5T9B) | GlpQ·G3P-2 (5T9C) | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 50.42, 60.04, 88.25 | 51.46, 60.23, 88.42 | 47.10, 58.24, 87.57 |
| a, b, c (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å)a | 44.12–1.53 (1.59–1.53) | 43.83–1.62 (1.68–1.62) | 48.49–1.48 (1.53–1.48) |
| CC½ | 0.999 (0.904) | 0.999 (0.854) | 0.999 (0.698) |
| Rpim | 0.017 (0.417) | 0.023 (0.607) | 0.025 (0.449) |
| Rmeas | 0.0457 (1.085) | 0.0624 (1.580) | 0.0549 (0.916) |
| I/σI | 21.6 (1.7) | 19.4 (1.6) | 15.8 (1.8) |
| Completeness (%) | 99.61 (96.43) | 99.92 (99.91) | 99.67 (98.61) |
| Redundancy | 7.2 (6.6) | 6.7 (6.7) | 4.7 (3.8) |
| Refinement | |||
| Resolution (Å) | 44.12–1.53 (1.59–1.53) | 43.83–1.62 (1.68–1.62) | 48.49–1.48 (1.53–1.48) |
| No. of reflections | 40,983 (3889) | 34,957 (3441) | 40,774 (3981) |
| Rwork/Rfreeb | 0.175/0.211 | 0.176/0.216 | 0.168/0.197 |
| No. of atoms | |||
| Protein | 2062 | 2071 | 2095 |
| Ligand/ion | 13 | 12 | 17 |
| Water | 238 | 193 | 165 |
| B factors (Å2)c | |||
| Protein | 39.4 | 40.6 | 29.0 |
| Ligand/ion | 29.2 | 29.3 | 24.7 |
| Calcium ion | 26.8 {0.95} | 24.5 {0.95} | 15.7 {1} |
| Sodium ion | 38.6 {0.71} | 47.3 {0.83} | 33.3 {1} |
| Bicine | 25.2–32.4 {0.97} | ||
| G3Pd | 22.0–35.4 {0.92} | 13.0–20.7 {1} | |
| Phosphate ion | 25.2–76.0 {0.85} | ||
| Water | 43.4 | 42.3 | 37.8 |
| Root mean square deviation | |||
| Bond lengths (Å) | 0.006 | 0.006 | 0.006 |
| Bond angles (°) | 0.98 | 0.92 | 1.01 |
| Ramachandran | |||
| % Favored | 99.2 | 98.4 | 98.5 |
| % Allowed | 0.8 | 1.6 | 1.5 |
| % Outliers | 0 | 0 | 0 |
| Molprobity | |||
| Clashscore | 0.48 | 2.64 | 2.84 |
a Values in parentheses represent the highest resolution shell.
b 5% of reflections were excluded from refinement and used to calculate Rfree.
c Values in braces indicate the occupancy.
d G3P is glycerol 3-phosphate.
GlpQ was found possessing a triose-phosphate isomerase (TIM) barrel domain (34) and a glycerophosphodiester phosphodiesterase insert (GDPD-I) domain (Fig. 6B) (35). The active site is centrally located in the TIM barrel and includes a residue from the GDPD-I domain (Fig. 6D). Glu-70, Asp-72, and Glu-152 form the metal-binding site where Ca2+ adopts a pentagonal bipyramidal coordination. In the GlpQ·G3P-1 structure (Fig. 6E), Gro hydroxyl groups are coordinated to Ca2+, and the phosphate is positioned by H-bonds to His-43 NE2, Arg-44 NH2, and His-85 NE2, of which highly conserved His-43 and His-85 are implicated in acid-base catalysis (36, 37). His-43 NE2 also formed an H-bond to the 2-hydroxyl of Gro, which is buried inside a pocket composed of Gln-188, Phe-279, Tyr-259, and Leu-210. The GlpQ·G3P-2 complex (Fig. 6F) featured a salt bridge between Lys-154 and the phosphate, altering the orientation of GroP and loop 11 relative to the GlpQ·G3P-1 complex. This movement decreased the distance between Ca2+ and the phosphorus in GroP from 4.8 to 3.6 Å, and between the phosphorus and 2-hydroxyl in Gro from 3.5 to 3.0 Å. As well, H-bonding between His-43 NE2 and 2-hydroxyl in Gro was lost, although a new H-bond formed between Glu-70 OE2 and the 1-hydroxyl.
The orientation of bound GroP, with the Gro moiety buried adjacent to the active site, suggests GlpQ is restricted from accessing internal regions of polymeric substrates, which supports the discerned exolytic mechanism of degradation. We predict that poly(GroP) would protrude to the exterior surface of loop 11 and encounter the negatively charged region adjacent to the GDPD-I (Fig. 6G), providing an efficient means for product release.
GlpQ and PhoD Appear Wholly Responsible for Degradation of Poly(GroP) WTA during Phosphate Starvation
Given the unique product profiles resulting from lipid φ.40 analog degradation by GlpQ or PhoD, we tested supernatants from phosphate-limited cultures for matching activity (Fig. 7). Incubation of 14C-lipid φ.40 analog with culture supernatant from the wild-type strain yielded products consistent with actions by both enzymes. GroP was the main product generated after incubation of the lipid φ.40 analog with culture supernatant from the ΔphoD strain; the Gro was present presumably due to activity by PhoA and/or PhoB. PhoD activity was not evident in culture supernatant from the ΔglpQ strain until after extended phosphate starvation. Importantly, culture supernatant from the double mutant was devoid of teichoicase activity, suggesting that GlpQ and PhoD are necessary for the depolymerization of poly(GroP) WTA.
FIGURE 7.

Extracellular teichoicase activity after phosphate-limited growth. Supernatants (25 ml) from phosphate-limited cultures of B. subtilis strains were recovered at 6 and 14 h past when the onset of post-exponential growth would occur in the wild-type strain and concentrated as described earlier (see Fig. 2B). Teichoicase activity was tested in a reaction comprising 50 mm Tris-HCl (pH 8.0), 1 mm CaCl2, 1 mm MgCl2, 40 μm lipid φ.40 analog, and 8 μl of concentrated supernatant (8 μl of buffer for the control). Reactions were incubated for 90 min at room temperature and quenched with urea. Chromatograms show radioactive products separated by HPAEX. Teichoicase activity was detected in the supernatants from the ΔglpQ and ΔphoD cultures by the presence of Gro and GroP and concomitant loss of starting substrate material. Incubation of lipid φ.40 analog with supernatant of the culture from ΔglpQ mutant recovered at 14 h resulted in a noticeable reduction in the abundance of starting material along with product emergence (indicated by the thick black arrow; see inset). The experiment was performed twice, resulting in similar profiles.
ΔglpQ and ΔphoD Mutants Exhibit Phenotypes Related to the Cell Envelope
Considering the importance of WTA to cell wall biogenesis, we wondered whether impaired WTA turnover would be manifest in effects on the cell envelope. Indeed, a rod to sphere transition is characteristic of B. subtilis cells depleted for WTA biosynthetic enzymes (5, 13). Interestingly, over the course of phosphate limitation, we noted that wild-type cells became shorter and wider, progressively losing their rod shape (Fig. 8). Both the ΔphoD and ΔglpQ strains showed a similar progression, but this was markedly delayed. Suppression of this rod to sphere transition under phosphate limitation was particularly striking in the double mutant (ΔglpQΔphoD), where cells maintained rod-like shape throughout phosphate-limited growth. In light of these observations, we predicted that the integrity and cell wall properties in deletion mutants would be impacted during phosphate starvation. Key to this integrity is the balance between PG synthesis and PG lysis, with both processes influenced by WTA. Because WTA synthesis was functional in the mutants, we suspected the latter would be dysregulated during phosphate starvation when PG synthesis slows significantly (38). In support of this hypothesis, phosphate-starved deletion mutants were found to exhibit accelerated rates of Triton X-100-induced autolysis compared with the wild type (Fig. 9A). Surprisingly, this was also evident with phosphate-replete growth (Fig. 9A), which may reflect a low constitutive transcription of genes within the phoPR operon that occurs independent of the PhoPR-mediated response (i.e. in the absence of phosphate stress) (39). Also, phosphate limitation was less inhibitory to cell lysis in the deletion mutants. Similar trends were noted for lysis in the absence of Triton X-100 (Fig. 9B), and together, these findings implicate glpQ and phoD in PG metabolism via WTA turnover-dependent modulation of autolytic activity.
FIGURE 8.

Deletion of glpQ and phoD impacts cell morphology during phosphate starvation. Strains were grown in phosphate-limited media and cells imaged along a growth curve toward phosphate starvation. Images show cell morphologies, for typical fields, of the various strains. Frequency distributions (n ≥200) for cell width are shown alongside micrographs; blue corresponds to 4 h and red to 24 h. Differences between groups were assessed for significance by analysis of variance. *, p < 0.001.
FIGURE 9.

Autolytic properties and antibiotic susceptibilities for glpQ and phoD deletion mutants. Autolytic profiles for B. subtilis strains in the presence (A) and absence (B) of Triton X-100. Means and S.E. (too small to be visible) from three independent experiments are shown. A and B, ●, WT; red square, ΔphoD; blue triangle, ΔglpQ; inverted pink triangle, ΔglpQΔphoD. Antibiotic susceptibilities of deletion mutants relative to the wild-type strain in phosphate-replete (C) and phosphate-deplete (D) media are shown. Fold change refers to the MIC in the mutant strain divided by the MIC in the wild-type strain. Fold changes <1 indicate increased sensitivity and >1 reduced sensitivity. MIC data are provided in supplemental Table S2.
To further investigate cell envelope effects from deficient WTA turnover, we determined the minimum inhibitory concentrations (MICs) of antibiotics against wild-type and mutant strains, focusing on aminoglycosides and β-lactams as these drug classes are known to be affected by teichoic acids (3, 40). Most notable was the dramatic de-sensitization of the double mutant to the highly charged aminoglycosides neomycin and paromomycin in both phosphate-replete and -deplete conditions (Fig. 9, C and D). For β-lactams, the ΔglpQ and the double deletion mutants were sensitized to cefuroxime and ceftizoxime under phosphate-replete conditions (Fig. 9C). This effect was reduced during phosphate-deplete growth; however, all mutant strains were in addition sensitized to ampicillin and methicillin (Fig. 9D). MICs were unaffected for spectinomycin, gentamycin (net charge <5), or phosphomycin and vancomycin (non-β-lactam cell wall-targeting drugs).
Discussion
Constituting half the cell wall mass, WTA embodies a valuable resource in B. subtilis faced with phosphate deficiency. In a biological setting, GlpQ and PhoD ostensibly encounter glycosylated d-alanylated WTA, yet these enzymes showed a preference for undecorated WTA in vitro. The PhoD catalytic mechanism is proposed to involve attack of phosphorus by Fe3+-bound hydroxide, followed by attack of the resulting phosphomonoester by the hydroxide bridging two active site metals coordinated by the phosphate (Fig. 10A) (24). This allows for endolytic degradation as observed here but implies that hydroxyl groups from glucose attached to Gro moieties flanking a phosphate may perturb coordination of active site metals. Based on our structural investigation, we propose a distinct mechanism for GlpQ that is similar to Thermoanaerobacter tengcongensis GlpQ (Fig. 10B) (36). Deprotonation of the 2-hydroxyl of a terminal GroP by His-43 enables nucleophilic attack of the phosphorus, aided by Lys-154-mediated substrate reorientation. His-85, stabilized by H-bonding with Asp-86, protonates the leaving group oxygen, leading to a cyclic phosphate intermediate and release of the alcohol leaving group, the WTA polymer with one less GroP unit. This explains the lack of activity with fully glycosylated lipid φ.40 analog, where all 2-hydroxyl groups in Gro moieties are occupied. Roles for the catalytic histidine residues are reversed to enable GroP release. Considering incomplete glycosylation of WTA and turnover of labile d-alanine esters in vivo (41), combined degradation by GlpQ and PhoD as shown here is sufficiently robust to be biologically relevant during phosphate-limited growth when doubling time exceeds 150 min (28). In the simplest scenario, WTA decoration may serve to control the rate of depolymerization, averting rapid losses that could be costly to the cell. However, our findings draw attention to the possibility of phosphate starvation-induced genes for as yet unspecified glycoside hydrolases or esterases that render the polymer more susceptible to degradation by GlpQ and PhoD.
FIGURE 10.

Simplified catalytic mechanisms of PhoD and GlpQ. A, PhoD is proposed to employ a catalytic mechanism akin to that described for purple acid phosphatases (67). Phosphodiester hydrolysis commences with nucleophilic attack by a Fe3+ coordinated hydroxide leading to ejection of the alcohol product, followed by processive nucleophilic attack of the phosphomonoester product by the bridging hydroxide. B, His-43 and His-85 act as general bases and acids in catalysis by GlpQ that involves the formation of a cyclic phosphate intermediate.
Despite efficient WTA turnover by GlpQ and PhoD in vitro, the corresponding single gene deletion mutants did not exhibit intermediate phenotypes in phosphate-limited growth. The discovery of different modes of depolymerization catalyzed by GlpQ and PhoD therefore implies that these enzymes act in a complementary fashion, with PhoD converting WTA into fragments, thereby elevating concentrations of substrates more efficiently turned over by GlpQ. Restriction of this activity to WTA attached to PG at the cell wall periphery, however, would leave a significant portion of WTA embedded within the PG matrix (41) and inaccessible. It is noteworthy that GlpQ is more highly secreted than PhoD during phosphate limitation (42), which coincides with our observations of low secreted teichoicase activity in the phosphate-starved ΔglpQ strain and the reported association of PhoD with the cell wall prior to slow processing and extracellular transport (43). Taking all into account, the most prudent explanation of the requirement for composite teichoicase activity entails action of PhoD at the cell wall, and to a lesser extent in the extracellular milieu, followed by extracellular GlpQ action on the WTA fragments generated.
Much of the current understanding on the correlation between WTA synthesis and PG assembly has come about though the study of bacterial strains impaired in WTA synthesis (3, 5, 6, 13, 44–47). These studies highlighted the sensitivity of PG synthesis to the abundance of the shared biosynthetic intermediate, undecaprenyl diphosphate, and to its perturbation by blocks in WTA synthesis. Furthermore, those efforts have described β-lactam sensitivity in S. aureus strains where WTA biosynthesis is prevented. Interestingly, the work reported here shows that such phenotypes, i.e. cell shape and β-lactam sensitivity, are also dependent on machinery dedicated to WTA depolymerization. Significantly, this influence is exerted with WTA synthetic machinery intact, and so the observations reported are likely not due to impacts on PG synthesis that arise from perturbations in WTA synthesis. Moreover, we found an impact of WTA turnover on autolytic activity. WTAs regulate such activity by directing the localization of autolysins (48, 49) and by promoting a low cell wall pH that is often inhibitory to their action (50). In B. subtilis, however, cell wall binding of the major secreted autolysin, N-acetylmuramoyl-l-alanine amidase is highly dependent on interactions with WTA (45, 51, 52). Accordingly, a net increase in WTA negative charge caused by an absence of d-alanine has been correlated with enhanced autolysis in B. subtilis (53). Our findings therefore support the view that an increased presence of WTA provides additional sites for interaction of this major B. subtilis autolysin, consequently causing an imbalance between slowed PG synthesis during phosphate starvation and PG lysis. Underscoring the effect from this altered PG landscape was the sensitization to β-lactams, which appear to be potentiated by the increased autolytic activity. Conversely, neomycin and paromomycin were antagonized, seemingly because these positively charged molecules become sequestered at a more negatively charged cell wall.
Herein, we provide substantial evidence that PhoP regulon genes glpQ and phoD encode teichoicases active during phosphate starvation in B. subtilis 168. Although teichoic acid hydrolytic activity was first described several decades ago, this is first genetic assignment of teichoicase activity in bacteria. Our findings illustrate a requirement of composite activity by mechanistically divergent teichoicases for WTA metabolism that extends vegetative growth after environmental phosphate becomes limiting. The widespread occurrence of glpQ homologs in the Bacillaceae and Staphylococcaceae families, and phoD homologs in Bacillaceae (Bacillus) and Streptomycetaceae, may signify a conserved function in WTA turnover for these genes. In addition, we show the significance of teichoicase activity to cell shape, autolytic susceptibility, and β-lactam sensitivity. Remarkably, phenotypes in glpQ or phoD deletion strains were present to lesser degrees during phosphate-replete growth, implying some relevance in the absence of phosphate stress. It thus conceivable that teichoicase activity in alternative contexts is of consequence to cell wall structure and properties. Continued discovery and characterization of such enzymes will provide unique perspectives for understanding the pathways and significance of cell wall turnover in Gram-positive bacteria and present possibilities for the manipulation of WTA degradation to probe drug susceptibility and virulence in WTA-synthesizing pathogens.
Experimental Procedures
Bacterial Strains and Growth Conditions
The strains, plasmids, and oligonucleotides used in this study are listed in supplemental Table S1. Genetic manipulation and propagation of strains were performed in LB medium with appropriate addition of antibiotics. The defined medium used for phosphate limitation studies was that of Grant (16); phosphate-replete and phosphate-deplete media contained 2.5 and 0.0625 mm KH2PO4, respectively. To assess phosphate-limited growth, strains were streaked on phosphate-replete agar plates, and single colonies emerging after overnight growth at 37 °C were used to inoculate 5-ml cultures in phosphate-replete liquid media. At the late log phase, cultures were used to inoculate (1:100) 50 ml of phosphate-deplete media, and growth was monitored at 37 °C with shaking at 250 pm. Growth supplementation experiments were carried out in 200 μl in a 96-well microplate; phosphate-deplete media was inoculated (1:50) with late-log phosphate-replete cells, and 190 μl of this cell suspension was mixed with 10 μl of 10 mg/ml WTA, 50 mm KH2PO4, or sterile ddH2O. The absorbance at 600 nm was monitored with a Sunrise microplate reader (Tecan), at 37 °C with aeration.
Generation of glpQ and phoD Deletion Mutants
Gene-targeting DNA fragments were generated by joining PCR of the following three fragments: an antibiotic resistance cassette and 1-kb 5′- and 3′-flanking sequences of the targeting gene. For preparation of antibiotic resistance cassettes, PAGE-purified primer pairs Ab-F and Ab-R (0.5 μm final each) were mixed with 10 ng of purified template plasmid (pDR240a for KanR and pDR242a for ErmR) and amplified by PCR under standard conditions using Phusion® Hot-Start DNA polymerase (New England Biolabs). Amplified KanR or ErmR fragments were purified by gel extraction. For preparation of flanking sequences of target genes, targeting gene-specific primer pairs, 5pL, 5pR and 3pL, 3pR (0.5 μm final each), were mixed with 20 ng of purified B. subtilis 168 genomic DNA and amplified by PCR under standard conditions. Amplified flanking DNA fragments were purified using the Agencourt AMPure XP (Beckman Coulter) magnetic beads. Antibiotic resistance cassette and 5′- and 3′-flanking DNA fragments (∼15 ng of each DNA) were mixed and subjected to the joining PCR in the presence of 5pL and 3pR (0.5 μm final each) under the following conditions: 1 min at 98 °C (10 s at 98 °C, 20 s at 55 °C, and 80 s at 72 °C) for 30 cycles; 5 min at 72 °C using detergent-free HF buffer and Phusion hot-start DNA polymerase. The joined PCR products were directly used for transformation. Competent cells were prepared by the following protocol: wild-type B. subtilis 168 cells were inoculated into 3 ml of MC medium (10.7 mg/ml K2HPO4, 5.2 mg/ml KH2PO4, 20 mg/ml glucose, 0.88 mg/ml trisodium citrate dihydrate, 0.022 mg/ml ferric ammonium citrate, 1 mg/ml casamino acids, 2.2 mg/ml potassium glutamate monohydrate, 20 mm MgSO4, 300 nm MnCl2, 20 mg/liter l-tryptophan) and incubated at 37 °C overnight with aeration. The overnight culture was diluted to an A600 of 0.1 in 30 ml of competence medium (10.7 mg/ml K2HPO4, 5.2 mg/ml KH2PO4, 20 mg/ml glucose, 0.88 mg/ml sodium citrate dihydrate, 0.022 mg/ml ferric ammonium citrate, 2.5 mg/ml potassium aspartate, 10 mm MgSO4, 150 nm MnCl2, 40 mg/liter l-tryptophan, 0.05% (w/v) yeast extract) and then grown in a 125-ml flask at 37 °C with shaking (250 rpm) until cells reached an A600 nm of ∼1.5. 120 μl of culture was then mixed with 10 μl of gene-targeting PCR fragments arrayed in a deep 96-well plate, covered with a breathable film, and incubated at 37 °C with shaking (900 rpm). After 2 h of incubation, cells were plated on LB agar containing selective antibiotics (7.5 μg/ml kanamycin or 1 μg/ml erythromycin, and 12.5 μg/ml lincomycin by activity). After overnight incubation, single colonies from each plate were purified by re-streaking on new selection plate. Deletion of target genes was confirmed by PCR carried out using different combinations of primers, the 3pR, and antibiotic resistance cassette-specific primers. Gene deletions were confirmed by ∼1.2-kb PCR product from the reaction.
To construct a double knock-out in glpQ and phoD, genomic DNA was isolated from the phoD::KanR strain and used to transform competent cells from the glpQ::ErmR strain according to established procedures (54). Successful transformants were identified by double selection on LB-agar containing kanamycin and erythromycin.
Genetic Complementation
Bacteria were grown in LB (supplemented with 0.2% (w/v) xylose when required) at 37 °C. Cultures were supplemented with chloramphenicol (10 μg/ml) or erythromycin (5 μg/ml), as required. DNA manipulations were performed according to established procedures (55). T4 DNA ligase, Antarctic phosphatase, and restriction endonucleases were from New England Biolabs. Transformation of E. coli NovaBlue was performed by electroporation. PCR was performed with Phusion DNA polymerase (Thermo Fisher Scientific) using B. subtilis 168 genomic DNA as a template.
The primers listed in supplemental Table S1 were used to place a consensus ribosome-binding site (56) upstream of the gene, and PCRs were optimized for each primer pair. Resulting PCR products were purified, double-digested with PacI and BamHI, and ligated into pSWEET-bgaB digested with the same restriction enzymes. The ligation mixture was electroporated into E. coli NovaBlue as described previously. The correct integration of sequences encoding phoD or glpQ into pSWEET was confirmed by PCR, and the sequence was verified by DNA sequencing (MOBIX Lab, McMaster University). Competent cells of the individual gene knock-out strains (B. subtilis 168 phoD::erm or glpQ::erm) were prepared as described previously (54, 57) with modifications. Mutant strains were inoculated into 5 ml of pre-transformation medium (8.968 ml of 1× T-Base, 1% (w/v) casamino acids, 1.2% (w/v) MgSO4, 25% (w/v) glucose, 10% (w/v) yeast extract, 0.1 m CaCl2, and deionized water to 10 ml). 1× T-Base was prepared as follows: 2 g of (NH4)2SO4, 18.3 g of K2HPO4·3H2O, 6 g of KH2PO4, 1 g of sodium citrate, deionized water to 1 liter, and incubated overnight at 37 °C with shaking at 250 rpm. Cultures were diluted 10-fold into warm transformation medium (17.672 ml of 1× T-Base, 1% (w/v) casamino acids, 1.2% (w/v) MgSO4, 25% (w/v) glucose, 10% (w/v) yeast extract, 0.1 m EGTA, and deionized water to 20 ml) and mixed with 1 μg of PstI-linearized pSWEET-phoD or pSWEET-glpQ, in 1-ml aliquots, and incubated for 90 min at 37 °C with shaking (250 rpm). Chloramphenicol was added to a final concentration of 1 μg/ml, and incubation was continued for 30 min. Cells were plated undiluted or after 10-fold dilution onto LB-agar supplemented with chloramphenicol. Chromosomal integration (via double recombination) downstream of the xylose promoter into the amyE locus was verified by PCR and confirmed by plating on LB-agar containing 1% (w/v) starch and incubation at 37 °C overnight, after which amylase activity was detected by adding Lugol's iodine solution. Lack of amylase activity indicated the disruption of amyE gene and chromosomal integration of the gene of interest.
Expression and Purification of GlpQ and PhoD
Synthetic DNA sequences codon-optimized for expression in E. coli (ThermoFisher Scientific) and encoding mature GlpQ (residues 27–293) or PhoD (residues 57–583) were cloned into the expression vector pDEST17 (ThermoFisher Scientific) with a tobacco etch virus protease-cleavable N-terminal hexahistidine tag. Plasmid constructs were transformed into E. coli BL21 DE.3 PlysS, and cells were cultured in LB broth supplemented with ampicillin (50 μg/ml) and chloramphenicol (34 μg/ml) at 37 °C, until an A600 nm of 0.6–0.8. Cultures were cooled to 16 °C, and protein expression was induced by addition of isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 0.5 mm. Expression was continued for 18 h at 16 °C, and the cells were harvested by centrifugation. Cell pellets were washed in 0.9% (w/v) saline and then resuspended in buffer A (50 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 25 mm imidazole). DNase and RNase were added, each at final concentrations of 10 μg/ml, as well as complete protease inhibitor (Roche Applied Science). Cells were lysed by disruption at 30,000 p.s.i., and the lysates were clarified by centrifugation (48,000 × g) prior to loading onto a 5-ml HisTrapHP column (GE Healthcare) pre-equilibrated with buffer A. His-tagged proteins were eluted over a linear gradient of 25–500 mm imidazole in buffer A, and fractions containing pure protein, as confirmed by SDS-PAGE, were pooled, and the buffer was exchanged for 50 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1 mm CaCl2, 1 mm MgCl2 using a 5-ml HiTrap desalting column (GE Healthcare). Proteins were divided into aliquots and stored at −80 °C.
Crystallography and Structural Solution GlpQ
GlpQ (residues 27–293; 5 mg/ml) was crystallized at room temperature by sitting-drop vapor diffusion using 1 μl of protein solution (5 mg/ml purified protein in 50 mm Tris-HCl (pH 7.4), 200 mm NaCl, and 1 mm CaCl2) mixed with 1 μl of reservoir solution (100 mm Bicine (pH 8.5) and 25% (w/v) PEG6000). X-ray diffraction data for the GlpQ·Bicine complex were collected on a single crystal flash-frozen in liquid N2, and structures of unique GlpQ·GroP complexes were acquired after soaking crystals in 100 mm HEPES (pH 8.4) containing 25% (w/v) PEG6000 and 25 mm GPG. All diffraction data were processed using Xia2 (58). Structures were solved by molecular replacement using the BALBES webserver (59), selecting the Bacillus anthracis GlpQ structure (Protein Data Bank code 4R70) as the starting model. Model building and refinement were performed with Phenix and Coot (60, 61). Coordinates and structure factors were deposited in the Protein Data Bank with accession codes 5T91 (GlpQ·Bicine), 5T9B (GlpQ·G3P-1), and 5T9C (GlpQ·G3P-2).
Isolation and PAGE of WTA
Cell wall was isolated from late-log cells by SDS extraction (30). WTA was released from PG by incubation in 0.1 m NaOH at room temperature for 16 h. The pH was adjusted to ∼7.0 with acetic acid, and insoluble material was removed by centrifugation (21,000 × g). The supernatant containing WTA was desalted over a PD MidiTrap G10 gel filtration column (GE Healthcare), eluting with ddH2O, and lyophilized.
PAGE was performed on samples containing ∼10 μg of WTA in 10 mm Tris-HCl (pH 8.0), 5% (v/v) glycerol, 1 mm EDTA, 0.05% (w/v) bromphenol blue. Samples were loaded onto pre-cast 15% TBE gels (Bio-Rad) and separated in Mini-Protean Tetra electrophoresis cell (Bio-Rad) using a constant power (100 V) and a running buffer of 89 mm Tris-HCl, 89 mm boric acid, and 2 mm EDTA (pH 8.3). Bands were visualized with Alcian blue/silver staining (32) using the Bio-Rad silver stain kit (Bio-Rad). Densitometric analysis was performed with ImageJ software, and image adjustments were limited to brightness, contrast, and background subtraction (62).
Cell Wall Phosphate Quantification
Cell wall preparations were further purified as described previously (63) and then resuspended in ddH2O to an absorbance of 0.7–1 and incubated in 10% (w/v) TCA at 80 °C for 16 h. Pi released was detected with the BioMol green reagent (Enzo Life Sciences) and quantified by absorbance at 630 nm and interpolation to a standard curve generated with KH2PO4.
Synthesis of WTA Oligomers
Phosphatidylglycerol or cardiolipin (100 mg; Avanti Polar Lipids) was dissolved in a 2:1 mixture of chloroform and methanol. An equimolar amount of NaOH, prepared in 100% ethanol, was added and the mixture incubated at room temperature for 2 h. Precipitate formed in the reaction was collected and dissolved in water and then extracted with chloroform/methanol (9:1). The aqueous layer containing the sodium salt of GPG or bGPG was recovered and lyophilized.
Synthesis of WTA Intermediate Analogs
[14C]GroP lipid φ.40 analog and CMP-poly(GroP) were synthesized as described previously (15).
WTA Degradation Assays
Degradation reactions of B. subtilis WTA were performed at room temperature in 50 mm Tris-HCl (pH 8.0) containing 1 mm MgCl2 and 1 mm CaCl2. Reactions were quenched with EDTA (50 mm), and activity was assessed by quantification of liberated Pi. Reactions containing GlpQ received 0.5 units of bovine APase (Sigma) 2 min prior to quenching.
Reactions with 14C-lipid φ.40 analog were performed as above but were quenched with urea (6.6 m). Radioactive products were separated by HPAEX (15) using a DNA PAC PA200 column (Thermo Fisher Scientific), with visualization by in-line scintillation counting. The amount of lipid φ.40 analog remaining or GroP produced was quantified by peak integration.
Assays for Hydrolysis of GPG and bGPG
Activity of GlpQ and PhoD with GPG or bGPG was assessed as described for B. subtilis WTA. Initial rates of Pi production were plotted as a function of substrate concentration, and the data were fit by non-linear regression (Prism 6.0, GraphPad) to Equation 1.
| (Eq. 1) |
Autolysis Assay
Strains were grown to late log phase (phosphate-replete media) or 6 h past the time when onset of post-exponential growth would occur in the wild-type strain (phosphate-deplete media). Cells were collected by centrifugation, washed twice in PBS, once in ice-cold ddH2O, and resuspended in PBS or PBS containing 0.05% (v/v) Triton X-100, to an absorbance at 600 nm of ∼1. Then 200 μl of cell suspension was transferred to a single well in a 96-well microplate, and the decreased absorbance at 600 nm was monitored at 37 °C with shaking.
Fluorescence Microscopy
Bacteria were imaged using the membrane dye FM 4-64 and a Nikon Ti-E inverted microscope (×1000 magnification). Cell width quantification was performed with ImageJ (62). Images were converted to 8-bit grayscale and background-subtracted using a 50 pixel rolling ball radius method. The Otsu thresholding algorithm was applied, and gaps in the resulting binary images were filled before quantification. The R statistical programming language (64) was used to examine the results, using a density function to generate frequency plots from images with sample sizes typically greater than 200 cells. The density distribution of image features, in this case cell width, is preferable over a mean value given the morphological variability existing even within a single culture.
Author Contributions
C. L. M., E. D. B., and N. C. J. S. designed the research. C. L. M., F. K. K. L., S. F., B. K., O. M. E., and C. A. G. performed the experiments. C. L. M., F. K. K. L., and S. F. analyzed the data. C. L. M. and E. D. B. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Robert Gale for helpful discussions, and we are grateful to the staff at the Canadian Light Source (Saskatoon, Canada) beamlines 08ID-1 and 08B1-1 for assistance with data collection.
This work was supported by Canadian Institutes of Health Research Foundation Grant FRN-143215, a Natural Sciences and Engineering Research Council of Canada Discovery Grant (to E. D. B.), the Canada Research Chairs Program (to E. D. B. and N. C. J. S.), the Canadian Institutes of Health Research, the Howard Hughes Medical Institute International Scholar Program, Canada Foundation for Innovation, British Columbia Knowledge Development Fund, and National Institutes of Health Grants R01 GM102790 and R35 GM118061 (to C. A. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1–S3 and Figs. S1 and S2.
The atomic coordinates and structure factors (codes 5T91, 5T9B, and 5T9C) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- WTA
- wall teichoic acid
- APase
- alkaline phosphatase
- bGPG
- bis-glycerophosphoglycerol
- Gro
- glycerol
- GroP
- glycerol 3-phosphate
- GPG
- glycerophosphoglycerol
- HPAEX
- high performance anion exchange chromatography
- LB
- Luria Bertani
- pNPC
- p-nitrophenylphosphorylcholine
- pNPP
- p-nitrophenyl phosphate
- PG
- peptidoglycan
- TIM
- triose-phosphate isomerase
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- ddH2O
- double distilled H2O
- MIC
- minimum inhibitory concentration.
References
- 1. Sewell E. W., and Brown E. D. (2014) Taking aim at wall teichoic acid synthesis: new biology and new leads for antibiotics. J. Antibiot. 67, 43–51 [DOI] [PubMed] [Google Scholar]
- 2. Winstel V., Kühner P., Salomon F., Larsen J., Skov R., Hoffmann W., Peschel A., and Weidenmaier C. (2015) Wall teichoic acid glycosylation governs Staphylococcus aureus nasal colonization. MBio. 6, e00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Farha M. A., Leung A., Sewell E. W., D'Elia M. A., Allison S. E., Ejim L., Pereira P. M., Pinho M. G., Wright G. D., and Brown E. D. (2013) Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β-lactams. ACS Chem. Biol. 8, 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee S. H., Wang H., Labroli M., Koseoglu S., Zuck P., Mayhood T., Gill C., Mann P., Sher X., Ha S., Yang S.-W., Mandal M., Yang C., Liang L., Tan Z., Tawa P., et al. (2016) TarO-specific inhibitors of wall teichoic acid biosynthesis restore β-lactam efficacy against methicillin-resistant staphylococci. Sci. Transl. Med. 8, 329ra32. [DOI] [PubMed] [Google Scholar]
- 5. D'Elia M. A., Millar K. E., Beveridge T. J., and Brown E. D. (2006) Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J. Bacteriol. 188, 8313–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Elia M. A., Pereira M. P., Chung Y. S., Zhao W., Chau A., Kenney T. J., Sulavik M. C., Black T. A., and Brown E. D. (2006) Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188, 4183–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park J. T., and Uehara T. (2008) How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol. Mol. Biol. Rev. 72, 211–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reith J., and Mayer C. (2011) Peptidoglycan turnover and recycling in Gram-positive bacteria. Appl. Microbiol. Biotechnol. 92, 1–11 [DOI] [PubMed] [Google Scholar]
- 9. Mauck J., Chan L., and Glaser L. (1971) Turnover of the cell wall of Gram-positive bacteria. J. Biol. Chem. 246, 1820–1827 [PubMed] [Google Scholar]
- 10. Wise E. M. Jr., Glickman R. S., and Teimer E. (1972) Teichoic acid hydrolase activity in soil bacteria. Proc. Natl. Acad. Sci. U.S.A. 69, 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kusser W., and Fiedler F. (1983) Teichoicase from Bacillus subtilis Marburg. J. Bacteriol. 155, 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chan Y. G., Frankel M. B., Dengler V., Schneewind O., and Missiakas D. (2013) Staphylococcus aureus mutants lacking the LytR-CpsA-Psr family of enzymes release cell wall teichoic acids into the extracellular medium. J. Bacteriol. 195, 4650–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawai Y., Marles-Wright J., Cleverley R. M., Emmins R., Ishikawa S., Kuwano M., Heinz N., Bui N. K., Hoyland C. N., Ogasawara N., Lewis R. J., Vollmer W., Daniel R. A., and Errington J. (2011) A widespread family of bacterial cell wall assembly proteins. EMBO J. 30, 4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiang Y., Leiman P. G., Li L., Grimes S., Anderson D. L., and Rossmann M. G. (2009) Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol. Cell 34, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Myers C. L., Ireland R. G., Garrett T. A., and Brown E. D. (2015) Characterization of wall teichoic acid degradation by the bacteriophage φ29 appendage protein GP12 using synthetic substrate analogs. J. Biol. Chem. 290, 19133–19145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grant W. D. (1979) Cell wall teichoic acid as a reserve phosphate source in Bacillus subtilis. J. Bacteriol. 137, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soldo B., Lazarevic V., Pagni M., and Karamata D. (1999) Teichuronic acid operon of Bacillus subtilis 168. Mol. Microbiol. 31, 795–805 [DOI] [PubMed] [Google Scholar]
- 18. Hulett F. M. (2002) in Bacillus subtilis and Its Closest Relatives from Genes to Cells (Sonseshein A. L., Losick R., and Hoch J. A., eds) pp. 193–201, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 19. Hulett F. M., Lee J., Shi L., Sun G., Chesnut R., Sharkova E., Duggan M. F., and Kapp N. (1994) Sequential action of two-component genetic switches regulates the PHO regulon in Bacillus subtilis. J. Bacteriol. 176, 1348–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allenby N. E., O'Connor N., Prágai Z., Carter N. M., Miethke M., Engelmann S., Hecker M., Wipat A., Ward A. C., and Harwood C. R. (2004) Post-transcriptional regulation of the Bacillus subtilis pst operon encoding a phosphate-specific ABC transporter. Microbiology 150, 2619–2628 [DOI] [PubMed] [Google Scholar]
- 21. Allenby N. E., O'Connor N., Prágai Z., Ward A. C., Wipat A., and Harwood C. R. (2005) Genome-wide transcriptional analysis of the phosphate starvation stimulon of Bacillus subtilis. J. Bacteriol. 187, 8063–8080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tjalsma H., Antelmann H., Jongbloed J. D., Braun P. G., Darmon E., Dorenbos R., Dubois J.-Y., Westers H., Zanen G., Quax W. J., Kuipers O. P., Bron S., Hecker M., and van Dijl J. M. (2004) Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 68, 207–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eder S., Shi L., Jensen K., Yamane K., and Hulett F. M. (1996) A Bacillus subtilis secreted phosphodiesterase/alkaline phosphatase is the product of a Pho regulon gene, phoD. Microbiology 142, 2041–2047 [DOI] [PubMed] [Google Scholar]
- 24. Rodriguez F., Lillington J., Johnson S., Timmel C. R., Lea S. M., and Berks B. C. (2014) Crystal structure of the Bacillus subtilis phosphodiesterase PhoD reveals an iron and calcium-containing active site. J. Biol. Chem. 289, 30889–30899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tommassen J., Eiglmeier K., Cole S. T., Overduin P., Larson T. J., and Boos W. (1991) Characterization of two genes, glpQ and ugpQ, encoding glycerophosphoryl diester phosphodiesterases of Escherichia coli. Mol. Gen. Genet. 226, 321–327 [DOI] [PubMed] [Google Scholar]
- 26. Ohshima N., Yamashita S., Takahashi N., Kuroishi C., Shiro Y., and Takio K. (2008) Escherichia coli cytosolic glycerophosphodiester phosphodiesterase (UgpQ) requires Mg2+, Co2+, or Mn2+ for its enzyme activity. J. Bacteriol. 190, 1219–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munson R. S. Jr., and Sasaki K. (1993) Protein D, a putative immunoglobulin D-binding protein produced by Haemophilus influenzae, is glycerophosphodiester phosphodiesterase. J. Bacteriol. 175, 4569–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Botella E., Devine S. K., Hubner S., Salzberg L. I., Gale R. T., Brown E. D., Link H., Sauer U., Codée J. D., Noone D., and Devine K. M. (2014) PhoR autokinase activity is controlled by an intermediate in wall teichoic acid metabolism that is sensed by the intracellular PAS domain during the PhoPR-mediated phosphate limitation response of Bacillus subtilis. Mol. Microbiol. 94, 1242–1259 [DOI] [PubMed] [Google Scholar]
- 29. Botella E., Hübner S., Hokamp K., Hansen A., Bisicchia P., Noone D., Powell L., Salzberg L. I., and Devine K. M. (2011) Cell envelope gene expression in phosphate-limited Bacillus subtilis cells. Microbiology 157, 2470–2484 [DOI] [PubMed] [Google Scholar]
- 30. Bhavsar A. P., Erdman L. K., Schertzer J. W., and Brown E. D. (2004) Teichoic acid is an essential polymer in Bacillus subtilis that is functionally distinct from teichuronic acid. J. Bacteriol. 186, 7865–7873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allison S. E., D'Elia M. A., Arar S., Monteiro M. A., and Brown E. D. (2011) Studies of the genetics, function, and kinetic mechanism of TagE, the wall teichoic acid glycosyltransferase in Bacillus subtilis 168. J. Biol. Chem. 286, 23708–23716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Min H., and Cowman M. K. (1986) Combined alcian blue and silver staining of glycosaminoglycans in polyacrylamide gels: application to electrophoretic analysis of molecular weight distribution. Anal. Biochem. 155, 275–285 [DOI] [PubMed] [Google Scholar]
- 33. Bhavsar A. P., Zhao X., and Brown E. D. (2001) Development and characterization of a Xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl. Environ. Microbiol. 67, 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagano N., Orengo C. A., and Thornton J. M. (2002) One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 321, 741–765 [DOI] [PubMed] [Google Scholar]
- 35. Santelli E., Schwarzenbacher R., McMullan D., Biorac T., Brinen L. S., Canaves J. M., Cambell J., Dai X., Deacon A. M., Elsliger M.-A., Eshagi S., Floyd R., Godzik A., Grittini C., Grzechnik S. K., et al. (2004) Crystal structure of a glycerophosphodiester phosphodiesterase (GDPD) from Thermotoga maritima (TM1621) at 1.60 Å resolution. Proteins 56, 167–170 [DOI] [PubMed] [Google Scholar]
- 36. Shi L., Liu J.-F., An X.-M., and Liang D.-C. (2008) Crystal structure of glycerophosphodiester phosphodiesterase (GDPD) from Thermoanaerobacter tengcongensis, a metal ion-dependent enzyme: Insight into the catalytic mechanism. Proteins 72, 280–288 [DOI] [PubMed] [Google Scholar]
- 37. Vuitika L., Chaves-Moreira D., Caruso I., Lima M. A., Matsubara F. H., Murakami M. T., Takahashi H. K., Toledo M. S., Coronado M. A., Nader H. B., Senff-Ribeiro A., Chaim O. M., Arni R. K., and Veiga S. S. (2016) Active site mapping of Loxosceles phospholipases D: biochemical and biological features. Biochim. Biophys. Acta 1861, 970–979 [DOI] [PubMed] [Google Scholar]
- 38. Bisicchia P., Lioliou E., Noone D., Salzberg L. I., Botella E., Hübner S., and Devine K. M. (2010) Peptidoglycan metabolism is controlled by the WalRK (YycFG) and PhoPR two-component systems in phosphate-limited Bacillus subtilis cells. Mol. Microbiol. 75, 972–989 [DOI] [PubMed] [Google Scholar]
- 39. Prágai Z., Allenby N. E., O'Connor N., Dubrac S., Rapoport G., Msadek T., and Harwood C. R. (2004) Transcriptional regulation of the phoPR operon in Bacillus subtilis. J. Bacteriol. 186, 1182–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Santa Maria J. P. Jr., Sadaka A., Moussa S. H., Brown S., Zhang Y. J., Rubin E. J., Gilmore M. S., and Walker S. (2014) Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc. Natl. Acad. Sci. U.S.A. 111, 12510–12515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neuhaus F. C., and Baddiley J. (2003) A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67, 686–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Antelmann H., Scharf C., and Hecker M. (2000) Phosphate starvation-inducible proteins of Bacillus subtilis: proteomics and transcriptional analysis. J. Bacteriol. 182, 4478–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Müller J. P., and Wagner M. (1999) Localisation of the cell wall-associated phosphodiesterase PhoD of Bacillus subtilis. FEMS Microbiol. Lett. 180, 287–296 [DOI] [PubMed] [Google Scholar]
- 44. Peschel A., Otto M., Jack R. W., Kalbacher H., Jung G., and Götz F. (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 274, 8405–8410 [DOI] [PubMed] [Google Scholar]
- 45. Yamamoto H., Kurosawa S.-I., and Sekiguchi J. (2003) Localization of the vegetative cell wall hydrolases LytC, LytE, and LytF on the Bacillus subtilis cell surface and stability of these enzymes to cell wall-bound or extracellular proteases. J. Bacteriol. 185, 6666–6677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. D'Elia M. A., Millar K. E., Bhavsar A. P., Tomljenovic A. M., Hutter B., Schaab C., Moreno-Hagelsieb G., and Brown E. D. (2009) Probing teichoic acid genetics with bioactive molecules reveals new interactions among diverse processes in bacterial cell wall biogenesis. Chem. Biol. 16, 548–556 [DOI] [PubMed] [Google Scholar]
- 47. Campbell J., Singh A. K., Santa Maria J. P. Jr, Kim Y., Brown S., Swoboda J. G., Mylonakis E., Wilkinson B. J., and Walker S. (2011) Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 6, 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schlag M., Biswas R., Krismer B., Kohler T., Zoll S., Yu W., Schwarz H., Peschel A., and Götz F. (2010) Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 75, 864–873 [DOI] [PubMed] [Google Scholar]
- 49. Kasahara J., Kiriyama Y., Miyashita M., Kondo T., Yamada T., Yazawa K., Yoshikawa R., and Yamamoto H. (2016) Teichoic acid polymers affect expression and localization of dl-endopeptidase LytE required for lateral cell wall hydrolysis in Bacillus subtilis. J. Bacteriol. 198, 1585–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Biswas R., Martinez R. E., Göhring N., Schlag M., Josten M., Xia G., Hegler F., Gekeler C., Gleske A.-K., Götz F., Sahl H.-G., Kappler A., and Peschel A. (2012) Proton-binding capacity of Staphylococcus aureus wall teichoic acid and its role in controlling autolysin activity. PLoS ONE 7, e41415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herbold D. R., and Glaser L. (1975) Interaction of N-acetylmuramic acid l-alanine amidase with cell wall polymers. J. Biol. Chem. 250, 7231–7238 [PubMed] [Google Scholar]
- 52. Lindsay B., and Glaser L. (1976) Characterization of the N-acetylmuramic acid l-alanine amidase from Bacillus subtilis. J. Bacteriol. 127, 803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wecke J., Madela K., and Fischer W. (1997) The absence of d-alanine from lipoteichoic acid and wall teichoic acid alters surface charge, enhances autolysis and increases susceptibility to methicillin in Bacillus subtilis. Microbiology 143, 2953–2960 [DOI] [PubMed] [Google Scholar]
- 54. Cutting S. M., and Vander Horn P. B. (1990) in Molecular Biological Methods for Bacillus (Cutting S. M., and Harwood C. R., eds) pp. 27–74, John Wiley & Sons Inc., New York [Google Scholar]
- 55. Sambrook J., and Russel D. W. (2012) Molecular Cloning, 4th Ed., Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 56. Vellanoweth R. L., and Rabinowitz J. C. (1992) The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol. Microbiol. 6, 1105–1114 [DOI] [PubMed] [Google Scholar]
- 57. Jenkinson H. F. (1983) Altered arrangement of proteins in the spore coat of a germination mutant of Bacillus subtilis. Microbiology 129, 1945–1958 [DOI] [PubMed] [Google Scholar]
- 58. Winter G., Lobley C. M., and Prince S. M. (2013) Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 69, 1260–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. de Jonge B. L., Chang Y. S., Gage D., and Tomasz A. (1992) Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 267, 11248–11254 [PubMed] [Google Scholar]
- 64. Ihaka R., and Gentleman R. (1996) R: A language for data analysis and graphics. J. Comp. Graph. Stat. 5, 299–314 [Google Scholar]
- 65. Nicholson W. L., and Setlow P. (1991) in Molecular Biological Methods for Bacillus (Cutting S. M., and Harwood C. R., eds) pp. 391–450, John Wiley & Sons Inc., New York [Google Scholar]
- 66. Blumenkrantz N., and Asboe-Hansen G. (1973) New method for quantitative determination of uronic acids. Anal. Biochem. 54, 484–489 [DOI] [PubMed] [Google Scholar]
- 67. Schenk G., Mitić N., Hanson G. R., and Comba P. (2013) Purple acid phosphatase: A journey into the function and mechanism of a colorful enzyme. Coord. Chem. Rev. 257, 473–482 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.