

Abstract
The relevance of mitochondrial phosphate carrier (PiC), encoded by SLC25A3, in bioenergetics is well accepted. However, little is known about the mechanisms mediating the cellular impairments induced by pathological SLC25A3 variants. To this end, we investigated the pathogenicity of a novel compound heterozygous mutation in SLC25A3. First, each variant was modeled in yeast, revealing that substituting GSSAS for QIP within the fifth matrix loop is incompatible with survival on non-fermentable substrate, whereas the L200W variant is functionally neutral. Next, using skin fibroblasts from an individual expressing these variants and HeLa cells with varying degrees of PiC depletion, PiC loss of ∼60% was still compatible with uncompromised maximal oxidative phosphorylation (oxphos), whereas lower maximal oxphos was evident at ∼85% PiC depletion. Furthermore, intact mutant fibroblasts displayed suppressed mitochondrial bioenergetics consistent with a lower substrate availability rather than phosphate limitation. This was accompanied by slowed proliferation in glucose-replete medium; however, proliferation ceased when only mitochondrial substrate was provided. Both mutant fibroblasts and HeLa cells with 60% PiC loss showed a less interconnected mitochondrial network and a mitochondrial fusion defect that is not explained by altered abundance of OPA1 or MFN1/2 or relative amount of different OPA1 forms. Altogether these results indicate that PiC depletion may need to be profound (>85%) to substantially affect maximal oxphos and that pathogenesis associated with PiC depletion or loss of function may be independent of phosphate limitation when ATP requirements are not high.
Keywords: confocal microscopy, energy metabolism, mitochondria, mitochondrial disease, mitochondrial transport, live cell imaging, mitochondrial dynamics
Introduction
The mitochondrial phosphate carrier (PiC),4 encoded by the nuclear gene SLC25A3, catalyzes the transport of inorganic phosphate (Pi) into the mitochondrial matrix (for a review, see Ref. 1); transport is electroneutral and either in symport with H+ or in exchange for OH−. PiC is a typical member of the family of mitochondrial inner membrane carriers with six transmembrane segments, 3-fold symmetry, and N and C termini projecting into the intermembrane space (2). SLC25A3 has alternatively spliced forms of exon 3 (referred to as exon 3A and 3B) giving rise to PiC-A and PiC-B isoforms. PiC-B is expressed ubiquitously, whereas PiC-A is confined to cardiac and skeletal muscle (3, 4). PiC-A has a 3-fold higher transport affinity for Pi, whereas PiC-B has a 3-fold higher maximal transport rate (5). Turnover rate has been estimated at 50,000/s at 25 °C, which is high among carriers (6).
The transport characteristics of PiC favor it as the most important, or only, means of supplying Pi for oxidative phosphorylation (oxphos) as well as for other Pi-dependent processes within the matrix such as Ca2+ buffering. The functional importance of the PiC was first demonstrated in yeast (Saccharomyces cerevisiae) that exhibited delayed growth or failed to grow on non-fermentable substrate when deleted for MIR1, the yeast homologue of PiC (7). That the yeast ΔMIR1 mutant fails to thrive specifically when grown on mitochondrial substrates supports the relevance of MIR1 for oxphos.
Fundamental aspects of Pi transport into mammalian mitochondria and its role in oxphos have been known for decades. However, only very recently has the role of PiC in oxphos and other aspects of mitochondrial function such as in control of the permeability transition pore been directly tested in mammals. This was achieved using mouse models of conditional heart-specific PiC depletion (8, 9) and depletion of PiC in HeLa cells (10) and through the discovery of pathologic variants in the human gene (11–13). These variants are both in exon 3A, specifically c.158–9A→G and c.215G→A, which each cause a frameshift and absence of protein. Functional analyses in skeletal muscle biopsy from homozygous individuals revealed greatly suppressed pyruvate and succinate oxidation rates despite elevated rates of maximal electron transport chain activity (11). Phenotypically, the individuals harboring these exon 3A mutations displayed muscle-related abnormalities soon after birth, namely elevated blood lactate, hypertrophic cardiomyopathy, and skeletal muscle hypotonia.
The first variants in SLC25A3 that potentially impact both PiC-A and PiC-B were recently described by one of us in a case report (13). An infant was diagnosed prenatally with cardiac hypertrophy, subsequently required a heart transplant, and is stable. There was no evidence of lactic acidosis or of skeletal muscle dysfunction. Whole-exome sequencing revealed a compound heterozygous mutation in SLC25A3: c.599T→G (p.L200W) and 22c.886_898delGGTAGCAGTGCTTinsCAGATAC23 (p.Gly296_Ser300delinsGlnIlePro). These represent two novel variants in SLC25A3 and impact both PiC isoforms. Protein modeling predicted both variants to be pathogenic (13).
Although the relevance of PiC in bioenergetics and in human health is now understood, little is known about its role at the cellular level and the specific mechanisms mediating the cellular impairments induced by pathological PiC variants (11, 12). For example, PiC depletion likely affects mitochondrial ATP production that is central to mitochondrial fusion and mitochondrial quality control. In the current report, we use yeast and mammalian cell models, including primary fibroblasts harboring the compound heterozygous mutation, to elucidate the pathogenicity of the novel variants, both individually and when co-expressed as heterozygous variants. In addition, we study the impact of PiC depletion on energy metabolism and mitochondrial dynamics.
Results
The PICQIP Mutant, but Not PICL200W, Is Non-functional
The identified human PiC mutations were each transformed into the Δmir1 yeast strain (Fig. 1A). Additionally, a humanized version of yeast Pic1p (PICHum) was included to delineate consequences of the mutation from that of the inherent differences between human and yeast proteins. The PICL200W missense variant, which models the point mutation, was similar to wild-type (WT) human PiC in terms of protein amount and functional growth (Fig. 1, A and B). Furthermore, there was no expression or growth defect observed for PICL200W even at elevated temperature (37 °C) (Fig. 1, C and D). In contrast, the PICQIP variant, which models the GSSAS → QIP mutation, resulted in a slightly decreased protein level (see Fig. 1A, compare QIP with Hum) and the inability to survive in respiratory medium (Fig. 1B), consistent with a defect in oxphos. These effects cannot be attributable to the humanized construct because reconstitution with PICHum, which expresses the wild-type GSSAS sequence, showed comparable expression and functionality to WT yeast at both optimal and higher temperatures (Fig. 1, A–E).
FIGURE 1.

Modeling the novel compound heterozygous mutation in human PiC using yeast. A, steady-state protein expression and growth analyses of PiC variants and humanized PiC in comparison with WT. Immunoblotting for Pic1p and the loading controls Tom70p and Atp2p was done using yeast whole cell extracts. B, 1:4 dilutions of the same set of strains were tested for functional growth using the indicated media. Plates were incubated at 30 or 37 °C for 3 days except for YPEG at 37 °C, which was cultured for 10 days. C, steady-state protein expression and growth analyses of WT PIC, PICL200W variant, and PICHum in the presence or absence of co-expressed PICQIP as in A. D, 1:4 dilutions of the same set of strains were tested for functional growth using the indicated media at the designated temperature. Plates were incubated for 3 days (YPD at 30 °C), 5 days (YPD at 37 °C and YPEG at 30 °C), or 13 days (YPEG at 37 °C). E, quantification of PIC or PICL200W protein amounts at 37 °C in the presence or absence of PICQIP (n = 9). Data are presented as means; error bars represent S.E. n.s., differences not significant. EV, empty vector; YPD, 1% yeast extract, 2% peptone, 2% dextrose; YPEG, 1% yeast extract, 2% peptone, 1% ethanol, 3% glycerol.
Because these novel PiC mutations were identified in a compound heterozygote, we used the yeast model to ask whether PICQIP could act in a dominant negative manner. To this end, pRS316PICQIP was transformed into Δmir1(pRS315), Δmir1(pRS315PIC1), Δmir1(pRS315PICL200W), or Δmir1(pRS315PICHum) strains. Growth on fermentable substrate was normal for all strains (Fig. 1D) and was hindered on non-fermentable substrate only when two copies of PICQIP were expressed. Thus, PICQIP is not sufficient to cause problems in expression and respiratory growth in yeast when a functional copy of PIC (WT, L200W, or humanized) is present (Fig. 1, C and D).
Delayed Proliferation and a Lower Functional Respiratory Capacity in PiC Compound Mutant Fibroblasts
We obtained primary skin fibroblasts from the individual expressing the compound heterozygous PiC mutation (PiC mutant) (13) as well as from four control (Ctrl) individuals. The tendency for a lower expression of PiC protein in the yeast cells transformed with the QIP mutation prompted us to evaluate PiC mRNA and protein expression in the fibroblasts. For quantitative PCR, primers recognizing mRNA for the PiC-A and PiC-B isoforms were validated using human skeletal myoblasts, which are known to predominantly express PiC-B and then to up-regulate PiC-A upon differentiation into myotubes (Fig. 2A). Quantitative PCR analysis using these validated primers revealed unchanged mRNA levels for both isoforms in PiC mutant fibroblasts (Fig. 2A; note that PiC-B is the predominant isoform in fibroblasts). Differently, immunoblot analysis using an antibody raised against the full-length PiC protein showed that PiC expression was ∼40% of the level in Ctrl fibroblasts as determined using fibroblasts from four healthy individuals (Fig. 2B; the representative blot shows two of the control fibroblast lines); the antibody typically revealed two bands, and only the upper band was quantified. Lower molecular weight fragments, which would indicate cleavage products, were not detected. In contrast to this lower PiC protein abundance, the protein expression level of many other mitochondrial proteins was similar between PiC mutant and Ctrl cells (Fig. 2B), pointing to a selective decrease in PiC protein in PiC mutant cells.
FIGURE 2.

Lower PiC protein expression in primary fibroblasts harboring the compound heterozygous PiC mutation. A, mRNA, as assessed by quantitative PCR analysis, of isoforms A and B was unchanged by the PiC mutant. Ctrl data were obtained from fibroblasts from three normal individuals; PiC mutant data were obtained from two different passages of the fibroblasts expressing the compound SLC25A3 mutation. As controls for identification of isoform A and B mRNA, mRNA was isolated from primary human skeletal myoblasts and myotubes (upper panels); myotubes were expected to show up-regulated PiC-A compared with myoblasts. B, left panels, example of an immunoblot from PiC mutant fibroblasts and Ctrl fibroblasts from two different individuals. Arrow, band used to quantify PiC abundance. Right panel, quantification from Ctrl fibroblasts from four normal individuals (each tested at three different passages) and from five separate passages (less than passage 10) of the PiC mutant fibroblasts. For each genotype, band intensity was first normalized to that of α-tubulin. Data are presented as means, and error bars represent S.E. except for the skeletal muscle cells, which are each from a single sample. C-I, C-II, and C-III, complexes I, II, and III; Cyto-c and cyt c, cytochrome c.
To investigate the effect of the PiC compound heterozygous mutation on proliferative capacity in a mammalian system, the primary skin fibroblasts were grown in parallel with Ctrl skin fibroblasts in different media. PiC mutant cells grown in glucose-rich medium appeared healthy but divided more slowly with a doubling time that was approximately twice the Ctrl rate (Fig. 3, upper and middle panels) whether cells were grown in 5 mm glucose or in 25 mm glucose. When cells were challenged with medium free of glucose and containing only mitochondrial substrates (β-hydroxybutyrate and glutamine), Ctrl fibroblasts continued to proliferate although at a much slower rate. In contrast, PiC mutant fibroblasts could not maintain proliferation when glucose was unavailable (Fig. 3, lower panel).
FIGURE 3.
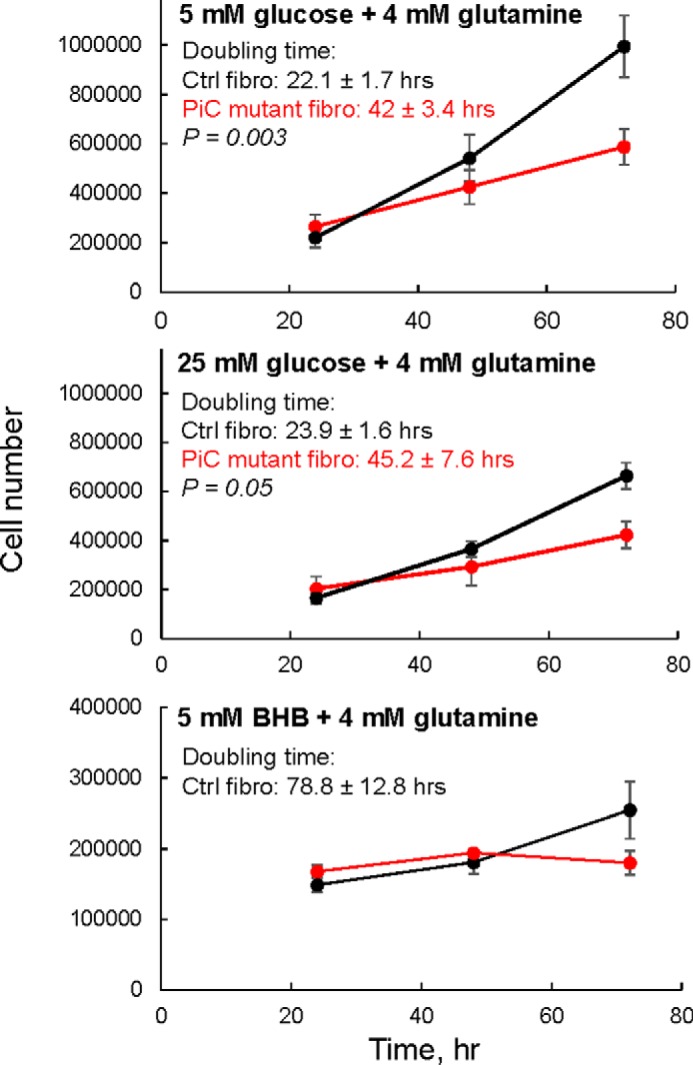
Slower proliferation rate in primary skin fibroblasts harboring the compound heterozygous PiC mutation. Cells tested in glucose plus glutamine had been growing in that substrate condition for two to three passages. Cells tested in β-hydroxybutyrate (BHB) plus glutamine (no glucose) were switched to that condition the day before cell counting was started. DMEM + 10% FBS was used as the base medium throughout. n = 3–6 growth curves/genotype derived from cells at different passages (less than passage 10). In the 25 mm glucose and no glucose conditions, Ctrl cells were from two different subjects (n = 3/subject). In the 5 mm glucose condition, Ctrl cells were from one subject. Data are presented as means; error bars represent S.E. Doubling time was significantly longer in PiC mutant cells; p values were calculated using an unpaired t test. fibro, fibroblasts.
Although the yeast models indicated the lack of a dominant negative effect of the GSSAS → QIP mutation, the patient-derived fibroblasts suggested that the presence of the mutation rendered the PiC protein unstable, leading to substantially lower levels of PiC protein, which could suppress oxphos. To determine whether this occurred, O2 consumption (JO2) was measured. Intact PiC mutant fibroblasts grown and studied in low or high glucose (plus 4 mm glutamine) had ∼50% of the basal mitochondrial JO2 (i.e. minus the rate measured after addition of antimycin to inhibit complex III of the electron transport chain) of the Ctrl cells (Fig. 4A shows raw O2 traces from an experiment, and Fig. 4B shows group averages). To indirectly determine whether a Pi limitation on oxphos could account for the lower basal JO2, we evaluated the leak-dependent mitochondrial JO2 (i.e. after addition of oligomycin to inhibit the ATP synthase) and the uncoupler-driven JO2 (using FCCP to evaluate electron transport chain capacity under the prevailing substrate conditions); if a Pi limitation on oxphos was the sole driver of the lower basal JO2 in PiC mutant cells, then leak-dependent and uncoupled JO2 should remain similar to that in Ctrl cells. Differently, both of these were lower in PiC mutant cells, pointing to a broader defect that results in not only lower basal JO2 but also decreased functional capacity of the electron transport chain.
FIGURE 4.
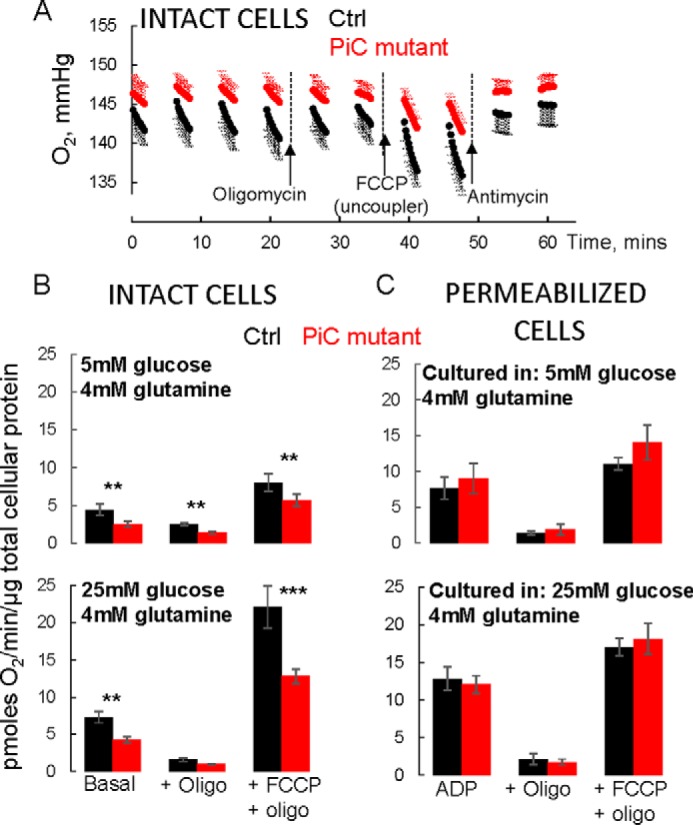
Suppressed bioenergetics in intact but not permeabilized PiC mutant skin fibroblasts. A, raw O2 traces from a single experiment where a Ctrl line and the PiC mutant cells were studied in parallel. Cells were grown and the experiment was performed in 5 mm glucose + 4 mm glutamine as substrates. Values are means ± S.D. with six technical replicates each. Oligomycin (Oligo) was injected to inhibit ATP synthase to measure leak-dependent respiration. Capacity of the electron transport chain under the prevailing substrate condition was determined using the chemical uncoupler FCCP. Finally, antimycin was injected to inhibit the electron transport chain to reveal non-mitochondrial O2 consumption. Note that protein abundance was similar for Ctrl and PiC mutant cells (∼10% higher for mutant cells). B and C, all values are mitochondrial O2 consumption rates calculated by subtracting the O2 consumption rate measured after antimycin. The basal rate is the rate measured in the absence of inhibitors or uncoupler. Cells were grown in the indicated substrate conditions for two to three passages before testing in those conditions. p values are as follows: ***, p < 0.01 from Tukey post hoc test after two-way ANOVA (p = 0.011 for genotype × JO2 condition interaction); **, p = 0.03 from two-way ANOVA (genotype × JO2 condition interaction not significant; p = 0.03 for genotype main effect; p < 0.001 for JO2 condition main effect). C, cells were cultured in the indicated media and then preincubated in those media for 45 min in 0 CO2 before being switched to intracellular medium containing 10 mm pyruvate, 2.5 mm malate, saturating ADP, and a permeabilizing reagent (see “Experimental Procedures” for further details). Permeabilization was confirmed by showing robust succinate-driven O2 consumption; succinate does not cross the plasma membrane (not shown). Data were normalized to total cellular protein from parallel plates of intact cells. n = 3/genotype from cells at different passages (less than passage 10). In B and C, data are presented as means, and error bars represent S.E. For the 5 mm glucose condition, n = 4/genotype from cells at different passages (less than passage 10). For the 25 mm glucose condition, four separate Ctrl fibroblast lines were tested, each in triplicate (three different passages); triplicates were averaged. Data from PiC mutant cells were split into parallel cultures for the different controls and then tested at three different passages each. Thus, n = 4/genotype for 25 mm glucose.
To further investigate the possibility of a Pi limitation on oxphos in PiC mutant fibroblasts, JO2 was measured in permeabilized cells supplied with either pyruvate + malate (Fig. 4B) or succinate + rotenone (not shown) and in the presence of saturating ADP (maximal phosphorylating JO2; state 3), followed by addition of oligomycin and then of FCCP. There were no differences between Ctrl and PiC mutant cells for either of the substrates or any of the conditions. Notably, state 3 JO2 was not different, indicating the availability of sufficient Pi to drive maximal oxidative phosphorylation in PiC mutant cells.
HeLa Cells with Substantial PiC Depletion Can Sustain Oxidative Phosphorylation
The continued presence in PiC mutant fibroblasts of oxphos, despite the ∼60% lower PiC protein abundance, suggested either that the capacity of PiC is far in excess of its requirement for oxphos in these cells or that alternative sources of Pi are available to the matrix. To further test whether lower PiC can be compatible with substantial oxphos, PiC was acutely depleted in HeLa cells using siRNA; two different siRNA duplexes were investigated (kd1 and kd2; Fig. 5). By 72 h post-siRNA introduction, PiC protein was ∼15% of control levels (Fig. 5A). In contrast, the protein expression level of other mitochondrial proteins was unchanged 72 h post-siRNA treatment (Fig. 5A). Cell proliferation, measured using cells seeded at 48 h post-siRNA addition, was delayed by PiC depletion (Fig. 5B). Measurements of JO2 in permeabilized cells revealed a 30–40% suppression of state 3 JO2 with either pyruvate + malate (Fig. 5C) or succinate (+rotenone; not shown), whereas leak-dependent JO2 and uncoupled JO2 were unchanged by PiC knockdown (Fig. 5C); these results are consistent with a Pi limitation on oxphos as the cause of the lower state 3 JO2. In intact cells, substantial PiC depletion had no effect on basal, leak-dependent, or uncoupler-driven JO2. We further investigated oxphos in HeLa cells that had a lesser acute depletion of PiC; cells were depleted by ∼60–65% (Fig. 5D), which is similar to the extent of the PiC decrease in PiC mutant fibroblasts (Fig. 2B). In contrast to permeabilized HeLa cells with ∼85% decrease in PiC, permeabilized HeLa cells expressing slightly more PiC had the same maximal phosphorylating JO2 as Ctrl HeLa cells. Altogether, the data from intact and permeabilized HeLa cells with different levels of PiC deletion further support that at least some oxphos can be maintained despite substantial PiC depletion.
FIGURE 5.

Acute PiC depletion in HeLa cells is still associated with substantial phosphorylating O2 consumption. A, immunoblots from HeLa cells exposed for 72 h to one of two different siRNA duplexes targeting PiC or scrambled controls. For each genotype, bands were normalized to either succinate dehydrogenase subunit A (SDHA) or α-tubulin. n = 3/genotype. B, growth curves in HeLa cells exposed to siRNA for 72 h and grown in DMEM + 25 mm glucose + 4 mm glutamine. p values (doubling times) are from an unpaired t test. n = 3/genotype. C, mitochondrial bioenergetics measured in cells exposed to one of two different siRNA duplexes for 72 h, grown and tested in either DMEM + 25 mm glucose + 4 mm glutamine (Intact), or permeabilized and supplied with pyruvate and malate (Permeabilized). See “Experimental Procedures” and Fig. 4 for further details. n = 3–5/genotype. D, immunoblots showing lesser depletion of PiC in HeLa cells exposed to siRNA for 48 h. Cells were grown in DMEM + 25 mm glucose + 4 mm glutamine, and mitochondrial bioenergetics were measured in that medium (Intact) or subsequently permeabilized and supplied with pyruvate and malate (Permeabilized). See Fig. 4 for further details. n = 3–5/genotype. In all panels, data are presented as means, and error bars represent S.E. For the immunoblots, p values are as follows: ***, p < 0.01; **, p = 0.02; *, p < 0.046 (unpaired t test). For the JO2 measurements, p values are as follows: ***, p < 0.001; *, p = 0.035 (Tukey post hoc tests following two-way ANOVA with significant genotype × JO2 condition interaction). Two different siRNA duplexes were used (kd1 and kd2), and controls were run with each (Ctrl 1 and Ctrl 2). Oligo, oligomycin; C-I, C-II, and C-III, complexes I, II, and III; cyto-c, cytochrome c.
Altered Mitochondrial Morphology in PiC Compound Mutant Fibroblasts and in HeLa Cells with Acute PiC Depletion
Emerging literature supports a mutual relationship between mitochondrial function and form (15). Indeed, visualization of mitochondrial morphology in fibroblasts expressing mitochondrial matrix-targeted DsRed1 (mtDsRed) revealed fewer elongated mitochondria in PiC mutant than in Ctrl fibroblasts (Fig. 6, A and B). Furthermore, in HeLa cells with ∼60% decrease in PiC a similar change in the mitochondrial form was obtained (Fig. 6C). A decrease in the fraction of elongated organelles can result from decreased mitochondrial fusion or increased mitochondrial fission.
FIGURE 6.

Mitochondrial morphology in primary PiC mutant fibroblasts and PiC-deficient HeLa cells. Mitochondrial morphology was scored as follows: fragmented, mainly small and round; partly fragmented, intermediate, mixture of round and shorter tubulated; normal, tubulated, long and higher interconnectivity; elongated, very long, tubulated. The percentage of cells with the indicated mitochondrial morphologies was determined as a percentage of the total number of DsRed-transfected cells counted (≥18 cells per experiment; n = 4 independent experiments). A, representative figures of the different mitochondrial forms in Ctrl and PiC mutant fibroblasts. B, distribution of the different type of mitochondria in primary fibroblasts. C, distribution of the different types of mitochondria after PiC silencing in HeLa cells. Scr., scrambled siRNA; PiC kd1 and kd2, two different PiC-specific duplexes.
Less Fusion Activity in PiC Compound Mutant Fibroblasts and in HeLa Cells with Acute PiC Depletion
To evaluate directly whether the distinct mitochondrial morphology in PiC patient and control fibroblasts can result from different levels of interorganellar continuity and fusion events, the cells were co-transfected with cDNA encoding mtDsRed and mitochondrial matrix-targeted photoactivatable GFP (mtPA-GFP). Using confocal microscopy, a time series of fluorescence images was recorded, and 25-μm2 square-shaped areas were illuminated by a pulsed laser to photoactivate mtPA-GFP (16, 17). Mitochondrial matrix continuity and connectivity are unveiled by the diffusion of the photoactivated PA-GFP to the regions outside the 2 photon illuminated area. Image time series indicated less mtPA-GFP diffusion in PiC mutant than in Ctrl fibroblasts (Fig. 7A). To quantify connectivity, the time course of the ratio of FmtPA-GFP and FmtDsRed was calculated for the region of photoactivation (RPA) (Fig. 7A). The slower decay of the fluorescence ratio in the RPA indicates a decrease in the combined activity of mitochondrial network formation (40 s) and mitochondrial fusion and mitochondrial movements (500 s) in the PiC mutant fibroblasts (Fig. 7B). To validate fusion events, reciprocal spreading of mtPA-GFP and mtDsRed among mitochondria that were not continuous at the time of mtPA-GFP photoactivation was sought. The fusion events were quantitatively analyzed by the progression of the distribution of mtPA-GFP fluorescence between the images collected within the first 24 s and 8 min after photoactivation. The result of manual counting indicates less mitochondrial fusion activity in the PiC mutant fibroblasts. The total fusion numbers were decreased significantly in the mutant fibroblasts (Fig. 7C). Although Fig. 7C shows fusion event number for only one control fibroblast line that was studied side by side with the PiC mutant, the fusion event numbers were very similar in other control fibroblast lines (7.3 ± 0.3 versus 6.5 ± 0.1, 7.5 ± 0.1, and 6.4 ± 0.3; n = 3–7). Conversely, the average duration of individual fusion events was similar in both PiC mutant fibroblasts and Ctrl (Fig. 7D). Furthermore, the distribution of different fusion types in terms of rapid reversal and orientations (Fig. 7E) did not show any alterations in PiC mutant fibroblasts as compared with the Ctrl. In HeLa cells with ∼60% decrease in PiC (attained by two different siRNA duplexes, kd1 and kd2), the mitochondrial connectivity and fusion activity were similarly decreased as in the mutant fibroblasts (Fig. 7, F–I).
FIGURE 7.

Mitochondrial continuity and fusion activity in primary fibroblasts. A, image time series showing a representative fibroblast before and after 2P photoactivation of mtPA-GFP (white squares) at 0 s (first step of the imaging), 24 s (first step directly after photoactivation), 40 s (the mtPA-GFP diffusion within the first 16 s of photoactivation), and 500 s (further spreading after 8 min). A region of interest for each condition (dashed white line) is magnified and shown as PA-GFP only (green) images for each time point. B, the time course of the ratio of FmtPA-GFP and FmtDsRed for the RPA (above) and the decay of the fluorescence ratio in the RPA at 40 and 500 s (below). Asterisks indicate significant differences (p < 0.05). C, fusion event rate in primary fibroblasts. D, average fusion times. E, distribution of different fusion types (left) and orientation (right). F–I, HeLa cells treated with scrambled siRNA or kd1 or kd2 duplexes targeting PiC. F, the time course of the normalized FmtPA-GFP for the region of photoactivation (RPA) (left) and the decay of the fluorescence ratio in the RPA at 500 s (right). Asterisks indicate significant differences (p < 0.05). Scr., scrambled siRNA-silenced cells; PiC kd1 and kd2, PiC siRNA-treated cells. G, fusion event rate in silenced HeLa cells. H, average fusion times in silenced HeLa cells. I, distribution of different fusion types (left) and orientation (right) in silenced HeLa cells. In B, C, D, F, G, and H, error bars represent S.E.
ATP produced by oxphos is required to maintain the fusion promoting activity of OPA1, the inner mitochondrial membrane fusion protein (18, 19). To test whether the decreased mitochondrial fusion activity can be attributed to a change in OPA1 or another mitochondrial fusion protein, immunoblotting was performed in whole cell lysates. The total abundance of OPA1 was unaltered in PiC mutant fibroblasts (Fig. 8A). A shift in OPA1 immunoreactivity from the high to the lower molecular weight bands indicates a decrease in the fusion-competent forms (as induced by FCCP; Fig. 8B). However, no change in the relative abundance of the high and low molecular weight bands appeared in the PiC mutant fibroblasts (Fig. 8B). Among the main outer membrane fusion proteins, mitofusin 1 (MFN1) was slightly but significantly decreased, whereas mitofusin 2 (MFN2) and the main fission protein DRP1 were unchanged (Fig. 8A). In HeLa cells with 60% PiC depletion, the abundance of none of these fusion and fission proteins was altered (Fig. 8C). Collectively, the results in two different paradigms of PiC depletion show suppressed mitochondrial fusion activity. However, the decreased fusion activity cannot be attributed to processing of OPA1 to fusion-incompetent forms or to substantial loss of another fusion protein.
FIGURE 8.

Abundance of mitochondrial fusion and fission proteins in PiC-deficient primary fibroblasts and HeLa cells. A, Western blotting of the main fusion and fission proteins in the Ctrl and PiC-deficient fibroblasts. Left, representative blots; right, normalized protein abundance of the percentage of the average protein levels of control fibroblast. B, OPA1 isoforms. FCCP (5 μm) was used as a positive control to show the mitochondrial uncoupling-induced loss of the long form of OPA1. C, Western blotting of the main fusion and fission proteins in HeLa cells. Scr., scrambled siRNA-silenced cells; PiC kd, PiC siRNA-treated cells. Left, representative blots; right, normalized protein abundance of the percentage of the average protein levels of scrambled siRNA-silenced cells. In A and C, error bars represent S.E.
Discussion
The novel compound heterozygous mutations in SLC25A3 described here and previously (13) were associated in vivo with respiratory distress and hypertrophic cardiomyopathy but with unremarkable function of other organs, including skeletal muscle (13). The pathological potential of the novel SLC25A3 mutations was not tested directly and was our aim. Here we report that the point mutation is likely to be functionally neutral, whereas the GSSAS → QIP variant has relevance compatible with a loss of function. Furthermore, the GSSAS → QIP variant does not obligatorily act as a dominant negative. However, in primary fibroblasts harboring the compound heterozygous PiC mutation and thus one potentially functional allele, PiC protein abundance was substantially lower, and the cells were not phenotypically normal; the most dramatic phenotype was a proliferation time that was approximately half the control rate and an inability to divide when supplied with only mitochondrial substrates. PiC mRNA was unaffected by the mutations. Thus the compound heterozygous PiC mutation has a functional impact in mammalian cells with instability of the PiC protein as a likely initiating factor. We also found in patient fibroblasts, as well as HeLa cells with different levels of acute PiC loss, that PiC protein could be substantially depleted before oxidative phosphorylation was suppressed. This suggests that the PiC can be expressed in excess of oxphos needs or that other PiC transport mechanisms can compensate. However, PiC mutant fibroblasts could not proliferate in mitochondrial substrate, suggesting that a partial depletion of PiC can become pathogenic when demand for mitochondrial ATP is high. Finally, PiC mutant fibroblasts, as well as HeLa cells with acute PiC loss, had a more fragmented mitochondrial network and slower rate of inner mitochondrial membrane fusion, raising the possibility that organ pathology might be contributed by dysfunctional mitochondrial processes other than, or in addition to, suppressed oxphos.
The Δmir1 yeast strain transformed with one or both of the newly identified human PiC variants was used to evaluate the functional relevance of these variants. Leu-200 is conserved among mammalian species and in yeast and is located within a region that is fully to strongly conserved between human and yeast. Based on the model of PiC determined by Bhoj et al. (13), Leu-200 resides in a small matrix helix that connects transmembrane helices 3 and 4 and interacts with adjacent hydrophobic residues near PX(D/E)XX(K/R), the consensus sequence of all inner mitochondrial membrane carrier proteins. It was predicted that substitution of leucine for tryptophan would not be well tolerated due to tryptophan's large hydrophobic side chain, which would be exposed to the solvent and could introduce steric clashes (13). However, the L200W substitution did not prevent growth on mitochondrial substrate even at elevated temperature, suggesting that this variant is neutral. In contrast, the GSSAS → QIP variant prevented growth only on mitochondrial substrate even in unchallenged yeast and despite only slightly decreased PiC protein abundance. This is in line with a loss-of-function mutation and a deleterious effect on oxphos. Based on the nomenclature of the ADP/ATP carrier, GSSAS occurs at matrix loop-connecting transmembrane helices 5 and 6. The variant replaces very small (Gly and Ala), flexible (Gly), and polar (Ser) residues for bulky (Gln), hydrophobic (Ile), and inflexible (Pro) residues. Thus, GSSAS → QIP was predicted to not be well tolerated (13), which is what we found here. Because previously confirmed pathological SLC25A3 variants affected only PiC-A (11, 12), GSSAS → QIP is the first variant that affects both isoforms.
PiC mutant fibroblasts with ∼60% less PiC protein had lower basal JO2, maximal leak-dependent JO2, and maximal electron transport chain activity. Changes in bioenergetics caused only by a Pi limitation on oxphos would be restricted to suppressed phosphorylating JO2 as we observed in permeabilized HeLa cells with >85% PiC loss. In contrast, permeabilized mutant fibroblasts had similar maximal phosphorylating JO2 as Ctrl fibroblasts, indicating that Pi did not limit oxphos. Moreover, lack of a difference in basal or uncoupled JO2 in permeabilized mutant fibroblasts suggests that the abundance of assembled respiratory complexes and ATP synthase as well as their ability to assemble into higher order complexes was unchanged in mutant fibroblasts. Rather, the decrease in not only basal but also leak-dependent JO2 and maximal electron transport chain activity in intact mutant fibroblasts is consistent with lower provisioning of reducing equivalents to the electron transport chain.
The pronounced decrease in JO2 in intact mutant fibroblasts contrasts with minimal decrease in intact HeLa cells with greater PiC depletion. This different response to PiC depletion may arise from long versus short term PiC loss. Another possibility is that mutant PiC protein in fibroblasts induced a stress response that might not occur in HeLa cells that were depleted of PiC through suppression of mRNA rather than mutation. In fact, lower PiC protein was not predicted by either of the mutations which are in-frame; indeed, mRNA expression of both PiC isoforms was unaffected. The lower PiC abundance in the mutation-harboring fibroblasts is also unlikely to reflect natural variability in translation efficiency or turnover because these PiC variant cells deviated similarly in their PiC abundance as compared with fibroblasts from four healthy donors. Rather, depletion of PiC protein in the mutant fibroblasts may reflect an inability of PiC to be properly translated or a tendency for mutant PiC to aggregate, to be misfolded, or to be incorrectly incorporated into complexes (which likely include dimers as well as other components such as the adenine nucleotide translocase (20–22)). Such disruptions in PiC protein folding might induce a mild stress response that is partially responsible for the mitochondrial phenotypes observed in mutant fibroblasts.
The substantial oxphos remaining in permeabilized mutant fibroblasts and PiC-depleted HeLa cells suggests that the abundance of PiC is in excess of oxphos needs or that alternate Pi transport pathways compensate for PiC loss. These possibilities are also suggested by observations made in mice with either ∼60 or >95% depletion of PiC in the heart (8, 9). Cardiac PiC depletion of ∼60% was associated with very mild cardiac hypertrophy and no discernible functional defect (9). Even when PiC was depleted by >95%, cardiac function was unaffected for at least 2 weeks, and ATP in the mitochondrial fraction was 50% of normal (8), suggesting compensation at the level of mitochondria or that very little PiC can sustain some level of oxphos. Using a fresh skeletal muscle biopsy from an individual with a homozygous c.215G→A mutation affecting only PiC-A, substrate oxidation was tested using a variety of conditions and was clearly suppressed (11). However, the extent of suppression varied considerably, ranging from as low as ∼10% of control (using the lower extent of the control range) to >30% of control. Because uncoupler-driven oxidation was within the control range, the variability in substrate oxidation would not be due to differences in the capacity to supply reducing equivalents to the electron transport chain or in the capacity of the electron transport chain itself. Rather, the variability in oxidation reported by Mayr et al. (11) could arise from variable access to alternate Pi transport pathways such as the dicarboxylate transporter, which exchanges succinate or malate for Pi (23). Generally the PiC has been assumed to be the Pi uptake mechanism in mitochondria. Although the present study and those discussed above cannot distinguish between excess PiC capacity and alternate Pi transport, the observations collectively suggest that this is an important question that needs to be addressed by future studies.
A shift of mitochondrial morphology to a less interconnected phenotype was present in both PiC mutant fibroblasts and HeLa cells with quantitatively similar acute PiC depletion. This shift can result from the observed decrease in mitochondrial fusion activity. A sensible mechanism to underlie decreased fusion could be a decrease in fusion-competent OPA1 that is dependent on oxphos-derived ATP (18, 19). However, neither total OPA1 nor the relative contribution of the different forms was altered in mutant fibroblasts or in the PiC-depleted HeLa cells. A decrease in the amount of the outer membrane fusion mediator MFN1 or MFN2 could also cause suppression of mitochondrial fusion (17, 24). Indeed, there was a lower level of MFN1 in the mutant fibroblasts, but it was unaltered in the PiC-deficient HeLa cells that displayed a similar mitochondrial fusion phenotype to the fibroblasts. Furthermore, MFN2 was unchanged in both PiC-deficient paradigms. Activity of the fusion proteins can also be affected by posttranslational modifications, but no specific mechanism appeared likely here and because of that was not tested. Although the specific molecular mechanism linking PiC activity to fusion requires further investigation, the present findings might illuminate a common cause of mitochondrial fragmentation observed in fixed skin fibroblasts derived from patients with various oxphos impairments with a general trend that fragmentation increases with severity of the defect (25–28). Based on the present results, live cell mitochondrial imaging in single cells might detect even a mild oxphos (or mitochondrial) defect that remains unnoticeable by JO2 measurements.
The patient harboring the compound heterozygous PiC mutation had severe hypertrophic cardiomyopathy that required heart transplantation but did not have skeletal myopathy, lactic acidosis, or other obvious organ dysfunction (13). In contrast, not only hypertrophic cardiomyopathy but also skeletal muscle hypotonia and lactic acidosis were symptoms in individuals with homozygous mutations leading to loss of PiC-A protein in muscle only (11, 12). PiC-B would still be expressed; however, there did not appear to be compensation, and the levels appeared low (12). Parenthetically, it is also not known to what extent PiC-B can functionally compensate for PiC-A. The compound heterozygous mutation affects both PiC-A and PiC-B, and we have shown here that the GSSAS → QIP variant is deleterious and that the variant(s) can destabilize PiC protein. However, our data also indicate that some PiC protein remains. Furthermore, we show that some oxphos capacity is retained even when PiC protein abundance is quite low. The latter is also supported by mouse models of PiC depletion in the heart (8, 9). However, we also show that oxphos capacity is sensitive to PiC abundance. We propose that these findings are consistent with confinement of the phenotype of the compound heterozygous mutation to cardiomyopathy because of the incessantly high demand for ATP of the heart.
Our study has revealed that PiC depletion is accompanied by defective cell proliferation and mitochondrial fusion and an oxphos defect that does not depend on Pi limitation. Thus, the pathogenesis associated with PiC depletion or loss of function may, at least in part, reflect these deficiencies. It will also be of interest to determine whether these deficiencies are a general feature of pathological PiC variants.
Experimental Procedures
Molecular Biology
Yeast MIR1 gene fragment, with 470-bp 5′-UTR and 50-bp 3′-UTR, was generated by PCR using GA74-1A genomic DNA as template and cloned into pRS315. The PIC1 mutants, L200W and QIP (replacing amino acid residues 247–258 of yeast Pic1p) as well as a humanized version (normal human amino acids GSSAS in place of residues 247–258 of yeast Pic1p) were created by overlap extension (29) and subcloned into pRS315. For the co-expression studies, PICQIP was subcloned into pRS316. Construct verification was performed by restriction digest analysis and/or sequencing.
Yeast Strains and Growth Conditions
All yeast strains used were derived from GA74-1A (MATa, his3–11,15, leu2, ura3, trp1, ade8 (rho+, mit+)). The open reading frame of MIR1 was replaced with HIS3MX6 by a PCR-based disruption method. The resulting strain was then transformed with pRS316PIC1 to prevent loss of mtDNA, and the desired strains were generated by plasmid shuffling. In brief, pRS315, pRS315PIC1, pRS315PICL200W, pRS315PICQIP, or pRS315PICHum was transformed into Δmir1(pRS316PIC1) followed by two selections in 5-fluoroorotic acid to generate clones lacking pRS316PIC1. Growth studies were performed to evaluate yeast strain functionality. Cultures were inoculated in synthetic complete dextrose without leucine (0.17% yeast nitrogen base, 0.5% ammonium sulfate, 0.2% synthetic dropout mixture minus leucine, 2% dextrose), grown overnight at either 30 or 37 °C, spotted in synthetic complete medium without leucine and supplemented with dextrose or 3% glycerol and 1% ethanol, and incubated at the indicated temperature and duration.
To assess whether the PICQIP allele exerts a dominant negative effect on WT PIC1 or the other PIC alleles, pRS316 or pRS316PICQIP was transformed into Δmir1(pRS315), Δmir1(pRS315PIC1), Δmir1(pRS315PICL200W), or Δmir1(pRS315PICHum). Growth analyses were done as described above except that synthetic complete dextrose without leucine and uracil and the same selection medium supplemented with either dextrose or 3% glycerol and 1% ethanol were utilized for inoculation and spotting, respectively.
Mammalian Cell Culture
Primary skin fibroblasts were obtained from the PiC mutant (13) as well as two age-matched controls and two controls from older individuals, resulting in fibroblasts from four healthy individuals that were used as controls. Fibroblasts were cultured in DMEM containing 4 mm glutamine and either 5 mm glucose or 25 mm glucose. For some growth curve experiments, cells were cultured in glucose-free DMEM containing 4 mm glutamine and supplemented with 5 mm β-hydroxybutyrate; this medium forced cells to rely on mitochondria for ATP production (30, 31). PiC mutant and Ctrl cells were only used for 10 passages and were always compared at the same passage number. HeLa cells (ATCC) were cultured in DMEM containing 25 mm glucose, 4 mm glutamine, and 1 mm pyruvate; HeLa cells were not used beyond passage 14. All cells were maintained in a 5% CO2, 21% O2 incubator at 37 °C.
Transfections with siRNA and Plasmid DNA
Before silencing, HeLa cells were plated in antibiotic-free medium, and the samples were silenced in serum-free culture medium with PiC-specific (Invitrogen and OriGene; two different siRNA duplexes) or, as a negative control, scrambled siRNAs (100 nm) using RNAiMAX (Life Technologies). After 4-h incubation, the medium was completed with antibiotic-free 30% FBS-containing medium. The next day, the medium was changed to normal growth medium, and the cells were grown for 72 h. The efficacy of the silencing was validated with Western blotting. For evaluation of mitochondrial morphology and fusion dynamics, the cells were transfected with mtDsRed (1 μg) and mtPA-GFP (1 μg) plasmid DNAs using Lipofectamine 2000 (Life Technologies) 24 h before the experiment.
Growth Curves
Cell growth curves were performed in fibroblasts and HeLa cells. For all conditions, cells were counted in duplicate wells for each time point. Cell counting was performed using a hemocytometer. Each growth curve was performed in cells from independent passages. For HeLa cells, growth curves were started 72 h after treatment with siRNA; continued knockdown for another 48 h was confirmed by Western blotting.
Mitochondrial Bioenergetics
JO2 was measured in adherent cells using the Seahorse XF24. Fibroblasts or HeLa cells were seeded at 40,000/well the day before the experiment. For experiments in intact cells, cells were washed two times in bicarbonate-free DMEM containing the amount of glucose in which the cells had been cultured (i.e. 5 or 25 mm) without or with 4 mm glutamine, pH 7.4 at 37 °C, and then 650 μl of that medium/well was added as the final assay medium. Injections of oligomycin (0.5 μg/ml final concentration), FCCP (see figure legend for final concentrations), and antimycin (1 μm final concentration) were prepared in the respective assay media. To equilibrate cells and degas the culture plate, cells were preincubated at 37 °C in environmental CO2 for 45 min.
For experiments in permeabilized cells, cells were washed and preincubated as for intact cells. After the 45-min preincubation, cells were washed again with mitochondrial assay solution (MAS) (32) (70 mm sucrose, 220 mm mannitol, 10 mm KH2PO4, 5 mm MgCl2, 2 mm HEPES, 1 mm EGTA and 0.2% (w/v) BSA, pH 7.2 at 37 °C), and then 450 μl/well MAS containing 10 mm pyruvate, 2 mm malate, 4 mm ADP (from a 1 m stock that had been adjusted to pH 7.2), and permeabilizing reagent (1 nm after optimization; Seahorse Biosciences) were added as the final assay medium. The final concentrations of ADP and substrates are expected to be saturating based on experiments in 5–10 μg of isolated mitochondria (32, 33). Oligomycin (2.5 μg/ml final) and FCCP (4 μm final) were injected sequentially. Some wells only received a final injection of oligomycin + FCCP to record phosphorylating JO2 for a longer time to monitor the stability of JO2; after an increase in JO2 over the first ∼5 min of the experiment, JO2 remained stable for at least 10 min prior to adding oligomycin and uncoupler. The mix-wait-measure protocol used was that recommended for isolated mitochondria (32) that has worked well in our hands (33). To verify that cells were permeabilized, in some experiments, a subset of wells contained succinate (5 mm) and rotenone (1 μm) instead of pyruvate and malate.
For both intact cell and permeabilized cell experiments, JO2 values were normalized to total cellular protein. Plates used for both types of experiment were always plated at the same time, allowing us to use the protein amount obtained from the intact cell experiment for the permeabilized cell experiment (measurement of protein from the latter was confounded by the presence of BSA in the MAS).
Mitochondrial Fusion Dynamics
Imaging measurements were performed as described previously (16, 17). Briefly, cells were incubated in a 0.25% BSA-containing extracellular medium consisting of 121 mm NaCl, 5 mm NaHCO3, 4.7 mm KCl, 1.2 mm KH2PO4, 1.2 mm MgSO4, 2 mm CaCl2, 10 mm glucose, and 10 mm Na-Hepes, pH 7.4, at 37 °C. Recordings of mtPA-GFP and mtDsRed (512 × 512 pixels) were performed using 488 and 561 nm laser lines at 0.25 s−1 data acquisition frequency using an LSM780 microscope and 63×/1.4 numerical aperture Apoplan oil objective. Experiments were run at 37 °C. To photoactivate PA-GFP (and photobleach Ds-Red) in 2P mode in 5 × 5-μm areas, a pulsed laser system (760 nm; Chameleon, Coherent, Inc.) was applied. Image analysis was done using either Spectralyzer (custom designed) or Zen2010 (Carl Zeiss).
Mitochondrial dynamics was evaluated by two different approaches. Spreading of PA-GFP from the area of photoactivation was evaluated by masking the 5 × 5-μm areas and quantifying the time-dependent decay in the fluorescence ratio of PA-GFP and DsRed. The ratio values in each region were normalized to the peak reached during 2P excitation. Fusion events were also counted manually (34). Image analysis was performed in Spectralyzer or Zen2010 imaging software.
Immunoblotting
For yeast studies, protein extraction from yeast cells, SDS-PAGE, and immunoblotting were carried out as described (35, 36). The antibodies used for the yeast studies have been described previously (37–39). Images were captured with the Fluorchem Q system (Cell Biosciences, Inc.), and relative protein levels were quantified using ImageJ. Statistical analyses were performed with StatPlus. For mammalian cell studies, cells were lysed in radioimmune precipitation assay buffer, and then lysates were clarified and assayed for protein content using the bicinchoninic acid assay. Immunoblotting was carried out as described previously (33, 14). The following antibodies were used: SLC25A3 (1:500; Sigma SAB1400208; rabbit), complex I 37-kDa subunit (1:1000; Abcam MS111; mouse), complex II subunit A (succinate dehydrogenase subunit A; 1:1000; Abcam MS204; mouse), complex III core 2 protein (1:1000; Abcam MS304; mouse), MFN1 (1:1000; rabbit polyclonal provided by Richard Youle, National Institutes of Health, Bethesda, MD), MFN2 (1:1000; mouse monoclonal generated against MFN2(710–757)-GST and N-terminal peptide CNSIVTVKKNKRIIM-OH provided by Heidi McBride, McGill University, Montreal, Canada), OPA1 (1:1000; BD Biosciences 612606; mouse), Drp1 (1:500; BD Biosciences 611738; mouse), prohibitin (1:1000; Abcam 28172; rabbit), α-tubulin (1:1000; Cell Signaling Technology 2125; rabbit), cytochrome c (1:1000; BD Pharmingen 556433), and GAPDH (1:10000; Advanced Immunochemicals 8-GAPDH-r; rabbit). Preliminary experiments, not shown, using different amounts of fibroblast and HeLa cell lysate were used to determine the amount of protein that provided a signal that was within the linear range of detection. In addition, proteins that were used as loading controls were chosen because they did not vary with PiC depletion.
Quantitative PCR
Total RNA was extracted from cells using Trizol® (Invitrogen). Purified RNA was treated with RQ1 DNase (Promega, Madison, WI) for 30 min at 37 °C. Total RNA concentration was measured with a Qubit® fluorometer (Invitrogen). RNA was reverse transcribed using oligo(dT)20 primers and SuperScript III (Invitrogen). Quantitative PCRs were done using ITaq SYBR Green Supermix with ROXTM Reference Dye (Bio-Rad) in 20-μl reactions (20 ng of cDNA/reaction) using an Eppendorf Mastercycler® ep realplex. Primers were designed using Eurofins Primer Design Tool: isoform A: forward primer, 5′-CGCCGTGGAAGAGCAG-3′; reverse primer, 5′-CAATGCTGTATGTGTTGTGCC-3′; isoform B: forward primer, 5′-CCGTGGAAGAGTACAGTTG-3′; reverse primer, 5′-AGCAGTGTGTGTAGACC-3′. Custom oligos were purchased from Eurofins MGW Operon (Huntsville, AL).
Statistics
The data are shown as the mean ± S.E. of cells recorded for at least three independent cultures unless it is specified differently. Significance of differences was determined by unpaired Student's t test or one- or two-way analysis of variance with Tukey post hoc tests as appropriate (see figure legends). p < 0.05 was taken as significant.
Author Contributions
E. L. S. and G. H. developed the project and designed the experiments in mammalian cells. S. M. C. designed experiments in yeast. A. G. conducted the mitochondrial morphology and fusion dynamics studies and performed related Western blotting and analyzed the corresponding results. M. G. A. conducted and analyzed experiments in yeast models. Q. L., L. A.-P., T. G., C. M., and E. L. S. conducted bioenergetics and cell proliferation experiments in mammalian cells, performed Western blotting of mammalian cell samples, and analyzed the corresponding results. N. S. procured and provided the human fibroblasts. E. L. S. and G. H. wrote the paper.
Acknowledgment
We thank Meredith Larose for contributing to analysis of the bioenergetics data.
This work was supported in part by United Mitochondrial Disease Foundation Grant 14-145R (to E. L. S.) and National Institutes of Health Grant R01HL108882 (to S. M. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PiC
- mitochondrial phosphate carrier
- oxphos
- oxidative phosphorylation
- PiC-A
- PiC isoform A
- PiC-B
- PiC isoform B
- Ctrl
- control
- JO2
- O2 consumption
- PA-GFP
- photoactivatable green fluorescent protein
- RPA
- region of photoactivation
- MFN1
- mitofusin 1
- MFN2
- mitofusin 2
- kd
- knockdown
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- mtDsRed
- mitochondrial matrix-targeted DsRed1
- mtPA-GFP
- mitochondrial matrix-targeted photoactivatable GFP
- PiC mutant
- individual expressing the compound heterozygous PiC mutation
- MAS
- mitochondrial assay solution
- Hum
- humanized
- ANOVA
- analysis of variance.
References
- 1. Seifert E. L., Ligeti E., Mayr J. A., Sondheimer N., and Hajnóczky G. (2015) The mitochondrial phosphate carrier: role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 464, 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Runswick M. J., Powell S. J., Nyren P., and Walker J. E. (1987) Sequence of the bovine mitochondrial phosphate carrier protein: structural relationship to ADP/ATP translocase and the brown fat mitochondria uncoupling protein. EMBO J. 6, 1367–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dolce V., Iacobazzi V., Palmieri F., and Walker J. E. (1994) The sequences of human and bovine genes of the phosphate carrier from mitochondria contain evidence of alternatively spliced forms. J. Biol. Chem. 269, 10451–10460 [PubMed] [Google Scholar]
- 4. Dolce V., Fiermonte G., and Palmieri F. (1996) Tissue-specific expression of the two isoforms of the mitochondrial phosphate carrier in bovine tissues. FEBS Lett. 399, 95–98 [DOI] [PubMed] [Google Scholar]
- 5. Fiermonte G., Dolce V., and Palmieri F. (1998) Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J. Biol. Chem. 273, 22782–22787 [DOI] [PubMed] [Google Scholar]
- 6. Ligeti E., Brandolin G., Dupont Y., and Vignais P. V. (1985) Kinetics of Pi-Pi exchange in rat liver mitochondria. Rapid filtration experiments in the millisecond time range. Biochemistry 24, 4423–4428 [DOI] [PubMed] [Google Scholar]
- 7. Zara V., Dietmeier K., Palmisano A., Vozza A., Rassow J., Palmieri F., and Pfanner N. (1996) Yeast mitochondria lacking the phosphate carrier/p32 are blocked in phosphate transport but can import preproteins after regeneration of a membrane potential. Mol. Cell. Biol. 16, 6524–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kwong J. Q., Davis J., Baines C. P., Sargent M. A., Karch J., Wang X., Huang T., and Molkentin J. D. (2014) Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 21, 1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gutiérrez-Aguilar M., Douglas D. L., Gibson A. K., Domeier T. L., Molkentin J. D., and Baines C. P. (2014) Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 72, 316–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varanyuwatana P., and Halestrap A. P. (2012) The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 12, 120–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mayr J. A., Merkel O., Kohlwein S. D., Gebhardt B. R., Böhles H., Fötschl U., Koch J., Jaksch M., Lochmüller H., Horváth R., Freisinger P., and Sperl W. (2007) Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 80, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mayr J. A., Zimmermann F. A., Horváth R., Schneider H. C., Schoser B., Holinski-Feder E., Czermin B., Freisinger P., and Sperl W. (2011) Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 21, 803–808 [DOI] [PubMed] [Google Scholar]
- 13. Bhoj E. J., Li M., Ahrens-Nicklas R., Pyle L. C., Wang J., Zhang V. W., Clarke C., Wong L. J., Sondheimer N., Ficicioglu C., and Yudkoff M. (2015) Pathologic variants of the mitochondrial phosphate carrier SLC25A3: two new patients and expansion of the cardiomyopathy/skeletal myopathy phenotype with and without lactic acidosis. JIMD Rep. 19, 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Csordás G., Golenár T., Seifert E. L., Kamer K. J., Sancak Y., Perocchi F., Moffat C., Weaver D., de la Fuente Perez S., Bogorad R., Koteliansky V., Adijanto J., Mootha V. K., and Hajnóczky G. (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pernas L., and Scorrano L. (2016) Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 78, 505–531 [DOI] [PubMed] [Google Scholar]
- 16. Weaver D., Eisner V., Liu X., Várnai P., Hunyady L., Gross A., and Hajnóczky G. (2014) Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol. Cell 54, 870–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eisner V., Lenaers G., and Hajnóczky G. (2014) Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J. Cell Biol. 205, 179–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mishra P., and Chan D. C. (2016) Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baricault L., Ségui B., Guégand L., Olichon A., Valette A., Larminat F., and Lenaers G. (2007) OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res. 313, 3800–3808 [DOI] [PubMed] [Google Scholar]
- 20. Ko Y. H., Delannoy M., Hullihen J., Chiu W., and Pedersen P. L. (2003) Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J. Biol. Chem. 278, 12305–12309 [DOI] [PubMed] [Google Scholar]
- 21. Chen C., Ko Y., Delannoy M., Ludtke S. J., Chiu W., and Pedersen P. L. (2004) Mitochondrial ATP synthasome: three-dimensional structure by electron microscopy of the ATP synthase in complex formation with carriers for Pi and ADP/ATP. J. Biol. Chem. 279, 31761–31768 [DOI] [PubMed] [Google Scholar]
- 22. Nůsková H., Mráček T., Mikulová T., Vrbacký M., Kovářová N., Koválčíková J., Pecina P., and Houštěk J. (2015) Mitochondrial ATP synthasome: expression and structural interaction of its components. Biochem. Biophys. Res. Commun. 464, 787–793 [DOI] [PubMed] [Google Scholar]
- 23. Palmieri F., Prezioso G., Quagliariello E., and Klingenberg M. (1971) Kinetic study of the dicarboxylate carrier in rat liver mitochondria. Eur. J. Biochem. 22, 66–74 [DOI] [PubMed] [Google Scholar]
- 24. Chen H., Chomyn A., and Chan D. C. (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280, 26185–26192 [DOI] [PubMed] [Google Scholar]
- 25. Huckriede A., Heikema A., Sjollema K., Briones P., and Agsteribbe E. (1995) Morphology of the mitochondria in heat shock protein 60 deficient fibroblasts from mitochondrial myopathy patients. Effects of stress conditions. Virchows Arch. 427, 159–165 [DOI] [PubMed] [Google Scholar]
- 26. Koopman W. J., Visch H. J., Verkaart S., van den Heuvel L. W., Smeitink J. A., and Willems P. H. (2005) Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am. J. Physiol. Cell Physiol. 289, C881–C890 [DOI] [PubMed] [Google Scholar]
- 27. Guillery O., Malka F., Frachon P., Milea D., Rojo M., and Lombès A. (2008) Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul. Disord. 18, 319–330 [DOI] [PubMed] [Google Scholar]
- 28. Burelle Y., Bemeur C., Rivard M. E., Thompson Legault J., Boucher G., LSFC Consortium, Morin C., Coderre L., and Des Rosiers C. (2015) Mitochondrial vulnerability and increased susceptibility to nutrient-induced cytotoxicity in fibroblasts from leigh syndrome French Canadian patients. PLoS One 10, e0120767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 30. Cotticelli M. G., Acquaviva F., Xia S., Kaur A., Wang Y., and Wilson R. B. (2015) Phenotypic screening for Friedreich ataxia using random shRNA selection. J. Biomol. Screen. 20, 1084–1090 [DOI] [PubMed] [Google Scholar]
- 31. Santra S., Gilkerson R. W., Davidson M., and Schon E. A. (2004) Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 56, 662–669 [DOI] [PubMed] [Google Scholar]
- 32. Anunciado-Koza R. P., Zhang J., Ukropec J., Bajpeyi S., Koza R. A., Rogers R. C., Cefalu W. T., Mynatt R. L., and Kozak L. P. (2011) Inactivation of the mitochondrial carrier SLC25A25 (ATP-Mg2+/Pi transporter) reduces physical endurance and metabolic efficiency in mice. J. Biol. Chem. 286, 11659–11671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moffat C., Bhatia L., Nguyen T., Lynch P., Wang M., Wang D., Ilkayeva O. R., Han X., Hirschey M. D., Claypool S. M., and Seifert E. L. (2014) Acyl-CoA thioesterase-2 facilitates mitochondrial fatty acid oxidation in the liver. J. Lipid Res. 55, 2458–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu X., Weaver D., Shirihai O., and Hajnóczky G. (2009) Mitochondrial 'kiss-and-run': interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Claypool S. M., Dickinson B. L., Yoshida M., Lencer W. I., and Blumberg R. S. (2002) Functional reconstitution of human FcRn in Madin-Darby canine kidney cells requires co-expressed human β2-microglobulin. J. Biol. Chem. 277, 28038–28050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Claypool S. M., McCaffery J. M., and Koehler C. M. (2006) Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 174, 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maccecchini M. L., Rudin Y., Blobel G., and Schatz G. (1979) Import of proteins into mitochondria: precursor forms of the extramitochondrially made F1-ATPase subunits in yeast. Proc. Natl. Acad. Sci. U.S.A. 76, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riezman H., Hay R., Gasser S., Daum G., Schneider G., Witte C., and Schatz G. (1983) The outer membrane of yeast mitochondria: isolation of outside-out sealed vesicles. EMBO J. 2, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whited K., Baile M. G., Currier P., and Claypool S. M. (2013) Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 22, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]