

Abstract
Prion diseases are devastating neurodegenerative disorders with no known cure. One strategy for developing therapies for these diseases is to identify compounds that block conversion of the cellular form of the prion protein (PrPC) into the infectious isoform (PrPSc). Most previous efforts to discover such molecules by high-throughput screening methods have utilized, as a read-out, a single kind of cellular assay system: neuroblastoma cells that are persistently infected with scrapie prions. Here, we describe the use of an alternative cellular assay based on suppressing the spontaneous cytotoxicity of a mutant form of PrP (Δ105–125). Using this assay, we screened 75,000 compounds, and identified a group of phenethyl piperidines (exemplified by LD7), which reduces the accumulation of PrPSc in infected neuroblastoma cells by >90% at low micromolar doses, and inhibits PrPSc-induced synaptotoxicity in hippocampal neurons. By analyzing the structure-activity relationships of 35 chemical derivatives, we defined the pharmacophore of LD7, and identified a more potent derivative. Active compounds do not alter total or cell-surface levels of PrPC, and do not bind to recombinant PrP in surface plasmon resonance experiments, although at high concentrations they inhibit PrPSc-seeded conversion of recombinant PrP to a misfolded state in an in vitro reaction (RT-QuIC). This class of small molecules may provide valuable therapeutic leads, as well as chemical biological tools to identify cellular pathways underlying PrPSc metabolism and PrPC function.
Keywords: drug discovery, drug screening, neurodegeneration, neurological disease, prion, small molecule
Introduction
Prion diseases are fatal neurodegenerative disorders that are due to the conversion of a normal, neuronal glycoprotein (PrPC)3 into an infectious isoform (PrPSc) that propagates itself by an autocatalytic templating process (1, 2). In addition to their intrinsic interest to biologists, prion diseases are of enormous medical and public health concern. A global epidemic of bovine spongiform encephalopathy, a prion disease of cattle, emerged in the 1980s and 1990s, resulting in contamination of food supplies and transmission of the disease to a small number of human beings (3, 4). Prion contamination has also increased the risk of blood transfusions, and organ transplants (5). Most recently, a prion-like process has been found to play a role in the CNS dissemination of misfolded proteins in more common neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, and tauopathies, and there is even evidence that these diseases can be spread between individuals by iatrogenic means (6, 7).
There are currently no cures for prion diseases. A great deal of effort has been invested over the past 25 years in identifying compounds that block the conversion of PrPC into PrPSc as a therapeutic strategy. Most of these efforts have used as a read-out a single kind of cellular assay system: N2a neuroblastoma cells that are chronically infected with scrapie prions (designated ScN2a cells). N2a cells are one of the few cell lines capable of propagating prions, and they can maintain a chronic state of infection with no outward signs of cytotoxicity (8, 9). A number of inhibitory compounds have been discovered with this assay, either by educated guessing, or by systematic screens of compound libraries consisting of up to several thousand entries (10, 11). Although some of these compounds are able to prolong the incubation time between prion exposure and the onset of symptoms in mice, none of them prevents the eventual occurrence of disease, and none has proven effective in limited human trials (12, 13). On a mechanistic level, there is evidence that many anti-prion compounds discovered in high-throughput screens of ScN2a cells do not interact with either PrPC or PrPSc, and presumably target non-PrP molecules (14). However, in virtually every case, the identity of these targets remains unknown, limiting the extension of target-based, drug discovery approaches beyond the PrP molecule itself.
In addition to blocking conversion of PrPC to PrPSc, another possible strategy for treating prion diseases would be to inhibit downstream neurotoxic signaling pathways. However, there is very little known about how the interaction of PrPSc with cell-surface PrPC results in pathogenic effects such as synapse loss (15–17). We and others have found that expression in transgenic mice of PrP molecules harboring deletions spanning the central domain of the protein (residues 105–125) cause spontaneous neurodegenerative phenotypes that can be suppressed by co-expression of wild-type PrP (18–20). These mice have been the object of considerable interest, because they are likely to provide insights into PrP-mediated neurotoxic pathways. Our laboratory created mice with the most toxic (and paradoxically the shortest) of these PrP deletions, Δ105–125 (referred to as ΔCR) (for deletion of the central region) (18). In the course of investigating what makes ΔCR and the other deletion mutants so neurotoxic, we discovered that they induce spontaneous ionic currents, recordable by patch-clamping techniques, when expressed in cultured cell lines and neurons (21, 22). Coincidentally, we also observed that these same mutant PrP molecules sensitize cells to the cytotoxic effects of certain antibiotics, including G418 and Zeocin, a phenomenon that may be related to enhanced uptake of these cationic antibiotics via the formation of membrane channels or pores (23, 24).
In the present study, we have used suppression of this antibiotic hypersensitivity phenotype as a cellular read-out in a high throughput screen of a diverse, small-molecule library. Using this approach, we have discovered a new class of lead compounds, phenethyl piperidines, which display a dual activity: they suppress the toxicity of the deleted forms of PrP, and they also inhibit PrPSc formation in ScN2a cells. The use of this novel cellular screening assay has allowed us to identify compounds that may have escaped detection in previous screens, and that may have value as therapeutic leads as well as chemical biological tools.
Results
Known Anti-prion Compounds Are Active in the DBCA
We previously described the drug-based cellular assay (DBCA) (23, 24), which employs the antibiotic hypersensitivity of HEK cells expressing certain internally deleted forms of PrP, and the use of this assay to study PrP-related toxic mechanisms (25–27). In the DBCA, HEK293 cells expressing a mutant form of PrP such as ΔCR (Δ105–125) are exposed to either of two cationic antibiotics, G418 or Zeocin, for 3 days, after which cell viability is assessed by MTT reduction. Although untransfected cells, or cells expressing wild-type PrP, are able to survive antibiotic exposure for 7–10 days, cells expressing ΔCR PrP are killed within 3 days. We previously showed that pentosan polysulfate (PPS), a known ligand for several regions within the flexible, N-terminal domain of PrPC (28, 29), suppressed the antibiotic hypersensitivity of ΔCR PrP-expressing HEK cells, and also blocked the spontaneous currents seen in these cells (21, 27). Because PPS is also a well known inhibitor of PrPSc formation in ScN2a cells (30), we were led to test whether other anti-prion compounds were inhibitory in the DBCA.
Like PPS, Congo Red (31) and certain tetrapyrroles (32, 33) have been shown previously to reduce PrPSc levels in ScN2a cells, effects that we confirm here. Consistent with these findings, we observed that PPS, Congo Red, and a cationic tetrapyrrole (TMPyP) (34) each inhibit accumulation of PrPSc in RML scrapie-infected ScN2a cells after 1 week of treatment, as judged by a reduction in the levels of protease K (PK)-resistant PrP (Fig. 1, A and B). Notably, we found that all three of these compounds also had inhibitory activity in the DBCA, as demonstrated by a decrease in G418-induced toxicity in HEK cells expressing ΔCR PrP (Fig. 1, C and D). The effect of TMPyP in the DBCA was recently reported by Massignan and colleagues (35). Given this correlation between the inhibitory activity of compounds in ScN2a cells and in the DBCA, we decided to use the DBCA as the read-out in a high-throughput screen to see if we could discover previously unknown molecules that might also have anti-prion activity.
FIGURE 1.

Known anti-prion compounds are active in the DBCA. A, RML-infected ScN2a cells were treated for a total of 1 week with DMSO vehicle, or with the indicated concentrations of Congo Red (CR), tetrapyrrole (TMPyP), or pentosan polysulfate (PPS). Cells were lysed, and PK-treated lysates were analyzed by Western blotting for PrPSc. Molecular size markers are given in kDa. B, quantification of PrPSc levels, expressed relative to DMSO control (mean ± S.D., n = 3 cultures). C and D, dose-response curves of the activity of CR, TMPyP, and PPS in the DBCA. Compounds were assayed for their ability to restore cell viability to ΔCR PrP-expressing HEK cells exposed to G418. Cell viability was measured by MTT reduction. Error bars represent mean ± S.D., n = 3 cultures.
Identification of Additional Small Molecules That Restore Cell Viability in the DBCA
We adapted the DBCA to a 384-well format to allow high-throughput screening of compound libraries (see “Experimental Procedures”). The optimized, high-throughput assay had a Z′ factor of 0.71 ± 0.09. As a source of small molecules, we used a large compound library compiled by the Harvard Laboratory for Drug Discovery in Neurodegeneration (LDDN), which consisted of commercial, NIH, and in-house sources. 75,000 compounds from the LDDN library were screened. A threshold of 50% inhibition of cell death was selected for identifying hits (>3 times the standard deviation from the mean of the DMSO controls).
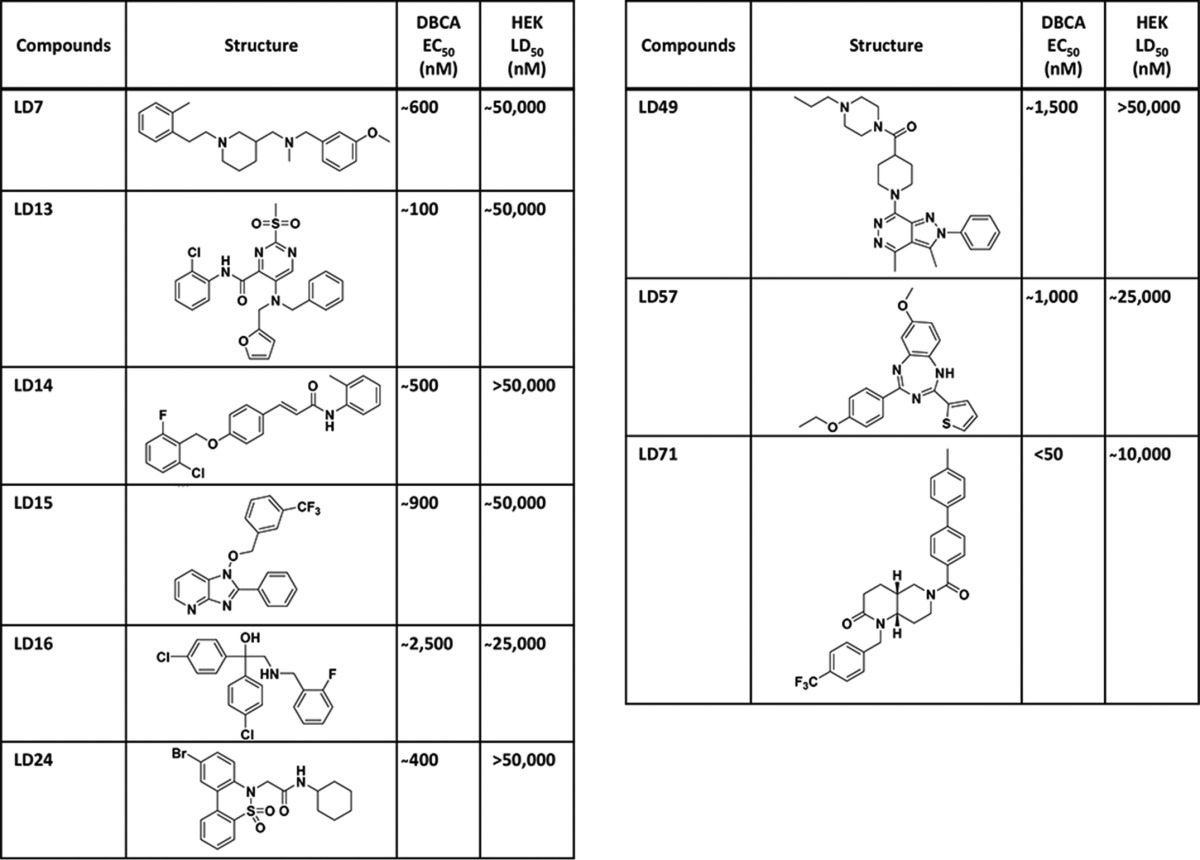
A total of 249 hits was identified using G418 as the antibiotic, representing 0.3% of the compounds tested. These 249 hits were then retested at five different concentrations against G418 and Zeocin, resulting in selection of 68 compounds (0.1%) that protected against both antibiotics. These molecules could be categorized into 9 distinct chemical groups, each representing a unique chemical scaffold. The fact that 7 of the groups contained multiple (between two and four), structurally related hits added to our confidence in the power of the screening assay. Representative small molecules from each class are displayed in Table 1, along with their EC50 in the DBCA, and their LD50 toward HEK cells, as measured in an MTT cell viability assay. None of the representative compounds, with the possible exception of LD49, displayed a significant physical interaction with recombinant PrP immobilized on the surface of a surface plasmon resonance (SPR) chip (supplemental Fig. S1).
TABLE 1.
Identification of additional small molecules that restore cell viability in the DBCA
Representative compounds from 9 chemical groups that restore viability in the DBCA, identified in a high-throughput screen of 75,000 compounds from the LDDN library. EC50 and LD50 values are derived from 12-point dose-response curves performed using the DBCA and the MTT viability assay, respectively. The number of related compounds in each group is given in partentheses: LD7 (3), LD13 (2), LD14 (2), LD15 (4), LD16 (1), LD24 (1), LD49 (4), LD57 (2), LD71 (2).
Hit Molecules from the DBCA Screen Reduce PrPSc Levels in ScN2a Cells
We tested representative compounds from the 9 groups for their ability to reduce PrPSc levels in N2a cells chronically infected with either the RML or 22L strains of scrapie prions (Fig. 2, A and B). Compounds were tested by treating cells for 1 week at concentrations below their limiting toxicity in N2a cells (5, 10, or 25 μm). Congo Red (5 μm) was used as a positive control. Three of the compounds (LD71, LD7, and LD49) reduced PrPSc levels by >50% in RML-infected ScN2a cells, whereas only LD7 had this effect in both RML- and 22L-infected ScN2a cells (Fig. 2C). We therefore decided to focus further efforts on LD7 (Fig. 3A). Based on its dose-response in ScN2a cells, LD7 had an EC50 of 5.8 μm with respect to inhibition of PrPSc accumulation, and an LD50 of 22.4 μm (Fig. 3, B and C). We also found that LD7 permanently cured RML-infected ScN2a cells after longer periods of exposure to the drug (Fig. 3D). After treating infected cells for one month and observing a rapid decline and the disappearance of PK-resistant PrPSc, we passed the cells for another month in the absence of LD7 and did not detect the reappearance of PK-resistant PrPSc.
FIGURE 2.

Hit molecules from the DBCA screen reduce PrPSc levels in ScN2a cells. ScN2a cells infected with RML (A) or 22L (B) scrapie prions were treated for a total of 1 week with DMSO vehicle, Congo Red (CR, 5 μm) as a positive control, or the indicated concentrations of LD compounds. Cells were lysed, and PK-treated lysates were analyzed by Western blotting for PrPSc. C, quantification of PrPSc levels, expressed relative to DMSO control (mean ± S.D., n = 9 cultures for RML, n = 3 cultures for 22L).
FIGURE 3.

LD7 reduces PrPSc levels in ScN2a cells, and permanently cures the cells. A, structure of LD7. B, ScN2a cells infected with RML (upper panel) or 22L (lower panel) scrapie prions were treated for a total of 1 week with DMSO vehicle or the indicated concentrations of LD7. Cells were lysed, and PK-treated lysates were analyzed by Western blotting for PrPSc. C, dose-response curves showing the amount of PrPSc remaining in ScN2a cells after 1 week of treatment with the indicated concentrations of LD7 (left ordinate), and viability of cells measured by MTT (right ordinate). Values in drug-treated cultures are expressed as a percentage of those in control cultures treated with DMSO. D, RML-ScN2a cells were treated for 31 days in the continued presence of LD7 (10 μm), followed by 31 days in the absence of LD7. Cells were split every 3–4 days, and at each split a portion of the cells was analyzed for PrPSc content by Western blotting. PrPSc levels are expressed relative to DMSO control (mean ± S.D., n = 3 cultures).
Structure-activity Relationships of LD7, and Identification of Active and Inactive Derivatives
Thirty-four chemical derivatives of LD7 (supplemental Fig. S2) were tested on RML-ScN2a cells at 7.5 μm to evaluate their anti-prion activity. The bar graph shown in Fig. 4A displays the compounds from the most potent to the least potent. Fourteen of the tested compounds decreased PrPSc levels by >50%, whereas nine reduced PrPSc levels by 0–50%. Twelve of the compounds had no effect, or increased PrPSc levels (maximum of 150%).
FIGURE 4.

Structure-activity relationships of LD7, and identification of active and inactive derivatives. A, effect of 34 derivatives of LD7 (structures shown in supplemental Fig. S2) on PrPSc levels in RML-ScN2a cells treated for 1 week with each compound at 7.5 μm, expressed relative to DMSO control. Bars represent mean ± S.D. (n = 3 cultures). B, pharmacophore of the LD7 class of compounds, based on the structure-activity data shown in panel A. C, dose-response curves showing the amount of PrPSc remaining in ScN2a cells after 1 week of treatment with the indicated concentrations of JZ-107 (left ordinate,) and viability of cells measured by MTT (right ordinate). The structure of JZ107 is shown in Fig. 5A. D, DBCA analysis of 10 LD7 derivatives: 5 compounds that were active in the ScN2a assay (green bars) and 5 compounds that were inactive in the ScN2a assay (red bars). Each compound was tested at concentrations of 5 μm (filled bars) and 10 μm (striped bars). Results are plotted as cell viability relative to cultures not exposed to G418. DMSO was used as a negative control and Congo Red (CR) as a positive control. E, ranking of the 10 LD7 derivatives based on their activity in the DBCA at 5 or 10 μm, from most active on the left to least active on the right. Compounds in the green and red boxes were active or inactive, respectively, in the ScN2a assay.
Based on these data, we were able to develop a preliminary structure-activity model for the phenyl piperidines (Fig. 4B). Two critical requirements for activity are the phenethyl amine at the piperidine nitrogen and the benzyl amine at the C3 position. Modification of either moiety resulted in loss of activity. We observed a number of additional attributes that modulated activity, including increased activity with ortho-substitution of the phenethyl amine and addition of hydrogen bond acceptors at meta and para positions of the benzyl amine. We also found that activity was greater when the benzylic amine was secondary, but activity persisted with a number of alkyl substituents. Interestingly, we did not observe a difference in activity between the R and S enantiomer of LD7 indicating that the absolute stereochemistry of the piperidine is not critical for activity. Based on this structure-activity study we synthesized the most potent compound, JZ107, which had an EC50 of 3.1 μm in both RML- and 22L-infected N2a cells, and an LD50 of 11.4 μm (Fig. 4C).
Effect of LD7 Derivatives in the DBCA
We tested the activity in the DBCA of 10 compounds from the LD7 structure-activity series: 5 that were active in the ScN2a assay, and 5 that were inactive. The results did not show a correlation between the activities of the compounds in these two assays (Fig. 4, D and E).
JZ107 Does Not Alter Total or Cell Surface Levels of PrPC
PrPSc proliferation requires PrPC as a substrate (36), so a possible mechanism of action of anti-prion compounds is the reduction of PrPC protein expression or surface localization. We therefore tested the effect of JZ107 (Fig. 5A) on these parameters. Treatment of N2a.3 cells with JZ107 did not cause a change in PrPC protein levels (Fig. 5, B and C) or PrPC surface expression (Fig. 5D, middle panel). As a positive control, PPS caused a dramatic reduction in surface PrPC (Fig. 5D, right panel), consistent with previously published results (37).
FIGURE 5.

JZ107 does not alter total or cell surface levels of PrPC. A, structure of JZ107, an active derivative. B, N2a.3 cells were treated for 1 week with the indicated concentrations of JZ107, after which cells were lysed and PrPC levels were evaluated by Western blotting. Actin was detected as a loading control. C, bars represent mean ± S.D. (n = 3 cultures) of PrPC levels, expressed relative to a DMSO control, and normalized for the content of actin. There was no statistically significant difference between any of the bars. D, unpermeabilized N2a.3 cells were stained for surface PrPC (red) after treatment for 1 week with DMSO vehicle, JZ107, or PPS. Nuclei were visualized by staining with Hoechst 33342 (blue). Scale bar = 10 μm. Images are representative of results from 3 independent experiments.
JZ107 Inhibits PrPSc-induced Synaptotoxicity
We have recently shown that PrPSc causes a rapid, PrPC-dependent retraction of dendritic spines in cultured hippocampal neurons, a phenomenon that we contend represents an early manifestation of the synaptotoxic effect of prion infection (38). As reported previously, incubation of hippocampal neurons with purified PrPSc, but not with mock-purified material from uninfected brain, caused a dramatic retraction of dendritic spines, as monitored by staining with Alexa-phalloidin, which stains F-actin in the spines (Fig. 6, A and B). We observed that preincubation of neurons with JZ107, but not the inactive compound, JZ103 (supplemental Fig. S2), blocked this effect (Fig. 6, C and D), despite the fact that JZ107 had a mild toxic effect on its own (Fig. 6E). Fig. 6G shows quantitation of spine numbers in this experiment. Although JZ107 by itself reduced spine number by ∼20% (compare “Mock” to “JZ107” bars), it was able to significantly protect against spine loss in the presence of PrPSc (compare “JZ107+PrPSc” to “PrPSc” bars), whereas JZ103 offered no significant protection (compare “JZ103+PrPSc to “PrPSc” bars). JZ103 by itself did not exhibit any toxic effect (compare “Mock” to “JZ103” bars).
FIGURE 6.
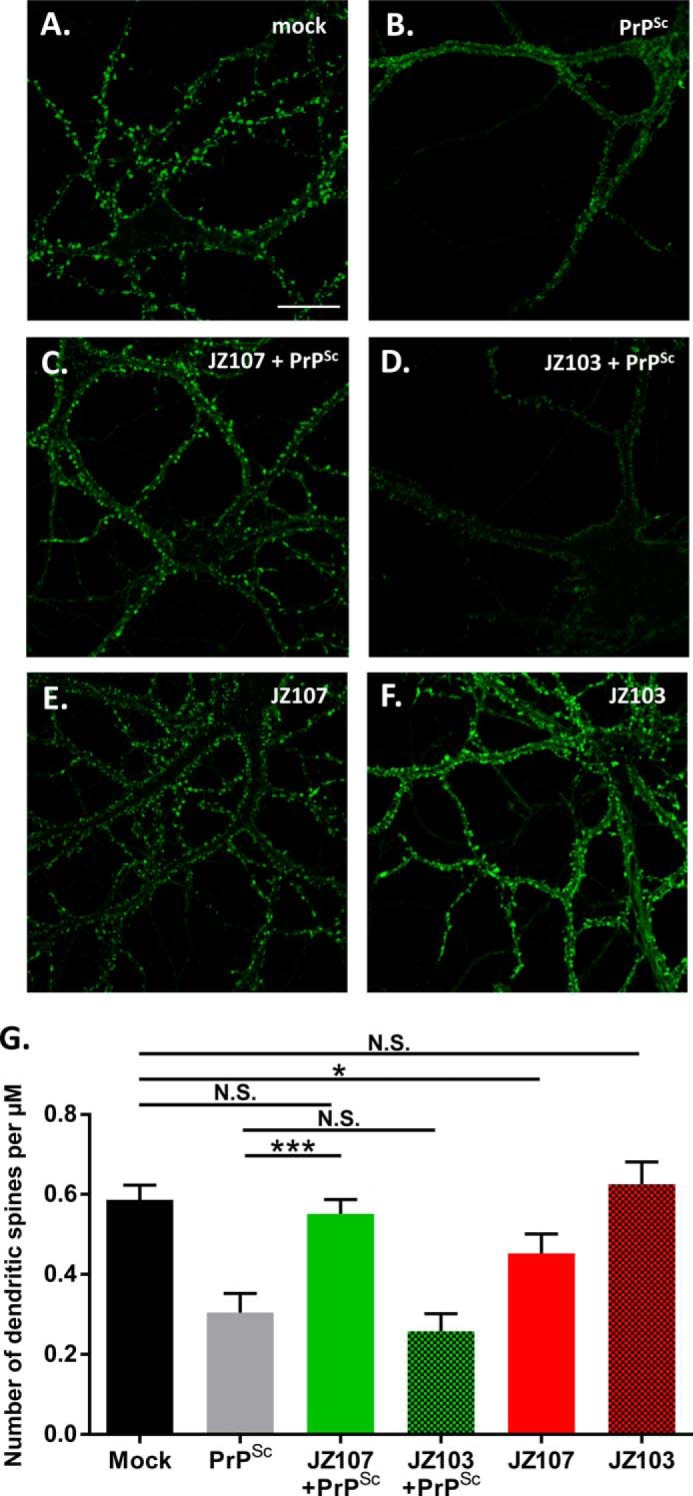
JZ107 inhibits PrPSc-induced synaptotoxicity. Cultures of hippocampal neurons from wild-type mice were preincubated for 2 h with 5 μm JZ107 (C and E) or JZ103 (D and F). Cultures were then treated for 24 h with mock-purified brain material (A), purified PrPSc (B–D), or neither (E and F). Neurons were then fixed and stained with Alexa 488-phalloidin to visualize actin in dendritic spines. Scale bar in panel A = 20 μm. G, pooled measurements of spine number were collected from 22 to 25 cells from 3 independent experiments (***, p < 0.001, *, p < 0.05, N.S., nonsignificant by Student's t test).
Inhibitory Activity of Compounds in the RT-QuIC Assay Correlates with Their Inhibitory Activity in the ScN2a Assay
As an adjunct to ScN2a cells for assaying the anti-prion effects of our compounds, we utilized RT-QuIC (real-time quaking-induced conversion), an in vitro assay for the seeding activity of PrPSc (39). In this assay, purified, recombinant PrPC is seeded with a small amount of brain-derived PrPSc under conditions of repetitive shaking, and conversion of the PrPC to a misfolded state is monitored by thioflavin T fluorescence. Although RT-QuIC is typically used as a diagnostic tool to quantitate small amounts of PrPSc in biological samples (39), we decided to use it here to measure the anti-prion activity of selected compounds.
We chose 10 different derivatives of LD7, five with inhibitory activity in the ScN2 assay, and five that were inactive. We preincubated the reaction mixture containing recombinant PrP with each compound at 200 μm for 20 min, and then initiated the conversion reaction by addition of scrapie-infected brain homogenate as a source of PrPSc seeds. We observed that all five of the compounds that were inhibitory in ScN2a cells significantly delayed conversion in the RT-QuIC assay, whereas the five inactive compounds had no effect (Fig. 7A). We then decreased the concentration of each compound to 150 or 100 μm. At 150 μm, three of five of the compounds that suppressed PrPSc levels in ScN2a cells delayed conversion in the RT-QuIC reaction, including JZ107 (Fig. 7C). At 100 μm, only one compound, JZ79, delayed RT-QuIC conversion (Fig. 7C). Notably, JZ107 and JZ79 were among the most efficacious in reducing PrPSc levels in ScN2a cells (Fig. 4A). Taken together, these results demonstrate a strong correlation between the inhibitory activity of LD7 derivatives in the RT-QuIC and ScN2a assays (Fig. 7D). We also observed that when we omitted the preincubation step, the efficacy of JZ79 at 200 μm in the RT-QuIC was diminished, because 2 of 5 wells eventually turned positive (Fig. 7B).
FIGURE 7.

Inhibitory activity of compounds in the RT-QuIC assay correlates with their inhibitory activity in the ScN2a assay. A, RT-QuIC conversion curves in the presence of DMSO (black), or 10 different LD-7 derivatives, each at 200 μm. Five of the derivatives were active in the ScN2a assay, and five were inactive. Curves are the average of 5 replicate wells. B, comparison of RT-QuIC reactions with and without 20 min preincubation of JZ79 (200 μm) with PrPC. The numbers next to each curve represent the number of wells (of a total of 5 wells for each condition) in which the ThT signal exceeded the threshold value by the end of the experiment. C, analysis of 3 different RT-QuIC experiments using the 10 LD7 derivatives at 100, 150, or 200 μm. Bars represents the time in hours (mean ± S.D., n = 5 wells) at which the thioflavin T signal reached a threshold value of 50,000 absorbance units. D, table showing whether each of the compounds significantly inhibited the RT-QuIC reaction (i.e. suppressed the conversion curves) at three different concentrations (100, 150, and 200 μm) (***, p < 0.001; *** or *, p < 0.05 by Student's t test, drug-treated versus DMSO control; −, no inhibitory effect).
Discussion
In this study, we have used a novel assay (DBCA) (23, 24), based on the cellular toxicity of a mutant form of PrP (Δ105–125, designated ΔCR), to perform a high-throughput screen of 75,000 compounds to identify those that significantly improved cell viability in the presence of two cationic antibiotics (G418 and Zeocin) normally toxic to ΔCR PrP-expressing cells. Surprisingly, several of the compounds identified in this screen also proved to be efficacious in reducing PrPSc levels in scrapie-infected N2a cells. Structure-activity relationships were established for one of these compounds, a phenethyl piperidine (LD7), based on analysis of 34 chemical derivatives, resulting in creation of a molecule, JZ107, that was more potent at reducing PrPSc levels. We showed that the mechanism of action of JZ107 does not involve decreasing total or cell-surface levels of the precursor molecule, PrPC. Our results using an in vitro PrPC-PrPSc seeding assay (RT-QuIC) raise the possibility that, although the compound does not bind with high affinity to recombinant PrP in SPR experiments, it might interact with PrPC in a cellular context, perhaps in conjunction with other cell-surface receptors. Altogether, our results establish a new assay, orthogonal to the standard ScN2a test, for discovering anti-prion compounds. Such molecules may have therapeutic potential in both prion and Alzheimer's disease, and may prove useful as tool compounds to further investigate PrP-mediated neurotoxic pathways.
Over the past 20 years, a number of different approaches to anti-prion therapy have been pursued. Perhaps the most effective has been to reduce the amount of PrPC substrate available for conversion into PrPSc. The validity of this approach is demonstrated by the key observation that mice in which the PrP gene has been knocked out are completely resistant to prion infection (36). A second major strategy has been the use of compounds that block the conversion of PrPC into PrPSc. By far, the most widely employed system for discovering and testing such compounds relies on ScN2a cells, which are chronically infected with scrapie prions. A number of inhibitory compounds have been identified using this assay (10, 11). Some of these compounds bind to PrPC, and may act by sterically occluding critical interactions, or by stabilizing the structure of the folded, C-terminal domain (e.g. glycosaminoglycans, Congo Red, and other sulfonated dyes, nucleic acids, cationic tetrapyrroles, anti-PrP antibodies). Other compounds appear to act by binding to PrPSc, destabilizing or otherwise altering its structure, and possibly enhancing its clearance (e.g. polyamine and phosphorus-containing dendrimers, polyoxometalates, and polythiophenes). Importantly, there is evidence that many anti-prion compounds discovered in high-throughput screens of ScN2a cells do not interact with either PrPC or PrPSc, and presumably target non-PrP molecules (14). However, in virtually every case, the identity of these targets remains unknown.
We predicted that the DBCA might represent an orthogonal assay that would be useful as a screening tool to identify novel anti-prion compounds, based on our observation that several molecules (PPS, Cong Red, and TMPyP) previously shown to reduce PrPSc levels in ScN2a cells were also active in the DBCA. For these compounds, there may be a mechanistic connection between their effects in the two assays. For example, both PPS and Congo Red bind to the flexible, N-terminal domain of PrPC, in particular the polybasic region encompassing residues 23–31 (28, 29, 40). This region is known to be essential both for the toxic activity of ΔCR PrP in the DBCA (22, 27), and for productive interaction between PrPC and PrPSc during the prion conversion process (41, 42). We found that three of the 68 confirmed hits from the DBCA screen, representing 3 of 9 distinct chemical scaffolds, possessed anti-prion activity when tested in ScN2a cells. It remains to be determined whether the parallel activities of these compounds in the two assays are mechanistically related. When we tested 10 compounds selected from the LD7 structure-activity series, we did not find a close correlation between their activities in the DBCA and ScN2a assays (Fig. 4, D and E). Regardless of the underlying mechanisms, however, we have shown that the DBCA does have the ability to identify new anti-scrapie compounds, and may therefore represent a useful preliminary filter in drug screening efforts.
What is the mechanism of action of the LD7/JZ107 class of compounds? JZ107 does not alter the levels of total or cell-surface PrPC, suggesting that it does not act by altering the amount or distribution of this precursor, as is the case for some other anti-prion drugs (43). LD7 and JZ107 do not bind to immobilized, recombinant PrP in SPR experiments, arguing that PrPC itself may not be a high-affinity target for these compounds. Interestingly, however, we found that JZ107 and other active derivatives suppressed PrPSc-seeded conversion of recombinant PrPC to a misfolded state in the RT-QuIC reaction. In contrast, derivatives that had no activity in the ScN2a assay had no effect on the RT-QuIC reaction. Although we cannot be certain whether the compounds are interacting with the PrPC substrate, the PrPSc seeds, or both, the fact that the inhibitory effect in the RT-QuIC reaction was partially dependent upon preincubation of the compounds with PrPC suggests the first possibility. Notably, the concentrations of the compounds required to observe an effect in the RT-QuIC reaction were quite high (100–200 μm), well above the EC50 of these molecules in the ScN2a assay (<10 μm). This discrepancy may be due to the high concentration of recombinant PrP (∼6 μm) present in the RT-QuIC reaction, compared with what would be present in a cellular context, or to modifications such as glycosylation and GPI membrane anchoring, which could enhance the affinity of cellular PrPC for these molecules. A third possibility is that PrPC functions as part of a complex with other membrane proteins that together constitute high-affinity binding targets that mediate the anti-prion activity of the compounds. Ongoing efforts are now directed toward definitively identifying the molecular target(s) for the LD7/JZ107 class of compounds using forward and reverse chemical genetic approaches.
There are several important implications of the work presented here. First, we have used a novel, cellular assay (the DBCA), based on a toxic activity of mutant PrP, to discover a new class of anti-prion compounds using high-throughput screening methods. These molecules may serve as a starting point for therapeutic applications in animals and humans, in conjunction with further optimization of their pharmacodynamic and pharmacokinetic properties. Second, these molecules are potentially valuable tool compounds, which may provide insights into pathways controlling PrPSc formation and degradation in cells. Finally, the compounds may provide clues to how PrPC mediates the toxic effects of PrPSc (15). The fact that these compounds were discovered using the DBCA, which we think is related in some way to alterations in a physiological activity of PrPC, makes it more likely that the compounds will affect processes beyond PrPSc metabolism. The combination of the DBCA, the ScN2a assay, and the PrPSc dendritic toxicity assay (38) should allow us to discover additional therapeutic leads that act via novel mechanisms.
Experimental Procedures
Materials
Congo Red was purchased from Sigma (catalog number V000600), pentosan polysulfate (average Mr = 4,500–5,000) from Biopharm Australia Pty Ltd., and Fe(III) meso-tetra(N-methyl-4-pyridyl)porphine pentachloride (TMPyP) from Frontier Scientific (catalog number T40322). The following LD7 derivatives were purchased from Chembridge: LD7A (catalog number 65785139), LD7B (catalog number 11354888), cpd224 (catalog number 22499542), cpd385 (catalog number 38560135), cpd467 (catalog number 46715271), cpd600 (catalog number 60060319), cpd648 (catalog number 64895529), cpd690 (catalog number 69061817), cpd783 (catalog number 78359110), and cpd788 (catalog number 78843565). All other compounds were derived from the LDDN library (see below), or were synthesized (supplemental Figs. S2 and S3). Brain homogenate stocks of RML and 22L strains of scrapie were obtained from the TSE Resource Center, Roslin Institute, University of Edinburgh, and were passaged in C57BL6 mice.
Chemical Syntheses
See supplemental Fig. S3.
Drug-based Cellular Assay
For low-throughput analyses (e.g. dose-response curves for selected compounds), the assay was carried out as previously described (23). Briefly, HEK cells stably expressing ΔCR PrP were seeded into 24-well plates and cultured for 24 h, after which the medium was replaced with medium containing the test compound, along with either G418 or Zeocin (Gibco) at 500 μg/ml. Twenty-four hours later, fresh medium containing G418 or Zeocin and test compound was added for another 24 h. Cells were then washed with PBS, after which MTT (500 μg/ml in PBS) was added and allowed to incubate for 30 min. Formazan product was solubilized in DMSO and the A570 was read on a Synergy H1 Plate Reader (Biotek). Cell viability was assessed relative to controls without G418 or Zeocin.
High-throughput Screening
We adapted the DBCA to a 384-well format to allow high-throughput screening of compound libraries. To arrive at the optimal assay, stably transfected HEK cells expressing ΔCR PrP were treated with drugs (G418 or Zeocin) over a range of concentrations and times, and toxicity was scored by measuring cell viability. After experimenting with several different read-outs, we settled on a luminescent cell viability assay (CellTiterGLO, Promega), which uses ATP generation to score viable cells. We compared different assay procedures in terms of their coefficient of variation, dynamic range, and signal-to-noise ratio. The optimized format had an average Z′ factor of 0.71 ± 0.09, which is in the desired range for high-throughput screening.
In the optimized assay, stably transfected HEK cells expressing PrP ΔCR are seeded in 384-well plates at a density of 5,000 cells per well. After 24 h, test compounds are added to the wells at a final concentration of 1 μm, followed by G418 (625 μg/ml). This concentration of antibiotic was sufficient to induce 50% cell death in ΔCR PrP-expressing HEK cells, and was not toxic to HEK cells expressing WT PrP or vector. Cells were maintained for 48 h after treatment prior to cell viability assays. Negative control wells contained cells expressing ΔCR PrP that were not treated with G418. As positive controls, some wells were treated with pentosan sulfate and G418. Compounds were scored as positive if they reduced cell death by at least 50% (after subtraction of negative control values).
Automation of the assay allowed a throughput of up to 50 384-well plates per run, with 3 runs per week. Cells were plated using blank tissue culture-treated plates using a multidrop dispenser (ThermoFisher). A Beckman NX liquid handling system with 384-well head was used to transfer test compounds, G418, and CellTiterGLO detection reagent to the assay plate containing cells. A threshold of 50% inhibition of cell death was selected for identifying hits, which is >3 times the mean ± S.D. of the DMSO controls.
Compounds that were positive in the primary screen using G418 were re-tested using Zeocin. EC50 and LD50 values were then calculated from dose-response curves using fresh stocks of each compound.
Small Molecule Library
The LDDN compound screening library consists of 159,488 compounds purchased or collected from a variety of sources, half of which were screened for this study. This library has been used successfully in a number of previous drug screening campaigns. The sources of the library compounds are as follows: ChemDiv (73628), ChemBridge (24130), Prestwick approved bioactives (640), Specs natural products (486), CEREP (4800), Maybridge (17462), Bionet (6909), Peakdale (3603), Advanced Syntech N-Ac tetrapeptides (4096), Enamine (6024), Life Chemicals (6153), Microsource NINDS collection (2000), as well as various academic institutions from the United States, France, and India (2442) and the LDDN (1563). Most of the compounds were selected based on adherence to Lipinski's rules (i.e. Mr < 500, log p < 5, H-bond donors < 5, H-bond acceptors < 10), low proportion of compounds containing known toxicophores or reactive functional groups (e.g. epoxides, alkyl halides, Michael acceptors) and predicted solubility. Attempts were also made to select compounds with predicted blood-brain barrier penetration (e.g. LogP = 2–4) and polar surface area ≤ 90 Å2. In addition, attempts were made to maximize molecular diversity by selecting a wide variety of structures from various sources and from a variety of molecular scaffolds (including saturated and unsaturated heterocycles, natural products, tetrapeptides etc.). Last, the collection includes a 2000-member fragment library, which is compliant with the “Rule of Three” criteria for fragment libraries: Mr ≤ 300, cLog p ≤ 3, H-bond acceptors ≤ 3, H-bond donors ≤ 3, rotatable bonds ≤ 3, and polar surface area ≤60 Å2 (44).
ScN2a Cell Assay
We previously isolated a sub-line of N2a cells (designated N2a.3) that were highly susceptible to scrapie infection. These cells were chronically infected by exposure of confluent cultures to RML or 22L brain homogenate (final concentration, 0.5%) for 24 h, followed by passaging twice per week in Opti-MEM (Gibco) supplemented with 10% FCS and penicillin/streptomycin. Cells were checked monthly by Western blotting for the continued presence of PrPSc, and when PrPSc levels decreased, a new vial of frozen cells was thawed. Cells were maintained in an atmosphere of 5% CO2, 95% air at 37 °C.
ScN2a cells infected with RML or 22L scrapie were plated in 6-well plates (Thermo Fisher) by splitting at a 1:5 ratio from a confluent flask. The cells were treated for 3 days with test compounds, or with DMSO vehicle as a negative control and Congo Red as a positive control. They were then split at a 1:5 ratio, and treatment with compounds in fresh medium was continued for 4 more days. At the end of this 7-day treatment period, cells were lysed in 300 μl of lysis buffer (0.5% Nonidet P-40, 0.5% deoxycholate, 10 mm Tris-HCl, pH 8, and 100 mm NaCl), and protein concentration was measured using the BCA assay (Thermo Fisher, catalog number 23225). Samples of 250 μl adjusted to contain equal amounts of protein were digested with 40 μg/ml of PK at 37 °C for 1 h. Digestion was stopped by addition of 25 μl of ×10 complete protease inhibitor mixture (Roche), and samples were centrifuged at 180,000 × g at 4 °C for 1 h. Pellets were dissolved in 20 μl of 1× Laemmli loading buffer (Bio-Rad) and were boiled for 3 min before loading on 12% SDS-PAGE precast Criterion gels (Bio-Rad). Western blotting was performed according to standard procedures. PrPSc was detected using the anti-prion antibody D18 (45) and HRP-coupled, anti-human secondary antibody (Jackson ImmunoResearch, catalog number 109-035-088).
To test the ability of LD7 to permanently cure infection, RML-ScN2a cells were plated as above, and then treated for 31 days with LD7 (10 μm) (or DMSO as a negative control), and then for another 31 days without compound. Every 3 or 4 days, each well was split into two new wells at a 1:5 ratio. One of these wells was carried onto the next passage, whereas the cells in the other well were lysed for analysis of PrPSc by Western blotting as described above.
Effect of Compounds on Cell Viability
RML-ScN2a cells or HEK cells expressing ΔCR PrP were plated in 12-well plates, and treated for 4 days with compounds over a range of concentrations. After 4 days, cells were washed with PBS, and then MTT was added (500 μl at 0.5 mg/ml) for 30 min at 37 °C. The MTT reagent was then replaced by 500 μl of DMSO, and wells were incubated for 10 min at 37 °C. 200 μl of the contents of each well were transferred into a 96-plate, and the A570 was read on a plate reader (Biotek Synergy-H1).
Assays of PrPC Levels and Localization
N2a.3 cells at 20% confluence were treated for 3 days with test compounds, and then treatment was continued for an additional 4 days after splitting at a 1:5 ratio into 6-well plates for analysis of PrPC levels by Western blotting, or into 8-well Lab-Tek II chamber slides (Thermo Fisher) for analysis of PrPC localization by immunofluorescence staining.
For Western blotting, cells were lysed in 300 μl of lysis buffer (0.5% Nonidet P-40, 0.5% deoxycholate, 10 mm Tris-HCl, pH 8, and 100 mm NaCl) containing the complete protease inhibitor mixture (Roche), and protein concentration was measured using the BCA assay (Thermo Fisher). The protein concentration was normalized for all samples in a final volume of 20 μl. Samples were then mixed with 20 μl of 2× Laemmli loading buffer (Bio-Rad), and boiled for 3 min before loading on 12% SDS-PAGE precast Criterion gels (Bio-Rad). Western blotting to reveal PrPC was performed using D18 primary antibody (45) and HRP-coupled anti-human secondary antibody. To further normalize for protein loading, actin was detected using an anti-actin antibody (Sigma, catalog number A2228) and HRP-coupled, anti-mouse secondary antibody (Sigma, catalog number A0168). Signals for PrPC and actin were quantitated using ImageJ.
For immunofluorescence staining of cell-surface PrPC, live cells were labeled with D18 antibody (45) for 30 min on ice, then washed with PBS prior to fixation for 10 min in 4% paraformaldehyde. After 3 more washes with PBS, cells were incubated with Alexa 594-conjugated goat anti-human secondary antibody (Life Technologies) for 1 h at room temperature. After 3 more washes, nuclei were stained by incubating cells for 10 min with Hoechst 33342 (Thermo Fisher, catalog number 62249) at 1 μg/ml. Cells were then washed 3 times with PBS, after which the chamber dividers were removed, and a coverslip applied with mounting medium.
Synaptotoxicity of PrPSc
Cultures of hippocampal neurons and purified PrPSc from infected brains were prepared as described previously (38). Neurons were preincubated for 2 h with 5 μm JZ107 or JZ103 (or DMSO vehicle for the controls), after which 4.4 μg/ml of PrPSc (or a mock-purified control sample from non-infected brains) was added for 24 h. Neurons were then fixed in 4% paraformaldehyde and stained with Alexa 488-phalloidin (Thermo Fisher) to visualize actin in dendritic spines. Images were acquired using a Zeiss 700 confocal microscope with a ×63 objective (N.A. = 1.4). The number of dendritic spines was determined using ImageJ software. The number of spines was normalized to the measured length of the dendritic segment to give the number of spines/μm. For each experiment, 15–24 neurons from 3 individual experiments were imaged and quantitated.
Surface Plasmon Resonance
Binding assays were performed on a ProteOn XPR36 Protein Interaction Array System (Bio-Rad). GLH chips (Bio-Rad) were prepared for protein coupling according to the manufacturer's directions. Briefly, cleaned chips were activated by injecting 200 mm 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 50 mm N-hydroxysuccinamide (EDC-NHS) in sterile water, followed by protein (5 μm recombinant mouse PrPC in 20 mm sodium acetate 4.5, prepared as described (46)), and finally deactivation with a solution of 1 m ethanolamine. All interactions were measured in PBS, pH 7.2, containing 0.05% Tween 20 (PBST). Compounds were diluted in PBST and injected over the PrPC-coupled surface. Background binding was subtracted by referencing against control spots with no protein and no compound.
RT-QuIC Assay
Recombinant hamster PrP was expressed and purified as described previously (39). The concentration of recombinant PrP was determined by measuring absorbance at 280 nm. Purity was ≥99%, as estimated by SDS-PAGE, immunoblotting (data not shown), and circular dichroism (data not shown). The protein was aliquoted and stored at −80 °C at a concentration of 0.3 mg/ml.
Ten percent (w/v) brain homogenates were prepared from RML-infected brain in homogenization buffer (1× PBS, 1 mm EDTA, 150 mm NaCl, 0.5% Triton X-100) containing protease inhibitor mixture (Roche, catalog number 11836170001). Homogenates were then clarified by centrifugation for 2 min at 2,000 × g, and the supernatants aliquoted and stored at −80 °C.
RT-QuIC reactions were performed in 96-well plates as described previously (39). Prior to loading samples into the wells, a master mix was prepared for each condition, with the following final concentrations of each component: 1× PBS, 1 mm EDTA, 10 μm ThT, 170 mm NaCl, 0.1% DMSO, 6 μm hamster PrP, and the test compound at the indicated concentration (100–200 μm). Samples were preincubated for 20 min at room temperature to allow the PrPC and test compound to interact, after which reactions were initiated by addition of RML brain homogenate (final concentration of 0.0002% w/v). In some experiments, preincubation was omitted. One hundred microliters of the final master mixture was then loaded into each of 5 replicate wells. Plates were sealed with a plate sealer film (Nalgene Nunc International, catalog number 265301), and were incubated in a BMG Polarstar plate reader at 42 °C for 116 h with cycles of 1 min of shaking (700 rpm double orbital) and 1 min of rest. ThT fluorescence measurements were taken every 15 min (450 ± 10 nm excitation and 480 ± 10 nm emission; bottom read; 20 flashes per well; manual gain of 1800; 20 μs integration time). To quantitate the effect of test compounds on conversion, we adopted a threshold method. For each replicate, we determined the time at which the ThT signal exceeded a 50,000-absorbance unit threshold value. For wells in which the conversion was totally inhibited, we used the last time point of the experiment (∼116 h). Data were analyzed using GraphPad Prism 6.
Author Contributions
D. A. H. conceived and coordinated the study. D. A. H. and T. I. wrote the paper. J. T. H. designed, performed, and analyzed the experiments and results shown in Table 1 and supplemental Fig. S1. J. T. H. and T. I. designed, performed, and analyzed the experiments shown in Fig. 1. T. I. designed, performed, and analyzed the experiments shown in Figs. 2–5 and 7, and supplemental S2. C. F. designed, performed, and analyzed the experiments shown in Fig. 6. A. B. B., H. Y., and J. Z. designed, synthesized, and analyzed the compounds shown in supplemental Figs. S2 and S3. C. B. R. provided technical assistance. M. G. coordinated the screening of compound libraries at the LDDN. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We acknowledge Tania Massignan and Emiliano Biasini (University of Trento, Italy) for participation in the initial phases of the project while in the Harris Laboratory. We thank Byron Caughey for providing bacteria expressing the recombinant hamster PrP(90–231) used in the RT-QuIC assay. NMR (CHE-0619339) and MS (CHE-0443618) facilities at Boston University are supported by the National Science Foundation.
This work was supported by National Institutes of Health Grants R01 NS065244 (to D. A. H.) and U24 NS049339 (to M. G.) and by the Harvard NeuroDiscovery Center (to M. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation.

This article contains supplemental Figs. S1–S3.
- PrPC
- cellular isoform of the prion protein
- PrP
- prion protein
- ΔCR
- Δ105–125 deletion mutant of the prion protein
- DBCA
- drug-based cellular assay
- DMSO
- dimethyl sulfoxide
- LD50
- lethal dose 50%
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PK
- proteinase K
- PPS
- pentosan polysulfate
- PrPSc
- scrapie isoform of the prion protein
- RML
- Rocky Mountain Laboratory
- RT-QuIC
- real-time quaking-induced conversion
- ScN2a
- N2 cells persistently infected with scrapie prions
- SPR
- surface plasmon resonance
- TMPyP
- Fe(III) meso-tetra(N-methyl-4-pyridyl)porphine pentachloride
- ThT
- thioflavin T
- LDDN
- Laboratory for Drug Discovery in Neurodegeneration.
References
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguzzi A., and Polymenidou M. (2004) Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327 [DOI] [PubMed] [Google Scholar]
- 3. Bruce M. E., Will R. G., Ironside J. W., McConnell I., Drummond D., Suttie A., McCardle L., Chree A., Hope J., Birkett C., Cousens S., Fraser H., and Bostock C. J. (1997) Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 389, 498–501 [DOI] [PubMed] [Google Scholar]
- 4. Wells G. A., and Wilesmith J. W. (1995) The neuropathology and epidemiology of bovine spongiform encephalopathy. Brain Pathol. 5, 91–103 [DOI] [PubMed] [Google Scholar]
- 5. Belay E. D., and Schonberger L. B. (2005) The public health impact of prion diseases. Annu. Rev. Public Health 26, 191–212 [DOI] [PubMed] [Google Scholar]
- 6. Jucker M., and Walker L. C. (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaunmuktane Z., Mead S., Ellis M., Wadsworth J. D., Nicoll A. J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A. S., Rudge P., Collinge J., and Brandner S. (2015) Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250 [DOI] [PubMed] [Google Scholar]
- 8. Race R. E., Caughey B., Graham K., Ernst D., and Chesebro B. (1988) Analyses of frequency of infection, specific infectivity, and prion protein biosynthesis in scrapie-infected neuroblastoma cell clones. J. Virol. 62, 2845–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Butler D. A., Scott M. R., Bockman J. M., Borchelt D. R., Taraboulos A., Hsiao K. K., Kingsbury D. T., and Prusiner S. B. (1988) Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J. Virol. 62, 1558–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trevitt C. R., and Collinge J. (2006) A systematic review of prion therapeutics in experimental models. Brain 129, 2241–2265 [DOI] [PubMed] [Google Scholar]
- 11. Sim V. L. (2012) Prion disease: chemotherapeutic strategies. Infect. Disord. Drug Targets 12, 144–160 [DOI] [PubMed] [Google Scholar]
- 12. Collinge J., Gorham M., Hudson F., Kennedy A., Keogh G., Pal S., Rossor M., Rudge P., Siddique D., Spyer M., Thomas D., Walker S., Webb T., Wroe S., and Darbyshire J. (2009) Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 8, 334–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haïk S., Marcon G., Mallet A., Tettamanti M., Welaratne A., Giaccone G., Azimi S., Pietrini V., Fabreguettes J. R., Imperiale D., Cesaro P., Buffa C., Aucan C., Lucca U., Peckeu L., et al. (2014) Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 13, 150–158 [DOI] [PubMed] [Google Scholar]
- 14. Poncet-Montange G., St. Martin S. J., Bogatova O. V., Prusiner S. B., Shoichet B. K., and Ghaemmaghami S. (2011) A survey of antiprion compounds reveals the prevalence of non-PrP molecular targets. J. Biol. Chem. 286, 27718–27728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Biasini E., Turnbaugh J. A., Unterberger U., and Harris D. A. (2012) Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mallucci G., Dickinson A., Linehan J., Klöhn P. C., Brandner S., and Collinge J. (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871–874 [DOI] [PubMed] [Google Scholar]
- 17. Mallucci G. R. (2009) Prion neurodegeneration: starts and stops at the synapse. Prion 3, 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li A., Christensen H. M., Stewart L. R., Roth K. A., Chiesa R., and Harris D. A. (2007) Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 26, 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baumann F., Tolnay M., Brabeck C., Pahnke J., Kloz U., Niemann H. H., Heikenwalder M., Rülicke T., Bürkle A., and Aguzzi A. (2007) Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 26, 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shmerling D., Hegyi I., Fischer M., Blättler T., Brandner S., Götz J., Rülicke T., Flechsig E., Cozzio A., von Mering C., Hangartner C., Aguzzi A., and Weissmann C. (1998) Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93, 203–214 [DOI] [PubMed] [Google Scholar]
- 21. Solomon I. H., Huettner J. E., and Harris D. A. (2010) Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J. Biol. Chem. 285, 26719–26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Solomon I. H., Khatri N., Biasini E., Massignan T., Huettner J. E., and Harris D. A. (2011) An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J. Biol. Chem. 286, 14724–14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Massignan T., Biasini E., and Harris D. A. (2011) A drug-based cellular assay (DBCA) for studying cytotoxic and cytoprotective activities of the prion protein: a practical guide. Methods 53, 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Massignan T., Stewart R. S., Biasini E., Solomon I. H., Bonetto V., Chiesa R., and Harris D. A. (2010) A novel, drug-based, cellular assay for the activity of neurotoxic mutants of the prion protein. J. Biol. Chem. 285, 7752–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Biasini E., Turnbaugh J. A., Massignan T., Veglianese P., Forloni G., Bonetto V., Chiesa R., and Harris D. A. (2012) The toxicity of a mutant prion protein is cell-autonomous, and can be suppressed by wild-type prion protein on adjacent cells. PLoS ONE 7, e33472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biasini E., Unterberger U., Solomon I. H., Massignan T., Senatore A., Bian H., Voigtlaender T., Bowman F. P., Bonetto V., Chiesa R., Luebke J., Toselli P., and Harris D. A. (2013) A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J. Neurosci. 33, 2408–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Westergard L., Turnbaugh J. A., and Harris D. A. (2011) A nine amino acid domain is essential for mutant prion protein toxicity. J. Neurosci. 31, 14005–14017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan T., Wong B. S., Liu T., Li R., Petersen R. B., and Sy M. S. (2002) Cell-surface prion protein interacts with glycosaminoglycans. Biochem. J. 368, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Warner R. G., Hundt C., Weiss S., and Turnbull J. E. (2002) Identification of the heparan sulfate binding sites in the cellular prion protein. J. Biol. Chem. 277, 18421–18430 [DOI] [PubMed] [Google Scholar]
- 30. Caughey B., and Raymond G. J. (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 67, 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caughey B., Ernst D., and Race R. E. (1993) Congo red inhibition of scrapie agent replication. J. Virol. 67, 6270–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caughey W. S., Raymond L. D., Horiuchi M., and Caughey B. (1998) Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. U.S.A. 95, 12117–12122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Priola S. A., Raines A., and Caughey W. S. (2000) Porphyrin and phthalocyanine antiscrapie compounds. Science 287, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 34. Nicoll A. J., Trevitt C. R., Tattum M. H., Risse E., Quarterman E., Ibarra A. A., Wright C., Jackson G. S., Sessions R. B., Farrow M., Waltho J. P., Clarke A. R., and Collinge J. (2010) Pharmacological chaperone for the structured domain of human prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 17610–17615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Massignan T., Cimini S., Stincardini C., Cerovic M., Vanni I., Elezgarai S. R., Moreno J., Stravalaci M., Negro A., Sangiovanni V., Restelli E., Riccardi G., Gobbi M., Castilla J., Borsello T., Nonno R., and Biasini E. (2016) A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein. Sci. Rep. 6, 23180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Büeler H., Aguzzi A., Sailer A., Greiner R. A., Autenried P., Aguet M., and Weissmann C. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347 [DOI] [PubMed] [Google Scholar]
- 37. Shyng S.-L., Lehmann S., Moulder K. L., and Harris D. A. (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J. Biol. Chem. 270, 30221–30229 [DOI] [PubMed] [Google Scholar]
- 38. Fang C., Imberdis T., Garza M. C., Wille H., and Harris D. A. (2016) A neuronal culture system to detect prion synaptotoxicity. PLoS Pathog. 12, e1005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilham J. M., Orrú C. D., Bessen R. A., Atarashi R., Sano K., Race B., Meade-White K. D., Taubner L. M., Timmes A., and Caughey B. (2010) Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6, e1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Caughey B., Brown K., Raymond G. J., Katzenstein G. E., and Thresher W. (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and Congo Red. J. Virol. 68, 2135–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Turnbaugh J. A., Unterberger U., Saá P., Massignan T., Fluharty B. R., Bowman F. P., Miller M. B., Supattapone S., Biasini E., and Harris D. A. (2012) The N-terminal, polybasic region of PrPC dictates the efficiency of prion propagation by binding to PrPSc. J. Neurosci. 32, 8817–8830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miller M. B., Geoghegan J. C., and Supattapone S. (2011) Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog. 7, e1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silber B. M., Gever J. R., Rao S., Li Z., Renslo A. R., Widjaja K., Wong C., Giles K., Freyman Y., Elepano M., Irwin J. J., Jacobson M. P., and Prusiner S. B. (2014) Novel compounds lowering the cellular isoform of the human prion protein in cultured human cells. Bioorg. Med. Chem. 22, 1960–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carr R. A., Congreve M., Murray C. W., and Rees D. C. (2005) Fragment-based lead discovery: leads by design. Drug Discov. Today 10, 987–992 [DOI] [PubMed] [Google Scholar]
- 45. Safar J. G., Scott M., Monaghan J., Deering C., Didorenko S., Vergara J., Ball H., Legname G., Leclerc E., Solforosi L., Serban H., Groth D., Burton D. R., Prusiner S. B., and Williamson R. A. (2002) Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat. Biotechnol. 20, 1147–1150 [DOI] [PubMed] [Google Scholar]
- 46. Fluharty B. R., Biasini E., Stravalaci M., Sclip A., Diomede L., Balducci C., La Vitola P., Messa M., Colombo L., Forloni G., Borsello T., Gobbi M., and Harris D. A. (2013) An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.