

Abstract
Blood-brain barrier (BBB) breakdown and the associated microvascular hyperpermeability followed by brain edema are hallmark features of several brain pathologies, including traumatic brain injuries (TBI). Recent studies indicate that pro-inflammatory cytokine interleukin-1β (IL-1β) that is up-regulated following traumatic injuries also promotes BBB dysfunction and hyperpermeability, but the underlying mechanisms are not clearly known. The objective of this study was to determine the role of calpains in mediating BBB dysfunction and hyperpermeability and to test the effect of calpain inhibition on the BBB following traumatic insults to the brain. In these studies, rat brain microvascular endothelial cell monolayers exposed to calpain inhibitors (calpain inhibitor III and calpastatin) or transfected with calpain-1 siRNA demonstrated attenuation of IL-1β-induced monolayer hyperpermeability. Calpain inhibition led to protection against IL-1β-induced loss of zonula occludens-1 (ZO-1) at the tight junctions and alterations in F-actin cytoskeletal assembly. IL-1β treatment had no effect on ZO-1 gene (tjp1) or protein expression. Calpain inhibition via calpain inhibitor III and calpastatin decreased IL-1β-induced calpain activity significantly (p < 0.05). IL-1β had no detectable effect on intracellular calcium mobilization or endothelial cell viability. Furthermore, calpain inhibition preserved BBB integrity/permeability in a mouse controlled cortical impact model of TBI when studied using Evans blue assay and intravital microscopy. These studies demonstrate that calpain-1 acts as a mediator of IL-1β-induced loss of BBB integrity and permeability by altering tight junction integrity, promoting the displacement of ZO-1, and disorganization of cytoskeletal assembly. IL-1β-mediated alterations in permeability are neither due to the changes in ZO-1 expression nor cell viability. Calpain inhibition has beneficial effects against TBI-induced BBB hyperpermeability.
Keywords: calpain, inflammation, inhibitor, interleukin 1 (IL-1), vascular biology
Introduction
The blood-brain barrier (BBB)2 plays an important role in maintaining the homeostasis of the brain. Blood-brain barrier breakdown and the associated hyperpermeability are hallmark features of several brain pathologies and injuries. The BBB is mainly composed of the cerebral endothelial cells and the tight junctions (TJs) between them (1). TJs between the neighboring endothelial cells include transmembrane TJs, i.e. occludin, claudins, junctional adhesion molecules, etc., and membrane-bound TJs, i.e. zonula occludens (1). Zonula occludens play an important role in regulating BBB permeability by binding to both transmembrane tight junctions and actin cytoskeleton intracellularly (2). Various mediators of inflammation are shown to modulate BBB breakdown and permeability in a variety of pathologies (3). Blood-brain barrier breakdown and the associated hyperpermeability is the leading cause of brain edema and elevated intracranial pressure followed by decreased perfusion pressure leading to poor clinical outcomes in traumatic brain injury (TBI) (4).
Inflammation that occurs as a consequence of brain injuries is carried out by various pro-inflammatory cytokines (5). IL-1β is the most implicated pro-inflammatory cytokine in various pathologies of the central nervous system, including TBI (6, 7). Interleukin-1 (IL-1) inhibition has beneficial effects as demonstrated in experimental models of brain damage (6). IL-1β induces BBB breakdown in rat brain endothelial cells and also increases human brain microvascular endothelial cell permeability (8). However, IL-1β-induced mechanisms that lead to barrier dysfunctions and hyperpermeability at the level of the BBB are not clearly known.
Calpains are thiol or cysteine proteases that are present in most of the mammalian cells. They are involved in a wide array of neurological pathologies like trauma, ischemia-reperfusion injury, spinal cord injury, and several non-neurological pathologies as well (9–12). Intracellular calcium levels and the endogenous inhibitor of calpains, namely calpastatin, tightly regulate calpain levels endogenously (9, 13). Calpains-1 and -2 are the predominant calpains in the central nervous system (14, 15). An increased calpain activity was observed following TBI in laboratory rodents (16, 17) and human patients (12). Calpain inhibitors protect the brain against various neurotraumas, including brain and spinal cord injury (18, 19). Calpain expression was found to be increased in the endothelial cells of the injured brain cortex following TBI in human patients compared with those who died from cardiac arrest (12). Calpain-dependent cleavage of intracellular cytoplasmic protein ZO-1 has been studied in human lung endothelial cells (13). However, their contribution in regulating BBB endothelial dysfunction and hyperpermeability is largely unknown.
Based on these observations, we hypothesized that calpain-mediated mechanisms play an important role in promoting IL-1β-induced BBB breakdown and hyperpermeability and that calpain inhibition will potentially down-regulate this pathway. Therefore, we studied the effect of calpain inhibition on BBB hyperpermeability in both cultured rat brain endothelial cells and a mouse model of TBI. The objectives and the specific questions that we addressed are as follows. What is the effect of calpain inhibition on IL-1β-induced BBB endothelial hyperpermeability, tight junctional integrity, and cytoskeletal organization? Does IL-1β treatment increase calpain activity in BBB endothelial cells? Does IL-1β treatment induce intracellular free calcium ([Ca2+]i) mobilization in BBB endothelial cells? Does IL-1β treatment influence ZO-1 expression at the mRNA and/or protein levels? What is the effect of IL-1β treatment on the total number of viable endothelial cells? What is the effect of calpain inhibition on TBI-induced BBB hyperpermeability in a mouse controlled cortical impact model of TBI?
Results
Calpain Inhibition Attenuates IL-1β-induced Endothelial Cell Hyperpermeability
RBMEC monolayers were pretreated with calpain inhibitor III or calpastatin to confirm the contribution of calpains in mediating IL-1β (10 ng/ml for 2 h)-induced endothelial cell hyperpermeability. Fig. 1 demonstrates that IL-1β treatment significantly increases endothelial cell hyperpermeability, whereas pretreatment with calpain inhibitor III (10 μm; 1 h; Fig. 1A; p < 0.05) and calpastatin (10 μm; 1 h; Fig. 1B; p < 0.05) significantly attenuated IL-1β-induced endothelial cell hyperpermeability. Calpain inhibitor III (10 μm; 1 h) and calpastatin (10 μm; 1 h) treatment alone did not alter rat brain endothelial cell hyperpermeability. Calpain inhibitor III (1, 10, and 50 μm) treatment decreased IL-1β (10 ng/ml)-induced monolayer hyperpermeability significantly (Fig. 3B; p < 0.05).
FIGURE 1.

Calpain inhibitor III and calpastatin pretreatment attenuates IL-1β treatment-induced monolayer hyperpermeability and calpain activity. Calpain inhibitor III (A, n = 4; p < 0.05) and calpastatin (B, n = 4; p < 0.05) pretreatment attenuates IL-1β-induced monolayer hyperpermeability significantly. Knockdown of calpain-1 by siRNA attenuates IL-1β treatment-induced monolayer hyperpermeability (C, n = 4; p < 0.05). Monolayer permeability is expressed as a percentage control of FITC-dextran-10 kDa fluorescence intensity, plotted on y axis. Calpastatin and calpain inhibitor III pretreatment attenuates IL-1β treatment-induced calpain activity significantly (D, n = 4; p < 0.05). Calpain activity is expressed as RFU, plotted on the y axis. Data are expressed as mean ± % S.E. *a indicates significant increase compared with the control group; *b indicates significant decrease compared with the IL-1β treatment group.
FIGURE 3.
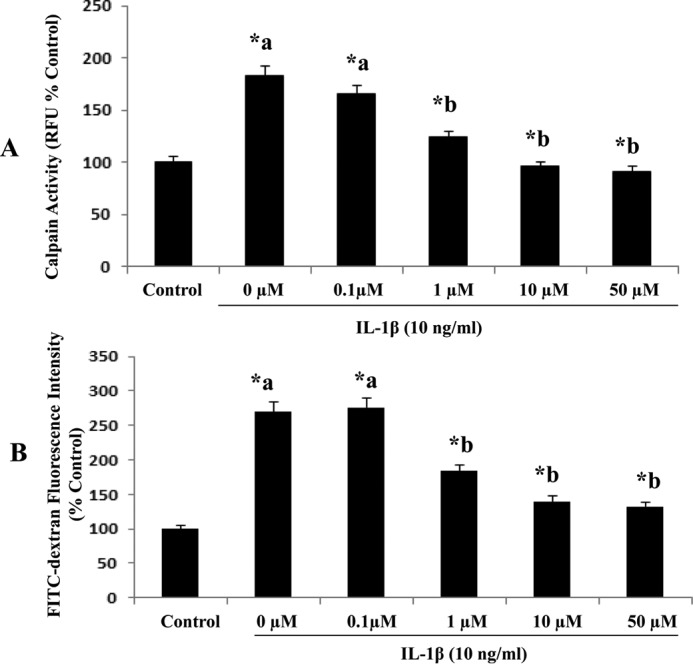
Calpain inhibitor III treatment of various concentrations (0. 1, 1, 10, and 50 μm) decreases IL-1β-induced calpain activity and monolayer hyperpermeability significantly (A and B, n = 5; p < 0.05) in RBMEC. Calpain activity is expressed as RFU, plotted on the y axis. Monolayer permeability is expressed as FITC-dextran fluorescence intensity (% control). Data are expressed as mean ± S.E. *a indicates significant increase compared with the control (0 μm) group). *b indicates significant decrease compared with the 0 μm group.
Significant contribution of calpain-1 is further supported by calpain-1 siRNA transfection studies. Fig. 1C demonstrates that IL-1β (10 ng/ml for 2 h) treatment-induced endothelial cell hyperpermeability was significantly reduced in calpain-1 knockdown (25 nm; 48 h) cells compared with IL-1β treatment alone (p < 0.05). Calpain-1 knockdown studies were performed using siRNA transfection technique as described earlier. Calpain-1 siRNA treatment alone did not induce any significant change in rat brain endothelial cell hyperpermeability compared with the control siRNA group. Calpain-1 siRNA-treated groups were compared with the control siRNA group, and the IL-1β alone-treated group was compared with the control group.
Calpain Inhibitors Attenuate IL-1β-induced Calpain Activity
Exposure of RBMEC to various concentrations of IL-1β (1, 10, and 100 ng/ml; 2 h) as well as exposure of IL-1β (10 ng/ml) for various durations of time (1, 2, and 4 h) increased calpain activity significantly (Fig. 2, A and B; p < 0.05). IL-1β (10 ng/ml; 2 h) treatment significantly increased calpain activity in RBMECs compared with the untreated control group, whereas pretreatment with calpain inhibitor III (10 μm; 1 h) and calpastatin (10 μm; 1 h) significantly attenuated IL-1β-induced calpain activity (Fig. 1D, p < 0.05). Calpastatin (10 μm; 1 h) and calpain inhibitor III (10 μm; 1 h) treatment alone did not alter rat brain endothelial cell hyperpermeability compared with the control untreated group. Treatment of RBMEC with calpain inhibitor III (1, 10, and 50 μm) resulted in a significant decrease in IL-1β-induced calpain activity (Fig. 3B; p < 0.05).
FIGURE 2.
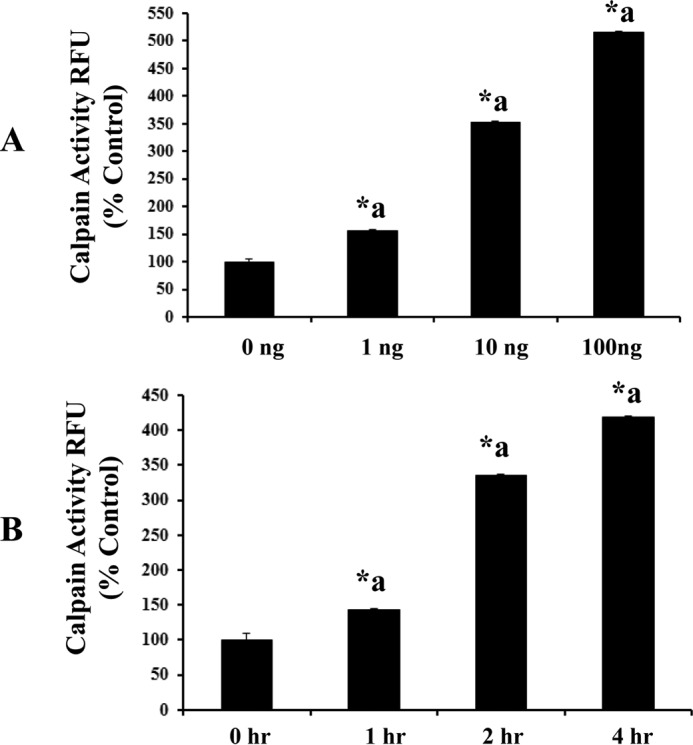
IL-1β treatment (at concentrations 1, 10, and 100 ng/ml) as well as various time points (1, 2, and 4 h) induces calpain activity significantly (A and B, n = 5; p < 0.05). Calpain activity is expressed as RFU, plotted on the y axis. Data are expressed as mean ± S.E. *a indicates significant increase compared with the control (0 h) group.
Calpain Inhibition Provides Protection against IL-1β-induced Loss of Tight Junction Integrity and F-actin Stress Fiber Formation
Cells were processed for immunofluorescence localization of tight junction protein, ZO-1. IL-1β (10 ng/ml; 2 h) treatment induced ZO-1 junctional discontinuity (Fig. 4, A white arrows, and C) compared with the control cells. Pretreatment with calpain inhibitor III (10 μm; 1 h) decreased IL-1β (10 ng/ml; 2 h)-induced ZO-1 junction disintegrity. Calpain inhibitor III (10 μm; 1 h) treatment alone did not alter ZO-1 tight junction integrity compared with the untreated control group (fluorescence analysis using ImageJ; Fig. 4C, p < 0.05). For assessing the cytoskeletal assembly, rhodamine phalloidin labeling technique was performed. Untreated cells served as control. IL-1β (10 ng/ml; 2 h) treatment-induced F-actin stress fiber formation (white arrows; Fig. 4, B and D) was reduced by pretreatment with calpain inhibitor III (10 μm; 1 h), whereas calpain inhibitor III (10 μm; 1 h) alone did not induce F-actin stress fiber formation compared with the control untreated group (fluorescence analysis using ImageJ; Fig. 4D, p < 0.05).
FIGURE 4.

IL-1β treatment-induced ZO-1 junctional disruption and F-actin stress fiber formation is reduced by pretreatment with calpain inhibitor III (A–D). IL-1β treatment-induced ZO-1 junctional disruption and F-actin stress fiber formation are shown by white arrows in A and B, respectively. ZO-1 junctional integrity and F-actin stress fiber formation were assessed using immunofluorescence localization and rhodamine phalloidin techniques, respectively (n = 4 for each study). The changes in ZO-1 localization and the formation of F-actin stress fibers were further determined using ImageJ software and presented as arbitrary units (C and D, respectively; p < 0.05). Under normal physiological conditions actin filaments are randomly distributed throughout the cell, but agents that induce endothelial hyperpermeability induce them to reorganize themselves into stress fibers that are linear parallel bundles across the cell interior (following IL-1β treatment as pointed by the arrows in B) and also exhibit increased binding to rhodamine phalloidin (as shown in B and D).
IL-1β Treatment Neither Induces ZO-1 mRNA Expression nor Alters ZO-1 Protein Expression
IL-1β (10 ng/ml; 2 h) treatment did not alter ZO-1 mRNA expression by RT-PCR studies (Fig. 5A). IL-1β treatment did not alter ZO-1 protein expression (Fig. 5B) by Western blotting studies. These studies suggest that the total ZO-1 gene and protein expression remains the same by IL-1β treatment (10 ng/ml; 2 h) compared with the control untreated cells, thus indicating that the loss of ZO-1 tight junction integrity is not due to the loss of the total ZO-1 protein expression in the cells, indicating a possibility of temporary relocalization of ZO-1.
FIGURE 5.
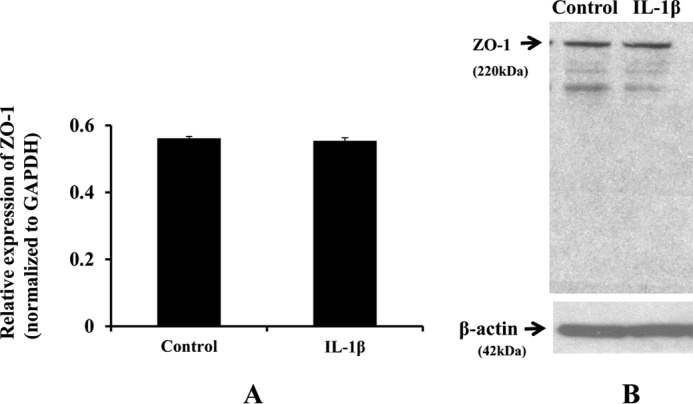
IL-1β treatment does not induce ZO-1 mRNA or protein expression. Fluorescence intensity from RT-PCR studies is expressed as relative expression of ZO-1 normalized to GAPDH, plotted on y axis (n = 3; A). Data are represented as mean ± S.E. ZO-1 protein expression did not change following IL-1β treatment (n = 4; B).
IL-1β Treatment Does Not Induce Cell Death in Endothelial Cells
IL-1β (10 ng/ml) treatment did not significantly decrease the number of viable cells compared with the control group up to 4 h, as shown in Fig. 6. IL-1β (10 ng/ml) treatment for 6 h significantly decreased the number of viable cells compared with the control group. 50, 25, and 10 mm concentrations of hydrogen peroxide were used as positive control at 2-, 4-, and 6-h treatments of IL-1β.
FIGURE 6.

IL-1β treatment does not induce cell death. IL-1β treatment did not alter cell viability up to 4 h. Treatment of hydrogen peroxide used as a positive control showed a significant decrease in cell viability compared with the untreated and IL-1β treatment groups (n = 5; p < 0.05). Data are expressed as mean ± % S.E. *a indicates significant decrease compared with the control group.
IL-1β Treatment Does Not Induce Intracellular Calcium Mobilization
To test whether IL-1β up-regulates the activity of calpains, calcium-dependent cysteine proteases, by increasing intracellular calcium concentration ([Ca2+]i), we directly measured [Ca2+]i in RBMECs by two different approaches. Initially, RBMECs were grown as monolayers and loaded with Fluo-4 AM (a Ca2+ indicator). Fluo-4 signals were measured from cells before (F0) and after (F) a 10-min incubation of cells with IL-1β (10 ng/ml). In parallel, control cells were treated with thapsigargin (TG; 2 μm), ionomycin (Iono; 1 μm), or DMSO as the vehicle control for TG and Iono. TG is a sarco/endoplasmic reticulum Ca2+-ATPase pump inhibitor, which passively depletes Ca2+ in the endoplasmic reticulum store in conjunction with triggering store-operated Ca2+ entry. Ionomycin is an ionophore that increases membrane permeability to Ca2+, thereby raising the intracellular level of Ca2+. As expected, both stimulations by TG and ionomycin triggered a significant increase of [Ca2+]i (Fig. 7A); however, there was only a negligible intracellular Ca2+ response to the IL-1β treatment (Fig. 7A).
FIGURE 7.
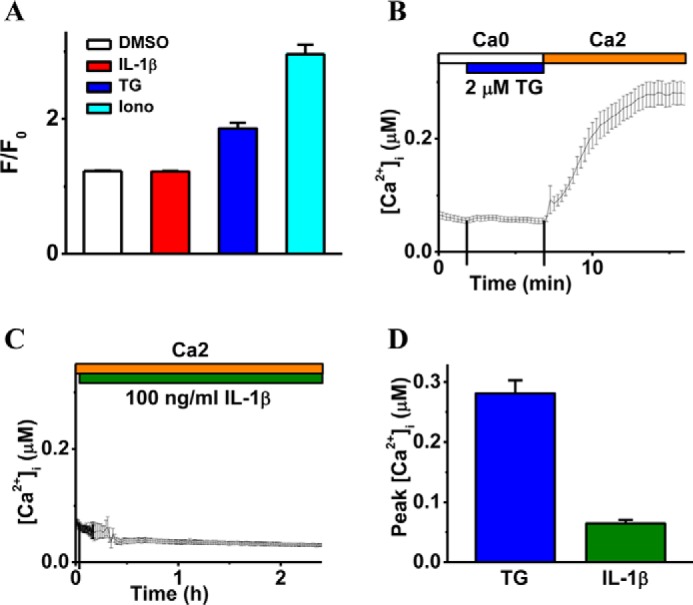
IL-1β treatment does not induce intracellular calcium mobilization. A demonstrates the effect of IL-1β treatment on intracellular calcium mobilization (F) relative to basal Ca2+ (F0) when measured 10 min after the application of DMSO (vehicle control), IL-1β (10 ng/ml), TG (2 μm), and Iono (1 μm) and displayed as mean ± S.E. (n = 4). B and C are the representative intracellular free calcium recordings ([Ca2+]i, mean ± S.E.) showing cytosolic Ca2+ levels coupled to TG (2 μm, B) or IL-1β (100 ng/ml, C) stimulation. The top bars indicate the type of extracellular solutions applied to the RBMECs, and the vertical lines on the x axis indicate the time of solution exchange. D displays averaged peak values of [Ca2+]i in response to TG (n = 40 cells) and IL-1β (n = 30 cells), respectively.
It is possible that IL-1β triggers a relatively transient [Ca2+]i response, and/or a small population of cultured RBMECs has a dramatic [Ca2+]i response to IL-1β, which could be masked during our Fluo-4-based [Ca2+]i measurement. Accordingly, RBMECs were then loaded with Fura-2 AM, a ratiometric Ca2+ indicator, and [Ca2+]i was continuously monitored at the single cell level. In the control experiments, TG could evoke a typical store-operated Ca2+ entry in RBMECs (Fig. 7B) that elevates [Ca2+]i from 65 ± 6 to 281 ± 22 nm (Fig. 7D). However, when RBMECs were incubated with 10 ng/ml IL-1β for 20 min, there were no cells showing significantly increased [Ca2+]i (data not shown). Cells were then treated with a higher dose (100 ng/ml) of IL-1β for 2 h, which still did not significantly promote intracellular calcium levels in any cells (Fig. 7, C and D). Finally, our results indicate that IL-1β treatment does not induce a dramatic [Ca2+]i mobilization in cultured RBMECs.
Calpain Inhibitor III Treatment Attenuates Mild TBI-induced BBB Hyperpermeability in Mice
Mice subjected to TBI demonstrated a significant increase in Evans blue leakage compared with the sham animals (p < 0.05). DMSO was used as a vehicle control. Vehicle + sham group did not show any significant increase in BBB permeability compared with the sham group, although the vehicle-treated group when subjected to mild TBI significantly induced BBB hyperpermeability. Pretreatment or post-treatment with calpain inhibitor III attenuated TBI-induced Evans blue leakage into the brain tissue significantly (Fig. 8, A and B; p < 0.05).
FIGURE 8.
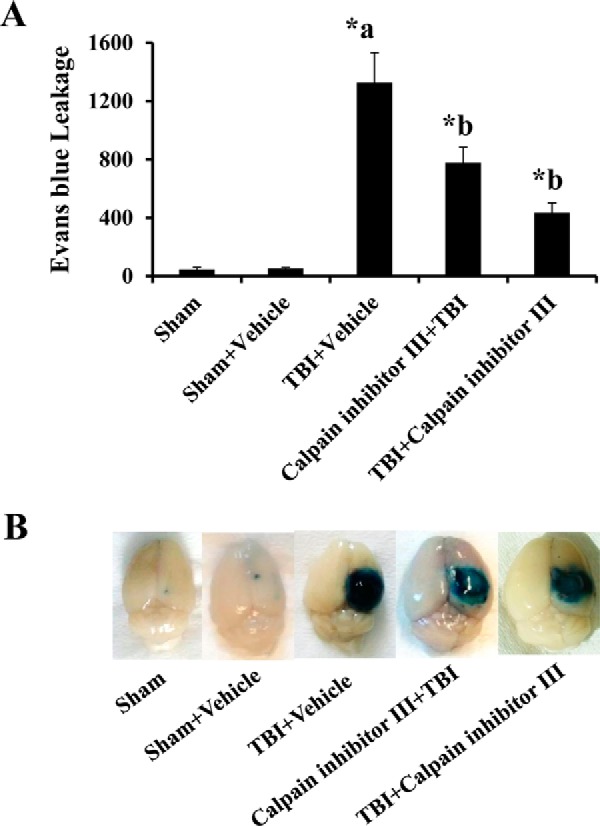
Calpain inhibitor III treatment attenuates TBI-induced BBB hyperpermeability studied by Evans blue dye extravasation method. Vehicle control group subjected to TBI demonstrated a significant increase in Evans blue leakage compared with the sham injury group (A, p < 0.05). Calpain inhibitor III (10 μg/g body weight of the animal) pre- or post-treatment attenuated TBI-induced Evans blue leakage into the extravascular tissue space significantly (A, p < 0.05). Pictorial representation of the brain tissue from various groups is shown in B. Each group consisted of five animals. Sham injury group is used as the baseline for all comparisons. Data are expressed as nanograms/brain cortex ± S.E. *a indicates significant increase compared with the sham injury group; *b indicates significant decrease compared with the vehicle + TBI group.
Intravital microscopy evaluation of brain pial vasculature of anesthetized mice showed BBB dysfunction and hyperpermeability following mild TBI compared with sham vehicle at 60 min post-trauma as evidenced by the leakage of FITC-dextran from intravascular space to the interstitium (Fig. 9, A and B, p < 0.05). Calpain inhibitor III treatment following TBI showed a decrease in hyperpermeability compared with the TBI group (p < 0.05).
FIGURE 9.
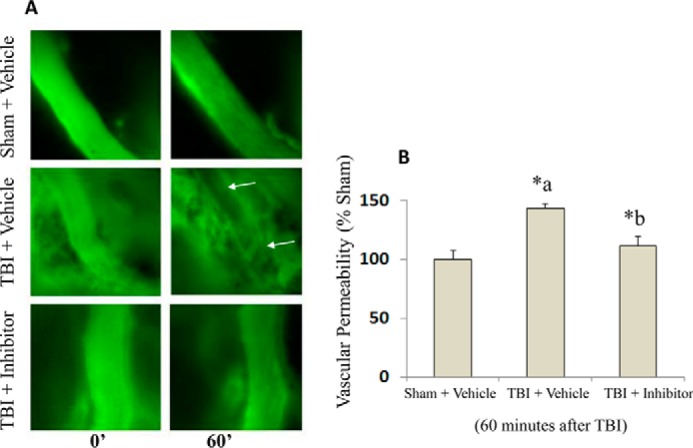
Intravital microscopy imaging of mouse brain demonstrates the protective effect of calpain inhibitor III against TBI-induced BBB hyperpermeability. Mice subjected to TBI demonstrated a significant increase in BBB permeability at 60 min as evidenced by enhanced FITC-dextran fluorescence compared with the sham injury group (A and B, n = 5 each group, p < 0.05). Calpain inhibitor III (10 μg/g body weight of the animal; n = 4) treatment 15 min following TBI attenuated TBI-induced BBB hyperpermeability as evidenced by the extravasation of FITC-dextran into the extravascular space compared with TBI + vehicle group (A and B, p < 0.05). Data are expressed as a change in relative fluorescence to the initial image (0 time point) in each group, and the sham injury group is used as the baseline for all comparisons. *a indicates significant increase compared with the sham injury group; *b indicates significant decrease compared with the vehicle + TBI group.
Discussion
The major findings of this study are as follows: 1) calpain(s) promote BBB dysfunction and hyperpermeability via disruption of the TJs in vitro; 2) IL-1β is an inducer of calpain-mediated BBB dysfunction and hyperpermeability in vitro; 3) inhibition of calpain activation provides protection against BBB hyperpermeability via preservation of TJ integrity in vitro; 4) calpain-mediated loss of barrier functions are independent of [Ca2+]i mobilization or loss of cell viability; and 5) pharmacological inhibition of calpains provide protection against BBB hyperpermeability in a mouse model of TBI.
In this study, the inhibition of calpains either by a pharmacological inhibitor or by its endogenous inhibitor, calpastatin, or by calpain-1 gene knockdown provided protection to the BBB. These observations strongly suggest a major role for calpains in modulating BBB integrity and permeability. Calpains are thiol or cysteine proteases that are present in most of the mammalian cells. Under physiological conditions, calpains possess low activity and play an important role in regulating kinases, transcription factors, and receptors apart from aiding in the cytoskeletal turnover (20). Calpain deficiencies as well as its over-activation are linked to a variety of diseases and pathological consequences (21). Because of their multifaceted nature, they control various irreversible signaling events and biological functions in the cell such as endothelial cell adhesion, differentiation, migration, proliferation, cell cycle control, cytoskeletal remodeling, embryonic development, and vesicular trafficking (12–14, 22–24). Our studies support a relationship between calpains and microvascular permeability at the level of the BBB.
Calpain-1 (also known as τ-calpain) and calpain-2 (also known as m-calpain) are predominantly expressed in the central and peripheral nervous systems (14, 15). Calpain-1 is activated under low calcium concentrations (3–50 μm), whereas calpain-2 is activated only under high concentrations (400–800 μm) of calcium in the cell (23, 24). Calpain-1 knockdown data from our studies demonstrate the contribution of calpain-1, but we do not disregard the contribution of calpain-2 in mediating BBB dysfunction and hyperpermeability, an area open for further investigation.
Calpastatin and intracellular Ca2+ levels tightly regulate the calpain levels endogenously (12, 21). This information was used to study the contribution of calcium and calpastatin in regulating IL-1β-induced endothelial hyperpermeability. To study the effect of IL-1β treatment on intracellular calcium, we performed calcium mobilization studies, which demonstrated that IL-1β treatment does not induce significant mobilization of intracellular calcium in RBMECs. However, we do not rule out the possible involvement of calcium in inducing calpains as established in several other conditions. We speculate that IL-1β-induced calpain activation may occur due to concentration changes in localized calcium (25).
Calpastatin is an endogenous inhibitor of calpains; it regulates the activity of calpains by binding to the active site cleft of both calpains-1 and -2. Our results support the significance of calpain/calpastatin interactions in regulating BBB functions. Although our studies demonstrate the ability of calpastatin in attenuating IL-1β-induced endothelial hyperpermeability, we have not further explored these studies in vivo. Understanding calpastatin-mediated regulation of BBB hyperpermeability in animal models can open new avenues for exploring endogenous therapeutic agents that can alter the secondary injuries that occur following brain injury.
Our studies demonstrate that pharmacological inhibition of calpains is effective in preserving barrier functions in vitro and in vivo. Calpain inhibitor III is a well studied cell-permeable pharmacological inhibitor of calpains (12), which was chosen for our in vitro and in vivo studies. Calpain inhibitor III crosses the BBB efficiently and binds specifically to calpains-1 and -2, unlike other inhibitors of calpains such as trypsin, plasmin, caspase-1, and cathepsin D (24). Cytoskeletal and neuroprotective properties of calpain inhibitor III were examined using the CCI model of TBI in male CF-1 mice, 24 h post-TBI (26). However, these studies do not aim to understand the effect of calpain inhibitor III on TBI-induced BBB hyperpermeability. Our study addresses this important question.
Studies by Tsubokawa et al. (27) indicate a link between calpain, cathepsin B, and matrix metalloproteinase-9 (MMP-9) up-regulation. MMP-2 and calpains have some common cleavage targets such as sarcomeric proteins troponin I, myosin light chain-1, and titin in heart cells (28). These studies support the possibility that calpain-induced BBB endothelial cell hyperpermeability may occur via MMP activation or vice versa. Understanding this relationship is important to selectively target the appropriate pathways for therapeutic purposes.
A calpain/MMP-9 interaction in endothelial cells of the BBB as well as the various other cell types of the neurovascular units is a possibility that regulates BBB functions. In addition to their presence in endothelial cells, calpains and MMPs are expressed in other cell types of the neurovascular unit such as astrocytes and pericytes. Pericytes are considered as one of the major sources of MMP-9 in the BBB (29). In a recent study, it was found that among the various cell types of the neurovascular unit, pericytes exhibited the highest level of MMP-9 secretion when challenged with thrombin that is known to induce barrier dysfunctions in endothelial cells (30). Furthermore, pericytes features such as contractility and cellular stiffness have been regulated by cellular calpains (31). Similarly, astrocytes are an integral component of the BBB that may be compromised by TBI or ischemic brain injury, and increased MMP-9 expression has been observed in astrocytes following trauma (32). Also, astrocyte-originated MMP-9 has been attributed to the pathogenesis of hemorrhagic brain edema (33). Our results conducted exclusively in BBB endothelial cells demonstrate that they are targeted by calpain-mediated and thus potentially MMP-9-regulated mechanisms of barrier dysfunction and hyperpermeability. Future studies that involve multiple co-culture models of all the major cell types of the neurovascular unit could provide more information toward this potential interaction.
Our results demonstrate changes in the cytoskeletal assembly as evidenced by increased F-actin stress fiber formation following IL-1β treatment-induced calpain activation and BBB hyperpermeability that was decreased following calpain inhibition. Actin filaments are linear polymers of filamentous actin formed by actin polymerization, and under normal physiological conditions they distributed randomly throughout the cell. Various hyperpermeability-inducing agents are known to induce the reorganization of actin filaments into stress fibers that are linear parallel bundles across the cell interior (34, 35). Increased F-actin stress fiber formation most often is associated with endothelial barrier dysfunction and hyperpermeability (35) The stress fiber formation that occurs following IL-1β treatment may also involve tight junction cleavage via the Ras homolog gene family member A (RhoA)-dependent mechanisms leading to BBB permeability. RhoA-mediated mechanisms also activate cell division control protein (Cdc42), which plays an important role in maintaining the tight junction integrity and actin cytoskeletal assembly (36).
Our studies on understanding the role of calpains were focused more on the tight junction protein, ZO-1, and the actin cytoskeleton. However, understanding the contribution of calpains in mediating BBB hyperpermeability via multiple other proteins is important as they target a wide range of proteins, which include the following: cytoskeletal proteins (α-spectrin, talin, filamin, paxillin, vinculin, ezrin, microtubule-associated proteins 2, myristoylated alanine-rich protein kinase C substrate, and neurofilament proteins); kinases (PP60 and proto-oncogene tyrosine-protein kinase Src); phosphatases (protein-tyrosine phosphatase 1B, focal adhesion kinase, talin, paxillin, protein-tyrosine phosphatase); membrane-associated proteins; junctional proteins (β-catenin, E-cadherin, and β-spectrin); and transcription factors such as c-Fos, c-Jun, and p53 (13, 21, 22, 37–40). Although the expression of some of these proteins in the BBB is not well established, it is important to know how calpains interact with them and how these molecules contribute to BBB dysfunction or trauma-induced brain damage directly or indirectly.
The cellular mechanisms by which IL-1β up-regulates calpain activity is not clearly known to us at this time. Recent studies indicate that calpain activation can occur independent of calcium via mechanisms such as mitogen-activated protein kinase (MAPK)-mediated phosphorylation in various cell types such as fibroblasts, neurons, HEK-TrkB cells, etc. and is calcium-independent (41, 42). Furthermore, in hippocampal neurons MAPK-mediated and calcium-independent calpain activation was associated with actin polymerization, which was prevented by calpain inhibition (42). Also, MAPKs have been shown to promote vascular endothelial permeability. However, further studies are required to delineate a potential IL-1β-MAPK-calpain activation pathway leading to barrier dysfunction in microvascular endothelial cells.
The use of calpain inhibitors for functional improvement has been investigated in various animal models of CNS pathologies. Calpain inhibitors has been found to provide neuroprotection in a mouse controlled cortical impact model of TBI and ischemic stroke (26, 43). Our results suggest that one of the mechanisms by which such neuroprotective effects occurred could be due to enhanced protection of the BBB that occurred following calpain inhibition. In subarachnoid hemorrhage, calpain inhibition reduced behavioral deficits and BBB permeability (44). Similarly, calpain inhibition has been found to be protective against neuropathology associated with Alzheimer's disease, Parkinson's disease and conditions, such as lissencephaly (45–47).
In conclusion, our studies demonstrate the novel role of calpains in promoting IL-1β-induced BBB dysfunction and hyperpermeability and how calpain inhibition regulates these pathways. Our results further demonstrate that calpain inhibition has the potential to be developed as a therapeutic target in controlling TBI-induced BBB hyperpermeability and edema when established in human patients.
Experimental Procedures
Materials
RBMECs and RBMEC medium were purchased from Cell Applications, Inc. (San Diego). Transwell® 24-well plates were obtained from Corning Costar (New York). Nunc Lab Tek II-CC, 8-well glass chamber slides, interleukin-1β human, fibronectin from bovine plasma, β-actin, albumin from bovine serum, Evans blue, trichloroacetic acid, and fluorescein isothiocyanate/dextran-10 kDa were purchased from Sigma. Rabbit anti-ZO-1 (catalog no. 617300), mouse anti-ZO-1 (catalog no. 339100), 0.25% trypsin (1×), Opti-MEM (1×)/reduced serum medium, Dulbecco's modified Eagle's medium (DMEM; with high glucose, HEPES, no phenol red (1×)), NuPAGE Novex® 10% BisTris protein gels, NuPAGE® MOPS SDS Running Buffer, NuPAGE® Transfer Buffer, HyClone Dulbecco's phosphate-buffered saline (PBS, without calcium, magnesium, or phenol red), TRIzol® reagent, SuperScript® IV first-strand synthesis system, Halt® protease inhibitor mixture (100×), PierceTM BCA protein assay kit, PierceTM ECL Western blotting substrate, and rhodamine phalloidin were purchased from Thermo Fisher Scientific (Carlsbad, CA). Goat anti-mouse IgG-HRP and donkey anti-rabbit IgG-FITC secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Calpain activity fluorometric assay kit (K240-100) and EZViableTM calcein AM cell viability fluorometric assay kit were bought from BioVision (Milpitas, CA). We also purchased Vector VECTASHIELD® mounting media with DAPI from Vector Laboratories (Burlingame, CA). RT2 qPCR primer assays for mouse GAPDH were purchased from Qiagen (Valencia, CA). Carbobenzoxy-valinyl-phenylalaninal, also called calpain inhibitor III or MDL 28170, was bought from Calbiochem. Cell lysis buffer (10×) was bought from Cell Signaling Technology, Inc. (Danvers, MA). Primers were purchased from Thermo Fisher Scientific (Carlsbad, CA). Calpain-1 siRNA and control siRNA (ON-TARGETplus siRNA) were purchased from Dharmacon, General Electric (Pittsburgh, PA).
In Vitro BBB Model
Primary cultures of RBMECs derived from the brain of adult Sprague-Dawley rats were purchased from the Cell Applications, Inc. (San Diego, CA). RBMECs were initially grown on 0.05% fibronectin-coated cell culture dishes, using the RBMEC medium in a cell culture incubator (95% O2, 5% CO2 at 37 °C). RBMECs were treated with 0.25% trypsin/EDTA for cell detachment. Detached cells were then grown on fibronectin-coated Transwell® inserts, chamber slides, or 100-mm dishes for experimental purposes. RBMEC passages 8–10 were chosen for all the experiments.
Animals and Surgeries
C57BL/6 mice (25–30 g) were obtained from Charles River Laboratories (Wilmington, MA). Animals were maintained at the Texas A&M University Health Science Center College of Medicine and Baylor Scott and White Health animal facility on a 12:12 h dark/light cycle, with free access to food and water. Room temperature was maintained at 25 ± 2 °C. Surgical and experimental procedures used in this study were conducted after approval by the Institutional Animal Care and Use Committee. The facility is approved by the Association for Assessment and Accreditation of Laboratory Animal Care International in accordance with the National Institutes of Health guidelines.
Craniotomy Procedure
The head of the animal was shaved, and the surgical site on the surface of the head was cleaned with an alcohol wipe. Lubricating ointment was applied to the eyes. A midline incision was made to remove the skin from top of the skull exposing the sagittal suture, bregma, and lambda. A circular craniotomy window, 3–4 mm in diameter, was made on the ipsilateral hemisphere between lambda and bregma using a microdrill. The resulting bone flap was removed. Sham animals received only craniotomy surgery, whereas TBI injury group receives brain injury via controlled cortical impactor following craniotomy procedure.
CCI
BenchMarkTM stereotaxic impactor from Leica was used for these studies. Following the craniotomy procedure, the animal was mounted on the stereotaxic frame. An impactor probe of 3 mm diameter was used to impact the exposed part of the brain. The depth of the injury was used to determine the intensity of the injury. Settings for mild TBI used in this study are as follows: 2 mm depth, 0.5 m/s velocity, and 100 ms contact time as described in Ref. 48.
Monolayer Permeability Assays
Briefly, RBMECs were grown on fibronectin-coated Transwell® inserts as monolayers for 72–96 h and regularly checked for confluency. Monolayers were initially exposed to phenol red-free DMEM for 45 min to an hour. DMEM-treated cells were then pretreated with calpain inhibitors and subsequently with IL-1β (10 ng/ml; 2 h). At the end of the treatments, FITC-labeled dextran-10 kDa (5 mg/ml; 30 min) was applied to the luminal compartment. Monolayer hyperpermeability was assessed fluorometrically at 485/520 nm using FITC dextran-10 kDa as described previously (49). Calpain inhibitor III (10 μm; 1 h) was used for pharmacological inhibition of calpains, and calpastatin (10 μm; 1 h) was used for endogenous inhibition of calpains. Calpain inhibitor III (MDL-28170; carbobenzoxy-valinyl-phenylalaninal) is a potent cell-permeable inhibitor of both calpain-1 and -2, and calpastatin is a cell-permeable peptide (27 amino acids in length) that inhibits both calpains-1 and -2. Untreated cells served as control. Each experiment was repeated four times. Fluorescence intensity was plotted on the y axis and represented as % control. Data were expressed as mean ± % S.E., and statistical differences among groups were determined by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test to determine the significant differences between specific groups. A value of p < 0.05 was considered statistically significant.
Calpain-1 Knockdown Studies
Briefly, RBMECs were transfected with control siRNA (25 nm; 48 h) or calpain-1 siRNA (25 nm; 48 h) on reaching 50% confluency. Transfection was performed according to manufacturer's instructions. Transfected monolayers were then exposed to IL-1β, and permeability was determined based on the leakage of FITC-dextran-10 kDa (5 mg/ml; 30 min) leakage from the luminal to the abluminal chamber. Monolayer permeability was assessed fluorometrically as described earlier. Untreated cells were used as control. Fluorescence intensity was plotted on the y axis and represented as % control. Data were expressed as mean ± % S.E., and statistical differences among groups were determined by one-way ANOVA followed by Bonferroni post hoc test to determine significant differences between specific groups. A value of p < 0.05 was considered statistically significant.
Calpain Activity Measurements
A calpain activity fluorometric assay kit was used to measure the calpain activity in the cells. For this assay, RBMECs were grown in Petri dishes until confluency was achieved. Cells were then trypsinized using lysis buffer, followed by resuspension in extraction buffer provided in the kit. The kit employs a synthetic calpain substrate, Ac-Leu-Leu-Tyr-7-amino-4-trifluoromethylcoumarin, to detect the calpain activity. An equal amount of protein lysates was taken and subsequently exposed to the substrate Ac-Leu-Leu-Tyr-7-amino-4-trifluoromethylcoumarin and incubated in the dark for an hour. Samples were then read using Fluoroskan AscentTM FL microplate fluorometer and luminometer at 400/505 nm (excitation/emission). Cells lysates were pretreated with calpain inhibitors (calpain inhibitor III and calpastatin at 10 μm for 1 h) and subsequently with IL-1β treatment. Untreated cells served as control. Each experiment was repeated five times. Calpain activity was expressed as relative fluorescence units (RFU) and plotted on the y axis. In a separate set of experiment, calpain activity was determined following exposure of the cells to various concentrations of IL-1β (0, 1, 10, and 100 ng/ml) for 2 h as well as IL-1β (10 ng/ml) for various durations of time (0, 1, 2, and 4 h). Data were expressed as mean ± S.E., and statistical differences among groups were determined by one-way ANOVA followed by Bonferroni post hoc test to determine significant differences between specific groups. A value of p < 0.05 was considered statistically significant.
Immunofluorescence Staining and Cytoskeletal Labeling
ZO-1 junctional localization and F-actin stress fiber formation were assessed. RBMECs were grown overnight on chamber slides. Pretreatment with calpain inhibitor III (10 μm; 1 h) was followed by IL-1β treatment (10 ng/ml; 2 h). Cells were then fixed in 4% paraformaldehyde in PBS for 10–15 min and permeabilized with 0.5% Triton X-100 in PBS for another 10–15 min. Cells were blocked with 2% bovine serum albumin (BSA) in PBS for an hour at room temperature. Cells were then incubated overnight with anti-rabbit primary antibodies against ZO-1 (617300) at 1:150 dilution, followed by incubation with anti-rabbit IgG-FITC-conjugated secondary antibody for an hour at room temperature. Cells were then washed and mounted. Following treatment study, cells were fixed, permeabilized, and blocked in 2% BSA/PBS as described earlier. Cells were then labeled with rhodamine phalloidin at 1:50 dilution in 2% BSA/PBS for 20 min. Chamber slides were then washed and mounted using VECTASHIELD® antifade mounting media with DAPI for nuclear staining. Cells were visualized and scanned at a single optical plane with an Olympus Fluoview 300 confocal microscope (Center Valley, PA) with a PLA PO ×60 water immersion objective. Untreated cells served as control. Each experiment was repeated four times. The changes in ZO-1 localization and the formation of F-actin stress fibers were analyzed using ImageJ software and presented as arbitrary units for statistical analysis (unpaired t test; p < 0.05) (34).
Quantitative Real Time-PCR
RBMECs were grown on 100-mm cell culture dishes; total RNA was then extracted from the cells using TRIzol® reagent according to the manufacturer's instructions. RNA concentration and quality were determined by employing the ratio of absorbance at 260/280 nm using Biotek Synergy Hybrid Spectrophotometer (Winooski, VT). Reverse transcription was performed using the SuperScript® IV first-strand synthesis system. Quantitative real time PCR was performed using the RT2 SYBR Green Fluor qPCR Mastermix with the following primer pairs: for ZO-1: forward primer, 5′-CCTCTGATCATTCCACACAGTC-3′, and reverse primer, 5′-TAGACATGCGCTCTTCCTCTCT-3′; for GAPDH: forward primer, 5′-AATGTATCCGTTGTGGATCT-3′, and reverse primer, 5′-CAAGAAGGTGGTGAAGCAGG-3′. Real time PCR detection was carried out using Stratagene Mx3000P qPCR system, Agilent Technologies (La Jolla, CA), using 1 μl of cDNA. Relative abundances of target genes were calculated by normalizing Ct values to endogenous control glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Cells were divided into two groups, control (untreated) and IL-1β (10 ng/ml; 2 h) treatment groups. Each experiment was repeated three times. Relative mRNA expression of ZO-1 was obtained by normalizing the Ct values to the endogenous control GAPDH for each repeat. Normalized Ct values were expressed as mean ± S.E. Statistical differences among groups were determined by one-way ANOVA followed by Bonferroni post hoc test to determine significant differences between specific groups. A value of p < 0.05 was considered statistically significant.
Western Blotting Assays
Western blottings were performed to investigate the expression of ZO-1 protein in RBMECs. Following treatment studies, cells were washed and incubated in ice-cold cell lysis buffer (1×) along with protease inhibitor mixture (1×) for 5 min in cell culture dishes. Cells were then scraped, sonicated, and centrifuged at 14,000 × g for 10 min at 4 °C. Supernatant was collected from the extracts, and protein concentration was determined using the protein assay kit. Equal amounts of total protein (50 μg) were separated by SDS-PAGE on 10% BisTris precast gels at a constant voltage (145 V) for 180 min. Proteins were then transferred onto the nitrocellulose membrane at a constant voltage (30 V) overnight, and the membranes were blocked using 5% nonfat dry milk in Tris-buffered saline (TBS) with 0.05% Tween 20 for 3 h and subsequently incubated with primary mouse monoclonal anti ZO-1 antibody (1:250 dilution). Membranes were washed three times in TBS-T and incubated with the goat anti-mouse IgG-HRP-conjugated secondary antibody. After washing, the immunoblots were visualized by ECL Western blotting substrate. Cells were divided into untreated (control) and IL-1β (10 ng/ml; 2 h) treatment groups. Each experiment was repeated four times. An equal amount of protein sample loading was verified by assessing β-actin protein expression.
Cell Viability Studies
An EZViableTM calcein AM cell viability assay kit (Fluorometric) was used to quantify the number of viable cells. Calcein AM is a non-fluorescent hydrophobic compound that easily penetrates intact and live cells. Equal numbers of cells were grown on sterile black 96-well plates. On reaching confluency, growth media were discarded, and cells were washed in PBS and pre-exposed to phenol red-free medium for an hour. Cells were divided into control (untreated) and IL-1β (10 ng/ml) treatment groups. Cells were treated with IL-1β for 2, 4, and 6 h with hydrogen peroxide at 50, 25, 10 mm, respectively, as a positive control. Following treatments, cells were then exposed to calcein buffer solution (calcein AM: calcein dilution buffer in 1:500 dilution) and incubated at 37 °C for 30 min, and a fluorometric reading was obtained. Each experiment was repeated five times. Fluorescence intensity was plotted on the y axis and represented as % control. Data were expressed as mean ± % S.E., and statistical differences among groups were determined by one-way ANOVA followed by Bonferroni post hoc test to determine significant differences between specific groups. A p value <0.05 was considered statistically significant.
Fluo-4 Based [Ca2+]i Measurements
For population measurements, RBMECs (monolayer) were incubated with 5 μm Fluo-4 AM (Thermo Fisher Scientific) at 37 °C for 40 min, and Fluo-4 fluorescence intensity was measured through a Biotek Synergy Hybrid Spectrophotometer (Winooski, VT). Iono (1 μm) and TG (2 μm) were used separately to increase [Ca2+]i as two positive controls. DMSO was used as a vehicle control. Each experiment was repeated four times. Data were presented as mean ± S.E.
Single Cell [Ca2+]i Imaging
RBMECs were grown on coverslips and incubated with 2 μm Fura-2 AM (ThermoFisher Scientific) in the culture medium at 37 °C for 40 min. Ratiometric [Ca2+]i imaging was performed on an IX-81 microscope (Olympus)-based system as described previously from individual cells (50). Data were analyzed using Metafluor software (Universal imaging) and OriginPro 8 software (Origin Lab) and expressed as mean ± S.E.
Evans Blue Leakage Studies
For this study, C57BL/6 mice (25–30 g) were anesthetized with urethane, i.p. injection (2 ml/kg body weight) followed by Evans blue dye, i.v. injection (2% w/v in saline; 4 ml/kg body weight). Evans blue was allowed to circulate in the animal for 30 min prior to performing sham surgery (only craniotomy) or TBI using CCI. Animals were grouped into sham (only craniotomy), vehicle + sham (DMSO injection followed by craniotomy), vehicle + TBI group (DMSO followed by mild TBI), calpain inhibitor III + TBI group (calpain inhibitor III (10 μg/g body weight of the animal) injection followed by mild TBI), and a TBI group followed by calpain inhibitor III treatment. Each group consisted of five animals. One-hour post-TBI, animals were transcardially perfused with sterile saline containing heparin (1000 units/ml) for at least 20 min. Briefly, brains were extracted, and brain cortex was carefully separated and weighed. Brain cortices were then homogenized in 1 ml of 50% (w/v) TCA in saline, and the homogenate was centrifuged at 6000 × g for 20 min at 4 °C. Supernatants were extracted and diluted in 3 parts of ethanol (1:3; 50% TCA, 95% ethanol). Samples were then quantitated fluorometrically at 620:680 nm (excitation/emission) using Biotek Synergy Hybrid H1 spectrophotometer (Winooski, VT). Evans blue concentration in the samples was evaluated using external standards for Evans blue ranging from 50 to 1000 ng/ml, prepared in same solvent (1:3; TCA, 95% ethanol). Evans blue amount in the samples was expressed as nanogram/brain cortex ± S.E. Statistical differences among groups were determined by one-way ANOVA followed by Bonferroni post hoc test to determine significant differences between specific groups. A value of p < 0.05 was considered statistically significant.
Intravital Microscopy
To further confirm the findings from the Evans blue dye leakage study described above, BBB integrity/permeability was determined using intravital microscopy imaging. For this study, C57BL/6 mice (25–30 g) were anesthetized with urethane, i.p. injection (2 ml/kg body weight) as described above and divided into three groups as follows: vehicle + sham (DMSO + craniotomy only group; n = 5), vehicle + TBI group (DMSO + mild TBI; n = 5), and TBI + calpain inhibitor III group (calpain inhibitor III (10 μg/g body weight of the animal) injection 15 min after mild TBI; n = 4). All animals received FITC-dextran (10 kDa; 50 mg/kg body weight) via tail vein. Following sham/TBI, animals were placed under an intravital microscope (Nikon intravital microscope) on a heating pad, and their pial microvasculature was observed (under a ×40 water immersion lens) for vascular permeability/BBB integrity. The animals were euthanized at the end of each study. The images were taken, and the fluorescent intensity within and outside the vessels was determined from multiple areas and analyzed using nuclear export signal element software. The images that show change in fluorescence intensity at 0 and 60 min (10 and 70 min after induction of trauma due to the setup time of the experiment and visualization of the vessels) from all the three groups are presented for comparison. Statistical analysis of the data at the 60-min time point was performed. Statistical differences among groups were determined by one-way ANOVA followed by Bonferroni post hoc test to determine significant differences between specific groups. A value of p < 0.05 was considered statistically significant.
Author Contributions
H. A. and B. T. conceived and coordinated the study and wrote the paper. H. A., S. L. Z., C. A. S., M. G., B. R., and K. P. V. designed and performed the experiments and analyzed the data. C. A. S. provided technical assistance and contributed to the preparation of the figures. M. R. B., K. P. V., C. P., and J. H. H. were involved in idea development, experimental design, and manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We acknowledge Glen Cryer for help with manuscript editing. We acknowledge the late Dr. Matthew L. Davis for his help and suggestion in this project.
This work was supported by Baylor Scott and White Health Academic Operations Research funds (to B. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- BBB
- blood-brain barrier
- RBMEC
- rat brain microvascular endothelial cells
- TJ
- tight junction
- RBMEC
- rat brain microvascular endothelial cell
- TBI
- traumatic brain injury
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ([Ca2+]i
- intracellular free calcium
- ANOVA
- analysis of variance
- CCI
- controlled cortical impact
- TG
- thapsigargin
- Iono
- ionomycin
- RFU
- relative fluorescence unit
- qPCR
- quantitative PCR
- MMP
- matrix metalloproteinase.
References
- 1. Alluri H., Wiggins-Dohlvik K., Davis M. L., Huang J. H., and Tharakan B. (2015) Blood-brain barrier dysfunction following traumatic brain injury. Metab. Brain Dis. 30, 1093–1104 [DOI] [PubMed] [Google Scholar]
- 2. Mayhan W. (2001) Regulation of blood–brain barrier permeability. Microcirculation 8, 1108–1110 [DOI] [PubMed] [Google Scholar]
- 3. Abbott N. J. (2000) Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 20, 131–147 [DOI] [PubMed] [Google Scholar]
- 4. Michinaga S., and Koyama Y. (2015) Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 16, 9949–9975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blamire A. M., Anthony D. C., Rajagopalan B., Sibson N. R., Perry V. H., and Styles P. (2000) Interleukin-1β-induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J. Neurosci. 20, 8153–8159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murray K. N., Parry-Jones A. R., and Allan S. M. (2015) Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Toulmond S., and Rothwell N. J. (1995) Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 13, 261–266 [DOI] [PubMed] [Google Scholar]
- 8. Wong D., Dorovini-Zis K., and Vincent S. R. (2004) Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp. Neurol. 190, 446–455 [DOI] [PubMed] [Google Scholar]
- 9. Pietsch M., Chua K. C., and Abell A. D. (2010) Calpains: attractive targets for the development of synthetic inhibitors. Curr. Top. Med. Chem. 10, 270–293 [DOI] [PubMed] [Google Scholar]
- 10. Loot A. E., Pierson I., Syzonenko T., Elgheznawy A., Randriamboavonjy V., Zivković A., Stark H., and Fleming I. (2013) Ca2+-sensing receptor cleavage by calpain partially accounts for altered vascular reactivity in mice fed a high-fat diet. J. Cardiovasc. Pharmacol. 61, 528–535 [DOI] [PubMed] [Google Scholar]
- 11. Miyazaki T., Taketomi Y., Takimoto M., Lei X. F., Arita S., Kim-Kaneyama J. R., Arata S., Ohata H., Ota H., Murakami M., and Miyazaki A. (2011) m-Calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-cadherin disorganization and atherosclerosis. Circulation 124, 2522–2532 [DOI] [PubMed] [Google Scholar]
- 12. Bralić M., and Stemberga V. (2012) Calpain expression in the brain cortex after traumatic brain injury. Coll. Antropol. 36, 1319–1323 [PubMed] [Google Scholar]
- 13. Wang T., Wang L., Moreno-Vinasco L., Lang G. D., Siegler J. H., Mathew B., Usatyuk P. V., Samet J. M., Geyh A. S., Breysse P. N., Natarajan V., and Garcia J. G. (2012) Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 9, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smith M. A., and Schnellmann R. G. (2012) Calpains, mitochondria, and apoptosis. Cardiovasc. Res. 96, 32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeong S. Y., Martchenko M., and Cohen S. N. (2013) Calpain-dependent cytoskeletal rearrangement exploited for anthrax toxin endocytosis. Proc. Natl. Acad. Sci. U.S.A. 110, E4007–E4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao X., Posmantur R., Kampfl A., Liu S. J., Wang K. K., Newcomb J. K., Pike B. R., Clifton G. L., and Hayes R. L. (1998) Subcellular localization and duration of μ-calpain and m-calpain activity after traumatic brain injury in the rat: a casein zymography study. J. Cereb. Blood Flow Metab. 18, 161–167 [DOI] [PubMed] [Google Scholar]
- 17. Saatman K. E., Creed J., and Raghupathi R. (2010) Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 7, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ray S. K., Hogan E. L., and Banik N. L. (2003) Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res. Rev. 42, 169–185 [DOI] [PubMed] [Google Scholar]
- 19. Bralic M., Stemberga V., and Stifter S. (2012) Introduction of calpain inhibitors in traumatic brain injury: a novel approach? Med. Hypotheses 79, 358–360 [DOI] [PubMed] [Google Scholar]
- 20. Kampfl A., Posmantur R. M., Zhao X., Schmutzhard E., Clifton G. L., and Hayes R. L. (1997) Mechanisms of calpain proteolysis following traumatic brain injury: Implications for pathology and therapy: a review and update. J. Neurotrauma 14, 121–134 [DOI] [PubMed] [Google Scholar]
- 21. Chakraborti S., Alam M. N., Paik D., Shaikh S., and Chakraborti T. (2012) Implications of calpains in health and diseases. Indian J. Biochem. Biophys. 49, 316–328 [PubMed] [Google Scholar]
- 22. Fei B., Yu S., and Geahlen R. L. (2013) Modulation by Syk of Bcl-2, calcium and the calpain–calpastatin proteolytic system in human breast cancer cells. Biochim. Biophys. Acta 1833, 2153–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prangsaengtong O., Senda K., Doki Y., Park J. Y., Jo M., Sakurai H., Shibahara N., Saiki I., and Koizumi K. (2012) Calpain 1 and -2 play opposite roles in cord formation of lymphatic endothelial cells via eNOS regulation. Hum. Cell 25, 36–44 [DOI] [PubMed] [Google Scholar]
- 24. Ma M. (2013) Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol. Dis. 60, 61–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hannah R. M., Dunn K. M., Bonev A. D., and Nelson M. T. (2011) Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J. Cereb. Blood Flow Metab. 31, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thompson S. N., Carrico K. M., Mustafa A. G., Bains M., and Hall E. D. (2010) A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J. Neurotrauma 27, 2233–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsubokawa T., Solaroglu I., Yatsushige H., Cahill J., Yata K., and Zhang J. H. (2006) Cathepsin and calpain inhibitor E64d attenuates matrix metalloproteinase-9 activity after focal cerebral ischemia in rats. Stroke 37, 1888–1894 [DOI] [PubMed] [Google Scholar]
- 28. Ali M. A., Stepanko A., Fan X., Holt A., and Schulz R. (2012) Calpain inhibitors exhibit matrix metalloproteinase-2 inhibitory activity. Biochem. Biophys. Res. Commun. 423, 1–5 [DOI] [PubMed] [Google Scholar]
- 29. Takata F., Dohgu S., Matsumoto J., Takahashi H., Machida T., Wakigawa T., Harada E., Miyaji H., Koga M., Nishioku T., Yamauchi A., and Kataoka Y. (2011) Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-alpha, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflammation 8, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Machida T., Takata F., Matsumoto J., Takenoshita H., Kimura I., Yamauchi A., Dohgu S., and Kataoka Y. (2015) Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood-brain barrier in vitro. Neurosci. Lett. 599, 109–114 [DOI] [PubMed] [Google Scholar]
- 31. Kotecki M., Zeiger A. S., Van Vliet K. J., and Herman I. M. (2010) Calpain- and talin-dependent control of microvascular pericyte contractility and cellular stiffness. Microvasc. Res. 80, 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ralay Ranaivo H., Hodge J. N., Choi N., and Wainwright M. S. (2012) Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J. Neuroinflammation 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tejima E., Zhao B. Q., Tsuji K., Rosell A., van Leyen K., Gonzalez R. G., Montaner J., Wang X., and Lo E. H. (2007) Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J. Cereb. Blood Flow Metab. 27, 460–468 [DOI] [PubMed] [Google Scholar]
- 34. Millán J., Cain R. J., Reglero-Real N., Bigarella C., Marcos-Ramiro B., Fernández-Martín L., Correas I., and Ridley A. J. (2010) Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan S. Y., and Rigor R. R. (2010) in Colloquium Series in Integrated Systems Physiology, Regulation of Endothelial Barrier Function (Granger N. D., and Granger J., series eds) pp. 39–54, Morgan & Claypool Life Sciences, San Rafael, CA: [PubMed] [Google Scholar]
- 36. ElAli A., Doeppner T. R., Zechariah A., and Hermann D. M. (2011) Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke 42, 3238–3244 [DOI] [PubMed] [Google Scholar]
- 37. Goll D. E., Thompson V. F., Li H., Wei W., and Cong J. (2003) The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 38. Hu H., Li X., Li Y., Wang L., Mehta S., Feng Q., Chen R., and Peng T. (2009) Calpain-1 induces apoptosis in pulmonary microvascular endothelial cells under septic conditions. Microvasc. Res. 78, 33–39 [DOI] [PubMed] [Google Scholar]
- 39. Pfaff M., Du X., and Ginsberg M. H. (1999) Calpain cleavage of integrin β cytoplasmic domains. FEBS Lett. 460, 17–22 [DOI] [PubMed] [Google Scholar]
- 40. Zhao Y., Malinin N. L., Meller J., Ma Y., West X. Z., Bledzka K., Qin J., Podrez E. A., and Byzova T. V. (2012) Regulation of cell adhesion and migration by Kindlin-3 cleavage by calpain. J. Biol. Chem. 287, 40012–40020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Glading A., Bodnar R. J., Reynolds I. J., Shiraha H., Satish L., Potter D. A., Blair H. C., and Wells A. (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol. 24, 2499–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zadran S., Jourdi H., Rostamiani K., Qin Q., Bi X., and Baudry M. (2010) Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J. Neurosci. 30, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anagli J., Han Y., Stewart L., Yang D., Movsisyan A., Abounit K., and Seyfried D. (2009) A novel calpastatin-based inhibitor improves postischemic neurological recovery. Biochem. Biophys. Res. Commun. 385, 94–99 [DOI] [PubMed] [Google Scholar]
- 44. Germanò A., Costa C., DeFord S. M., Angileri F. F., Arcadi F., Pike B. R., Bramanti P., Bausano B., Zhao X., Day A. L., Anderson D. K., and Hayes R. L. (2002) Systemic administration of a calpain inhibitor reduces behavioral deficits and blood-brain barrier permeability changes after experimental subarachnoid hemorrhage in the rat. J. Neurotrauma 19, 887–896 [DOI] [PubMed] [Google Scholar]
- 45. Toba S., Tamura Y., Kumamoto K., Yamada M., Takao K., Hattori S., Miyakawa T., Kataoka Y., Azuma M., Hayasaka K., Amamoto M., Tominaga K., Wynshaw-Boris A., Wanibuchi H., Oka Y., et al. (2013) Post-natal treatment by a blood-brain-barrier permeable calpain inhibitor, SNJ1945 rescued defective function in lissencephaly. Sci. Rep. 3, 1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Medeiros R., Kitazawa M., Chabrier M. A., Cheng D., Baglietto-Vargas D., Kling A., Moeller A., Green K. N., and LaFerla F. M. (2012) Calpain inhibitor A-705253 mitigates Alzheimer's disease-like pathology and cognitive decline in aged 3xTgAD mice. Am. J. Pathol. 181, 616–625 [DOI] [PubMed] [Google Scholar]
- 47. Samantaray S., Knaryan V. H., Shields D. C., Cox A. A., Haque A., and Banik N. L. (2015) Inhibition of calpain activation protects MPTP-induced nigral and spinal cord neurodegeneration, reduces inflammation, and improves gait dynamics in mice. Mol. Neurobiol. 52, 1054–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Y., Mao H., Yang K. H., Abel T., and Meaney D. F. (2014) A modified controlled cortical impact technique to model mild traumatic brain injury mechanics in mice. Front. Neurol. 5, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alluri H., Stagg H. W., Wilson R. L., Clayton R. P., Sawant D. A., Koneru M., Beeram M. R., Davis M. L., and Tharakan B. (2014) Reactive oxygen species-caspase-3 relationship in mediating blood-brain barrier endothelial cell hyperpermeability following oxygen-glucose deprivation and reoxygenation. Microcirculation 21, 187–195 [DOI] [PubMed] [Google Scholar]
- 50. Zhou M. H., Zheng H., Si H., Jin Y., Peng J. M., He L., Zhou Y., Muñoz-Garay C., Zawieja D. C., Kuo L., Peng X., and Zhang S. L. (2014) Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 289, 29446–29456 [DOI] [PMC free article] [PubMed] [Google Scholar]