

Abstract
Nucleotide signaling networks are key to facilitate alterations in gene expression, protein function, and enzyme activity in response to diverse stimuli. Cyclic di-adenosine monophosphate (c-di-AMP) is an important secondary messenger molecule produced by the human pathogen Staphylococcus aureus and is involved in regulating a number of physiological processes including potassium transport. S. aureus must ensure tight control over its cellular levels as both high levels of the dinucleotide and its absence result in a number of detrimental phenotypes. Here we show that in addition to the membrane-bound Asp-His-His and Asp-His-His-associated (DHH/DHHA1) domain-containing phosphodiesterase (PDE) GdpP, S. aureus produces a second cytoplasmic DHH/DHHA1 PDE Pde2. Although capable of hydrolyzing c-di-AMP, Pde2 preferentially converts linear 5′-phosphadenylyl-adenosine (pApA) to AMP. Using a pde2 mutant strain, pApA was detected for the first time in S. aureus, leading us to speculate that this dinucleotide may have a regulatory role under certain conditions. Moreover, pApA is involved in a feedback inhibition loop that limits GdpP-dependent c-di-AMP hydrolysis. Another protein linked to the regulation of c-di-AMP levels in bacteria is the predicted regulator protein YbbR. Here, it is shown that a ybbR mutant S. aureus strain has increased acid sensitivity that can be bypassed by the acquisition of mutations in a number of genes, including the gene coding for the diadenylate cyclase DacA. We further show that c-di-AMP levels are slightly elevated in the ybbR suppressor strains tested as compared with the wild-type strain. With this, we not only identified a new role for YbbR in acid stress resistance in S. aureus but also provide further insight into how c-di-AMP levels impact acid tolerance in this organism.
Keywords: bacterial signal transduction, pH regulation, phosphodiesterases, Staphylococcus aureus (S. aureus), stress, YbbR
Introduction
The Gram-positive bacterium Staphylococcus aureus is persistently carried by ∼30% of the human population (1). This colonization is often asymptomatic, but S. aureus can become pathogenic and cause various diseases ranging from skin and soft tissue infections to pneumonia and bacteremia (2). This microorganism has also developed resistance to many antibiotics, which has reduced the number of therapeutic choices available (3, 4). Although methicillin-resistant S. aureus (MRSA)4 is mostly associated with the hospital setting, community-associated MRSA (CA-MRSA) strains have spread rapidly in some countries (3).
Bacteria have a remarkable ability to readily adapt to fluctuating environmental conditions and cope with periods of stress. Signal transduction pathways are crucial to achieving this flexibility, where stimuli are sensed and signals relayed within the cell to trigger the induction of an appropriate response. Signaling nucleotides, functioning as secondary messenger molecules, are known to regulate a diverse array of cellular networks in all forms of life. Cyclic adenosine monophosphate (cAMP) and cyclic di-guanine monophosphate (c-di-GMP) are among the best studied of these molecules and regulate a number of physiological processes including carbon catabolism and the switch between a sessile and motile lifestyle, respectively (5). The functions of the stringent response alarmones guanosine tetraphosphate and pentaphosphate ((p)ppGpp) have also been extensively investigated. These nucleotides are produced under various stress conditions including nutrient limitation. They act to decrease the expression of genes required for active growth and increase the expression of amino acid synthesis and transporter genes (6). Although more recently discovered, an impressive effort has been made to elucidate the function of cyclic di-adenosine monophosphate (c-di-AMP) in bacteria.
The signaling molecule c-di-AMP is predominantly produced by Gram-positive bacteria belonging to the Firmicutes and Actinobacteria phyla but can also be synthesized by some Gram-negative bacteria (7–14). c-di-AMP is synthesized from two molecules of ATP via a condensation reaction requiring the activity of a diadenylate cyclase (DAC), and hydrolyzed by a phosphodiesterase (PDE) into 5′-phosphadenylyl-adenosine (pApA), or two molecules of AMP (7, 8, 10, 15–17). S. aureus produces a single diadenylate cyclase enzyme called DacA, which is encoded by the first gene in the dacA-ybbR-glmM operon (7, 11). The third gene of this operon encodes the phosphoglucosamine mutase GlmM, a cytoplasmic enzyme that converts glucosamine-6-phosphate into glucosamine-1-phosphate (18–20). This product is subsequently used for the production of an early peptidoglycan synthesis intermediate. The second gene in the operon, ybbR, encodes a predicted membrane protein that has been shown to interact with, and influence the activity of DacA (21–23). However, there are conflicting reports as to whether YbbR stimulates or inhibits the activity of the cyclase. Therefore, a conclusive role for YbbR is still lacking.
GdpP (also named YybT or Pde1) was the first c-di-AMP-hydrolyzing enzyme identified in Bacillus subtilis (15). GdpP contains two N-terminal transmembrane helices followed by a cytoplasmic Per-Arnt-Sim (PAS) domain, a modified GGDEF domain, and a DHH/DHHA1 domain, which possesses phosphodiesterase activity (15). GdpP homologs have since been discovered in a range of microorganisms including Streptococcus pneumoniae, Listeria monocytogenes and S. aureus, and function to specifically degrade c-di-AMP into pApA (7, 17, 24, 25). The absence of GdpP in S. aureus leads to an ∼10-fold increase in cellular c-di-AMP levels, a reduced bacterial cell size, and production of a peptidoglycan polymer with increased cross-linking (7). Further studies have also correlated levels of c-di-AMP in S. aureus with resistance to β-lactam antibiotics (7, 26–28).
A cytoplasmic protein containing a stand-alone DHH/DHHA1 domain, but lacking the sensory domains found in GdpP, has also been associated with the metabolism of c-di-AMP (14, 24, 29). In Borrelia burgdorferi the DHH/DHHA1 domain containing protein DhhP has been described as a c-di-AMP PDE and essential for growth (14). S. pneumoniae produces a homolog of DhhP named Pde2, which can hydrolyze c-di-AMP and pApA as well as 3′-phosphoadenosine-5′-phosphate (pAp) to AMP and is essential for pathogenesis (24, 29). The Mycobacterium tuberculosis and Mycobacterium smegmatis homologs, MtbPDE and MtPDE, respectively, have been shown to degrade both c-di-AMP and pApA (30–33). A third class of c-di-AMP PDE found in c-di-AMP-producing microorganisms is an HD-domain containing enzyme (17, 34). Similarly to GdpP, this PDE is an integral membrane protein and degrades c-di-AMP into pApA (17, 34).
In this study we further investigated the c-di-AMP metabolism in S. aureus by examining the function of the DHH/DHHA1 PDE Pde2 and the membrane protein YbbR. We show that the S. aureus PDE Pde2 preferentially hydrolyzes pApA over c-di-AMP, leading in its absence to an accumulation of pApA. We also show that deleting ybbR in S. aureus does not significantly affect c-di-AMP production or growth under standard growth conditions. However, its absence leads to increased acid sensitivity, which can be compensated by the production of an altered DacA variant.
Results
An S. aureus pde2 Mutant Strain Displays a Growth Defect in the Early Growth Phase
Although HD domain-containing PDEs are not found in Staphylococcus, a stand-alone DHH/DHHA1 domain protein and Pde2 ortholog is present in S. aureus. This protein corresponds to SAUSA300_1650 in S. aureus strain FRP5737 and will be from here on out referred to as Pde2. To assess the function of Pde2 in S. aureus, strain LAC*Δpde2::erm was constructed in which the pde2 gene was replaced by an erythromycin marker. When grown in tryptic soya broth (TSB) medium, LAC*Δpde2::erm exhibited a growth defect in the early stages of growth similar to that observed for the gdpP mutant strain LAC*ΔgdpP::kan (Fig. 1A). In contrast to the gdpP mutant, LAC*Δpde2::erm reached a similar density as the WT LAC* control culture at later time points (Fig. 1A). This growth defect could be complemented by expressing pde2 from its native promoter from the single site integration vector in strain LAC*Δpde2::erm pCL55-pde2 (Fig. 1B). Strain LAC*ΔgdpP::kanΔpde2::erm, deleted for both gdpP and pde2, exhibited a greater growth defect as compared with the single mutants at the early time points, with the culture reaching a similar density as the gdpP mutant strain at the 8-h time point (Fig. 1A). This growth analysis revealed that the pde2 and gdpP mutants have distinct growth phenotypes, and the additive growth defect observed in the double mutant suggests that Pde2 might also be involved in the c-di-AMP metabolism of S. aureus.
FIGURE 1.

An S. aureus pde2 mutant has a slight growth defect and increased cellular c-di-AMP levels. A and B, bacterial growth curve. A, cultures of WT LAC* and the indicated mutant strains were diluted to an A600 of 0.05, grown to an A600 of 1, and back-diluted again to an A600 of 0.05 with this time point set to time = 0. Growth was subsequently monitored over an 8-h period, and average A600 readings and standard deviations from three independent experiments are plotted. B, overnight culture of LAC* (WT) and the pde2 mutant strain containing the vector pCL55 as well as the pde2 complementation strain were diluted to an A600 of 0.01, and growth was monitored over 10 h. Average A600 readings and standard deviations from three independent experiments are plotted. C, determination of intracellular c-di-AMP levels by LC-MS/MS. LAC* (WT) and the indicated mutants were grown overnight in TSB medium, cell extracts were prepared, and c-di-AMP levels were determined by LC-MS/MS. The nucleotide levels are reported as ng of c-di-AMP/mg cell dry weight, and the average values and standard deviations from three independent experiments are plotted. D, determination of intracellular c-di-AMP levels by ELISA. The same strains as in C were grown overnight in TSB medium, cell extracts were prepared, and c-di-AMP levels were determined by ELISA. The nucleotide levels are reported as ng of c-di-AMP/mg of protein, and the average values and standard deviations from three independent experiments are plotted. E, determination of intracellular c-di-AMP levels by ELISA. Cell extracts were prepared from LAC* (WT), and the pde2 mutant strain containing the vector pCL55 as well as the pde2 complementation strain and c-di-AMP levels were determined by ELISA. The nucleotide levels are reported as ng of c-di-AMP/mg protein, and the average values and standard deviations from four independent experiments are plotted. Statistical analysis was performed using an unpaired Student's t test where one asterisk (*), two asterisks (**), or three asterisks (***) indicate differences between WT and mutant strains with p values of <0.05, <0.01, or <0.001, respectively.
Deletion of pde2 in S. aureus Leads to an Increase in Cellular c-di-AMP Levels and Increased Oxacillin Resistance
To directly assess the contribution of Pde2 to the c-di-AMP metabolism in S. aureus, cytosolic extracts were prepared from WT LAC*, the isogenic pde2, gdpP single mutants, and the gdpP/pde2 double mutant strains. As an additional control, strain LAC*dacAG206S was constructed, which produces a DacA variant with a glycine-to-serine change at amino acid residue 206. S. aureus strains containing this dacAG206S allele have been reported to produce lower amounts of c-di-AMP (26). Next, the cellular c-di-AMP concentration was determined by LC-MS/MS as well as by an ELISA assay. As expected, decreased or increased amounts of c-di-AMP were found in extracts derived from the dacAG206S and gdpP mutant strains, respectively, when compared with the WT LAC* control strain (Fig. 1, C and D). Using both c-di-AMP detection methods (LC-MS/MS and ELISA), a slight but significant increase in the cellular c-di-AMP concentration was also observed for the pde2 mutant when compared with WT LAC*, with the levels in the double mutant found to be even greater than in the gdpP single mutant (Fig. 1, C and D). Finally, as assessed by ELISA, the c-di-AMP levels were restored to wild-type levels in the pde2 complementation strain LAC*Δpde2::erm pCL55-pde2 (Fig. 1E). Altogether, these data suggest that Pde2 contributes to the c-di-AMP metabolism in S. aureus. A correlation between c-di-AMP levels and oxacillin resistance of MRSA strains has been observed in previous studies where strains with high and low dinucleotide levels exhibit increased and decreased resistance, respectively (7, 26). To assess the contribution of Pde2 to oxacillin resistance, M.I.C. EvaluatorTM strips were used to determine the susceptibility of the different strains to this antibiotic. The oxacillin MIC for WT LAC* was 4 μg/ml, and as expected, the gdpP and dacAG206S mutants showed higher and lower resistances, respectively, with MICs of >256 μg/ml and 0.12 μg/ml. Similar to the gdpP mutant, the pde2 single and gdpP/pde2 double mutant strains also displayed an increased resistance to oxacillin with MICs of >256 μg/ml. It is, however, of note that although growth was observed along all concentrations of the antibiotic strip for the gdpP and pde2 single mutant strains, a halo, and area of reduced growth, was visible at concentrations above 4 μg/ml but not visible for the gdpP/pde2 double mutant strain (data now shown). Taken together, these results indicate that Pde2 contributes to oxacillin resistance and the metabolism of c-di-AMP in S. aureus.
S. aureus Pde2 Preferentially Hydrolyzes pApA over c-di-AMP
To determine the substrate specificity of the S. aureus Pde2 enzyme, an N-terminally His-tagged version of the protein was expressed and purified from Escherichia coli, and used in in vitro enzyme activity assays. 1 μm recombinant Pde2 was incubated for 2 h with 100 μm ATP, c-di-AMP, or pApA (spiked with nm concentrations of the respective radiolabeled nucleotide), and reaction products were analyzed by TLC. As a control, reactions were also set up with recombinant GdpP, known to specifically cleave c-di-AMP into pApA (7, 35). As expected, GdpP converted c-di-AMP into a single product, pApA, but did not hydrolyze ATP or pApA (Fig. 2). Pde2 was incapable of hydrolyzing ATP, whereas 35% of the input c-di-AMP and 100% of the input pApA was hydrolyzed and, based on analogy with other Pde2 enzymes, likely converted to AMP (Fig. 2). To confirm that c-di-AMP and pApA were converted to AMP, 100 μm unlabeled c-di-AMP or pApA substrates were incubated with Pde2, and the reaction products were analyzed by LC-UV. This analysis confirmed that Pde2 converted both c-di-AMP and pApA into AMP (data not shown). These initial experiments also indicated that the S. aureus Pde2 enzyme preferentially hydrolyzes pApA over c-di-AMP. To investigate this further, a time-course experiment was performed. Complete hydrolysis of 200 μm pApA was observed within 1 h using 4 nm Pde2 (Fig. 3, A and B). Using this low enzyme concentration, no hydrolysis of c-di-AMP was observed within the 1-h incubation time (Fig. 3, A and B), suggesting that the native substrate of the S. aureus Pde2 enzyme is pApA rather than c-di-AMP.
FIGURE 2.

Pde2 degrades both c-di-AMP and pApA. A, GdpP and Pde2 enzyme activity assays. 100 μm ATP, c-di-AMP, or pApA spiked with a low nm concentration of the respective radiolabeled nucleotides were incubated for 2 h at 37 °C in the absence of enzyme or with 1 μm purified GdpP or Pde2. The reaction products were separated by TLC, and radioactive signals were detected using a phosphorimager. Substrate nucleotides are shown on top, and nucleotide products are listed along the left of the figure. A representative image from four independent experiments is shown. B, quantification of GdpP and Pde2 nucleotide hydrolysis activity. The % c-di-AMP or pApA substrate hydrolysis by GdpP and Pde2 was determined by quantification of the radioactive signal of TLC plates shown in A, and the average value and standard deviations from four experiments are plotted.
FIGURE 3.
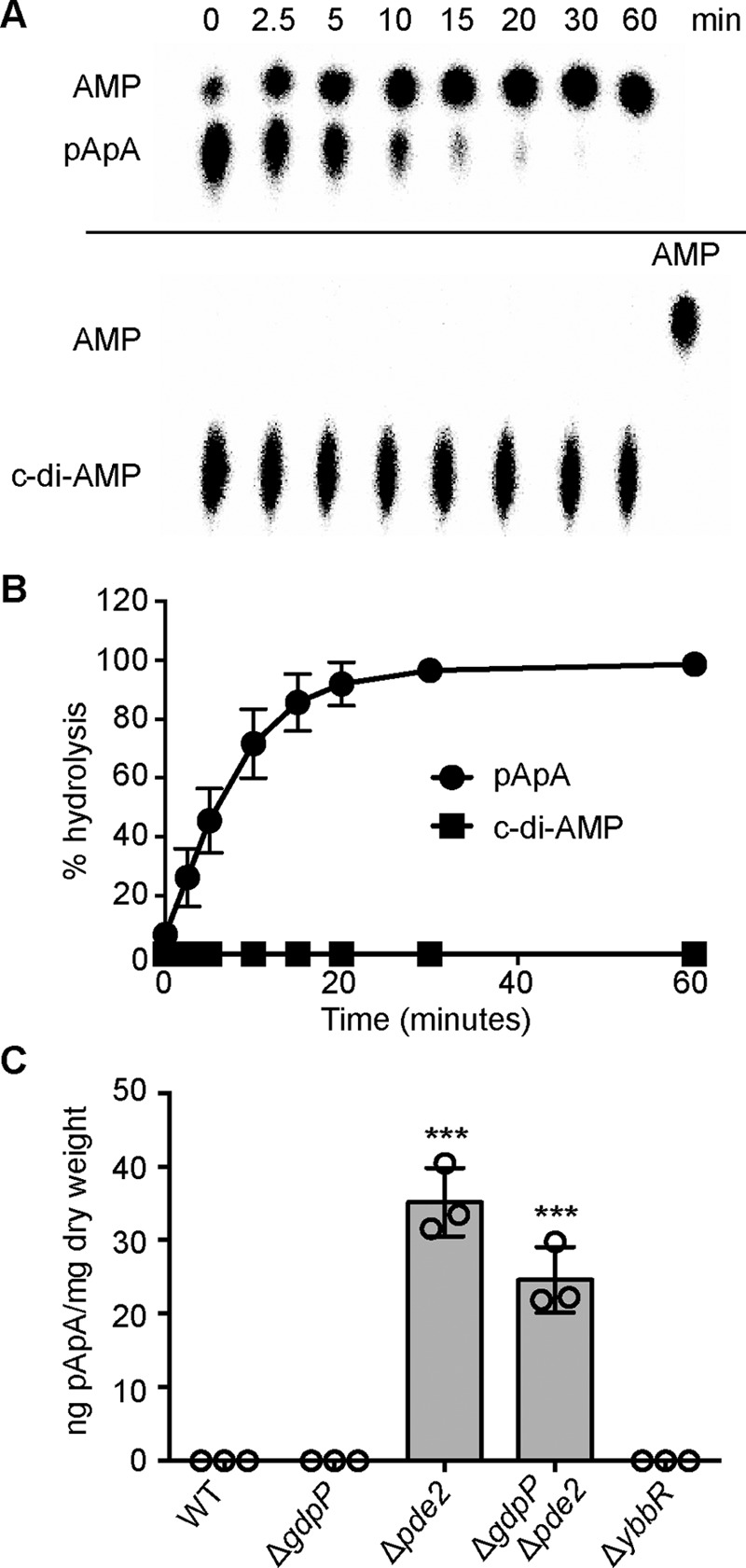
Pde2 preferably hydrolyzes pApA over c-di-AMP and its deletion leads to an accumulation of pApA. A, Pde2 enzyme activity time course experiment. 200 μm pApA or c-di-AMP spiked with a small concentration of the respective radiolabeled nucleotide was incubated with 4 nm concentrations of purified Pde2 at 37 °C. Samples were removed at timed intervals and analyzed by TLC as described in Fig. 2. B, quantification of c-di-AMP and pApA hydrolysis by Pde2. The radioactive signals of the input nucleotide were quantified, the % c-di-AMP or pApA substrate hydrolysis was calculated, and the average values and standard deviations from six independent experiments are plotted. C, determination of intracellular pApA levels by LC-MS/MS. WT LAC* and the indicated mutants were grown overnight in TSB medium, cell extracts were prepared, and pApA levels were determined by LC-MS/MS as described in the “Experimental Procedure” section. The nucleotide levels are reported as ng of pApA/mg cell dry weight, and the average values and standard deviations from three independent experiments are plotted. Statistical analysis was performed using an unpaired Student's t test where three asterisks (***) indicate differences between WT and mutant strains with a p value of <0.001.
Deletion of pde2 from S. aureus Results in Increased Cellular Levels of pApA
We reasoned that if Pde2 also degrades pApA in vivo in S. aureus, it is likely that upon deletion of pde2, cellular levels of pApA would be elevated. Therefore, cellular extracts were prepared from the WT LAC* strain, gdpP and pde2 single mutants, and the gdpP/pde2 double mutant strains, and pApA levels were determined by LC-MS/MS analysis. Although pApA was not detectable in WT LAC* or the gdpP mutant, the nucleotide was readily detected in the pde2 deletion strain (Fig. 3C). This result supports the in vitro enzyme activity assays and suggests that the degradation of pApA is the main cellular function of Pde2 in S. aureus. Surprisingly, pApA could also be detected in extracts prepared from the gdpP/pde2 double mutant strain (Fig. 3C), suggesting the presence of yet another enzyme that can convert c-di-AMP to pApA in S. aureus or the existence of an alternative cellular pathway leading to the generation of pApA.
pApA Inhibits GdpP Phosphodiesterase Activity, whereas Pde2 Activity Is Inhibited by ppGpp
The hydrolysis of c-di-AMP by Pde2 is extremely inefficient in vitro and may not be of physiological relevance. However, an increase in cellular c-di-AMP levels was detected in a pde2 mutant S. aureus strain along with an increase in pApA levels (Fig. 1, C–E, and Fig. 3C). It is possible that high levels of pApA may inhibit the activity of GdpP, thereby leading to an elevation of c-di-AMP levels in the cell, even if Pde2 is unable to degrade c-di-AMP in vivo. To establish whether pApA can indeed inhibit the PDE activity of GdpP, enzyme reactions were set up with purified GdpP enzyme and 50 μm c-di-AMP (spiked with a nm concentration of radioactive c-di-AMP) and increasing concentrations of pApA. The reactions were incubated for 1 h, and the c-di-AMP hydrolysis activity of GdpP was assessed by TLC. This analysis revealed that pApA could indeed inhibit the GdpP-dependent conversion of c-di-AMP in a dose-dependent manner (Fig. 4A), and an IC50 value of 78 ± 2.34 μm was established (Fig. 4B). These data suggest that an accumulation of pApA in the cell might lead to a negative feedback loop that acts to reduce the rate of c-di-AMP hydrolysis by GdpP in S. aureus. This inhibition is similar to that observed for the Pseudomonas aeruginosa c-di-GMP PDE RocR by 5′-phosphoguanylyl-guanosine (pGpG) (36, 37).
FIGURE 4.

GdpP phosphodiesterase activity is inhibited by pApA and Pde2 activity by the alarmone ppGpp. A, GdpP enzyme activity in the presence of increasing concentrations of pApA. 50 μm c-di-AMP spiked with a nm amount of radiolabeled c-di-AMP was incubated for 1 h at 37 °C with 0.3 μm GdpP and increasing concentrations of pApA ranging from 0 to 0.6 mm. The reaction products were separated by TLC, and the radioactive signal was detected using a phosphorimager. Experiments were performed in triplicate, and a representative TLC plate is shown. B, quantification of c-di-AMP hydrolysis by GdpP in the presence of increasing amounts of pApA. The radioactive signal from panel A was quantified, the % c-di-AMP hydrolysis was calculated, and the average values and standard deviations from three independent experiments are plotted. The data were fitted, and the IC50 was determined using the dose response inhibition analysis in GraphPad Prism. C, Pde2 enzyme activity assay and TLC analysis. 200 μm pApA spiked with radiolabeled pApA was incubated for 1 h at 37 °C with 4 nm Pde2 and increasing concentrations of ppGpp ranging from 0 to 0.8 mm. TLC plates were analyzed as described in panel A, and a representative image from three independent experiments is shown. D, quantification of pApA hydrolysis by Pde2 in the presence of increasing concentrations of ppGpp. The radioactive signal from panel C was quantified, the % pApA hydrolysis was determined, and the average values and standard deviations from three independent experiments are plotted. The data were fitted, and IC50 were determined using the dose response inhibition analysis.
It has previously been shown that the c-di-AMP PDEs YybT and GdpP of B. subtilis and S. aureus, respectively, and the HD-domain PDE PgpH of L. monocytogenes are inhibited by ppGpp (15, 34, 35). To establish if S. aureus Pde2 is also inhibited by ppGpp, degradation of pApA by Pde2 was assessed in the presence of an increasing concentration of ppGpp. This analysis revealed that Pde2 is inhibited by ppGpp in a concentration-dependent manner (Fig. 4C), and an IC50 of 139 ± 5.6 μm was determined (Fig. 4D). These data indicate that the alarmone ppGpp can inhibit the activities of both the membrane-bound c-di-AMP PDE GdpP and the cytoplasmic pApA PDE Pde2 and suggests an importance in limiting c-di-AMP and pApA hydrolysis under nutrient limiting and other stress conditions.
Deletion of ybbR Does Not Affect Growth, Oxacillin Resistance, or c-di-AMP Levels in S. aureus under Standard Growth Conditions
Besides Pde2, another protein that has been linked to the metabolism of c-di-AMP in various organisms is YbbR. To investigate the function of YbbR in S. aureus, the ybbR mutant strain LAC*ΔybbR was generated. The growth of this mutant was compared with that of the WT LAC* strain under standard growth conditions in TSB medium; however, no difference in growth was noted (Fig. 5A). To investigate if the deletion of ybbR might affect the stability of the DacA cyclase, DacA levels were assessed by Western blotting at the 2- and 6-h time points. However, no significant difference in DacA protein levels was found between the WT and the ybbR mutant strain (Fig. 5B). Of note, the absence of YbbR in the mutant strain was confirmed by Western blotting analysis using a YbbR-specific antibody (Fig. 5B). Next, to establish whether or not YbbR affects c-di-AMP or pApA levels in S. aureus, cytoplasmic extracts were prepared from overnight cultures of the WT and LAC*ΔybbR mutant strains and subjected to LC-MS/MS and ELISA analysis. Levels of c-di-AMP, however, remained unchanged in the absence of YbbR (Fig. 1, C and D), and similar to the WT control strain, no pApA was detected in the mutant (Fig. 3C). Consistent with this, the ybbR mutant also had an identical oxacillin MIC of 4 μg/ml as observed for the WT LAC* strain. Taken together, these data show that under standard growth conditions, YbbR affects neither DacA protein levels nor the cellular levels of c-di-AMP or pApA or oxacillin resistance in S. aureus.
FIGURE 5.

Deletion of ybbR does not affect growth or DacA protein production under standard growth conditions. A, bacterial growth curves. Overnight cultures of WT LAC* and the ybbR mutant strain were diluted to an A600 of 0.01, grown to an A600 of 1, and again back-diluted to an A600 of 0.01 (time = 0). Growth was subsequently monitored over an 8-h period, and average A600 values and standard deviations from three independent experiments are plotted. B, detection of DacA and YbbR proteins by Western blotting. Samples were removed at the 2-h and 6-h time points from cultures grown as described in panel A, and whole cell protein lysates were prepared. YbbR and DacA proteins were detected by Western blotting using YbbR and DacA specific antibodies. A representative blot of three independent experiments is shown.
The Absence of YbbR Results in Increased Acid Sensitivity
The S. aureus ybbR mutant grew identically to the WT LAC* strain in TSB medium (Fig. 5A). To identify a growth phenotype and potentially unmask a function for YbbR in S. aureus, the WT LAC* strain and ybbR mutant were subjected to a phenotypic microarray and grown on several BioLog plates. A significant difference in respiration was observed between the WT and ybbR mutant when cultured in rich medium containing urea, and the pH lowered to 4.5. Although it was not possible to obtain information on the exact urea and medium composition within this well, the data suggested a potential difference in the growth between the strains under acid conditions. To investigate this further, serial 10-fold dilutions of LAC* and LAC*ΔybbR cultures were spotted onto standard TSA pH 7.3 and TSA pH 4.5 plates. Interestingly, disruption of ybbR led to a decreased resistance to acid stress (Fig. 6A). To confirm that this acid sensitivity phenotype is due to the deletion of ybbR, the single site integration vector piTET-ybbR, which allows for ybbR expression from the anhydrotetracyline (Atet)-inducible promoter, was integrated into the ybbR mutant strain to produce the complementation strain LAC*ΔybbR piTET-ybbR (ΔybbR compl.). The empty piTET vector was also introduced into WT LAC* and LAC*ΔybbR yielding control strains LAC* piTET and LAC*ΔybbR piTET. Production of YbbR in the presence of Atet was confirmed by Western blotting using a YbbR-specific antibody (Fig. 6B). It is of note, however, that even upon full induction with 200 ng/ml Atet, the YbbR protein level was reduced in the complementation strain as compared with the LAC* piTET control strain (Fig. 6B). The acid sensitivity phenotype of the ybbR mutant was partially complemented in the LAC*ΔybbR piTET-ybbR strain upon supplementing the medium with Atet (Fig. 6A), all together indicating that deletion of ybbR does indeed lead to the acid sensitivity phenotype.
FIGURE 6.

An S. aureus ybbR mutant has increased acid sensitivity. A, bacterial growth on TSA pH 7.3 versus TSA pH 4.5 plates. WT LAC* and the ybbR mutant as well as the complementation strain LAC*ΔybbR piTET-ybbR (ΔybbR compl.) and the empty vector containing control strains ΔybbR piTET and WT piTET were grown overnight in TSB medium. Cultures were then diluted to an A600 of 5- and 10-fold serial dilutions ranging from 10−1 to 10−7 prepared and spotted onto TSA pH 7.3 or pH 4.5 plates containing 200 ng/ml Atet. B, detection of YbbR by Western blotting. A single colony of the same S. aureus strains as described in panel A was used to inoculate TSB containing 200 ng/ml Atet, and these starter cultures were incubated for 4 h at 37 °C. Cultures were diluted to an A600 of 0.05 and grown overnight at 37 °C in TSB 200 ng/ml Atet before preparing whole cell lysates. The YbbR protein and the loading control protein, SrtA, were detected by Western blotting using YbbR- and SrtA-specific antibodies, respectively.
The Acid Sensitivity of a ybbR Mutant Strain Can Be Bypassed by Compensatory Mutations at Multiple Chromosomal Locations
In an attempt to further understand the contribution of YbbR to acid resistance in S. aureus, suppressor strains were raised by plating high cell density aliquots of LAC*ΔybbR cultures on TSA, pH 4.5, plates. Colonies that had grown within 24 h were then further characterized. Increased acid tolerance was confirmed for 10 of these suppressor strains, referred to as LAC*ΔybbR-S1 to S5 and LAC*ΔybbR-S7 to S11, by spotting serial dilutions of cultures on TSA pH 4.5 plates (Fig. 7). As observed for the WT LAC* strain, but in contrast to the ybbR mutant strain, a similar number of colonies was obtained for the suppressor strains on both plates (Fig. 7). Whole genome sequencing was then employed to identify potential compensatory mutations in these strains. Good sequence coverage was obtained for eight different LAC*ΔybbR suppressor strains, and their genome sequences were compared with that of the WT LAC* and the LAC*ΔybbR mutant strain. This analysis revealed two high frequency and good quality sequence changes in strain LAC*ΔybbR when compared with strain LAC* (Table 1). These mutations were also present in all suppressor strains, indicating that these mutations must have been acquired during the construction of strain LAC*ΔybbR (Table 1). The different LAC*ΔybbR suppressor strains contained one or two additional high frequency and good quality changes. Interestingly, suppressor strains LAC*ΔybbR-S1 and S2 contained a mutation in dacA, resulting in a glycine-to-valine amino acid substitution at amino acid position 78 in the encoded protein. This amino acid is predicted to be located in the third transmembrane helix of the c-di-AMP cyclase DacA. Strains LAC*ΔybbR-S3 and S7 contained a mutation in proP, coding for a predicted proline/betaine transporter. LAC*ΔybbR-S4 contained a mutation in glyA and escA coding for the serine hydroxymethyltransferase GlyA and an ABC transporter ATP-binding protein, respectively. LAC*ΔybbR-S9 contained a mutation in a PilT domain-containing protein, and finally strains LAC*ΔybbR-S10 and S11 contained mutations in a putative membrane-associated zinc metalloprotease (Table 1). In summary, this suppressor mutant analysis indicates that the acid-sensitive phenotype observed in the ybbR mutant can be bypassed in multiple ways, one of which likely affects the function of the c-di-AMP cyclase DacA.
FIGURE 7.
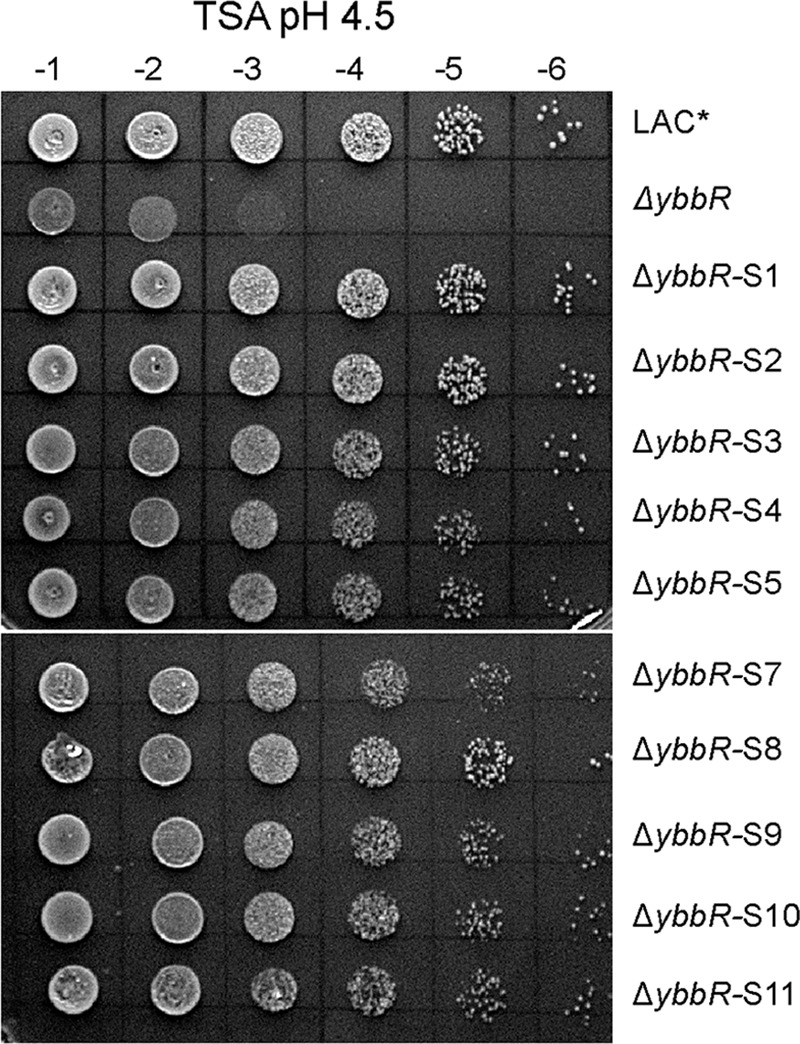
Identification of LAC*ΔybbR suppressor strains with improved acid resistance. WT LAC*, ΔybbR, and ΔybbR suppressor strains (ΔybbR-S1 to S5 and S7 to S11) were grown overnight at 37 °C, cultures were normalized to an A600 of 5, and 10-fold dilutions ranging from 10−1 to 10−6 were spotted on TSA pH 4.5 plates.
TABLE 1.
Single nucleotide variants and deletions identified in strain LAC*ΔybbR (ANG3301) and eight different LAC*ΔybbR suppressor strains compared to strain LAC*
Shown in bold are two sequence changes found in LAC*ΔybbR and all suppressor strains when compared to strain LAC*, indicating that these mutations were acquired during the construction of strain LAC*ΔybbR.
| Strain number | Reference positiona | Typeb | Ref.c | Alleled | Freq.e | Av. qualityf | Annotations | AA changeg |
|---|---|---|---|---|---|---|---|---|
| ANG3301 (ΔybbR) | 1706409 | SNV | G | A | 100 | 37.33 | SAUSA300_1561, iron transporter of the NRAMP family, MntH | A367V |
| 1671814 | SNV | T | C | 85.71 | 35.67 | SAUSA300_1525, glycyl-tRNA synthetase GlyS | ||
| ANG3817 (ΔybbR-S1) | 2284056 | SNV | A | C | 95.83 | 35.3 | SAUSA300_2113, c-di-AMP cyclase DacA | V78G |
| ANG3818 (ΔybbR-S2) | 2284056 | SNV | A | C | 91.67 | 34.67 | SAUSA300_2113, c-di-AMP cyclase DacA | V78G |
| ANG3855 (ΔybbR-S3) | 631381 | SNV | G | A | 100 | 31.73 | SAUSA300_0558, putative proline/betaine transporter ProP | G26D |
| ANG3819 (ΔybbR-S4) | 2225642 | SNV | T | A | 96.43 | 36.78 | SAUSA300_2067, serine hydroxymethyl-transferase GlyA | Y61F |
| 1963962 | SNV | C | A | 95 | 35.42 | SAUSA300_1786, ABC transporter ATP-binding protein EcsA | Glu-159* | |
| ANG3820 (ΔybbR-S7) | 631695 | SNV | C | T | 100 | 35.43 | SAUSA300_0558, putative proline/betaine transporter ProP | Gln-131* |
| ANG3822 (ΔybbR-S9) | 573037 | SNV | T | A | 90.9 | 35.4 | SAUSA300_0512, PilT domain-containing protein | Leu-33* |
| ANG3823 (ΔybbR-S10) | 1262490 | DEL | A | - | 95.45 | 37 | SAUSA300_1155, putative membrane-associated zinc metalloprotease RseP | Lys-239fs |
| ANG3824 (ΔybbR-S11) | 257175 | SNV | A | T | 100 | 38.15 | SAUSA300_0217, AraC family DNA binding response regulator | D99E |
| 1262490 | DEL | A | - | 100 | 33.06 | SAUSA300_1155, putative membrane-associated zinc metalloprotease RseP | K239fs |
a Reference position is based on the USA300 FPR3757 genome sequence (NC_007793.1).
b Type of mutation with SNV indicating single nucleotide variant, DEL nucleotide deletion, and INS nucleotide insertions.
c Ref. indicates base in reference genome.
d Allele indicates base at the same position in sequenced strain.
e Freq. denotes frequency at which base changes were found in the sequence data.
f Average quality score.
g Where applicable, the resulting amino acid change in the protein found in the sequenced strain as compared to the reference strain; fs indicates frame shift and * indicates a stop codon.
An S. aureus Strain Producing a High Level of c-di-AMP Has Increased and a Stain Producing Low Levels Has Decreased Acid Resistance
It seemed likely that the DacAG78V variant found in two of the suppressor strains has altered cyclase activity, leading to a change in the cellular c-di-AMP levels. Elevated levels of the dinucleotide have previously been linked to increased resistance to acid stress in B. subtilis and Lactococcus lactis (15, 38). To possibly unmask a correlation between c-di-AMP levels and acid resistance in S. aureus, WT LAC* and the high and low level c-di-AMP producing strains LAC*ΔgdpP::kan and LAC*dacAG206S as well as strain LAC*ΔybbR were cultured in TSB pH 7.3 or TSB pH 4.5 and growth was monitored over 8 h. Whereas LAC*, LAC*ΔybbR, and LAC*dacAG206S grew equally well in TSB pH 7.3 strain LAC*ΔgdpP::kan demonstrated the expected growth defect (Fig. 8A). When cultured in TSB, pH 4.5, however, strain LAC*ΔgdpP::kan grew better than WT, reaching a higher optical density at the 6- and 8-h time points (Fig. 8B). In contrast, the LAC*ΔybbR and LAC*dacAG206S strains struggled to grow in TSB pH 4.5 (Fig. 8B). Of note, LAC*dacAG206S exhibited a clumping phenotype within 1.5 h under acid stress, LAC*ΔybbR at 2 h, and WT at 4 h. No clumping was observed for strain LAC*ΔgdpP::kan. To confirm that the observed acid resistance and sensitivity phenotypes were due to a mutation in the respective genes, a complementation analysis was performed using strains LAC*dacAG206S pCL55-dacA (Fig. 8C), LAC* gdpP compl. (Fig. 8D), and LAC*ΔybbR piTET-ybbR (Fig. 8E) along with the appropriate control strains. Full complementation was observed for the gdpP complementation strain, whereas partial complementation was observed for strain LAC*dacAG206S pCL55-dacA (Fig. 8C). The fact that the wild-type copy of dacA is expressed in addition to the mutated dacAG206S gene could explain why only partial complementation was observed. Partial complementation was also observed for the ybbR complementation strain (Fig. 8E), but as shown earlier, reduced YbbR protein levels are produced by this strain as compared with WT LAC* (Fig. 6C). All together, these results suggest that high levels of c-di-AMP lead to increased acid tolerance, whereas low levels of c-di-AMP result in acid sensitivity.
FIGURE 8.

S. aureus strain LAC*ΔybbR phenocopies the low c-di-AMP producing LAC*dacAG206S strain when challenged with low pH. A, bacterial growth curves in TSB pH 7.3. Cultures of WT LAC* and the indicated mutant were diluted to an A600 of 0.05 and grown for 2.5 h. All cultures were then again back-diluted to an A600 of 0.05 (time = 0), and bacterial growth was monitored for 8 h by determining A600 readings at timed intervals. The average values and standard deviations from three independent experiments are plotted. B–E, bacterial growth curves in TSB, pH 4.5. Overnight cultures of WT LAC* and indicated mutant or complementation strains were back-diluted to an A600 of 0.05 in TSB pH 4.5 medium and grown for 3.5 h. All cultures were diluted again to an A600 of 0.05 (time = 0), and growth was monitored and plotted as described above.
c-di-AMP Levels Increase in WT as well as ybbR Mutant S. aureus Strains upon Exposure to Acid Stress
As described above, we observed a correlation between c-di-AMP levels and acid resistance in S. aureus; that is, a strain with increased levels of c-di-AMP is more resistant, whereas a low level strain has increased sensitivity to acid stress. Therefore, we set out to test if acid stress conditions would induce c-di-AMP production in WT S. aureus. This would point to the possible scenario where induction is blocked in the ybbR mutant strain, leading to increased acid sensitivity that can be rescued by the ybbR suppressor strain producing the DacAG78V variant. The WT S. aureus strain LAC* was grown in TSB pH 7.3, TSB pH 4.5, and TSB pH 4.25, a pH at which the WT strain had a similar growth defect as the LAC*ΔybbR when grown in TSB pH 4.5. Cytoplasmic extracts were then prepared, and c-di-AMP levels were measured by ELISA. Cellular c-di-AMP levels were indeed observed to increase when S. aureus was propagated in low pH medium (Fig. 9A). Next, we assessed c-di-AMP levels in the WT, the ybbR mutant, and the ybbR-S1 suppressor strain producing the DacAG78V variant after growth in TSB pH 7.3 or TSB pH 4.5 medium. When grown in TSB pH 7.3 medium, the c-di-AMP levels in the suppressor strain ybbR-S1 were slightly but significantly increased as compared with the wild-type and ybbR mutant strains. Surprisingly, the c-di-AMP levels were found to increase in both the ybbR mutant and suppressor strain when grown in TSB pH 4.5 as compared with TSB pH 7.3 (Fig. 9B). These results indicate that the induction of c-di-AMP production is not blocked in the ybbR mutant strain. However, because a small but significant increase in the “basal” c-di-AMP level was noted for the LAC*ΔybbR-S1 suppressor strain when grown in TSB pH 7.3 medium (Fig. 9B), this might suggest that the dacAG206S variant has increased cyclase activity. Finally, to address if the increase in c-di-AMP production observed in the ybbR suppressor strain with a mutation in dacA is also observed in other suppressor strains, we determined the intracellular c-di-AMP levels of several other ybbR suppressor strains after their growth in TSB pH 7.3 or pH 4.5 medium. Interestingly the c-di-AMP levels in all suppressor strains were slightly increased as compared to the WT LAC* strain as well as the ybbR mutant strain in TSB pH 7.3 medium (Fig. 9C), and under acid stress conditions, the c-di-AMP levels remained slightly elevated in all suppressor strains (Fig. 9D). Taken together, these data highlight an important role for c-di-AMP in acid stress resistance in S. aureus.
FIGURE 9.

c-di-AMP levels increase when S. aureus is exposed to low pH and are raised in the ybbR suppressor strains in standard growth medium. A–D, determination of intracellular c-di-AMP levels by ELISA. A, cultures of WT LAC* were diluted to an A600 of 0.05 in TSB pH 7.3, pH 4.5, and pH 4.25. B, cultures of WT LAC* and the indicated mutant strains were diluted to an A600 of 0.05 in TSB pH 7.3 or pH 4.5. C, cultures of WT LAC* and the indicated mutant strains were diluted to an A600 of 0.05 in TSB pH 7.3 D, cultures of WT LAC* and the indicated mutant strains were diluted to an A600 of 0.05 in TSB pH 4.5. All cultures were grown to an A600 of 0.7, bacteria were harvested, and cell extracts were prepared. Lysates were normalized for protein concentration, cellular c-di-AMP levels were determined by ELISA, and values are reported as ng of c-di-AMP/mg protein. The average values and standard deviations from three independently prepared cell extracts are plotted. Statistical analysis was performed for panels A and B using an unpaired Student's t test where one (*) and three asterisks (***) indicate differences between WT and mutant strains or strains grown in TSB pH 7.3 versus low pH medium with a p value of <0.05 and <0.001, respectively. ns, not significant.
Discussion
The work performed in this study revealed a function of the second DHH/DHHA1 domain protein in S. aureus, which we have termed Pde2, as a c-di-AMP- and pApA-hydrolyzing PDE. Although Pde2 can degrade both c-di-AMP and pApA in vitro, biochemical assays and measurements of intracellular nucleotide levels indicated that pApA is the preferred substrate (Figs. 1–3). In the absence of Pde2, the intracellular pApA concentration increased drastically, and to the best of our knowledge, this is the first time that this dinucleotide has been detected in a bacterial cell (Fig. 3C). As part of this study, we also established that pApA can inhibit the c-di-AMP hydrolyzing activity of GdpP in a dose-dependent manner (Fig. 4, A and B, Fig. 10). Hence, Pde2 plays an important role in controlling intracellular pApA concentrations in S. aureus and prevents an increase in c-di-AMP levels either directly through its weak c-di-AMP hydrolase activity or indirectly through the pApA-dependent inhibition of GdpP. It is, however, likely that the enzyme has additional functions and nucleotide hydrolase activities in the cell that will be discussed.
FIGURE 10.
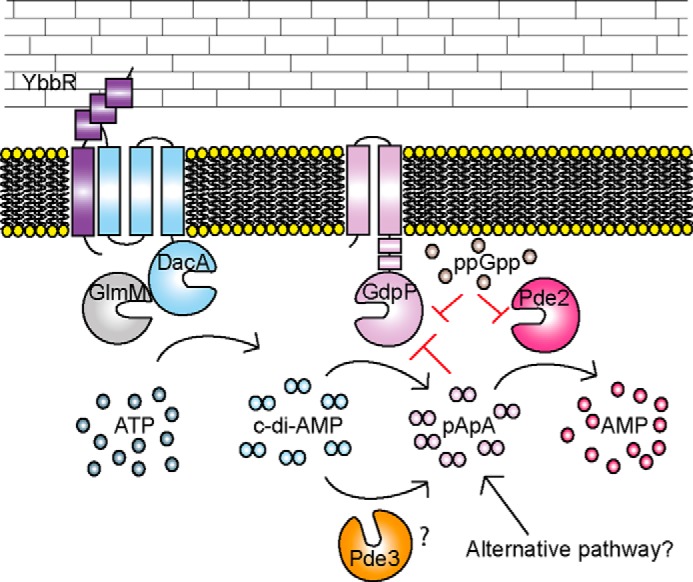
Schematic representation of proteins involved in the c-di-AMP metabolism of S. aureus. c-di-AMP is produced by the di-adenylate cyclase enzyme DacA, which is thought to form a complex with YbbR and GlmM. c-di-AMP is degraded by GdpP to pApA, which is further converted by the Pde2 enzyme to AMP. pApA can, in a feedback inhibition loop, reduce the activity of GdpP, and the activities of both GdpP and Pde2 are inhibited by the stringent response alarmone ppGpp. The results presented in this work suggest that yet another PDE Pde3, capable of degrading c-di-AMP to pApA, is present in S. aureus or that an alternative pathway for the production of pApA exists. The S. aureus DacA, GpdP, YbbR, and GlmM proteins have all been detected by Western blotting under standard laboratory growth conditions, and based on the observed phenotype of the pde2 mutant strain it is expected that these proteins are produced and present at the same time.
Through recent studies it has become apparent that stand-alone DHH/DHHA1-domain enzymes can act on a variety of different nucleotide substrates ranging from nanoRNAs (RNA molecules ≤5 nucleotides), pApA, pGpG, cyclic-di-nucleotides, and pAp, which is a byproduct of sulfur assimilation and of acyl carrier protein production (39, 40). The S. aureus Pde2 enzyme shares a high degree of sequence similarity with the B. subtilis nanoRNase A NrnA (also referred to as YtqI), M. tuberculosis Rv2837c (also referred to as MtbPDE or CnpB), and S. pneumoniae Pde2 (also referred to as PapP). The latter two enzymes have been shown to degrade c-di-AMP and pApA to AMP, but all three enzymes also have confirmed nanoRNase and pAp-phosphatase activities (24, 29, 30, 32, 33, 39, 41). Based on these findings, it seems likely that S. aureus Pde2 may also degrade pAp and function as a general nanoRNA ribonuclease in the cell.
Although the S. pneumoniae and S. aureus Pde2 enzymes hydrolyze pApA more efficiently than c-di-AMP (Fig. 2 and Fig. 3, A and B) (24), this is not the case for all stand-alone DHH/DHHA1 enzymes involved in the metabolism of c-di-AMP characterized to date. Similar turnover rates toward c-di-AMP and pApA have been reported for the M. tuberculosis Rv2837c enzyme (32). In addition, this enzyme can degrade c-di-GMP and pGpG and was, therefore, not only implicated in c-di-AMP but also in c-di-GMP metabolism in M. tuberculosis (32). Detailed enzymatic in vitro studies combined with investigations into the effect that inactivation of these enzymes has on cellular nucleotide and ribonucleotide pools are, therefore, crucial in determining the actual physiological activities of DHH/DHHA1 domain enzymes.
The detection of pApA in S. aureus extracts derived from the pde2 mutant strain (Fig. 3C) provides the first experimental support that this dinucleotide is a physiologically relevant substrate for this class of enzyme and present in the cell. Although we did not determine the cell size and exact μm amount of pApA in the pde2 mutant strain, based on a comparison with the previously determined c-di-AMP amounts in a wild-type S. aureus strain grown under similar conditions, we estimate that pApA reaches a cellular concentration of ∼25 μm in the pde2 mutant. It is possible that under certain conditions when Pde2 function is inhibited, pApA levels may also be high in a wild-type strain. This is an interesting possibility, as it has previously been speculated that the c-di-AMP breakdown product pApA could potentially act as a nanoRNA source in bacteria (42), an increase that can result in changes in gene expression through differential priming of transcription (43). Surprisingly, an accumulation of pApA was also observed in a S. aureus mutant strain lacking both GdpP and Pde2 (Fig. 3C), highlighting that besides these two enzymes, another c-di-AMP PDE might be present (Fig. 10). As GdpP and Pde2 are the only DHH/DHHA1 domain proteins annotated in the S. aureus proteome, an enzyme with another domain architecture must be responsible for this activity. Alternatively, pApA could be derived as a byproduct from a different cellular pathway (Fig. 10), an interesting possibility that will be investigated in future studies.
We show here that pApA can inhibit the activity of the c-di-AMP PDE GdpP (Fig. 4, A and B). This might be an alternative explanation as to why, upon deletion of pde2, an increase in the concentration of c-di-AMP is evident in S. aureus (Fig. 3C). The regulation of GdpP is even more complex, as its activity is also inhibited, at least in vitro, by the stringent response nucleotide ppGpp (15, 35). Interdependencies between cellular (p)ppGpp and c-di-AMP levels have been reported in several studies (35, 44). Although the molecular mechanisms behind this have not yet been well studied, one direct link between these two signaling molecules appears to be the c-di-AMP hydrolyzing enzymes such as GdpP and, as shown in this study, the pApA-degrading enzyme Pde2 (Fig. 4, C and D, and Fig. 10). The link between c-di-AMP and (p)ppGpp-controlled pathways is, however, a lot more complex and is not solely controlled by the inhibition of c-di-AMP- and pApA-metabolizing enzymes by (p)ppGpp as it has also been shown that the absence of c-di-AMP leads to a toxic increase in (p)ppGpp levels (44).
Our study has also provided further insight into the cellular function of YbbR (also referred to as CdaR). YbbR homologs have been shown to directly interact with DacA homologs (also referred to as CdaA) in other bacteria (21–23). In addition, an interaction between CdaA and GlmM has been reported, leading to the hypothesis that DacA, YbbR, and GlmM form a three-protein complex (22, 32) (Fig. 10). We have identified a new role for YbbR in acid-stress resistance in S. aureus and also provide further information on how c-di-AMP levels impact acid tolerance in this organism (Figs. 6 and 9).
In this study we show that a S. aureus gdpP mutant strain that produces high levels of c-di-AMP displays enhanced acid resistance (Fig. 8, B and D), as has been reported for L. lactis, L. monocytogenes, and B. subtilis gdpP mutant strains (15, 25, 38). Expanding on this, we further show that a dacAG206S mutant S. aureus strain, which produces low levels of c-di-AMP, demonstrates decreased acid resistance (Fig. 8, B and C). Furthermore, c-di-AMP levels increased in a wild-type S. aureus strain when grown in acidified medium (Fig. 9A), a response that likely helps this organism cope with this stress condition. S. aureus can colonize a variety of sites within the human host that vary in pH between 4.2 and 7.6 (45). It is, therefore, vital that the bacterium mounts a suitable response to survive these challenging conditions, and this work highlights that c-di-AMP likely plays an important role in this. There are a number of characterized mechanisms that bacteria utilize in an effort to survive acidic environments. These include expelling cytoplasmic protons through the F1F0-ATPase, producing alkaline compounds to neutralize the cytoplasmic pH, and importing osmolytes such as choline or proline (46, 47). Microarray data from S. aureus cultured in medium acidified to pH 4.5 revealed that besides genes encoding urease and proton efflux pumps, oxidative stress response genes and the kdp and opuC operons, encoding the c-di-AMP receptor proteins KdpD and OpuCA, are up-regulated (45, 48–51). Although an intriguing correlation, understanding the mechanism through which elevated c-di-AMP levels contributes to the acid tolerance in S. aureus or other microorganisms awaits future studies.
The ybbR mutant strain exhibited decreased acid resistance (Fig. 8), and therefore, we expected this strain to produce low concentrations of c-di-AMP under acid stress conditions. Surprisingly, cellular c-di-AMP concentrations were increased in the ybbR mutant strain as compared with wild-type when grown in low pH medium (Fig. 9B). Whole genome sequencing of LAC*ΔybbR suppressor strains that had acquired the ability to grow on acidified medium offered some insight into the mechanisms used by these strains to survive in the absence of YbbR. All of these suppressor mutations caused a slight increase in the basal c-di-AMP levels in the cell when the bacteria were grown under our standard growth conditions in TSB pH 7.3 medium (Fig. 9C), and we believe that this increase in c-di-AMP levels might help these bacteria to cope with the acid stress. For a number of the suppressor strains it is harder to speculate on the connection between their genomic alterations, the increase in c-di-AMP production under standard laboratory conditions, and how the mutation leads to a reversion in acid sensitivity. However, it is interesting to note that a premature stop codon was identified in escA, a gene encoding an ABC transporter ATP-binding protein, and a frameshift mutation in the predicted membrane-associated zinc metalloprotease RseP (Table 1). B. subtilis orthologs of these two proteins have been shown to function in the same cellular pathway, although a connection with the acid stress response is at this point not clear. It has been shown that the ABC transporter EcsAB is required for the activity of the intramembrane cleaving protease RasP (the RseP ortholog in B. subtilis), and escA and rasP mutants show similar signal sequence processing and protein secretion defects (52, 53). Two independent mutations in an annotated proline/glycine betaine transporter were identified, one of which resulted in a premature stop codon. Osmolyte uptake is known to be important in maintaining pH homeostasis and for bacterial osmoprotection (47, 54). It is possible that deactivation or decreased proline/glycine betaine transporter activity allows for the accumulation of a more efficient compatible solute. In fact, deletion of a putative glycine betaine transporter from Salmonella enterica serovar Typhimurium led to increased resistance to salt stress and low pH as long as trehalose was produced (55). Perhaps two of the most interesting LAC*ΔybbR suppressor strains obtained in two independent screens contained the same valine-to-glycine substitution at amino acid position 79 in DacA. It is possible that this substitution may result in a change in the conformation of DacA, affect DacA dimerization, its interaction with GlmM, or its ability to sense stimuli. Based on cellular c-di-AMP measurements, the ultimate consequence of this appears to be an increase in cyclase activity under “non-stressed” standard growth conditions (Fig. 9B). This increase in c-di-AMP activity may now allow a ybbR mutant strain to survive under acid stress conditions. Altogether, this work provides not only new insights into the c-di-AMP metabolism of S. aureus but also opens up a number of exciting possibilities for future investigations.
Experimental Procedures
Bacterial Strains and Culture Conditions
E. coli strains were cultured in Luria Bertani (LB) medium and S. aureus strains in TSB at 37 °C with aeration unless otherwise stated. All bacterial strains used in this study are listed in Table 2 along with the relevant antibiotics and inducers used for their propagation. For growth under acidic conditions, the pH of TSB, which usually has a pH of 7.3, was lowered to pH 4.5 or pH 4.25 with HCl, and for growth on plates, 30 g of agar per liter TSB was added before lowering the pH.
TABLE 2.
Bacterial strains used in this study
Antibiotics were used at the following concentrations: for E. coli cultures: 100 μg/ml ampicillin (AmpR), 10 μg/ml chloramphenicol (CamR), and 30 μg/ml kanamycin (KanR); for S. aureus cultures: chloramphenicol (CamR), 10 μg/ml for plasmids and 7.5 μg/ml for integrated plasmids; 10 μg/ml erythromycin (ErmR), and 90 μg/ml kanamycin (KanR). The inducer anhydrotetracycline (Atet) was used at a concentration of 200 ng/ml in agar and 50 ng/ml in broth.
| Strain name | Description | Reference |
|---|---|---|
| Escherichia coli | ||
| XL1-Blue | Cloning strain, TetR, ANG127 | Stratagene |
| BL21(DE3) | Protein expression strain, ANG191 | Novagen |
| DC10B | Cloning strain, dcm, ANG2151 | (58) |
| DH10B pIMAY | DH10B pIMAY; CamR, ANG2154 | (58) |
| IM08B | E. coli strain expressing a S. aureus DNA modification system; ANG3724 | (62) |
| ANG243 | XL1-Blue pCL55; S. aureus single-site integration vector: AmpR | (63) |
| ANG284 | XL1-Blue piTET; AmpR | (64) |
| ANG1824 | XL1-Blue pET28b; KanR | Novagen |
| ANG1861 | BL21 (DE3) pET28b-gdpP84–655; KanR | (7) |
| ANG2262 | XL1-Blue pET28b-ybbRnoTM; KanR | This study |
| ANG2281 | BL21 (DE3) pET28b-ybbRnoTM; KanR | This study |
| ANG3048 | BL21 (DE3) pET28b-disABT-His; KanR | (65) |
| ANG3472 | XL1-Blue piTET-ybbR; AmpR | This study |
| ANG3480 | XL1-Blue pET28b-His-cabPSP; KanR | This study |
| ANG3481 | BL21 (DE3) pET28b-His-cabPSP; KanR | This study |
| ANG3511 | H10B pIMAYΔpde2::erm; CamR | This study |
| ANG3544 | XL1-Blue pIMAYΔpde2::erm; CamR | This study |
| ANG3732 | IM08B pCL55; AmpR | (51) |
| ANG3898 | XL1-Blue pET28b-His-pde2; KanR | This study |
| ANG3899 | BL21 (DE3) pET28b-His-pde2; KanR | This study |
| ANG3606 | XL1-Blue pIMAY-dacAG206A; CamR | This study |
| ANG3962 | XL1-Blue pIMAY-gdpP; CamR | This study |
| ANG3963 | IM08B pIMAY-gdpP; CamR | This study |
| ANG3990 | XL1-Blue pIMAYΔybbR; CamR | This study |
| ANG3991 | DC10B pIMAYΔybbR; CamR | This study |
| ANG4123 | XL1-Blue pCL55-pde2; AmpR | This study |
| ANG4124 | XL1-Blue pCL55-dacA; AmpR | This study |
| ANG4125 | IM08B pCL55-pde2; AmpR | This study |
| ANG4126 | IM08B pCL55-dacA; AmpR | This study |
| Staphylococcus aureus | ||
| RN4220 | Restriction-deficient derivative of 8325-4 | (66) |
| LAC* | CA-MRSA LAC strain; ErmS; ANG 1575 | (67) |
| ANG290 | RN4220 piTET | This study |
| ANG1961 | LAC*ΔgdpP::kan; KanR | (7) |
| ANG3301 | LAC*ΔybbR | This study |
| ANG3516 | RN4220 pIMAYΔpde2::erm; CamR ErmR 28 °C | This study |
| ANG3547 | RN4220 piTET-ybbR; CamR | This study |
| ANG3548 | LAC*ΔybbR piTET-ybbR; CamR | This study |
| ANG3559 | LAC* Δpde2::erm; ErmR | This study |
| ANG3652 | RN4220 pIMAY-dacAG206S; CamR 28 °C | This study |
| ANG3659 | LAC* pIMAY-dacAG206S; CamR 28 °C | This study |
| ANG3664 | LAC*dacAG206S | This study |
| ANG3726 | LAC*ΔgdpP::kan Δpde2::erm; KanR ErmR | This study |
| ANG3817 | LAC*ΔybbR-S1 (suppressor strain 1 ) | This study |
| ANG3818 | LAC*ΔybbR-S2 (suppressor strain 2) | This study |
| ANG3819 | LAC*ΔybbR-S4 (suppressor strain 4) | This study |
| ANG3820 | LAC*ΔybbR-S5 (suppressor strain 5) | This study |
| ANG3821 | LAC*ΔybbR-S7 (suppressor strain 7) | This study |
| ANG3822 | LAC*ΔybbR-S8 (suppressor strain 8) | This study |
| ANG3823 | LAC*ΔybbR-S9 (suppressor strain 9) | This study |
| ANG3824 | LAC*ΔybbR-S10 (suppressor strain 10) | This study |
| ANG3825 | LAC*ΔybbR-S11 (suppressor strain 11) | This study |
| ANG3855 | LAC*ΔybbR-S3 (suppressor strain 3) | This study |
| ANG3869 | LAC*ΔybbR piTET; CamR | This study |
| ANG3795 | LAC* pCL55 | (51) |
| ANG3988 | LAC* piTET | This study |
| ANG4003 | RN4220 pIMAYΔybbR; CamR, 28 °C | This study |
| ANG4040 | LAC*gdpP compl. | This study |
| ANG4045 | LAC* pIMAYΔybbR; CamR, 28 °C | This study |
| ANG4127 | LAC*Δpde2 pCL55; CamR | This study |
| ANG4128 | LAC*Δpde2 pCL55-pde2; CamR | This study |
| ANG4129 | LAC*dacAG206S pCL55; CamR | This study |
| ANG4130 | LAC*dacAG206S pCL55-dacA; CamR | This study |
Strain Construction
E. coli and S. aureus strains used in this study are listed in Table 2, and the primers used are in Table 3. Plasmid pET28b-His-pde2 was constructed for the expression and purification of the N-terminally His-tagged S. aureus Pde2 protein. To this end, the pde2 gene was amplified by PCR from LAC* chromosomal DNA using primers ANG2214 and ANG2215. The product was digested with NheI and EcoRI and ligated with the similarly digested vector pET28b. Plasmid pET28b-His-pde2 was initially recovered in E. coli XL1-Blue and then introduced into E. coli BL21 (DE3) for protein production, yielding strains ANG3898 and ANG3899, respectively. Plasmid pET28-His-cabPSP was constructed for the expression and purification of the His-tagged S. pneumoniae c-di-AMP receptor protein CabP (56). To this end, the cabP gene was amplified from S. pneumoniae strain D39 chromosomal DNA using primers ANG2034 and ANG2035. The resulting PCR product was digested with NdeI and HindIII and ligated with the similarly digested pET28b. The plasmid was then transformed into E. coli XL1-Blue yielding strain ANG3480 and subsequently introduced into E. coli strain BL21(DE3) for protein expression, giving strain ANG3481. Plasmid pET28b-ybbRnoTMwas generated for the expression and purification of the soluble YbbR protein domains, a protein that was subsequently used for antibody production. The ybbR gene (minus the predicted transmembrane region) was amplified from LAC* DNA using primers ANG1370 and ANG1371 and digested with BamHI and NcoI. The product was ligated into similarly digested pET28b and transformed into XL1-Blue yielding strain ANG2262 and subsequently into BL21(DE3) yielding strain ANG2281.
TABLE 3.
Primers used in this study
Restriction site sequences are underlined.
| Primer | Name | Sequence |
|---|---|---|
| ANG1370 | R_YbbRnoTM | CGGGATCCCGTTTTACATTTATATAAGCCTTCGTTTCAC |
| ANG 1371 | F_YbbRnoTM | CATGCCATGGCTGGTAATCTTGGTCAAAAGTCTAGTAAAAC |
| ANG1765 | F_EcoRI_dacA | GCGCGAATTCTATAAAGTATATTTTGCTTTTTGC |
| ANG1848 | F_KpnI_YbbRdel1 | GCGCGGTACCAATTGTAACGAGTATCCTTG |
| ANG1814 | R_YbbRdel1 | GCCTTCGTTTCAAATCTCAAGCCCCATTTACTTTC |
| ANG1815 | F_YbbRdel2 | GGCTTGAGATTTGAAACGAAGGCTTATATAAATGTA |
| ANG1816 | R_EcoRI_YbbRdel2 | GCGCGAATTCAACGATATGTCCAGATTGTTCTCC |
| ANG1817 | F_YbbRdel_check | GCAATGATTATTCAAGGCACGAAG |
| ANG1818 | R_YbbRdel_check | CCCATATCGCGTGTTAAATATGC |
| ANG1908 | F_AvrII_YbbR | GCGCCCTAGGCATTGGTTTGGCACACGCTTTC |
| ANG1909 | R_SacII_YbbR | GCGCCCGCGGTTATTTTACATTTATATAAGCCTTCG |
| ANG2034 | F_NdeI_CabP_Nhis | TTTCATATGTCAGATCGTACGATTGGAATTTTGGGC |
| ANG2035 | R_HindIII_CabP | TTTAAGCTTACGAATTCAATGCTACTAGGGTATCC |
| ANG2040 | F_EcoRI_1650del1 | GCGCGAATTCGTTCAAGATATAATGACGCCATTAG |
| ANG2041 | R_1650del1 | GAAAAAGGAAGAGTCAAAGTACTAATCATTTTCATACTCCC |
| ANG2042 | F_1650del2 | GATTAGTACTTTGACTCTTCCTTTTTCAATATTATTGAAGC |
| ANG2043 | R_1650del2 | GTTAAGTTTTGTTTATTTCCTCCCGTTAAATAATAGATAAC |
| ANG2044 | F_1650del3 | CGGGAGGAAATAAACAAAACTTAACTAATAGAAAGGGGCC |
| ANG2045 | F_KpnI_1650del3 | GCGCGGTACCCCAATAAATAACTGACCAGTGAGCC |
| ANG2086 | F_1650del_check | CGGAAATCGTGAAGATGTGCAGATTG |
| ANG2087 | R_1650del_check | CCAACATCTCTAGCAACTGCTCTTGC |
| ANG2104 | F_EcoRI_dacAG206A1 | CGGAATTCCCAGGCACTGGTACTAGAGTTTTGGGTGGTC |
| ANG2106 | R_XmaI_dacAG206A2 | TCCCCCCGGGCATTTATATAAGCCTTCGTTTCACTTGGTTG |
| ANG2109 | R_dacAG206A1 | GCTCTATGTCTTGTcgaCAAACTTTTAGATATCTTAGGACTATCAGAC |
| ANG2110 | F_dacAG206A2 | CTAAAAGTTTGtcgACAAGACATAGAGCTGCGGTTGGTATTTCAG |
| ANG2214 | F_NheI_Pde2_Nhis | GCGCGCTAGCATGATTAGTACTTTGAATGAAATTATG |
| ANG2215 | R_EcoRI_Pde2 | GCGCGAATTCTTAGTTAAGTTTTGTGCGTAAAGCTGTAGC |
| ANG2254 | F_KpnI_gdpP | GACTCGGTACCTTCAATTAAATGAAATAGAAGCATACAATC |
| ANG2255 | R_NotI_gdpP | GAATAGCGGCCGCCTCTTCAGCTGTTTCATACACTTGTC |
| ANG2472 | F_EcoRI_1650 | GCGCGAATTCGAACTTAGCAGAGAAAATAGGCATAAG |
| ANG2473 | R_BamHI_1650 | GCGCGGATCCTTAGTTAAGTTTTGTGCGTAAAGCTG |
| ANG2474 | R_BamHI_dacA | GCGCGGATCCTTATTTCACACCTTTCTTTTGAAAGCGTG |
To generate the erm-marked pde2 mutant strain LAC*Δpde2::erm (ANG 3559), 1-kb DNA fragments up- and downstream of the pde2 gene were amplified by PCR from LAC* genomic DNA using primer pairs ANG2040/2041 and ANG2044/2045. The erythromycin antibiotic resistance gene ermC was amplified from plasmid pCN49 (57) using primers ANG2042 and ANG2043. The upstream DNA and ermC fragments were fused by splice overlap extension (SOE) PCR using primers ANG2040 and ANG2043, and the resulting PCR product was then fused to the downstream DNA fragment using primers ANG2040 and ANG2045. This PCR product was digested with EcoRI and KpnI and cloned into pIMAY digested with the same restriction enzymes, yielding plasmid pIMAYΔpde2::erm. The plasmid was initially recovered in E. coli strain XL-1 Blue and subsequently introduced into the dcm mutant E. coli strain DC10B, yielding strains ANG3544 and ANG3511. The pIMAYΔpde2::erm construct was then electroporated into S. aureus RN4220 yielding strain ANG3516 and subsequently into S. aureus LAC*, which was propagated at 28 °C. Allelic exchange was performed as described by Monk et al. (58), finally yielding strain LAC*Δpde2::erm (ANG3559). ThegdpP/pde2 double mutant strain LAC*ΔgdpP::kanΔpde2::erm (ANG3726) was generated by transducing the gdpP::kan mutation from strain ANG1961 into strain ANG3359 using phage Φ85, and successful transduction was confirmed by PCR using primers ANG2086 and ANG2087. The complementation strain LAC* Δpde2 pCL55-pde2 (ANG4128) was generated by first amplifying the pde2 gene (including its promoter) from LAC* genomic DNA using primers ANG2472 and ANG2473. The PCR product was digested with EcoRI and BamHI and ligated with the similarly digested plasmid pCL55. The resulting plasmid pCL55-pde2 was recovered in E. coli XL1-Blue and subsequently introduced into E. coli strain IM08B, yielding strains ANG4123 and ANG4125, respectively. The plasmid was isolated from strain ANG4125 and introduced and integrated into the geh locus of S. aureus LAC*Δpde2::erm (ANG3559), yielding strain LAC*Δpde2::erm pCL55-pde2 (ANG4128). As control strains, the empty pCL55 vector was isolated from strain IM08B pCL55 (ANG3732) (51) and introduced into strains LAC* and LAC*Δpde2::erm, yielding strains LAC* pCL55 (ANG3795) (51) and LAC*Δpde2::erm pCL55 (ANG4127), respectively. The gdpP complementation strain LAC*gdpP compl. was generated by replacing the kanamycin-marked deletion of LAC*ΔgdpP::kan (ANG1961) with a complete version of gdpP gene. First, the entire gdpP gene including 1-kb up- and downstream fragments was amplified from LAC* genomic DNA using primers ANG2254 and ANG2255. The resulting PCR product was digested with KpnI and NotI, inserted into the similarly digested pIMAY plasmid, and recovered in E. coli XL-Blue to yield strain ANG3962. The plasmid was then transformed into E. coli IM08B and subsequently introduced into LAC*ΔgdpP::kan (ANG1961). Allelic exchange was performed as described by Monk et al. (58) yielding strain LAC*gdpP compl. (ANG4040). Repair of the gdpP locus was confirmed by PCR and sequencing. To generate the S. aureus ybbR deletion strain LAC*ΔybbR (ANG3301), 1-kb DNA fragments up- and downstream of ybbR were amplified from LAC* genomic DNA using primer pairs ANG1848/ANG1814 and ANG1815/ANG1816. The PCR products were fused by SOE PCR using primers ANG1848 and ANG1816, digested with EcoRI and KpnI, and ligated with similarly digested pIMAY, yielding plasmid pIMAYΔybbR. The plasmid was recovered in E. coli XL1-Blue (giving strain ANG3990), shuttled through DC10B (strain ANG3991), and subsequently electroporated into RN4220 and LAC*, yielding strains ANG4003 and ANG4045, respectively. The allelic exchange was performed as described above, yielding strain LAC*ΔybbR (ANG3301), and the ybbR deletion was confirmed by PCR using primers ANG1817 and ANG1818. The plasmid piTET-ybbR for Atet-inducible expression of ybbR in S. aureus was generated by amplifying the ybbR gene from LAC* chromosomal DNA using primers ANG1908 and ANG1909. The PCR product and plasmid piTET were digested with AvrII and SacII, ligated, and then transformed into E. coli XL1-Blue yielding strain XL1-Blue piTET-ybbR (ANG3472). The plasmid was subsequently introduced and integrated into the geh locus of S. aureus strain RN4220, yielding strain RN4220 piTET-ybbR (ANG3547), and subsequently moved by phage transduction using phage Φ85 into strain LAC*ΔybbR to generate the ybbR complementation strain LAC*ΔybbR piTET-ybbR (ANG3548). As controls, strains LAC* piTET (ANG3988) and LAC*ΔybbR piTET (ANG3869), containing the empty piTET vector were produced. To this end, plasmid piTET was first integrated into the geh locus of S. aureus strain RN4220, yielding strain RN4220 piTET (ANG290), and subsequently moved by phage transduction using phage Φ85 into strain LAC* and LAC*ΔybbR, respectively.
An S. aureus strain producing a DacA variant in which the glycine at amino acid position 206 is replaced with a serine (from here on out referred to as dacAG206S allele) has been shown to produce reduced levels of c-di-AMP (26). To generate the low c-di-AMP level S. aureus strain LAC*dacAG206S, primer pairs ANG2104/ANG2109 and ANG2106/2110 were used to amplify 1-kb fragments upstream and downstream from the dacA Gly-206 codon using LAC* genomic DNA in PCRs, and the resulting products were fused by SOE PCR using primers ANG2104 and ANG2110. Primer ANG2109 and ANG2110 were designed to introduce the Gly-206-to-Ser substitution. The resulting PCR product was then digested with EcoRI and XmaI and ligated into pIMAY digested with the same enzymes. Plasmid pIMAYdacAG206S was initially recovered into E. coli strain XL1-Blue to yield ANG3606, and the plasmid was shuttled through S. aureus RN4220 to generate strain RN4220 pIMAY-dacAG206S (ANG3652) and subsequently introduced into strain LAC* to produce LAC* pIMAY-dacAG206S (ANG3659) that was propagated at 28 °C. The allelic exchange was performed as described above yielding strain LAC*dacAG206S (ANG3664). The substitution of the glycine to serine codon in dacA as well as all other plasmid inserts was verified by fluorescence-automated sequencing. To generate the complementation strain LAC*dacAG206S pCL55-dacA, the dacA gene (including its promoter) was amplified from LAC* genomic DNA using primers ANG1765 and ANG2474. The product was digested with EcoRI and BamHI and inserted into the similarly digested plasmid pCL55. Plasmid pCL55-dacA was recovered in E. coli XL1-Blue and subsequently introduced into E. coli IM08B, resulting in the strains ANG4124 and ANG4126, respectively. The plasmid was then electroporated into strain LAC*dacAG206S (ANG3664) yielding strain LAC*dacAG206S pCL55-dacA (ANG4130).
Growth Curves
S. aureus strains were grown overnight in TSB medium containing the relevant antibiotic. Overnight cultures of WT LAC* and the indicated mutant strains were diluted in fresh TSB medium to an A600 of 0.01 or diluted to an A600 of 0.05 if strain LAC*ΔgdpP::kan was included in the growth curve. Cultures were incubated at 37 °C with aeration until an A600 of 1 was reached. Fresh TSB was inoculated with the starter culture to an A600 of 0.01 or 0.05 as before, and growth was monitored for 8 h by taking A600 readings every 2 h. Growth curves in TSB pH 4.5 medium were performed as follows. S. aureus strains were grown overnight in TSB medium before diluting in TSB pH 4.5 to an A600 of 0.05. Cultures were incubated at 37 °C with aeration for 3.5 h before diluting again into fresh TSB pH 4.5 medium to an A600 of 0.05. Growth was then monitored for 8 h by taking A600 readings every 2 h. Alongside this, the same strains were cultured in TSB medium and treated similarly as a control. The first dilution was made into TSB medium, but cultures were grown for 2.5 h before diluting into fresh TSB medium once more. For the pde2 growth complementation assay in TSB 7.3 medium, overnight cultures of the indicated strains were diluted to an A600 of 0.01, and growth was monitored for 10 h by taking A600 readings every 2 h. All growth curves were performed in triplicate with average values and standard deviations plotted.
Oxacillin Minimum Inhibitory Concentrations
Overnight cultures of WT LAC* and LAC* mutant strains were diluted to an A600 of 0.1 before spreading on Mueller-Hinton agar plates containing 2% NaCl. Oxacillin M.I.C. EvaluatorTM (ThermoScientific) strips were then applied to the agar, and the plates were incubated at 35 °C for 24 h. MIC values represent an average of three independent experiments.
Protein Purifications
His-tagged S. aureus GdpP, Pde2, and YbbRnoTM proteins and the His-tagged Bacillus thuringiensis DisA protein (DisABT) were purified from 1 or 2 liters of E. coli BL21(DE3) cultures. After overnight growth at 30 °C, strains were diluted 1:20 and cultured to an A600 of 0.5–0.8. Protein expression was then induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. Strains were incubated overnight at 16 °C for GdpP and YbbRnoTM overexpression at 37 °C for 4 h in the case of Pde2 and overnight at 18 °C for DisABT. Proteins were purified by nickel affinity and size exclusion chromatography as previously described (7). His-tagged CabP from S. pneumonia was expressed and purified essentially as described by Bai et al. (56). Briefly, 1 liter of BL21(DE3) pET28b-His-cabPSP culture was grown at 37 °C to an A600 of 0.6, and protein expression was induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 24 °C. Bacteria were collected by centrifugation and suspended in 20 ml of 50 mm Tris, pH 7.5, 500 mm NaCl, 10% glycerol lysis buffer containing 10 mm imidazole, and cells were lysed by French pressing. The His-CabPSP protein was purified by nickel affinity chromatography, where the column was washed after protein loading with 10 column volumes lysis buffer containing 10 mm, 20 mm, and finally 30 mm imidazole and subsequently eluted in 4 ml of lysis buffer containing 250 mm imidazole. The protein was subsequently dialyzed at 4 °C twice for 2 h against 1 liter of phosphate-buffered saline (PBS), pH 7.4, buffer and finally overnight against 1 liter of PBS, pH 7.4, buffer containing 10% glycerol. Protein concentrations were determined using the Pierce BCA kit, snap-frozen, and stored at −80 °C.
Western Blotting
Bacteria from 0.5–1-ml cultures of the different S. aureus strains were harvested by centrifugation and suspended in TM buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2) containing 100 μg/ml lysostaphin and 20 μg/ml DNase. Samples were normalized for A600 readings of the original cultures; that is, cells collected from a 1-ml culture with an A600 of 1 were suspended in 15 μl of TM buffer. Samples were incubated at 37 °C for 30 min and subsequently mixed with an equal volume of 2× SDS protein sample buffer. This was followed by boiling the samples for 10 min and centrifugation for 5 min. Samples were then separated on 12% SDS-polyacrylamide gels and transferred to PVDF membranes. DacA and YbbR were detected using rabbit polyclonal anti-DacA (35) or anti-YbbR antibodies (produced at Covalab as part of this study) at 1:10,000 and 1:20,000 dilutions, respectively, and SrtA was detected using a rabbit polyclonal anti-SrtA antibody (59) at a dilution of 1:20,000. An HRP-conjugated anti-rabbit IgG secondary antibody (Cell Signaling Technologies, catalogue no. 7074) was used at a 1:10,000 dilution. Blocking and antibody incubation steps were performed in the presence of 10 μg/ml human IgG to reduce unspecific antibody binding to S. aureus Spa and Spi proteins. Blots were developed using ClarityTM Western ECL Blotting Substrate (Bio-Rad) and imaged using the ChemiDocTM Touch Imaging System (Bio-Rad). Western blottings were performed in triplicate, and a representative result is shown.
Quantification of c-di-AMP and pApA by LC-MS/MS
Starter cultures of the WT LAC* S. aureus strain and the different mutant strains were prepared in 5 ml of TSB medium and grown for 4 h at 37 °C with aeration. The cultures were back-diluted to an A600 of 0.05 in 5 ml if TSB medium and grown overnight in the same conditions. Cells were harvested from the overnight cultures, and bacterial cell extracts were prepared and analyzed as described previously using 0.58 μm 13C,15N isotope-labeled c-di-AMP as an internal standard (7). At the same time the dry weight of the bacteria from a culture aliquot was determined for normalization purposes. For pApA measurements, 0.58 μm 13C,15N isotope-labeled c-di-AMP was added to the samples after extraction and before the LC-MS/MS analysis to account for internal ion suppression. c-di-AMP and pApA concentrations within the samples were determined based on a standard curve produced with known concentrations of c-di-AMP and pApA, respectively. Values are reported as ng of c-di-AMP/mg cell weight or ng of pApA/mg cell weight, and the averages values and standard deviations from three samples were plotted. A two-tailed two-sample equal variance Student's t test was used to determine statistically significant differences between c-di-AMP and pApA levels in WT versus mutant strains.
Detection and Quantification of Nucleotides by LC-UV
100 μm unlabeled c-di-AMP or pApA (BioLog) were mixed with 1 μm purified Pde2 enzyme in 50 mm Tris-HCl, pH 7.5, 0.1 mm MnCl2, and 20 mm KCl buffer and incubated at 37 °C for 2 h. Reactions were stopped by boiling for 10 min, and precipitated proteins were subsequently removed by centrifugation. The LC-UV analysis of the reaction products was performed as previously described (60) with the following modifications. Instead of mass spectrometric analysis, a Dionex HPLC-UV system with a constant flow rate of 0.6 ml/min and a wavelength at 259 nm was applied. Solutions with known concentrations of AMP (retention time 3.0 min), c-di-AMP (retention time 8.6 min), and pApA (retention time 9.0 min) were used as calibrators.
Synthesis of Radiolabeled c-di-AMP and pApA
c-di-AMP used in in vitro nucleotide conversion assays was synthesized by incubating 2-mm unlabeled ATP spiked with 9.99 nm 32P-labeled ATP (PerkinElmer Life Sciences) with 10 μm DisABT in 40 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm MgCl2 buffer. This reaction was incubated at 45 °C for 24 h and deactivated by heating to 95 °C for 10 min, and the protein was removed by centrifugation at 13,000 rpm for 5 min. 1 mm c-di-AMP was converted to pApA using 3 μm GdpP in 50 mm Tris-HCl, pH 7.5, 0.1 mm MnCl2, and 20 mm KCl buffer, and the samples were incubated at 37 °C for 2 h. Reactions were stopped as described above. Conversions were analyzed by spotting 1 μl of each reaction on a PEI-cellulose F TLC plate (Merck Millipore). The nucleotides were separated using a 1:1.5 (v/v) saturated NH4SO4, 1.5 m KH2PO4, pH 3.6 buffer, and visualized with a FLA 7000 Typhoon PhosphorImager.
In Vitro GdpP and Pde2 Enzyme Activity Assays
GdpP and Pde2 activity assays were performed using 50 mm Tris-HCl, pH 7.5, 0.1 mm MnCl2, and 20 mm KCl buffer, and incubated at 37 °C. Reactions were stopped at the indicated times by heating the samples to 95 °C for 10 min and subsequently removing the proteins by centrifugation at 13,000 rpm for 5 min. The activity of Pde2 was first assessed through hydrolysis reactions using ATP, c-di-AMP, and pApA as substrates. 100 μm concentrations of each substrate spiked with a low nm concentration of radiolabeled substrate were incubated with 1 μm GdpP or Pde2 for 2 h. This was performed twice with two separate preparations of purified Pde2 (four times in total). Time course experiments were initiated by incubating 200 μm c-di-AMP or pApA (again spiked with a low concentration of the respective radiolabeled substrate) with 4 nm Pde2 for 1 h. 5 μl samples were removed from each reaction at times 0, 2.5, 5, 10, 15, 20, 30, and 60 min. This was performed twice with two separate preparations of purified Pde2 (four times in total). GdpP inhibition assays were performed by incubating 50 μm c-di-AMP (spiked with radiolabeled c-di-AMP) with 0.3 μm GdpP in the presence of pApA (Biolog) ranging from 0 to 0.6 mm for 1 h. Pde2 inhibition assays were performed by incubating 200 μm pApA (spiked with a low concentration of radiolabeled pApA) with 4 nm Pde2 in the presence of ppGpp (tebu-bio) ranging from 0 to 0.8 mm. Inhibition assays were performed in triplicate. After heat inactivation and centrifugation, 1 μl from each reaction was spotted onto PEI-cellulose F TLC plates (Merck Millipore) and run in 1:1.5 (v/v) saturated NH4SO4 and 1.5 m KH2PO4, pH 3.6 buffer. Conversions were visualized using a FLA 7000 Typhoon phosphorimager and quantified using the ImageQuantTL software. Values were calculated based on an average from at least three individual experiments and plotted with standard deviations.
Acid Susceptibility Assay and Generation of Suppressor Strains
The indicated S. aureus strains were grown overnight at 37 °C in 5 ml of TSB. Cultures were normalized to an A600 of 5, 10-fold serial dilutions prepared, and 5 μl of the 10−1-10−7 dilutions were spotted onto TSA or TSA pH 4.5 plates containing 200 ng/ml Atet where indicated. To raise acid-resistant suppressor mutants, cultures of strain LAC*ΔybbR were grown overnight in TSB, and 100 μl of 10−1 and 10−2 dilutions were spread onto TSA pH 4.5 plates. To confirm that the obtained colonies were indeed acid-resistant ybbR suppressor strains, several colonies were picked and grown overnight in TSB, and the next day 5 μl of 10−1-10−6 dilutions were spotted onto TSA pH 4.5 plates. A strain that was able to plate at the 10−6 dilution was considered a bona fide suppressor strain, and in this manner strains LAC*ΔybbR-S1 to S5 and LAC*ΔybbR-S7 to S11 were generated.
Whole Genome Sequencing
WT LAC*, LAC*ΔybbR, and LAC*ΔybbR suppressor strains were cultured overnight at 37 °C, cells were harvested, and genomic DNA was extracted. Genome sequencing for the S. aureus strain LAC* was performed by MicrobesNG using an Illumina Hi-Seq platform and a 250-bp paired end read kit. All other strains were sequenced at the Department of Microbiology and Immunobiology at the Harvard Medical School using an Illumina Mi-Seq platform and a 150-bp paired end read kit. Sequence analysis was performed using the CLC Genomics work bench software package. First the LAC* reads were aligned against the published USA300 FPR3757 genome sequence (RefSeq accession number NC_007793.1) and assembled into a reference contig, and the USA300 FPR3757 annotation and base numbering was transferred onto the LAC* sequence. Next, the LAC*ΔybbR and LAC*ΔybbR suppressor strain Illumina reads were mapped onto the assembled LAC* sequence, and high frequency (>65%) and good quality base changes were identified using the CLC Genomics Workbench software package; the data are summarized in Table 1. The Illumina short reads are deposited in the European Nucleotide Archive under accession numbers PRJEB14759 for strain LAC* and PRJEB14723 for all other strains.
c-di-AMP Quantification by Competitive Enzyme-linked Immunosorbent assay (ELISA)
5 ml of TSB was inoculated with a single colony of WT LAC* or the indicated S. aureus mutant strains and incubated for 4 h at 37 °C with aeration. Next, the cultures were diluted to an A600 of 0.05 in 5 ml of TSB and incubated overnight under the same conditions. For growth in acidified medium (pH 4.5 and pH 4.25), 5 ml of TSB was inoculated with single colonies of WT and mutant strains and incubated overnight at 37 °C with aeration. Cultures were diluted to an A600 of 0.05 in 5 ml of acidified TSB (and TSB pH 7.3 as a control) and grown to an A600 of 0.7. Cells were harvested from overnight or exponential phase cultures and washed 3 times with 1 ml of PBS, pH 7.4. Pellets were suspended in 1 ml of lysis buffer (50 mm Tris, pH 8, containing 20 μg of lysostaphin) and then incubated at 37 °C for 15 min. Lysates were added to 0.5-ml glass beads and lysed using a Fast-Prep instrument (FP120, MP Biomedicals, LLC) at setting 6 for 30 s. This was repeated twice, placing the samples on ice for 2 min between runs. Glass beads were removed by centrifugation at 17,000 × g at 4 °C for 5 min, and the cleared cell lysate was transferred to a fresh tube. Lysates were boiled for 10 min, debris were removed by centrifugation, and the supernatants were transferred to a new tube. The protein concentration of each sample was determined using the BCA kit (Pierce), and cell lysates were normalized to a concentration ranging from 0.3 to 0.75 mg/ml. The ELISAs were performed as previously described (61). Briefly, the wells of a 96-well NUNC Maxisorp plate were coated with 100 μl of a 10 μg/ml CabP protein solution in coating buffer (50 mm Na2CO3, 50 mm NaHCO3, pH 9.6), and plates were incubated overnight at 4 °C. Wells were washed 3 times with PBS, pH 7.4, containing 0.05% Tween 20 (PBST) and blocked with 5% BSA in PBS. Next the control samples and S. aureus extracts (in triplicate) were added to the plate. To prepare the control samples, c-di-AMP (BioLog) was diluted 2-fold in 50 mm Tris, pH 8 buffer, from 200 to 3.125 nm. The same volume of biotinylated c-di-AMP (BioLog) was added to each sample to give a final concentration of 25 nm. Next, 100 μl of these control and S. aureus extracts samples were added to the coated ELISA plate. 100 μl of lysis buffer only was added as a background reading control, and the plate was then incubated at room temperature for 2 h. After washing the wells 3 times with 200 μl of PBST, 100 μl of the PierceTM High Sensitivity Streptavidin solution (ThermoFisher Scientific) diluted 1:5,000 or 1:10,000 in PBS was added to the wells, and the plate was incubated at room temperature for 1 h. Wells were washed again 3 times with 200 μl of PBST. 10 mg of o-phenylenediamine dihydrochloride (Sigma) was dissolved in 20 ml of citrate buffer, pH 5, containing 20 μl of H2O2, and 100 μl was added as substrate to each well. The plate was incubated at room temperature for 30 min, and the reaction was stopped by the addition of 100 μl of 2 m H2SO4. The A490 was measured on a plate reader, buffer only readings were subtracted from controls, and buffer/lysostaphin readings were subtracted from samples. The absorbance readings for the c-di-AMP standard control samples were used to generate a second-order polynomial calibration curve, and the obtained quadratic equation was used to determine the c-di-AMP levels in the S. aureus extracts. nm of c-di-AMP were converted into ng of c-di-AMP/mg protein, and average values and standard deviations from three samples plotted. A two-tailed two sample equal variance Student's t test was used to determine statistically significant differences between c-di-AMP levels in WT versus mutant strains.
Author Contributions
L. B. and A. G. designed the study and wrote the paper. L. B., M. S. Z., C. F. S., and A. G. performed the experiments. V. K. designed the nucleotide mass spectrometry and HPLC-UV experiments, and all authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Andrew Ulijasz for providing the S. pneumoniae D39 DNA and Thomas Bernhardt and Jackson Buss for help with the Illumina sequencing of the S. aureus LAC*ΔybbR mutant and suppressor strains as well as help with the genome sequence analysis. The Illumina sequencing of the LAC* strain was performed by MicrobesNG, which is supported by the Biotechnology and Biological Sciences Research Council Grant BB/L024209/1). We also thank Sanika Hakim for optimization of bacterial growth conditions for MIC determinations, Charlie Millership for purification of the DisABT protein, Mikaila Jayaweera Bandara for construction of the E. coli YbbR overexpression strain, and Annette Garbe for the nucleotide analysis by mass spectrometry. Finally, we thank Manal AbuQun for the phenotype microarray experiments of the LAC* and LAC*ΔybbR strains on Biolog plates.
This work was supported by European Research Council Grant 260371 and Wellcome Trust Grant 100289 (to A. G.). The authors declare that they have no conflicts of interest with the contents of this article.
The Illumina short reads have been deposited in the European Nucleotide Archive under accession numbers PRJEB14759 for strain LAC* and PRJEB14723 for all other strains.
- MRSA
- methicillin-resistant S. aureus
- c-di-GMP
- cyclic di-guanine monophosphate
- (p)ppGpp
- guanosine tetraphosphate and pentaphosphate
- c-di-AMP
- cyclic di-adenosine monophosphate
- DAC
- diadenylate cyclase
- pApA
- 5′-phosphadenylyl-adenosine
- PDE
- phosphodiesterase
- pAp
- 3′-phosphoadenosine-5′-phosphate
- TSB
- tryptic soya broth
- MIC
- minimal inhibitory concentration
- Atet
- anhydrotetracyline
- compl.
- complementation strain
- SOE
- splice overlap extension.
References
- 1. van Belkum A., Melles D. C., Nouwen J., van Leeuwen W. B., van Wamel W., Vos M. C., Wertheim H. F., and Verbrugh H. A. (2009) Co-evolutionary aspects of human colonisation and infection by in Staphylococcus aureus. Infect. Genet. Evol. 9, 32–47 [DOI] [PubMed] [Google Scholar]
- 2. Thammavongsa V., Kim H. K., Missiakas D., and Schneewind O. (2015) Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 13, 529–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chambers H. F., and Deleo F. R. (2009) Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kluytmans J., van Belkum A., and Verbrugh H. (1997) Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 10, 505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kalia D., Merey G., Nakayama S., Zheng Y., Zhou J., Luo Y., Guo M., Roembke B. T., and Sintim H. O. (2013) Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 42, 305–341 [DOI] [PubMed] [Google Scholar]
- 6. Hauryliuk V., Atkinson G. C., Murakami K. S., Tenson T., and Gerdes K. (2015) Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 13, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corrigan R. M., Abbott J. C., Burhenne H., Kaever V., and Gründling A. (2011) c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7, e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Witte G., Hartung S., Büttner K., and Hopfner K. P. (2008) Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30, 167–178 [DOI] [PubMed] [Google Scholar]
- 9. Römling U. (2008) Great times for small molecules: c-di-AMP, a second messenger candidate in bacteria and Archaea. Sci. Signal. 1, pe39. [DOI] [PubMed] [Google Scholar]
- 10. Woodward J. J., Iavarone A. T., and Portnoy D. A. (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corrigan R. M., and Gründling A. (2013) Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 11, 513–524 [DOI] [PubMed] [Google Scholar]
- 12. Bai Y., Yang J., Zhou X., Ding X., Eisele L. E., and Bai G. (2012) Mycobacterium tuberculosis Rv3586 (DacA) is a diadenylate cyclase that converts ATP or ADP into c-di-AMP. PLoS ONE 7, e35206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barker J. R., Koestler B. J., Carpenter V. K., Burdette D. L., Waters C. M., Vance R. E., and Valdivia R. H. (2013) STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio 4, e00018–00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye M., Zhang J. J., Fang X., Lawlis G. B., Troxell B., Zhou Y., Gomelsky M., Lou Y., and Yang X. F. (2014) DhhP, a cyclic di-AMP phosphodiesterase of Borrelia burgdorferi, is essential for cell growth and virulence. Infect. Immun. 82, 1840–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rao F., See R. Y., Zhang D., Toh D. C., Ji Q., and Liang Z. X. (2010) YybT is a signaling protein that contains a cyclic dinucleotide phosphodiesterase domain and a GGDEF domain with ATPase activity. J. Biol. Chem. 285, 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Commichau F. M., Dickmanns A., Gundlach J., Ficner R., and Stülke J. (2015) A jack of all trades: the multiple roles of the unique essential second messenger cyclic di-AMP. Mol. Microbiol. 97, 189–204 [DOI] [PubMed] [Google Scholar]
- 17. Huynh T. N., and Woodward J. J. (2016) Too much of a good thing: regulated depletion of c-di-AMP in the bacterial cytoplasm. Curr. Opin. Microbiol. 30, 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glanzmann P., Gustafson J., Komatsuzawa H., Ohta K., and Berger-Bächi B. (1999) glmM operon and methicillin-resistant glmM suppressor mutants in Staphylococcus aureus. Antimicrob. Agents Chemother. 43, 240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jolly L., Wu S., van Heijenoort J., de Lencastre H., Mengin-Lecreulx D., and Tomasz A. (1997) The femR315 gene from Staphylococcus aureus, the interruption of which results in reduced methicillin resistance, encodes a phosphoglucosamine mutase. J. Bacteriol. 179, 5321–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mengin-Lecreulx D., and van Heijenoort J. (1996) Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J. Biol. Chem. 271, 32–39 [DOI] [PubMed] [Google Scholar]
- 21. Mehne F. M., Gunka K., Eilers H., Herzberg C., Kaever V., and Stülke J. (2013) Cyclic di-AMP homeostasis in Bacillus subtilis: both lack and high level accumulation of the nucleotide are detrimental for cell growth. J. Biol. Chem. 288, 2004–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gundlach J., Mehne F. M., Herzberg C., Kampf J., Valerius O., Kaever V., and Stülke J. (2015) An essential poison: synthesis and degradation of cyclic di-AMP in Bacillus subtilis. J. Bacteriol. 197, 3265–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rismondo J., Gibhardt J., Rosenberg J., Kaever V., Halbedel S., and Commichau F. M. (2015) Phenotypes associated with the essential diadenylate cyclase CdaA and its potential regulator CdaR in the human pathogen Listeria monocytogenes. J. Bacteriol. 198, 416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bai Y., Yang J., Eisele L. E., Underwood A. J., Koestler B. J., Waters C. M., Metzger D. W., and Bai G. (2013) Two DHH subfamily 1 proteins in Streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J. Bacteriol. 195, 5123–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Witte C. E., Whiteley A. T., Burke T. P., Sauer J. D., Portnoy D. A., and Woodward J. J. (2013) Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. MBio 4, e00282–00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dengler V., McCallum N., Kiefer P., Christen P., Patrignani A., Vorholt J. A., Berger-Bächi B., and Senn M. M. (2013) Mutation in the C-di-AMP cyclase dacA affects fitness and resistance of methicillin resistant Staphylococcus aureus. PLoS ONE 8, e73512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Griffiths J. M., and O'Neill A. J. (2012) Loss of function of the gdpP protein leads to joint beta-lactam/glycopeptide tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 579–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pozzi C., Waters E. M., Rudkin J. K., Schaeffer C. R., Lohan A. J., Tong P., Loftus B. J., Pier G. B., Fey P. D., Massey R. C., and O'Gara J. P. (2012) Methicillin resistance alters the biofilm phenotype and attenuates virulence in Staphylococcus aureus device-associated infections. PLoS Pathog. 8, e1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuipers K., Gallay C., Martínek V., Rohde M., Martínková M., van der Beek S. L., Jong W. S., Venselaar H., Zomer A., Bootsma H., Veening J. W., and de Jonge M. I. (2016) Highly conserved nucleotide phosphatase essential for membrane lipid homeostasis in Streptococcus pneumoniae. Mol. Microbiol. 101, 12–26 [DOI] [PubMed] [Google Scholar]
- 30. Manikandan K., Sabareesh V., Singh N., Saigal K., Mechold U., and Sinha K. M. (2014) Two-step synthesis and hydrolysis of cyclic di-AMP in Mycobacterium tuberculosis. PLoS ONE 9, e86096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang Q., Luo Y., Zheng C., Yin K., Ali M. K., Li X., and He J. (2015) Functional Analysis of a c-di-AMP-specific Phosphodiesterase MsPDE from Mycobacterium smegmatis. Int. J. Biol. Sci. 11, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Q., Wang F., Liu S., Zhu D., Cong H., Gao F., Li B., Wang H., Lin Z., Liao J., and Gu L. (2016) Structural and biochemical insight into the mechanism of Rv2837c from Mycobacterium tuberculosis as a c-di-NMP phosphodiesterase. J. Biol. Chem. 291, 14386–14387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang J., Bai Y., Zhang Y., Gabrielle V. D., Jin L., and Bai G. (2014) Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol. Microbiol. 93, 65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huynh T. N., Luo S., Pensinger D., Sauer J. D., Tong L., and Woodward J. J. (2015) An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc. Natl. Acad. Sci. U.S.A. 112, E747–E756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corrigan R. M., Bowman L., Willis A. R., Kaever V., and Gründling A. (2015) Cross-talk between two nucleotide-signaling pathways in in Staphylococcus aureus. J. Biol. Chem. 290, 5826–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cohen D., Mechold U., Nevenzal H., Yarmiyhu Y., Randall T. E., Bay D. C., Rich J. D., Parsek M. R., Kaever V., Harrison J. J., and Banin E. (2015) Oligoribonuclease is a central feature of cyclic diguanylate signaling in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 112, 11359–11364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orr M. W., Donaldson G. P., Severin G. B., Wang J., Sintim H. O., Waters C. M., and Lee V. T. (2015) Oligoribonuclease is the primary degradative enzyme for pGpG in Pseudomonas aeruginosa that is required for cyclic-di-GMP turnover. Proc. Natl. Acad. Sci. U.S.A. 112, E5048–E5057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rallu F., Gruss A., Ehrlich S. D., and Maguin E. (2000) Acid- and multistress-resistant mutants of Lactococcus lactis: identification of intracellular stress signals. Mol. Microbiol. 35, 517–528 [DOI] [PubMed] [Google Scholar]
- 39. Postic G., Danchin A., and Mechold U. (2012) Characterization of NrnA homologs from Mycobacterium tuberculosis and Mycoplasma pneumoniae. RNA 18, 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan K. X., Wirtz M., Phua S. Y., Estavillo G. M., and Pogson B. J. (2013) Balancing metabolites in drought: the sulfur assimilation conundrum. Trends Plant Sci. 18, 18–29 [DOI] [PubMed] [Google Scholar]
- 41. Mechold U., Fang G., Ngo S., Ogryzko V., and Danchin A. (2007) YtqI from Bacillus subtilis has both oligoribonuclease and pAp-phosphatase activity. Nucleic Acids Res. 35, 4552–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nickels B. E., and Dove S. L. (2011) NanoRNAs: a class of small RNAs that can prime transcription initiation in bacteria. J. Mol. Biol. 412, 772–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldman S. R., Sharp J. S., Vvedenskaya I. O., Livny J., Dove S. L., and Nickels B. E. (2011) NanoRNAs prime transcription initiation in vivo. Mol. Cell 42, 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whiteley A. T., Pollock A. J., and Portnoy D. A. (2015) The PAMP c-di-AMP is essential for Listeria monocytogenes growth in rich but not minimal media due to a toxic increase in (p)ppGpp. Cell Host Microbe 17, 788–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weinrick B., Dunman P. M., McAleese F., Murphy E., Projan S. J., Fang Y., and Novick R. P. (2004) Effect of mild acid on gene expression in in Staphylococcus aureus. J. Bacteriol. 186, 8407–8423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cotter P. D., and Hill C. (2003) Surviving the acid test: responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 67, 429–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kitko R. D., Wilks J. C., Garduque G. M., and Slonczewski J. L. (2010) Osmolytes contribute to pH homeostasis of Escherichia coli. PLoS ONE 5, e10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bore E., Langsrud S., Langsrud Ø., Rode T. M., and Holck A. (2007) Acid-shock responses in Staphylococcus aureus investigated by global gene expression analysis. Microbiology 153, 2289–2303 [DOI] [PubMed] [Google Scholar]
- 49. Moscoso J. A., Schramke H., Zhang Y., Tosi T., Dehbi A., Jung K., and Gründling A. (2015) Binding of cyclic di-AMP to the Staphylococcus aureus sensor kinase KdpD occurs via the universal stress protein domain and downregulates the expression of the Kdp potassium transporter. J. Bacteriol. 198, 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huynh T. N., Choi P. H., Sureka K., Ledvina H. E., Campillo J., Tong L., and Woodward J. J. (2016) Cyclic di-AMP targets the cystathionine beta-synthase domain of the osmolyte transporter OpuC. Mol. Microbiol. 102, 233–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schuster C. F., Bellows L. E., Tosi T., Campeotto I., Corrigan R. M., Freemont P., and Gründling A. (2016) The second messenger c-di-AMP inhibits the osmolyte uptake system OpuC in in Staphylococcus aureus. Sci. Signal. 9, ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leskelä S., Wahlström E., Hyyryläinen H. L., Jacobs M., Palva A., Sarvas M., and Kontinen V. P. (1999) Ecs, an ABC transporter of Bacillus subtilis: dual signal transduction functions affecting expression of secreted proteins as well as their secretion. Mol. Microbiol. 31, 533–543 [DOI] [PubMed] [Google Scholar]
- 53. Heinrich J., Lundén T., Kontinen V. P., and Wiegert T. (2008) The Bacillus subtilis ABC transporter EcsAB influences intramembrane proteolysis through RasP. Microbiology 154, 1989–1997 [DOI] [PubMed] [Google Scholar]
- 54. Moses S., Sinner T., Zaprasis A., Stöveken N., Hoffmann T., Belitsky B. R., Sonenshein A. L., and Bremer E. (2012) Proline utilization by Bacillus subtilis: uptake and catabolism. J. Bacteriol. 194, 745–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pilonieta M. C., Nagy T. A., Jorgensen D. R., and Detweiler C. S. (2012) A glycine betaine importer limits Salmonella stress resistance and tissue colonization by reducing trehalose production. Mol. Microbiol. 84, 296–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bai Y., Yang J., Zarrella T. M., Zhang Y., Metzger D. W., and Bai G. (2014) Cyclic di-AMP impairs potassium uptake mediated by a cyclic di-AMP binding protein in Streptococcus pneumoniae. J. Bacteriol. 196, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Charpentier E., Anton A. I., Barry P., Alfonso B., Fang Y., and Novick R. P. (2004) Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 70, 6076–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Monk I. R., Shah I. M., Xu M., Tan M. W., and Foster T. J. (2012) Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio 3, e00277–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mazmanian S. K., Liu G., Jensen E. R., Lenoy E., and Schneewind O. (2000) Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U.S.A. 97, 5510–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Burhenne H., and Kaever V. (2013) Quantification of cyclic dinucleotides by reversed-phase LC-MS/MS. Methods Mol. Biol. 1016, 27–37 [DOI] [PubMed] [Google Scholar]
- 61. Underwood A. J., Zhang Y., Metzger D. W., and Bai G. (2014) Detection of cyclic di-AMP using a competitive ELISA with a unique pneumococcal cyclic di-AMP binding protein. J. Microbiol. Methods 107, 58–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Monk I. R., Tree J. J., Howden B. P., Stinear T. P., and Foster T. J. (2015) Complete bypass of restriction systems for major Staphylococcus aureus lineages. MBio 6, e00308–00315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee C. Y., Buranen S. L., and Ye Z. H. (1991) Construction of single-copy integration vectors for in Staphylococcus aureus. Gene 103, 101–105 [DOI] [PubMed] [Google Scholar]
- 64. Gründling A., and Schneewind O. (2007) Genes required for glycolipid synthesis and lipoteichoic acid anchoring in in Staphylococcus aureus. J. Bacteriol. 189, 2521–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Campeotto I., Zhang Y., Mladenov M. G., Freemont P. S., and Gründling A. (2015) Complex structure and biochemical characterization of the Staphylococcus aureus cyclic diadenylate monophosphate (c-di-AMP)-binding protein PstA, the founding member of a new signal transduction protein family. J. Biol. Chem. 290, 2888–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kreiswirth B. N., Löfdahl S., Betley M. J., O'Reilly M., Schlievert P. M., Bergdoll M. S., and Novick R. P. (1983) The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712 [DOI] [PubMed] [Google Scholar]
- 67. Boles B. R., Thoendel M., Roth A. J., and Horswill A. R. (2010) Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS ONE 5, e10146. [DOI] [PMC free article] [PubMed] [Google Scholar]