

Abstract
GIV (aka Girdin) is a guanine nucleotide exchange factor that activates heterotrimeric G protein signaling downstream of RTKs and integrins, thereby serving as a platform for signaling cascade cross-talk. GIV is recruited to the cytoplasmic tail of receptors upon stimulation, but the mechanism of activation of its G protein regulatory function is not well understood. Here we used assays in humanized yeast models and G protein activity biosensors in mammalian cells to investigate the role of GIV subcellular compartmentalization in regulating its ability to promote G protein signaling. We found that in unstimulated cells GIV does not co-fractionate with its substrate G protein Gαi3 on cell membranes and that constitutive membrane anchoring of GIV in yeast cells or rapid membrane translocation in mammalian cells via chemically induced dimerization leads to robust G protein activation. We show that membrane recruitment of the GIV “Gα binding and activating” motif alone is sufficient for G protein activation and that it does not require phosphomodification. Furthermore, we engineered a synthetic protein to show that recruitment of the GIV “Gα binding and activating” motif to membranes via association with active RTKs, instead of via chemically induced dimerization, is also sufficient for G protein activation. These results reveal that recruitment of GIV to membranes in close proximity to its substrate G protein is a major mechanism responsible for the activation of its G protein regulatory function.
Keywords: bioluminescence resonance energy transfer (BRET), G protein-coupled receptor (GPCR), guanine nucleotide exchange factor (GEF), heterotrimeric G protein, receptor tyrosine kinase
Introduction
Heterotrimeric G proteins regulate virtually all physiological functions in humans, and their dysregulation is the cause of many diseases (1, 2). They cycle between inactive (GDP-bound) and active (GTP-bound) states to control the flow of information from extracellular cues to intracellular effectors (1, 3). Under resting conditions the nucleotide-binding α subunit (Gα) is loaded with GDP and forms a complex with Gβγ. According to the classic paradigm, heterotrimeric G protein activation in response to extracellular stimuli is carried out via G protein-coupled receptors (GPCRs)2 located at the plasma membrane. GPCRs are guanine nucleotide exchange factors (GEFs) that promote the exchange of GDP for GTP on Gαβγ (1, 3). The consequence of this is the dissociation of the Gαβγ heterotrimer into Gα-GTP and free Gβγ, which are the two active molecular species that signal to downstream effectors.
Despite the well established mechanism of heterotrimeric G protein activation by GPCRs, there is mounting evidence indicating that heterotrimeric G proteins also mediate signaling via other types of receptors. This is documented best for receptor tyrosine kinases (RTKs), for which the role of heterotrimeric G protein signaling is supported by experiments in isolated cells, as well as transgenic and knock-out mice (4–10). However, the molecular mechanism of G protein activation by RTKs to support this functional evidence has remained rather obscure and controversial because neither RTKs nor their best characterized adaptors have GEF activity toward heterotrimeric G proteins. A plausible explanation for this quandary is provided by the existence of non-receptor GEFs (11–15), which could be activated by RTKs to relay signals to downstream effectors via heterotrimeric G proteins. Recent work indicates that GIV (Gα-interacting, vesicle-associated protein, also known as Girdin) fulfills such role (reviewed in Ref. 16). GIV contains a ∼30–35-amino acid sequence, namely the Gα binding and activating (GBA) motif, conserved across functionally and evolutionarily divergent non-receptor GEF proteins (17–21). Among Gα subunits, GIV binds predominantly to Gαi (equally to all three members of mammals: Gαi1, Gαi2, and Gαi3), although it also interacts weakly with Gαo and Gαs (17, 22–24). The GIV GBA motif activates Gαi proteins in vitro via acceleration of nucleotide exchange (17, 23), and it also promotes G protein activation in cells, as determined by readouts for either Gαi-GTP (e.g. conformation-specific antibodies, cAMP dampening) (25–27) or free Gβγ (e.g. PI3K-Akt signaling, resonance energy transfer-based biosensors) (17, 26, 28). Importantly, GIV-mediated G protein activation operates downstream of receptor types different from GPCRs, like RTKs and integrins (16, 25, 26, 29–31) and is crucial for the role of GIV in cancer progression (32–38) by promoting tumor cell migration, actin remodeling, and activation of the oncogenic PI3K-Akt pathway (10, 39, 40). Additional evidence indicates that the GIV GBA motif is also important in a wide range of cellular processes (e.g. autophagy, protein trafficking, or epithelial junction integrity) (22, 41–43) and diseases (e.g. liver fibrosis, diabetes, or kidney failure) (27, 30, 44, 45).
A major gap in the understanding of GIV is the lack of clear mechanistic insights into how its G protein regulatory function becomes activated. Because GIV is not a transmembrane protein and its role is restricted to ligands that do not cross the plasma membrane, its activation must be regulated via receptor-mediated activation. The molecular events linked to GIV-mediated G protein activation are characterized best in the context of RTKs. GIV directly binds to the phosphorylated tail of RTKs (25, 26), and this is required for different signaling functions of GIV, e.g. G protein-dependent (25, 26) and tyrosine phosphorylation-dependent signaling (46). In addition, stimulation of EGFR, a prototypical RTK, has been recently shown to lead to the phosphorylation of GIV at serine 1674, a residue adjacent to its GBA motif, which enhances the GEF activity (47). However, this enhancement is unlikely to fully account for all GIV-mediated G protein activation because it is modest (∼1.5-fold enhancement of GEF activity in vitro) and dependent on a particular kinase (cyclin-dependent kinase 5), which might not be relevant for all RTKs or other receptor types, like integrins, that rely on GIV for signaling. Here we set out to elucidate the mechanism responsible for activating the G protein regulatory function of GIV in a broader context. We reasoned that relocalization of GIV to membranes would be sufficient to trigger G protein activation based on several observations: (i) GIV is recruited to the plasma membrane in response to different stimuli (10, 16, 29, 34, 40), (ii) the G protein substrate (Gαi) is constitutively attached to membranes (48), and (iii) GIV without modifications (i.e. bacterially expressed) possesses intrinsic GEF activity (17, 22, 23). By using synthetic biology approaches and G protein activity biosensors, here we provide evidence that the spatial relocalization of GIV to membranes is a mayor mechanism responsible for the activation of its G protein regulatory function.
Results
GIV Does Not Co-fractionate with Gαi3 on Cell Membranes
GIV has been shown to localize in different subcellular compartments under different conditions (10, 22, 24, 42, 49) and, more specifically, to become enriched at the plasma membrane in response to different stimuli (10, 16, 29, 34, 40). Acute ligand stimulation also induces the co-localization of GIV with Gαi3 at the plasma membrane (10, 29). However, the overall distribution of GIV in non-stimulated cells and how it relates to that of Gαi subunits has not been thoroughly investigated. For this, we performed biochemical fractionation of mammalian cells under standard culture conditions. We focused on Gαi3 for this and for subsequent parts of this study because the vast majority of prior biochemical and cell biological work in relation to GIV has been carried out with Gαi3. However, all three Gαi subunits of mammals (Gαi1, Gαi2, and Gαi3) share identical membrane localization signals and bind equally to GIV (17, 24), suggesting that observations with Gαi3 in this context are very likely to apply to Gαi subunits in general. In HEK293T, endogenous Gαi3 is almost exclusively found in the P100 fraction (Fig. 1A), which is consistent with its well documented constitutive association with membranes (48). In contrast, endogenous GIV shows the opposite distribution, localizing predominantly (∼90–95%) in the cytosolic S100 fraction (Fig. 1A). Because the P100 is a crude particulate fraction containing not only cell membranes, we separated it into detergent-soluble (TX-S) and detergent-insoluble (TX-I) fractions to further characterize the distribution of Gαi3 and GIV. As expected for a membrane protein, Gαi3 was efficiently extracted by detergents from the P100 into the TX-S fraction (Fig. 1A). In contrast, all GIV present in the P100 fraction remained in the TX-I fraction (Fig. 1A), which is consistent with a previously described pool of GIV that associates with the actin cytoskeleton (40). Equivalent fractionation experiments looking at endogenous GIV and Gαi3 were carried out in two additional cell lines: HeLa (Fig. 1B) and MDA-MB-231 (Fig. 1C). The results were similar to those obtained in HEK293T cells in that a large pool of GIV was cytosolic (S100: ∼75% in HeLa and ∼50% in MDA-MB-231), and the remaining pool of GIV in the P100 was detergent-insoluble (Fig. 1, B and C). Gαi3 distribution was almost identical in the three cell lines. These results indicate that, regardless of the relative partition of GIV between the cytosolic and detergent-insoluble fractions in the three different cell lines, GIV is excluded from the detergent-soluble P100 fraction where Gαi3 is predominantly found. Similar results were also obtained with the three cells lines using a buffer of different composition,3 indicating that our observations are consistent across different experimental conditions.
FIGURE 1.

GIV does not fractionate with Gαi3 in cell membranes. A–C, HEK293T (A), HeLa (B), or MDA-MB-231 (C) cells were homogenized in the absence of detergents, and the PNS was separated into supernatant (S100) and pellet (P100) after 100,000 × g centrifugation. P100 fractions were resuspended in buffer containing Triton X-100 and centrifuged to obtain the detergent soluble (TX-S) and insoluble (TX-I) fractions. Immunoblots (IB) of equal aliquots of each fraction from one experiment representative of three is shown. TfR and tubulin were used as membrane and cytosol markers, respectively. D, detergent lysates of MDA-MB-231 cells were centrifuged in a discontinuous sucrose gradient (5-35-45%) to separate low density detergent-resistant membranes (“lipid rafts”) from other cell fractions. Equal gradient fractions were immunoblotted with the indicated antibodies. Caveolin-1 was used as lipid raft marker, whereas β1-integrin and tubulin were membrane and cytosolic markers, respectively, of non-lipid raft fractions. One experiment representative of three is shown.
A possible interpretation of the TX-I fraction of GIV is that it corresponds to a pool that localizes in detergent-resistant domains of cell membranes, the so-called lipid rafts (50). If GIV localizes in lipid rafts, it could potentially interact with Gαi3 because it has been previously described that Gαi proteins partially localize in these membrane microdomains (51, 52). To investigate this possibility, we fractionated MDA-MB-231, the cell line with the largest proportion of GIV in the TX-I fraction (Fig. 1C), using a protocol that separates detergent-insoluble lipid rafts from other cell fractions by ultracentrifugation in a sucrose density gradient. Consistent with previous reports, some Gαi3 was found in low density, detergent-insoluble fractions enriched in the lipid raft marker caveolin-1 (Fig. 1D). However, GIV was exclusively detected in the high density fractions along with other non-lipid raft markers (Fig. 1D), ruling out the possibility that the TX-I fraction of GIV corresponds to a pool localized in lipid rafts. This supports the alternative interpretation that GIV present in the TX-I fraction corresponds to a previously described pool of the protein that associates with the actin cytoskeleton (40). Taken together, these results indicate that in the absence of an acute stimulus GIV is not localized in the cell membranes where Gαi3 resides.
Constitutive Membrane Targeting of GIV Enhances Gi3 Signaling in Yeast
The results presented above show that in unstimulated cells GIV is spatially segregated from its substrate G protein Gαi3 that is located in cell membranes. As a first approach to investigate the impact of targeting GIV to cell membranes on G protein activation, we used a yeast-based system with a modified mating pheromone pathway (53) (Fig. 2A). Briefly, the genetically engineered yeast strain used in this system lacks pheromone-responsive GPCRs, and its endogenous yeast Gα protein Gpa1 is replaced by mammalian Gαi3. Thus, only an exogenous G protein activator can trigger a signaling pathway that is normally activated as a mating pheromone response, which leads to an increase in Fus3 phosphorylation and to transcriptional activation of the Fus1 promoter. For these experiments we used a construct consisting of the C-terminal region of GIV (aa 1660–1870). This fragment expresses well in yeast, and it has been previously shown (17, 23) to bind and activate Gαi3 as efficiently as larger fragments of GIV. Moreover, a thorough characterization of GIV (aa 1660–1870) in two studies (26, 28) has validated that it recapitulates the main biological functions of the full-length protein, including the regulation of receptor-mediated signaling via heterotrimeric G proteins. This domain of GIV lacks any known membrane targeting sequence but contains the GBA motif necessary for G protein activation. We designed a modified version of this construct by fusing it to the first 9 amino acids of Gpa1 (N9Gpa1), which are sufficient to target proteins to cell membranes in yeast (54). Unmodified GIV and N9Gpa1-GIV were expressed at similar levels, and as expected, GIV was found almost exclusively in the cytosolic soluble (S) fraction, whereas N9Gpa1-GIV was predominantly recovered in the particulate insoluble (P) fraction (Fig. 2B), validating that the N9Gpa1 sequence efficiently targets GIV to membranes. Expression of membrane-anchored N9Gpa1-GIV led to an increase in G protein activation much larger (∼25-fold) than that observed when expressing cytosolic GIV, as determined by the β-galactosidase activity of a LacZ reporter under the control of the Fus1 promoter (Fig. 2C). Membrane-anchored GIV was also more potent than cytosolic GIV in two other independent assays that reflect G protein signaling in this system, i.e. Fus3 phosphorylation and cell growth in the absence of histidine (Fig. 2D). None of these G protein-dependent responses induced by cytosolic or membrane-anchored GIV were reproduced by constructs bearing a mutation in the GBA motif (F1685A, FA) that disrupts GEF activity (17, 22) (Fig. 2, C and D). The FA mutation does not affect the subcellular fractionation of GIV or N9Gpa1-GIV (Fig. 2B). These findings indicate that association of GIV with membranes is sufficient to enhance G protein activation via its GBA motif.
FIGURE 2.

Membrane association of GIV enhances its ability to activate G protein signaling in yeast. A, schematic diagram of the yeast-based assays used to monitor G protein signaling. A genetically engineered strain lacking endogenous GPCRs and with human Gαi3 replacing the endogenous Gα Gpa1 was used to determine the levels of G protein activation upon expression of exogenous GIV. G protein activation was determined by immunoblotting (IB) for Fus3 phosphorylation or two gene reporters controlled by the Fus1 promoter (β-galactosidase activity or histidine auxotrophy). B, yeast cells expressing unmodified GIV (aa 1660–1870) or fused to a membrane-targeting sequence corresponding to the first 9 aa of Gpa1 (N9Gpa1-GIV) were homogenized in the absence of detergents, and the PNS was separated into supernatant (S) and pellet (P) fractions after 100,000 × g centrifugation. Immunoblots of equal aliquots of each fraction from one experiment representative of four are shown. C, membrane-targeted N9Gpa1-GIV enhances G protein-dependent β-galactosidase activity ∼25-fold more than cytosolic GIV. The results are the averages of n = 4, and error bars are the S.E. D, membrane-targeted N9Gpa1-GIV induces more Fus3 phosphorylation (ppERK/ppFus3; top, immunoblot panels) and more rapid cell growth in media lacking histidine (bottom, spot growth panels) than cytosolic GIV. One experiment representative of four is shown.
Acute Translocation of GIV to Membranes Induces Rapid G Protein Activation in Mammalian Cells
A limitation of the experiments described above is that they reflect the long term consequences of keeping GIV constitutively anchored to membranes, which does not directly relate to the rapid kinetics (seconds to minutes) of the signaling events regulated by GIV (10, 16, 26, 29, 31). To regulate the spatial localization of GIV and mimic its rapid recruitment to cell membranes in response to receptor stimulation (16, 29), we used a chemically induced dimerization (CID) system in mammalian cells (Fig. 3A) (55, 56). Briefly, this system consists of two domains (FRB and FKBP) that dimerize in the presence of rapamycin. FRB was fused to the first 11 aa of Lyn (Lyn11-FRB), which targets it to the plasma membrane (56), whereas FKBP was fused to C-terminal region of GIV (aa 1660–1870, FKBP-GIV). We chose to work with GIV (aa 1660–1870) because (i) it recapitulates the signaling functions of full-length GIV in mammalian cells, including receptor-mediated signaling via its GBA motif (26, 28), (ii) it has been validated that bulky tags (i.e. GFP) can be tagged to its N terminus without compromising function in mammalian cells (26), (iii) it binds and activates G proteins as efficiently as larger fragments (17, 23), and (iv) it provides continuity with the previous yeast experiments. Because FKBP-GIV lacks any membrane targeting sequence, FRB/FKBP dimerization by rapamycin induces the translocation of FKBP-GIV to the location of Lyn11-FRB, i.e. the plasma membrane (Fig. 3A). To monitor the effect of this translocation on G protein activation, we used a bioluminescence resonance energy transfer (BRET)-based assay (57, 58) that measures G protein activation more directly than the yeast-based assays above, which rely on measuring the activity of downstream effectors/reporters. In this BRET system, dissociation of G protein Gαβγ heterotrimers upon activation leads to the association of free Venus-Gβγ (BRET acceptor) with the C-terminal domain of its effector GRK3 fused to an enhanced luciferase (Nanoluciferase, BRET donor), which leads to an increase in BRET (Fig. 3A). Expression of FKBP-GIV WT did not cause an increase in BRET in the absence of rapamycin compared with control cells expressing the GEF-deficient FA mutant (Fig. 3B). However, addition of rapamycin induced an increase in BRET in cells expressing FKBP-GIV WT but not FKBP-GIV FA (Fig. 3B), indicating that translocation of GIV to cell membranes induces G protein activation via its GBA motif. The amplitude of G protein activation was dependent on the dose of FKBP-GIV (Fig. 3, C–E) and the kinetics were comparable with those previously described (56) for rapamycin-induced FRB/FKBP membrane recruitment (Fig. 3C), suggesting that G protein activation occurs rapidly upon CID-mediated translocation of GIV to cell membranes.
FIGURE 3.

Acute translocation of GIV to membranes induces rapid G protein activation in mammalian cells. A, translocation of GIV to membranes is controlled by CID using the FKBP-rapamycin-FRB system. FKBP-fused GIV is recruited to membranes upon rapamycin-induced binding of FKBP to the FRB domain that is fused to a membrane-targeting sequence (Lyn11). G protein activation is determined by BRET. Dissociation of Gαi3:Gβγ trimers upon G protein activation leads to the release of Venus-tagged Gβγ (V-Gβγ), which binds to the C-terminal domain of GRK3 fused to nanoluciferase (GRK3-Nluc) and causes an increase of BRET signal. B, rapamycin-induced translocation of FKBP-GIV (aa 1660–1870, 0.25 μg of plasmid DNA) WT but not the GEF-deficient F1685A (FA, 1 μg of plasmid DNA) mutant leads to G protein-dependent BRET. HEK293T cells expressing all the required assay components were treated with rapamycin (0.5 μm, Rapa) or ethanol (0.005%, Ctrl) for 2 min before BRET measurements. The results are presented as the increases in BRET (ΔBRET) compared with cells not expressing GIV (average of n = 3). The error bars are the S.E. *, p < 0.05 using Student's t test. C–E, dose-dependent G protein activation induced by membrane recruitment of GIV. Representative traces of kinetic BRET measurements of cells transfected with the indicated amounts of FKBP-GIV plasmids and stimulated with rapamycin are shown in C. Quantification of the average increase in BRET (ΔBRET) after 2 min of stimulation with rapamycin (0.5 μm, Rapa) compared with ethanol (0.05%, Ctrl) from five independent experiments is shown in D. The error bars are the S.E. *, p < 0.05; **, p < 0.01 using Student's t test. Representative immunoblots of the cells used in these experiments are shown in E. F–H, membrane targeting of FRB is required FKBP-GIV-mediated G protein activation upon rapamycin stimulation. Mutation of the myristoylation site in the Lyn11 sequence (Lyn11G2A) is predicted to preclude rapamycin-induced translocation of FKBP-GIV and subsequent G protein activation (F). Kinetic BRET measurements of cells transfected with the indicated Lyn11-FRB and FKBP-GIV (2 μg) constructs and stimulated with rapamycin at the indicated time (arrow) are shown in G, and representative immunoblots of the cells used in these experiments are shown in H. The results are the average of three independent experiments, and the error bars are the S.E. (shown only at 5-s intervals for clarity).
The time scale and dependence on the GIV GBA motif suggest that the increase in G protein activation is a consequence of the action of rapamycin on Lyn11-FRB/GIV-FKBP dimerization rather than on other cellular targets (e.g. mTOR). To confirm this, we carried out control experiments in which Lyn11-FRB was mutated to prevent its association with membranes (59). More specifically, we created a mutant (Lyn11G2A-FRB) disrupting the lipidation site of Lyn required for its association with membranes (Fig. 3F). Although cells expressing Lyn11-FRB and FKBP-GIV led to a robust increase in BRET upon rapamycin stimulation, cells expressing the same amount of Lyn11G2A-FRB mutant and FKBP-GIV failed to show any measurable response (Fig. 3, G and H). We conclude that translocation of GIV to membranes is sufficient to promote rapid G protein activation in mammalian cells and that it requires an intact GBA motif.
GIV Translocation and GPCR Stimulation Induce G Protein Responses of Similar Magnitude
Next, we benchmarked the G protein response triggered by GIV upon membrane translocation by comparing it with the response observed after GPCR stimulation. For this, we used the M4 muscarinic receptor (M4R), a prototypical Gi-coupled receptor, stimulated with saturating concentrations of its synthetic ligand carbachol. We found that the maximal amplitude of the GIV-mediated BRET response within the first minutes of stimulation was of similar magnitude to that elicited by the M4R (Fig. 4, A and B). Similar results were obtained with two other GPCRs, i.e. adenosine A1 receptor (A1R) and endothelin ETA receptor (ETAR), stimulated with physiological concentrations of their natural ligands (Fig. 4, C and D). These results indicate that translocation of GIV to cell membranes via synthetic CID triggers a G protein response similar to the natural G protein activation induced by ligand stimulation of GPCRs.
FIGURE 4.

The amplitude and kinetics of G protein activation induced by the recruitment of GIV to membranes is comparable with GPCR-mediated activation. HEK293T cells were transfected with plasmids for all the components required for CID (1 μg FKBP-GIV WT/FA) and for BRET-based G protein activity measurements as described in Fig. 3 and different GPCRs (M4R in A and B and the A1R and ETAR in C and D). The cells were stimulated with rapamycin (0.5 μm), carbachol (100 μm), adenosine (0.1 μm), or ET-1 (3 μm) at the indicated times (arrows). BRET results (A and C) are the average of three to five independent experiments (baseline corrected by subtraction of the BRET values before stimulation), and the error bars are the S.E. (shown only at 5-s intervals for clarity). Representative immunoblots of the cells used in these experiments are shown in B and D.
The GBA Motif of GIV Is Sufficient to Promote G Protein Signaling upon Membrane Translocation
Our results with the F1685A mutant indicate that the GBA motif of GIV is required for the observed G protein signaling responses. However, the C-terminal region of GIV used in these experiments (i.e. amino acids 1660–1870) contains additional domains and mediates other interactions with important signaling functions. In particular, this region contains an SH2-like domain that associates with active RTKs (25, 26) and tyrosine phosphorylation sites (46); two signaling events have been shown to work coordinately with the GBA motif to mediate certain downstream responses (25, 31, 46). Thus, translocation of GIV to membranes may promote some of these signaling-related events, which in turn might influence how the GBA motif engages and modulates G proteins. We found that this is likely not the case because a truncated FKBP-fused GIV construct containing almost exclusively the GBA motif sequence (FKBP-GIV-GBA) induced a G protein activation response equivalent to that observed with the entire C-terminal region (aa 1660–1870) upon CID-mediated membrane translocation (Fig. 5). This result suggests that the GBA motif is not only necessary but also sufficient to activate G protein signaling when recruited to membranes.
FIGURE 5.
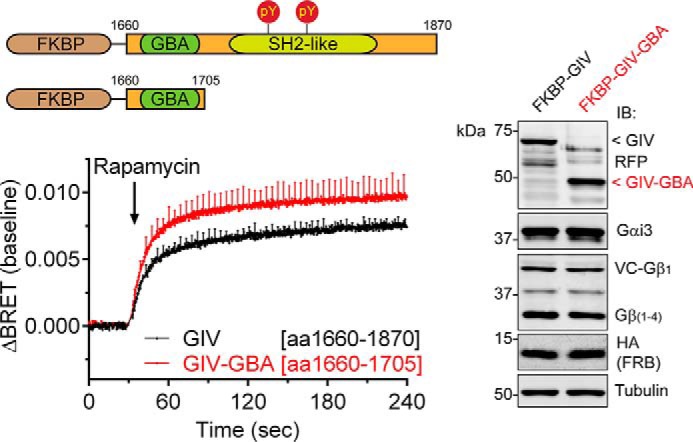
Membrane recruitment of the GBA motif of GIV is sufficient to activate G proteins. HEK293T cells were transfected with plasmids for all the components required for CID (0.5 μg of FKBP-GIV or 0.5 μg of FKBP-GIV-GBA (aa 1660–1705)) and BRET-based G protein activity measurements as described in Fig. 3. The cells were stimulated with rapamycin (0.5 μm) at the indicated time (arrow). BRET results (graph) are the averages of three independent experiments (baseline corrected), and the error bars are the S.E. (shown only at 5-s intervals for clarity). Representative immunoblots (IB) of the cells used in these experiments are shown on the right.
Membrane Recruitment of GIV Is More Potent than Phosphorylation at Ser1674 in Activating G Proteins
It has been previously reported that after receptor stimulation, GIV is phosphorylated at Ser1674, a residue adjacent to the GBA motif, and that a mutant that mimics this phosphorylation (S1674D) enhances G protein binding and activation in vitro, as well as some signaling outputs in cells (47). Because the experiments reported in the current manuscript were carried out under conditions in which GIV is presumably not phosphorylated (e.g. absence of serum or receptor stimulation), Ser1674 phosphorylation does not seem to be a requirement for G protein activation upon GIV recruitment to membranes. Next, we compared the impact of both mechanisms (Ser1674 phosphorylation and membrane recruitment) on GIV-mediated G protein activation and also investigated their possible cooperation. For this, we first performed experiments in the yeast-based system described in Fig. 2. When the phosphomimicking S1674D mutation was introduced in the cytosolic version of GIV, it induced larger G protein activation than GIV WT, as determined by the levels of Fus3 phosphorylation (Fig. 6A) and Fus1 promoter-dependent β-galactosidase activity (Fig. 6B). However, the enhancement mediated by S1674D was very modest compared with the effect of targeting GIV to membranes (N9Gpa1-GIV; Fig. 6, A and B); in the β-galactosidase reporter assay (Fig. 6B), N9Gpa1-GIV was ∼10 times more potent that GIV S1674D (∼25-fold versus ∼2.5-fold increase compared with GIV WT for N9Gpa1-GIV and GIV S1674D, respectively). Moreover, when the S1674D mutation was introduced in the membrane-anchored N9Gpa1-GIV construct, it did not cause any further increase in G protein activation in neither of the two assay readouts (Fig. 6, A and B). Although this suggests that the activation induced by GIV recruitment to membranes overcomes any possible modulatory effect of S1674D on G protein activation, it is also possible that such modulatory effect is beyond the sensitivity of these assays because of the very high responses induced by membrane-associated GIV.
FIGURE 6.

Membrane recruitment of GIV is a mechanism of G protein activation more potent than its phosphorylation at Ser1674. A and B, membrane targeting of GIV enhances G protein activation in yeast much more potently than the phosphomimicking S1674D mutation and S1674D fails to further enhance G protein activation induced by membrane-anchored GIV. G protein activation in yeast was determined by ppFus3 immunoblotting (A, IB) or β-galactosidase reporter activity (B) in the strain described for Fig. 2 expressing cytoplasmic (GIV) or membrane-anchored (N9Gpa1-GIV) versions of GIV. SD, phosphomimicking S1674D mutant. One experiment representative of four is shown for the immunoblotting results (A) and the average of n = 4–5 for the β-galactosidase results (B). C–E, expression of FKBP-GIV-GBA S1674D at the same concentration as WT (D) does not affect basal (C) or GIV-stimulated (E) G protein activity as determined by BRET in mammalian cells. HEK293T cells were transfected with plasmids for all the components required for CID (0.125 μg of FKBP-GIV-GBA; aa 1660–1705) and BRET-based G protein activity measurements as described for Fig. 3. BRET results (C and E) are the averages of three independent experiments, and the error bars are the S.E. The data corresponding to FKBP-GIV-GBA WT 0.5 μg from Fig. 5 is presented here for comparison. The bar graph in E corresponds to the ΔBRET values 2 min after rapamycin addition. Representative immunoblots of the cells used in these experiments are shown in D. The error bars represent the S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant using Student's t test.
To overcome this possible limitation and further compare the two mechanisms, we performed additional experiments via an alternative approach, i.e. the CID/BRET system described in Fig. 3. In this format, it is possible to titrate the amount of GIV to obtain submaximal G protein activation response and therefore be able to interrogate a possible modulatory effect of S1674D. Expression of equivalent amounts of FKBP-GIV-GBA WT or S1674D did not alter the basal BRET levels in the absence of rapamycin (Fig. 6, C and D), indicating that S1674D cannot enhance G protein signaling per se under conditions in which FKBP-GIV-GBA is not associated with membranes. As expected, rapamycin induced a rapid increase in BRET in cells expressing FKBP-GIV-GBA WT (Fig. 6E), indicative of G protein signaling activation upon membrane recruitment. When FKBP-GIV-GBA S1674D was tested in parallel, the BRET response was almost identical to WT (Fig. 6E). This cannot be attributed to a lack of sufficient dynamic range in the assay because FKBP-GIV-GBA WT and S1674D were tested at concentrations that led to approximately half of the maximal BRET response observed when larger amounts of FKBP-GIV-GBA WT are transfected (Fig. 6E). Also, the lack of difference between WT and S1674D cannot be attributed to constitutive phosphorylation of Ser1674 in FKBP-GIV-GBA WT because the experiments were performed under conditions (i.e. absence of serum or other growth factors) in which GIV phosphorylation at Ser1674 is nearly absent (60) and because even when GIV is phosphorylated at Ser1674, the stoichiometry is low (47). In summary, we conclude that membrane recruitment of GIV is a mechanism of G protein signaling activation more potent than phosphorylation at Ser1674 and that it masks the G protein modulatory effect of this phosphoevent.
EGFR-mediated Recruitment of the GBA Motif of GIV to Membranes Is Sufficient to Trigger G Protein Signaling
Our results so far indicate that translocation of the GIV GBA motif to membranes in the proximity of its target, Gαi3, is sufficient to activate G protein signaling. Next we asked whether membrane translocation in response to a physiological input, instead of via synthetic CID, is also sufficient for G protein activation. For this we used activation of the prototypical RTK EGFR, which is a natural input for GIV-mediated G protein activation. It has been previously shown that full-length GIV or GIV 1660–1870 binds to phosphotyrosines in the C-terminal tail of active EGFR at the plasma membrane (25, 26, 29), and such binding is required for Gαi activation (25, 26, 29). However, it is unclear whether physical relocalization of GIV to membrane-bound receptors is sufficient by itself for directly activating G proteins or only required, because it might need to work in coordination with other GIV-related functions to eventually lead to G protein activation. To test for sufficiency, we generated a chimeric protein (namely Grb2-GBA) consisting of the GBA motif of GIV (thus lacking any other domain possibly involved in signaling) fused to Grb2 (as a module for binding to active EGFR) (Fig. 7A). Grb2 is a well characterized adaptor protein that translocates from the cytosol to the plasma membrane by directly binding to phosphotyrosines in the C-terminal tail of active EGFR. One of the major biological functions of this translocation is to recruit SOS (Son of Sevenless), a constitutive binding partner or Grb2 and a GEF for Ras, from the cytosol to the plasma membrane to activate membrane-bound Ras (61–63). Thus, Grb2-GBA should mimic the recruitment of native GIV in response to a stimulus that normally activates its G protein regulatory function but lacking any non-GBA contribution to the outcome. We found that EGF stimulation triggers G protein activation in cells expressing Grb2-GBA (Fig. 7B). This response was dependent on the amount of Grb2-GBA expressed and not reproduced by the expression of equivalent amounts of the GEF-deficient mutant Grb2-GBA FA or in the absence of EGFR (Fig. 7, B and C). These results indicate that relocalization of the GBA motif of GIV from the cytosol to membrane-bound receptors in response to a natural stimulus is sufficient for G protein activation.
FIGURE 7.

Recruitment of the GBA motif of GIV to activated EGFR is sufficient to induce G protein activation. A, diagram depicting how Grb2 fused to the GBA motif (aa 1660–1705) of GIV (Grb2-GBA) is recruited from the cytosol to tyrosine phosphorylated EGFR at the plasma membrane upon activation. Grb2-mediated binding to EGFR brings the GBA motif close to membrane-bound Gi3. B and C, HEK293T cells were transfected with plasmids for all the components required BRET-based G protein activity measurements as described for Fig. 3, Grb2-GBA (WT or FA) and EGFR. EGF (50 ng/μl) was added at the indicated time (arrow in B). BRET results (B) are the averages of three or four independent experiments, and the error bars are the S.E. (shown only at 5-s intervals for clarity). Representative immunoblots of the cells used in these experiments are shown in C.
Discussion
The main finding of this work is the identification of GIV recruitment to membranes as a major mechanism responsible for the activation of its G protein regulatory function. The data presented here together with previously published evidence (16, 25, 29, 40) support a model in which GIV and its substrate G protein, Gαi, are spatially segregated from each other under resting conditions but are brought in close proximity when GIV is recruited to the plasma membrane upon cell stimulation. Our results using synthetic or physiological stimuli demonstrate that such physical relocalization of GIV is sufficient to promote G protein signaling. This mechanism operates in the context of GPCR-independent G protein activation and provides novel insights into a long standing gap in the understanding of signaling cross-talk and G protein transactivation by RTKs. We believe that the mechanism described here shares striking similarity to one of the best characterized paradigms in RTK signaling, i.e. GEF-mediated activation of the small G protein Ras (Fig. 8). It has been appreciated for over two decades that the primary mechanism, albeit not the only one (64), by which the GEF SOS activates Ras is its translocation to membranes upon RTK stimulation (61–63). Much like Ras, Gαi is constitutively associated with membranes, and much like SOS, GIV is not. Both SOS and GIV have intrinsic GEF activity without modifications, and both of them can be relocated to the proximity of their respective target G proteins on membranes by recruitment to activated RTKs. The mechanism underlying the GEF recruitment also shares similarities because in both cases it relies on phosphotyrosines on RTK tails that serve as docking sites for SH2 domains. The difference is that SOS is recruited indirectly via the SH2 domain-containing adaptor Grb2 (61–63), to which SOS binds constitutively, whereas GIV does it directly via an SH2-like domain located in its C-terminal region (25) (Fig. 8). Therefore, it appears that non-canonical activation of heterotrimeric G proteins by RTKs can be achieved with a set of signaling elements (constitutively active GEF, SH2 domains) and events (spatial segregation, recruitment via RTK-pY) analogous to those used for canonical activation of small G proteins by RTKs. Interestingly, recent evidence4 suggests that the structural basis for the GEF action of GIV on Gαi3 also shares similarities with the mechanism used by SOS on Ras, i.e. both GIV and SOS bind to a region that overlaps with the effector binding site on the G protein and perturb primarily structural elements responsible for binding the nucleotide phosphates.
FIGURE 8.

Model depicting the parallelism between the mechanisms of activation of Gαi and Ras by their respective cytoplasmic GEFs, GIV, and SOS, upon RTK stimulation. A, under resting conditions, SOS is primarily located in the cytosol along with Grb2, whereas its substrate G protein Ras is constitutively anchored to the plasma membrane, thereby precluding SOS action. Upon RTK stimulation, Grb2-SOS complexes translocate to the plasma membrane via binding of Grb2 SH2 domains to tyrosine phosphorylated EGFR. This change of localization brings SOS in physical proximity to Ras, thereby promoting G protein activation. B, under resting conditions, GIV is primarily located in the cytosol, whereas its substrate G protein Gαi is constitutively anchored to the plasma membrane, thereby precluding GIV action. Upon RTK stimulation, GIV translocates to the plasma membrane via binding of its SH2-like domain to tyrosine phosphorylated EGFR. This change of localization brings GIV in physical proximity to Gαi, thereby promoting G protein activation.
Muntean and Martemyanov (65) recently reported that the activity of a different type of G protein regulators, i.e. RGSs (regulators of G protein signaling), might also be determined by membrane localization. Much like in some of our experiments, they combined chemogenetic control of spatial localization with real time BRET-based biosensors to show that membrane association of RGS proteins of the R7 family is sufficient for their GTPase-accelerating protein activity. These observations not only emphasize the importance of subcellular localization for the activity of G protein regulators but also showcase the power of combining synthetic biology approaches and biosensors to simultaneously manipulate and monitor G protein signaling with high spatiotemporal resolution.
GIV translocation to membranes might also be important to regulate G proteins in a context different from receptor signaling at the plasma membrane. GIV has been reported to regulate G protein signaling in membranous compartments of the cell different from the plasma membrane, like endosomes (22), autophagosomes (41), or the Golgi apparatus (42). Because these compartments are also accessible to cytosolic GIV, it is tempting to speculate that its translocation to organelle membranes might also serve as a mechanism of G protein regulation therein. However, it is unclear how GIV localization would be regulated in these scenarios. Although it is conceivable that GIV might traffic along early steps of the endocytic pathway still associated with tyrosine phosphorylated RTKs, this could not explain the action of GIV on autophagosomes. As for the association with Golgi membranes, it has been recently reported that active Arf1, a Golgi-resident small G protein, directly recruits GIV to the Golgi apparatus, where it activates heterotrimeric G proteins to regulate trafficking (42). In light of the findings reported here, it is possible that Arf1-mediated recruitment of GIV serves as the mechanism that enables its G protein regulatory function on Golgi membranes.
Yet another mechanism of regulation for the association of GIV with membranes could be provided by its previously described phosphoinositide 4-phosphate (PI4P) binding function (40). Recent evidence indicates that, contrary to previous belief, PI4P is not only found on the Golgi apparatus but also at the plasma membrane and endosomes (66, 67). Based on results presented here (Fig. 1) or previously published by others (40), full-length GIV does seem to use this PI4P-binding region to associate with membranes under unstimulated conditions. Nevertheless, it is possible that it enhances GIV membrane association achieved by other means, like receptor-mediated recruitment. An interesting speculation in this regard is raised by the fact that PI4P binding is abolished upon GIV phosphorylation at Ser1416 by Akt (40). Because it is well established that the GEF activity of GIV leads to downstream Akt activation (16–18, 29, 31), we speculate that GIV phosphorylation at Ser1416 might serve as a negative feedback regulation for GIV-mediated G protein activation by favoring its dissociation from membranes. However, additional work will be required to establish the impact of PI4P binding on the association of GIV with membranes and its possible role as a negative feedback regulation mechanism.
We have recently identified another non-receptor GEF of the same family as GIV, namely DAPLE (20). DAPLE contains a GBA motif that regulates heterotrimeric G proteins in the context of Wnt signaling. DAPLE shares other domains in common with GIV, including the PI4P-binding region, central coiled-coil, and N-terminal HOOK domain, but differs in the C-terminal region where the SH2-like domain of GIV is located (20). Interestingly, the corresponding C-terminal region of DAPLE is responsible for its association with Wnt receptors of the Frizzled family, which occurs only upon ligand stimulation and leads to DAPLE redistribution from the cytosol to the plasma membrane (20). This suggests that membrane translocation upon recruitment to receptors might be a mechanism of activation common for different GBA-motif containing GEFs but via a different set of molecular interactions.
Along the same lines, it is also possible that membrane translocation is a mechanism to activate GIV downstream of receptors different from RTKs. More specifically, it has been recently described that GIV also triggers G protein signaling in response to activation of integrins (16, 31). Interestingly, three independent studies have shown that GIV directly binds to integrins after stimulation (16, 68, 69). Our results presented here suggest that such recruitment to integrins might be sufficient to induce the observed GIV-dependent G protein signaling.
Although here we report membrane recruitment as a major regulatory mechanism for GIV-mediated G protein activation, we cannot rule out that other mechanisms come into play to modulate the GEF function of GIV. In particular, it will be important to address in the future how phosphorylation of Ser1674 impinges on GIV-mediated signaling. Although our results clearly indicate that membrane translocation is more potent than Ser1674 phosphorylation in activating GIV, previous evidence has established that this phosphorylation regulates events downstream of GIV (47). One possibility is that Ser1674 phosphorylation fine tunes G protein activation and that the methods used in the present work cannot detect such fine alterations. Another possibility is that Ser1674 works in coordination with other events to control the fate of the G protein regulatory function of GIV via alteration of its subcellular localization or other mechanisms. In support of the latter, it has been recently reported that sequential phosphorylation of GIV at Ser1674 and Ser1689 determines its preferential coupling to different G protein subtypes in different subcellular locations (60).
In summary, our findings provide novel mechanistic insights into how G protein activation by GIV is achieved in cells, which also has important implications for the understanding of G protein transactivation by non-GPCR receptors. It remains to be investigated whether this mechanism also applies to other GIV-related GEFs, like DAPLE, and how it is integrated with other possible regulatory events, like phosphorylation or phosphoinositide binding.
Experimental Procedures
Reagents and Antibodies
Unless otherwise indicated all reagents were of analytical grade and obtained from Fisher Scientific or Sigma. Cell culture media and goat anti-rabbit and goat anti-mouse Alexa Fluor 680 antibodies were from Life Technologies. Fluorescein di-β-d-galactopyranoside was from Marker Gene Technologies, and the protein inhibitor mixture (catalog no. S8830) from Sigma. Human EGF, leupeptin, pepstatin, and aprotinin were from Gold Biotechnology. All restriction endonucleases were from Thermo Scientific, and Escherichia coli strain DH5α was purchased from New England Biolabs. Pfu ultra DNA polymerase was purchased from Agilent. Rabbit antibodies raised against GIV (T-13), TfR (CD71, H68.4), Gαi3 (C10), and Gβ (M-14) were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies raised against Myc tag (9E10), α-tubulin (Sigma catalog no. T6074), and HA tag (12CA5) were from the Developmental Studies Hybridoma Bank (University of Iowa), Sigma, and Roche, respectively. Rabbit antibodies for ppERK (catalog no. 4370) and integrin β1 (catalog no. D2E5) and mouse antibodies for Myc (catalog no. 9B11) were from Cell Signaling. Mouse monoclonal antibodies for Caveolin-1 (catalog no. 610406) were from BD Transduction Laboratories. Rabbit polyclonal antibody for RFP was from Abcam (catalog no. 62341). Goat anti-rabbit and goat anti-mouse IRDye 800 F(ab′)2 were from Li-Cor Biosciences (Lincoln, NE).
Plasmids
GIV (1660–1870) was PCR-amplified with BglII and XhoI flanking sites and inserted in the BamHI/XhoI sites of pYES2 (2 μm URA) to generate pYES2-GIV. The sequence corresponding to the first 9 amino acids of Saccharomyces cerevisiae Gpa1 (MGCTVSTQT) was inserted upstream of GIV in the KpnI site of pYES2-GIV to generate pYES2-N9Gpa1-GIV. The centromeric plasmid (CEN TRP) encoding for the LacZ gene under the control of the Fus1 promoter (PFus1::LacZ) was kindly provided by M. Cismowski (Nationwide Children's Hospital) and has been described previously (53). Lyn11-FRB-HA was generated by introducing a stop codon after the HA sequence of the Lyn11-FRB-CFP plasmid (Addgene catalog no. 38003). Lyn11-FRB-Myc was created by introducing the sequence corresponding to the first 11 amino acids of Lyn (MGCIKSKGKDS) into the KpnI and EcoRI sites of the FRB-Myc plasmid (Addgene catalog no. 20228) by primer annealing and ligation. The G2A of Lyn11-FRB-Myc was similarly generated by inserted a sequence in which the myristoylation site mutated (MASIKSKGKDS). The plasmid encoding for FKBP-GIV was generated by replacing the pseudojanin cassette of Addgene plasmid 37999 with GIV (1660–1870) (NruI/BamHI sites). The final FKBP-GIV construct was composed of (N terminus to C terminus) mRFP, FKBP, and GIV (1660–1870). FKBP-GIV-GBA (1660–1705) was generated by introducing a stop codon right after GIV amino acid 1705 in the FKBP-GIV plasmid. Grb2-GBA was generated by replacing the mRFP and FKBP sequences of FKBP-GIV-GBA (1660–1705) with the human Grb2 sequence (amplified from Addgene plasmid catalog no. 70383) preceded by a Myc tag (NheI/NruI sites). pcDNA6A-EGFR was obtained from Addgene (catalog no. 42665). Cloning of rat Gαi3 into pcDNA3 has been described previously (10, 17). pcDNA3.1-A1R, pcDNA3.1-Venus(1–155)-Gγ2 (VN-Gγ2), pcDNA3.1-Venus(155–239)-Gβ1 (VC-Gβ1), pcDNA3.1-masGRK3ct-Nluc, and pcMin-ETAR have been described previously (58). pcDNA3.1-M4 muscarinic receptor (M4R, HA-tagged) was purchased from the cDNA Resource Center (Bloomsburg University). Point mutations were generated using following the manufacturer's instructions (QuikChange II; Agilent). All constructs were checked by DNA sequencing.
Fractionation of Mammalian Cells
HEK293T, HeLa, or MDA-MB-231 cells cultured in DMEM plus 10% FBS and at ∼90% confluency were scraped in PBS and pelleted by centrifugation. The cells corresponding to 0.5 (HEK293T) or 1.5 (HeLa or MDA-MB-231) 10-cm dishes were resuspended with 250 μl of detergent-free homogenization buffer (20 mm HEPES, pH 7.2, 5 mm Mg(CH3COO)2, 125 mm K(CH3COO), 1 mm DTT, and protease inhibitor mixture) in ice. All subsequent steps were carried out in ice or at 4 °C. The cells were homogenized by 30 passages though a 30-gauge needle and subsequently centrifuged at 1,000 × g for 10 min. An aliquot of the supernatant was set aside (post-nuclear supernatant (PNS) fraction), and the rest was centrifuged in a fixed angle rotor TLA-55 in a Beckman Coulter Optima MAX-E tabletop centrifuge for 45 min at 100,000 × g. An aliquot of the supernatant was set aside (S100 fraction). The pellet was resuspended to the starting volume with homogenization buffer, and an aliquot was set aside (P100 fraction). Triton X-100 was added to the P100 fraction (0.4% v/v final) and after a 30-min incubation centrifuged for 45 min at 100,000 × g as described above. The supernatant containing the Triton X-100-soluble material (TX-S fraction) was saved, and the pellet containing the Triton X-100-insoluble material resuspended to the starting volume (TX-I). Equal volumes of each fraction were added Laemmli sample buffer and boiled before protein separation by SDS-PAGE. Identical experiments were carried out with an alternative homogenization buffer (10 mm HEPES, pH 7.4, sucrose 250 mm, 10 mm KCl, 1.5 mm MgCl2, 1 mm DTT, and protease inhibitor mixture), obtaining similar results.
Lipid Raft Isolation
This assay was performed as previously described (70) with some modifications. MDA-MB-231 cells cultured in DMEM plus 10% FBS and at ∼90% confluency were scraped in PBS and pelleted by centrifugation. Cells corresponding to two 10-cm dishes were resuspended with 1 ml of lysis buffer (20 mm HEPES, pH 7.2, 5 mm Mg(CH3COO)2, 125 mm K(CH3COO), 0.4% (v/v) Triton X-100, 1 mm DTT, and protease inhibitor mixture) in ice. All subsequent steps were carried out in ice or at 4 °C. The lysate was incubated in ice for 30 min and subsequently mixed with an equal volume of 90% (w/v) sucrose (prepared in lysis buffer). A discontinuous sucrose gradient was prepared by layering 2 ml of 35% sucrose and 1.25 ml of 5% sucrose on top of 1.5 ml of the 45% sucrose lysate mixture. This gradient was centrifuged at 200,000g for 16h in a SW50.1 rotor using a Beckman Coulter Optima LE-80K ultracentrifuge. Fractions (0.5 ml) were carefully collected from the top. Equal volumes of each fraction were added Laemmli sample buffer and boiled before protein separation by SDS-PAGE.
Fractionation of Yeast Cells
This procedure was performed as described previously (54) with minor modifications. Pellets corresponding to 15 OD600 of exponentially growing cells resuspended in 200 μl of cold homogenization buffer (40 mm triethanolamine, pH 7.2, 2 mm EDTA, 2 mm DTT, 1 μm leupeptin, 2.5 μm pepstatin, 0.2 μm aprotinin and 1 mm PMSF). All subsequent steps were carried out in ice or at 4 °C. Approximately 50 μl of glass beads were added to the tubes and vortexed for 10 min. The homogenate was recovered by poking a whole in the bottom of the tubes and centrifugation onto new tubes. Beads were washed with 100 μl of homogenization buffer and the recovered volume pooled with initial homogenate. After centrifugation at 500g for 10 min, an aliquot of the supernatant was set aside (PNS fraction) and the rest was centrifuged at 100,000 as described above in “Fractionation of Mammalian Cells” to obtain the “S” and “P” fractions. Equal volumes of each fraction were added Laemmli sample buffer and boiled before protein separation by SDS-PAGE.
Yeast Strains and Manipulations
The previously described (53) S. cerevisiae strain CY7967 [MATα GPA1(1–41)-Gαi3 far1Δ fus1p-HIS3 can1 ste14:trp1:LYS2 ste3Δ lys2 ura3 leu2 trp1 his3] (kindly provided by James Broach, Penn State University) was used for all experiments. The main features of this strain are that the only pheromone responsive GPCR (Ste3) is deleted, the endogenous Gα-subunit GPA1 is replaced by a chimeric GPA1(1–41)-human Gαi3 (36–354) and the cell cycle arrest-inducing protein far1 is deleted. In this strain, the pheromone response pathway can be up-regulated by the ectopic expression of activators of human Gαi3 and does not result in the cell cycle arrest that occurs in the native pheromone response (53). Plasmid transformations were carried out using the lithium acetate method. CY7967 cells were transformed with a plasmid encoding for the PFus1::LacZ reporter and subsequently with plasmids pYES2, pYES2-GIV or pYES2-N9Gpa1-GIV (see “Plasmids”). Double transformants were selected in Synthetic Defined (S.D.)-TRP-URA media. Individual colonies were inoculated into 3 ml of SDGalactose-TRP-URA and incubated overnight at 30 °C to induce the expression of the proteins of interest under the control of a galactose-inducible promoter of pYES2. For the spot growth assays, the overnight cultures were washed and resuspended in water at an OD600 of 1 and equal volumes of each strain (6 μl) were spotted on SDGal-TRP-URA or SDGal-TRP-URA-HIS (plus 10 mm 3-amino-triazole) agar plates and incubated at 30 °C for 4 days. Plates were scanned at days 2, 3 and 4 using the same settings. For the preparation of samples for immunoblotting and β-galactosidase activity assays, the overnight cultures were used to inoculate 20 ml of SDGalactose-TRP-URA at 0.3 OD600. Pellets of exponentially growing cells (∼0.7–0.8 OD600, 4–5 h) corresponding to 5 OD600 were washed with PBS + 0.1% BSA (w/v) and used to prepare immunoblotting samples as described previously (21). Briefly, washed pellets were resuspended in 150 μl of lysis buffer (10 mm Tris-HCl, pH 8.0, 10% (w/v) TCA, 25 mm NH4OAc, 1 mm EDTA). 100 μl of glass beads were added to each tube and vortexed at 4 °C for 5 min. Lysates were separated from glass beads by poking a hole in the bottom of the tubes followed by centrifugation onto a new set of tubes. The process was repeated after the addition of 50 μl of lysis buffer to wash the glass beads. Proteins were precipitated by centrifugation (10 min, 20,000 × g) and resuspended in 60 μl of solubilization buffer (0.1 m Tris-HCl, pH 11.0, 3% SDS). Samples were boiled for 5 min, centrifuged (1 min, 20,000 × g), and 50 μl of the supernatant was transferred to new tubes containing 12.5 μl of Laemmli sample buffer and boiled for 5 min (∼15–20 μl were typically loaded per lane). For the β-galactosidase assays, pellets of exponentially growing cells corresponding to 0.5 A600 were washed with PBS + 0.1% BSA (w/v) and resuspended in 200 μl assay buffer (60 mm Na2PO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgCl2, 0.25% (v/v) β-mercaptoethanol, 0.01% (w/v) SDS, 10% (v/v) chloroform) and vortexed. 100 μl were transferred to 96-well plates, and reactions were started by the addition of 50 μl of the fluorogenic β-galactosidase substrate fluorescein di-β-d-galactopyranoside (100 μm final). Fluorescence (excitation, 485 ± 10 nm; emission, 528 ± 10 nm) was measured every 2 min for 90 min at 30 °C in a Biotek H1 synergy plate reader. Enzymatic activity was calculated from the slope of fluorescence (arbitrary units) versus time (min). Each independent clone was measure in duplicate, and the results are expressed as raw arbitrary fluorescent units/min or normalized (%) to the activity of strains carrying pYES2-GIV.
Cell Culture, Transfections, and BRET Assay
HEK293T, HeLa and MDA-MB-231 cells were grown at 37 °C in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 1% l-glutamine and 5% CO2. HEK293T cells were transfected using the calcium phosphate method in gelatin-coated 6-well plates with the following plasmids (quantities in parenthesis): masGRK3ct-Nluc (0.1 μg), VC-Gβ1 (0.2 μg), VN-Gγ2 (0.2 μg), A1R (0.2 μg), ETAR (0.2 μg), M4R (0.2 μg), Gαi3 (1.0 μg), Lyn11-FRB-HA (3.0 μg), Lyn11-FRB-Myc (3.0 μg), FKBP-GIV (0.025–2.0 μg, as indicated in the figures), FKBP-GIV-GBA (0.125–0.5 μg, as indicated in the figures), Grb2-GBA (0.5–2 μg, as indicated in the figures), and pcDNA3.1(+) (to equalize the amount of DNA per well). G protein activity was monitored by BRET using a system based on a sensor for free Gβγ that was originally described in Ref. 57 and optimized in Ref. 71 with a new generation luciferase (Nluc). BRET experiments were carried out and analyzed as described previously (58) with minor modifications. Briefly, 22–28 h after transfection, the cells were gently scrapped in PBS, centrifuged, and resuspended in Tyrode's solution (140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 0.37 mm NaH2PO4, 24 mm NaHCO3, 10 mm HEPES, pH 7.4, 0.1% glucose) at a density of 106 cell/ml. Twenty-five μl of cell suspensions were added to a white opaque 96-well plate (Opti-Plate®; Perkin Elmer) and mixed with 3 volumes of the Nanoluciferase substrate Nano-Glo (Promega, diluted 1:150 in Tyrode's solution). After 2 min of incubation, luminescence was measured at room temperature in a Synergy H1 (Biotek) or a POLARstar Omega (BMG Labtech) plate reader at 460 ± 20 and 528 ± 10 nm. BRET signals were calculated as the ratio of the emission intensity at 528 ± 10 nm divided by the emission intensity at 460 ± 20 nm. For the single point measurements, the cells were treated with rapamycin (0.5 μm) or an equal volume of vehicle (ethanol, 0.005% final concentration), and BRET was measured after 2 min. For the kinetic experiments, BRET measurements were done every second or every 0.24 s. The BRET baseline was recorded for 30 s, after which 5 μl of rapamycin (0.5 μm final), adenosine (0.1 μm final), endothelin (ET-1, 3 μm final), or EGF (50 ng/ml final) were added to the well. For clarity, most experiments are presented as increase in BRET (ΔBRET) relative to control conditions, i.e. baseline signal in kinetic experiments or unstimulated controls for single time point measurements (as indicated in the figures).
Immunoblotting
Yeast protein samples were prepared as described above (see “Yeast Strains and Manipulations”). Protein samples from BRET experiments were prepared by centrifugation of HEK293T cells and resuspension in lysis buffer (20 mm HEPES, pH 7.2, 5 mm Mg(CH3COO)2, 125 mm K(CH3COO), 0.4% Triton X-100, 1 mm DTT, and protease inhibitor mixture). After clearing by centrifugation at 14,000 × g at 4 °C for 10 min, protein concentration was determined by Bradford. Samples were supplemented with Laemmli and boiled for 5 min. Proteins were separated by SDS-PAGE and transferred to PVDF membranes, which were sequentially incubated with primary and secondary antibodies (goat anti-rabbit or anti-mouse coupled to Alexa Fluor 680 or IRDye 800, 1:10,000). The primary antibodies were used at the following dilutions: GIV 1:500, Gαi3 1:250, TfR 1:500, β1 integrin 1:1,000, caveolin-1 1:2,500, pan-Gβ 1:250, α-tubulin 1:2,500, ppERK (which recognizes yeast ppFus3) 1:2,500, HA, 1:1,000, Myc 1:1,000, and RFP 1:1,000. Infrared imaging of immunoblots was performed according to the manufacturer's protocols using an Odyssey Infrared Imaging System (Li-Cor Biosciences). Images were processed using ImageJ software (National Institutes of Health) and assembled for presentation using Photoshop and Illustrator software (Adobe).
Statistical Analyses
Each experiment was performed at least three times. The data shown are presented as means with error bars representing the S.E. or as one representative result of each biological replicate. Statistical significance between various conditions was assessed with Student's t test. p < 0.05 was considered significant.
Author Contributions
K. P.-S., A. L., V. D., A. M., S. B., and M. G.-M. performed experiments; K. P.-S. and M. G.-M. analyzed the data; and M. G.-M. designed research and wrote the paper.
Acknowledgments
We thank N. Lambert (Georgia Regents University), K. Martemyanov (Scripps, Florida), P. Polgar (Boston University), and M. Cismowski (Nationwide Children's Hospital) for providing plasmids and James Broach (Penn State University) for providing the CY7967 yeast strain.
This work was supported by National Institutes of Health Grants R01GM108733 and R01GM112631, American Cancer Society Grant RSG-13-362-01-TBE, and funds from the Karin Grunebaum Research Foundation (to M. G.-M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
K. Parag-Sharma and M. Garcia-Marcos, unpublished observations.
K. Parag-Sharma, V. DiGiacomo, A. Marivin, A. Leyme, and M. Garcia-Marcos, unpublished observations.
- GPCR
- G protein-coupled receptor
- GEF
- guanine nucleotide exchange factor
- GBA
- Gα binding and activating
- CDK5
- cyclin-dependent kinase 5
- RTK
- receptor tyrosine kinase
- PNS
- post-nuclear supernatant
- EGFR
- EGF receptor
- Grb2
- growth factor receptor-bound 2
- CID
- chemically induced dimerization
- aa
- amino acid(s)
- FA
- F1685A mutant
- BRET
- bioluminescence resonance energy transfer
- M4R
- M4 muscarinic receptor
- A1R
- adenosine A1 receptor
- ETAR
- endothelin ETA receptor
- PI4P
- phosphoinositide 4-phosphate
- TfR
- transferrin receptor.
References
- 1. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 2. Farfel Z., Bourne H. R., and Iiri T. (1999) The expanding spectrum of G protein diseases. N. Engl. J. Med. 340, 1012–1020 [DOI] [PubMed] [Google Scholar]
- 3. Oldham W. M., and Hamm H. E. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 [DOI] [PubMed] [Google Scholar]
- 4. Marty C., and Ye R. D. (2010) Heterotrimeric G protein signaling outside the realm of seven transmembrane domain receptors. Mol. Pharmacol 78, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liang M. N., and Garrison J. C. (1991) The epidermal growth factor receptor is coupled to a pertussis toxin-sensitive guanine nucleotide regulatory protein in rat hepatocytes. J. Biol. Chem. 266, 13342–13349 [PubMed] [Google Scholar]
- 6. Moxham C. M., and Malbon C. C. (1996) Insulin action impaired by deficiency of the G-protein subunit Giα2. Nature 379, 840–844 [DOI] [PubMed] [Google Scholar]
- 7. Song X., Zheng X., Malbon C. C., and Wang H. (2001) Gαi2 enhances in vivo activation of and insulin signaling to GLUT4. J. Biol. Chem. 276, 34651–34658 [DOI] [PubMed] [Google Scholar]
- 8. Gohla A., Klement K., Piekorz R. P., Pexa K., vom Dahl S., Spicher K., Dreval V., Häussinger D., Birnbaumer L., and Nürnberg B. (2007) An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc. Natl. Acad. Sci. U.S.A. 104, 3003–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cao C., Huang X., Han Y., Wan Y., Birnbaumer L., Feng G. S., Marshall J., Jiang M., and Chu W. M. (2009) Gαi1 and Gαi3 are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci. Signal. 2, ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghosh P., Garcia-Marcos M., Bornheimer S. J., and Farquhar M. G. (2008) Activation of Gαi3 triggers cell migration via regulation of GIV. J. Cell Biol. 182, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cismowski M. J., Ma C., Ribas C., Xie X., Spruyt M., Lizano J. S., Lanier S. M., and Duzic E. (2000) Activation of heterotrimeric G-protein signaling by a ras-related protein: implications for signal integration. J. Biol. Chem. 275, 23421–23424 [DOI] [PubMed] [Google Scholar]
- 12. Tall G. G., Krumins A. M., and Gilman A. G. (2003) Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 [DOI] [PubMed] [Google Scholar]
- 13. Lee M. J., and Dohlman H. G. (2008) Coactivation of G protein signaling by cell-surface receptors and an intracellular exchange factor. Curr. Biol. 18, 211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Natochin M., Campbell T. N., Barren B., Miller L. C., Hameed S., Artemyev N. O., and Braun J. E. (2005) Characterization of the Gαs regulator cysteine string protein. J. Biol. Chem. 280, 30236–30241 [DOI] [PubMed] [Google Scholar]
- 15. Sato M., Blumer J. B., Simon V., and Lanier S. M. (2006) Accessory proteins for G proteins: partners in signaling. Annu. Rev. Pharmacol. Toxicol. 46, 151–187 [DOI] [PubMed] [Google Scholar]
- 16. Leyme A., Marivin A., Perez-Gutierrez L., Nguyen L. T., and Garcia-Marcos M. (2015) Integrins activate trimeric G proteins via the nonreceptor protein GIV/Girdin. J. Cell Biol. 210, 1165–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garcia-Marcos M., Ghosh P., and Farquhar M. G. (2009) GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 3178–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garcia-Marcos M., Kietrsunthorn P. S., Pavlova Y., Adia M. A., Ghosh P., and Farquhar M. G. (2012) Functional characterization of the guanine nucleotide exchange factor (GEF) motif of GIV protein reveals a threshold effect in signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 1961–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garcia-Marcos M., Kietrsunthorn P. S., Wang H., Ghosh P., and Farquhar M. G. (2011) G Protein binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of Gαi-regulatory motifs. J. Biol. Chem. 286, 28138–28149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aznar N., Midde K. K., Dunkel Y., Lopez-Sanchez I., Pavlova Y., Marivin A., Barbazán J., Murray F., Nitsche U., Janssen K. P., Willert K., Goel A., Abal M., Garcia-Marcos M., and Ghosh P. (2015) Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. Elife 4, e07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coleman B. D., Marivin A., Parag-Sharma K., DiGiacomo V., Kim S., Pepper J. S., Casler J., Nguyen L. T., Koelle M. R., and Garcia-Marcos M. (2016) Evolutionary conservation of a GPCR-independent mechanism of trimeric G protein activation. Mol. Biol. Evol. 33, 820–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beas A. O., Taupin V., Teodorof C., Nguyen L. T., Garcia-Marcos M., and Farquhar M. G. (2012) Gαs promotes EEA1 endosome maturation and shuts down proliferative signaling through interaction with GIV (Girdin). Mol. Biol. Cell 23, 4623–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garcia-Marcos M., Ghosh P., Ear J., and Farquhar M. G. (2010) A structural determinant that renders Gαi sensitive to activation by GIV/girdin is required to promote cell migration. J. Biol. Chem. 285, 12765–12777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Le-Niculescu H., Niesman I., Fischer T., DeVries L., and Farquhar M. G. (2005) Identification and characterization of GIV, a novel Gαi/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J. Biol. Chem. 280, 22012–22020 [DOI] [PubMed] [Google Scholar]
- 25. Lin C., Ear J., Midde K., Lopez-Sanchez I., Aznar N., Garcia-Marcos M., Kufareva I., Abagyan R., and Ghosh P. (2014) Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol. Biol. Cell 25, 3654–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Midde K. K., Aznar N., Laederich M. B., Ma G. S., Kunkel M. T., Newton A. C., and Ghosh P. (2015) Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proc. Natl. Acad. Sci. U.S.A. 112, E937–E946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopez-Sanchez I., Dunkel Y., Roh Y. S., Mittal Y., De Minicis S., Muranyi A., Singh S., Shanmugam K., Aroonsakool N., Murray F., Ho S. B., Seki E., Brenner D. A., and Ghosh P. (2014) GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat. Commun. 5, 4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma G. S., Aznar N., Kalogriopoulos N., Midde K. K., Lopez-Sanchez I., Sato E., Dunkel Y., Gallo R. L., and Ghosh P. (2015) Therapeutic effects of cell-permeant peptides that activate G proteins downstream of growth factors. Proc. Natl. Acad. Sci. U.S.A. 112, E2602–E2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghosh P., Beas A. O., Bornheimer S. J., Garcia-Marcos M., Forry E. P., Johannson C., Ear J., Jung B. H., Cabrera B., Carethers J. M., and Farquhar M. G. (2010) A Gαi-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol. Biol. Cell 21, 2338–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang H., Misaki T., Taupin V., Eguchi A., Ghosh P., and Farquhar M. G. (2015) GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J. Am. Soc. Nephrol. 26, 314–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leyme A., Marivin A., and Garcia-Marcos M. (2016) GIV/Girdin (Gα-interacting, vesicle-associated protein/Girdin) creates a positive feedback loop that potentiates outside-in integrin signaling in cancer cells. J. Biol. Chem. 291, 8269–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garcia-Marcos M., Jung B. H., Ear J., Cabrera B., Carethers J. M., and Ghosh P. (2011) Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB J. 25, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghosh P., Garcia-Marcos M., and Farquhar M. G. (2011) GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh. Migr. 5, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang P., Enomoto A., Jijiwa M., Kato T., Hasegawa T., Ishida M., Sato T., Asai N., Murakumo Y., and Takahashi M. (2008) An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 68, 1310–1318 [DOI] [PubMed] [Google Scholar]
- 35. Jun B. Y., Kim S. W., Jung C. K., Cho Y. K., Lee I. S., Choi M. G., Choi K. Y., and Oh S. T. (2013) Expression of girdin in human colorectal cancer and its association with tumor progression. Dis. Colon. Rectum. 56, 51–57 [DOI] [PubMed] [Google Scholar]
- 36. Ling Y., Jiang P., Cui S. P., Ren Y. L., Zhu S. N., Yang J. P., Du J., Zhang Y., Liu J. Y., and Zhang B. (2011) Clinical implications for girdin protein expression in breast cancer. Cancer Invest. 29, 405–410 [DOI] [PubMed] [Google Scholar]
- 37. Liu C., Xue H., Lu Y., and Chi B. (2012) Stem cell gene Girdin: a potential early liver metastasis predictor of colorectal cancer. Mol. Biol. Rep. 39, 8717–8722 [DOI] [PubMed] [Google Scholar]
- 38. Liu C., Zhang Y., Xu H., Zhang R., Li H., Lu P., and Jin F. (2012) Girdin protein: a new potential distant metastasis predictor of breast cancer. Med. Oncol. 29, 1554–1560 [DOI] [PubMed] [Google Scholar]
- 39. Anai M., Shojima N., Katagiri H., Ogihara T., Sakoda H., Onishi Y., Ono H., Fujishiro M., Fukushima Y., Horike N., Viana A., Kikuchi M., Noguchi N., Takahashi S., Takata K., et al. (2005) A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J. Biol. Chem. 280, 18525–18535 [DOI] [PubMed] [Google Scholar]
- 40. Enomoto A., Murakami H., Asai N., Morone N., Watanabe T., Kawai K., Murakumo Y., Usukura J., Kaibuchi K., and Takahashi M. (2005) Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 9, 389–402 [DOI] [PubMed] [Google Scholar]
- 41. Garcia-Marcos M., Ear J., Farquhar M. G., and Ghosh P. (2011) A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol. Biol. Cell 22, 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lo I. C., Gupta V., Midde K. K., Taupin V., Lopez-Sanchez I., Kufareva I., Abagyan R., Randazzo P. A., Farquhar M. G., and Ghosh P. (2015) Activation of Gαi at the Golgi by GIV/Girdin imposes finiteness in Arf1 signaling. Dev. Cell 33, 189–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sasaki K., Kakuwa T., Akimoto K., Koga H., and Ohno S. (2015) Regulation of epithelial cell polarity by PAR-3 depends on Girdin transcription and Girdin-Gαi3 signaling. J. Cell Sci. 128, 2244–2258 [DOI] [PubMed] [Google Scholar]
- 44. Ma G. S., Lopez-Sanchez I., Aznar N., Kalogriopoulos N., Pedram S., Midde K., Ciaraldi T. P., Henry R. R., and Ghosh P. (2015) Activation of G proteins by GIV-GEF is a pivot point for insulin resistance and sensitivity. Mol. Biol. Cell 26, 4209–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hartung A., Ordelheide A. M., Staiger H., Melzer M., Häring H. U., and Lammers R. (2013) The Akt substrate Girdin is a regulator of insulin signaling in myoblast cells. Biochim. Biophys. Acta 1833, 2803–2811 [DOI] [PubMed] [Google Scholar]
- 46. Lin C., Ear J., Pavlova Y., Mittal Y., Kufareva I., Ghassemian M., Abagyan R., Garcia-Marcos M., and Ghosh P. (2011) Tyrosine phosphorylation of the Gα-interacting protein GIV promotes activation of phosphoinositide 3-kinase during cell migration. Sci. Signal. 4, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bhandari D., Lopez-Sanchez I., To A., Lo I. C., Aznar N., Leyme A., Gupta V., Niesman I., Maddox A. L., Garcia-Marcos M., Farquhar M. G., and Ghosh P. (2015) Cyclin-dependent kinase 5 activates guanine nucleotide exchange factor GIV/Girdin to orchestrate migration-proliferation dichotomy. Proc. Natl. Acad. Sci. U.S.A. 112, E4874–E4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mumby S. M., Heukeroth R. O., Gordon J. I., and Gilman A. G. (1990) G-protein α-subunit expression, myristoylation, and membrane association in COS cells. Proc. Natl. Acad. Sci. U.S.A. 87, 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mao J. Z., Jiang P., Cui S. P., Ren Y. L., Zhao J., Yin X. H., Enomoto A., Liu H. J., Hou L., Takahashi M., and Zhang B. (2012) Girdin locates in centrosome and midbody and plays an important role in cell division. Cancer Sci. 103, 1780–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pike L. J. (2003) Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667 [DOI] [PubMed] [Google Scholar]
- 51. Lisanti M. P., Scherer P. E., Vidugiriene J., Tang Z., Hermanowski-Vosatka A., Tu Y. H., Cook R. F., and Sargiacomo M. (1994) Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J. Cell Biol. 126, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang C., Hepler J. R., Chen L. T., Gilman A. G., Anderson R. G., and Mumby S. M. (1997) Organization of G proteins and adenylyl cyclase at the plasma membrane. Mol. Biol. Cell 8, 2365–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cismowski M. J., Takesono A., Ma C., Lizano J. S., Xie X., Fuernkranz H., Lanier S. M., and Duzic E. (1999) Genetic screens in yeast to identify mammalian nonreceptor modulators of G-protein signaling. Nat. Biotechnol. 17, 878–883 [DOI] [PubMed] [Google Scholar]
- 54. Song J., Hirschman J., Gunn K., and Dohlman H. G. (1996) Regulation of membrane and subunit interactions by N-myristoylation of a G protein α subunit in yeast. J. Biol. Chem. 271, 20273–20283 [DOI] [PubMed] [Google Scholar]
- 55. Bayle J. H., Grimley J. S., Stankunas K., Gestwicki J. E., Wandless T. J., and Crabtree G. R. (2006) Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem Biol. 13, 99–107 [DOI] [PubMed] [Google Scholar]
- 56. Inoue T., Heo W. D., Grimley J. S., Wandless T. J., and Meyer T. (2005) An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hollins B., Kuravi S., Digby G. J., and Lambert N. A. (2009) The C-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell Signal. 21, 1015–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marivin A., Leyme A., Parag-Sharma K., DiGiacomo V., Cheung A. Y., Nguyen L. T., Dominguez I., and Garcia-Marcos M. (2016) Dominant negative Gα subunits as a novel mechanism of trimeric G protein signaling dysregulation in human disease. Sci. Signal. 9, ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kovárová M., Tolar P., Arudchandran R., Dráberová L., Rivera J., and Dráber P. (2001) Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fcϵ receptor I aggregation. Mol. Cell. Biol. 21, 8318–8328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gupta V., Bhandari D., Leyme A., Aznar N., Midde K. K., Lo I. C., Ear J., Niesman I., López-Sánchez I., Blanco-Canosa J. B., von Zastrow M., Garcia-Marcos M., Farquhar M. G., and Ghosh P. (2016) GIV/Girdin activates Gαi and inhibits Gαs via the same motif. Proc. Natl. Acad. Sci. U.S.A. 113, E5721–E5730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quilliam L. A., Huff S. Y., Rabun K. M., Wei W., Park W., Broek D., and Der C. J. (1994) Membrane-targeting potentiates guanine nucleotide exchange factor CDC25 and SOS1 activation of Ras transforming activity. Proc. Natl. Acad. Sci. U.S.A. 91, 8512–8516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aronheim A., Engelberg D., Li N., al-Alawi N., Schlessinger J., and Karin M. (1994) Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 78, 949–961 [DOI] [PubMed] [Google Scholar]
- 63. Kholodenko B. N., Hoek J. B., and Westerhoff H. V. (2000) Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 10, 173–178 [DOI] [PubMed] [Google Scholar]
- 64. Findlay G. M., and Pawson T. (2008) How is SOS activated?: let us count the ways. Nat. Struct. Mol. Biol. 15, 538–540 [DOI] [PubMed] [Google Scholar]
- 65. Muntean B. S., and Martemyanov K. A. (2016) Association with the plasma membrane is sufficient for potentiating catalytic activity of regulators of G protein signaling (RGS) proteins of the R7 subfamily. J. Biol. Chem. 291, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hammond G. R., Fischer M. J., Anderson K. E., Holdich J., Koteci A., Balla T., and Irvine R. F. (2012) PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hammond G. R., Machner M. P., and Balla T. (2014) A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 205, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lopez-Sanchez I., Kalogriopoulos N., Lo I. C., Kabir F., Midde K. K., Wang H., and Ghosh P. (2015) Focal adhesions are foci for tyrosine-based signal transduction via GIV/Girdin and G proteins. Mol. Biol. Cell 26, 4313–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weng L., Enomoto A., Miyoshi H., Takahashi K., Asai N., Morone N., Jiang P., An J., Kato T., Kuroda K., Watanabe T., Asai M., Ishida-Takagishi M., Murakumo Y., Nakashima H., et al. (2014) Regulation of cargo-selective endocytosis by dynamin 2 GTPase-activating protein girdin. EMBO J. 33, 2098–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. García-Marcos M., Pochet S., Tandel S., Fontanils U., Astigarraga E., Fernández-González J. A., Kumps A., Marino A., and Dehaye J. P. (2006) Characterization and comparison of raft-like membranes isolated by two different methods from rat submandibular gland cells. Biochim. Biophys. Acta 1758, 796–806 [DOI] [PubMed] [Google Scholar]
- 71. Masuho I., Ostrovskaya O., Kramer G. M., Jones C. D., Xie K., and Martemyanov K. A. (2015) Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal. 8, ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]