

Abstract
Hepatocyte growth factor (HGF) signaling via c-Met is known to promote endothelial cell motility and angiogenesis. We have previously reported that HGF stimulates lamellipodia formation and motility of human lung microvascular endothelial cells (HLMVECs) via PI3K/Akt signal transduction and reactive oxygen species generation. Here, we report a role for HGF-induced intracellular sphingosine-1-phosphate (S1P) generation catalyzed by sphingosine kinase 1 (SphK1), S1P transporter, spinster homolog 2 (Spns2), and S1P receptor, S1P1, in lamellipodia formation and perhaps motility of HLMVECs. HGF stimulated SphK1 phosphorylation and enhanced intracellular S1P levels in HLMVECs, which was blocked by inhibition of SphK1. HGF enhanced co-localization of SphK1/p-SphK1 with actin/cortactin in lamellipodia and down-regulation or inhibition of SphK1 attenuated HGF-induced lamellipodia formation in HLMVECs. In addition, down-regulation of Spns2 also suppressed HGF-induced lamellipodia formation, suggesting a key role for inside-out S1P signaling. The HGF-mediated phosphorylation of SphK1 and its localization in lamellipodia was dependent on c-Met and ERK1/2 signaling, but not the PI3K/Akt pathway; however, blocking PI3K/Akt signaling attenuated HGF-mediated phosphorylation of Spns2. Down-regulation of S1P1, but not S1P2 or S1P3, with specific siRNA attenuated HGF-induced lamellipodia formation. Further, HGF enhanced association of Spns2 with S1P1 that was blocked by inhibiting SphK1 activity with PF-543. Moreover, HGF-induced migration of HLMVECs was attenuated by down-regulation of Spns2. Taken together, these results suggest that HGF/c-Met-mediated lamellipodia formation, and perhaps motility is dependent on intracellular generation of S1P via activation and localization of SphK1 to cell periphery and Spns2-mediated extracellular transportation of S1P and its inside-out signaling via S1P1.
Keywords: cell signaling, hepatocyte growth factor/scatter factor (HGF/SF), lipid signaling, Sphingosine kinase (SphK), sphingosine-1-phosphate (S1P), Spns2, trans
Introduction
Migration of vascular endothelial cells (ECs)2 is a complex process involving protrusion, adhesion, contraction, and retraction, which is important for a variety of physiologic and pathologic conditions such as vasculogenesis, angiogenesis, wound healing, and atherogenesis (1, 2). Emerging evidence suggests that formation of protruding structures, termed lamellipodia, generated at the leading edge of migrating ECs in response to a variety of growth factors, are primarily involved in cell motility (3). Earlier studies have demonstrated the involvement of actin microfilaments, microtubules, and intermediate filaments in the generation of lamellipodia and EC motility (4). The driving force for EC motility is fueled by continuous growth of actin filaments and rearrangement of the actin binding protein cortactin in the lamellipodia. The lamellipodia formation is mediated by Rac and Cdc42, which regulate Arp2/3 complex through the Wiskott-Aldrich protein, while Rho GTPase regulates actomyosin contractility via stress actin fiber formation and focal adhesions (5).
Hepatocyte growth factor (HGF), also known as scatter factor, promotes EC migration, barrier enhancement, and tumorigenesis through ligation to its receptor, c-Met (6–10). c-Met is a receptor tyrosine kinase and ligand binding triggers autophosphorylation at multiple tyrosine sites that serves as a docking platform for recruitment of several adapter proteins such as Grb2, SHC, Crk/CrkL, and Gab1. These adapter proteins in turn recruit several signal transducing proteins to form an intricate signaling complex (11). HGF binding to c-Met triggers downstream signaling cascade such as PI3K, Akt, and ERK1/2 activation and promotes EC migration (11). HGF-induced EC migration is also up-regulated by enhanced expression of inducible nitric-oxide synthase, but not endothelial NOS, and partially abrogated by PI3K inhibition (12). We have recently demonstrated that HGF stimulated c-Met phosphorylation at Tyr1234/1235, Tyr1349, Tyr1003, and Tyr1313, as well as Ser985 and Akt phosphorylation at Thr308 and Ser473, and potentiated lamellipodia formation in HLMVECs (13). Further, HGF-stimulated NADPH oxidase-dependent reactive oxygen species (ROS) production in lamellipodia and inhibition of c-Met/PI3K/Akt signaling axis and NADPH oxidase attenuated lamellipodia formation and motility of lung ECs (13).
During our investigation into mechanism(s) of EC lamellipodia formation, we observed that down-regulation of sphingosine kinase (SphK) 1 or sphingosine-1-phosphate (S1P) transporter, Spns2, partially abrogated HGF-induced lamellipodia formation. Further, inhibiting c-Met tyrosine kinase with SU11274 attenuated intracellular S1P levels in lung ECs. These results suggested a cross-talk and potential interaction between HGF/c-Met and SphK/S1P/S1P receptor (S1PR) signaling axis in lamellipodia formation and EC motility; however, the mechanism(s) of regulation of HGF-mediated lamellipodia formation and EC migration by SphK/S1P/S1PR signaling axis is unclear. Here, we determined the role of SphK1, Spns2, and S1PR in HGF-induced lamellipodia formation in lung ECs. Our results demonstrated that HGF/c-Met signaling stimulated phosphorylation of ERK1/2, which in turn phosphorylated SphK1. Both ERK1/2 and SphK1, but not SphK2, were localized in lamellipodia. Further, HGF stimulation of lung ECs increased intracellular S1P levels via activation of SphK1 and blocking SphK1 or Spns2 abrogated HGF-induced lamellipodia formation. Additionally, knocking down of S1P1, but not S1P2 or S1P3, with siRNA attenuated HGF-induced lamellipodia formation. Collectively, these results identify cross-talk between HGF/c-Met and SphK1/Spns2/S1P signaling as a novel pathway of lamellipodia formation and perhaps EC motility.
Results
HGF Induces Phosphorylation of SphK1 and S1P Generation in HLMVECs
In our recent study, we have shown a significant role for HGF-induced c-Met/PI3K/Akt signaling in NADPH oxidase activation and ROS generation in lamellipodia formation and motility of human lung ECs (13). HGF binding to c-Met triggers a cascade of signal transduction pathways including transactivation of G protein-coupled S1P receptors (8); however, the role of this transactivation in lamellipodia formation and cell motility is unclear. We therefore determined the role of HGF/c-Met signaling axis in SphK-mediated S1P generation in lamellipodia formation and EC motility. HLMVECs expressed both the isoforms of SphK, SphK1 and SphK2; however, stimulation with HGF (20 ng/ml) resulted in a time-dependent phosphorylation of SphK1 (Ser225) with no detectable phosphorylation of SphK2 (Thr578) (Fig. 1, A and B). The HGF-induced activation of SphK1 resulted in enhanced accumulation of intracellular S1P levels, which was blocked by PF-543 (1 μm), a potent and specific inhibitor of SphK1 (14) (Fig. 1C). HGF enhanced intracellular S1P generation as early as 2 min post-challenge and peaked at 10 min followed by a gradual decline in accumulation (Fig. 1C). Further, cell lysates from HGF-stimulated HLMVECs exhibited enhanced SphK1 activity compared with control cells in vitro, which was blocked by the SphK1 inhibitor PF-543 (Fig. 1D). These results suggest that phosphorylation and activation of SphK1 by HGF enhances accumulation of S1P in HLMVECs.
FIGURE 1.

Effect of HGF on activation of SphK1 and S1P generation. Serum-deprived HLMVECs (∼90% confluence) were exposed to 20 ng/ml HGF for 2–30 min. A, cell lysates (30 μg of protein) were subjected to SDS-PAGE Western blotted for phosphorylation of SphK1 and SphK2 and total SphK1 and SphK2. Shown is a representative blot of three independent experiments in triplicate. Note the increase in SphK1 phosphorylation at 10, 15, and 30 min of HGF stimulation. B, graph represents quantification of the Western blots, and the data are means ± S.E. and expressed as the ratios of phospho-SphK1 or SphK2 to total protein. *, significantly different from cells not exposed to HGF (p < 0.01). C, HLMVECs (∼90% confluence) were pretreated with SphK1 inhibitor PF-543 in DMSO (1 μm) or DMSO (1: 2000) for 60 min and then challenged with HGF (20 ng/ml) for varying periods of time. Intracellular S1P was measured by LC-MS/MS as described under “Experimental Procedures.” In addition to 0 time, basal S1P levels at 15 and 30 min without HGF treatment were measured, and they were not significantly different from the 0 min. Therefore, basal S1P level at 0 time without HGF addition is shown. The data are means ± S.E. of three independent experiments. *, p < 0.05 in cells exposed to HGF; #, p < 0.05 in cells pretreated with PF-543 and exposed to HGF. PF-543, a SphK1 inhibitor, blocked HGF-induced SphK1 phosphorylation. D, HLMVECs grown to ∼90% confluence were pretreated with vehicle (DMSO 1:2000) or PF-543 (1 μm) for 60 min and challenged with HGF (20 ng/ml) for 15 min. SphK activity in cell lysates was carried out as described under “Experimental Procedures.” *, p < 0.05 compared with vehicle control without sphingosine supplement; **, p < 0.001 compared with vehicle control with sphingosine supplement; #, p < 0.05 compared with HGF treatment without sphingosine supplement; ##, p < 0.001 compared with HGF treatment in the presence of sphingosine.
HGF Enhances Co-localization of SphK1 and p-SphK1 with Actin and Cortactin in Lamellipodia of Lung ECs
We have earlier demonstrated that HGF stimulated reorganization and co-localization of actin and cortactin in lamellipodia of HLMVECs (13); however, the role of SphK1 in HGF-mediated lamellipodia formation is not well defined. Therefore, we hypothesized that HGF-induced lamellipodia formation, in part, is dependent on SphK1 activation and its redistribution to lamellipodia and co-localization with actin and cortactin cytoskeleton at cell periphery. Cells challenged with vehicle revealed diffused SphK1, actin, and cortactin staining; however, HGF stimulated F-actin (red) and SphK1 (green) (Fig. 2, A and B) and cortactin (green) and SphK1 (red) (Fig. 2, C and D) redistribution and co-localization (merge, yellow) to cell periphery. A similar co-localization of p-SphK1 (Ser225) with actin and cortactin in lamellipodia after HGF treatment was observed in HLMVECs (Fig. 3, A–D). These results suggest that HGF-induced phosphorylation of SphK1 may play a role in lamellipodia formation in human lung ECs.
FIGURE 2.
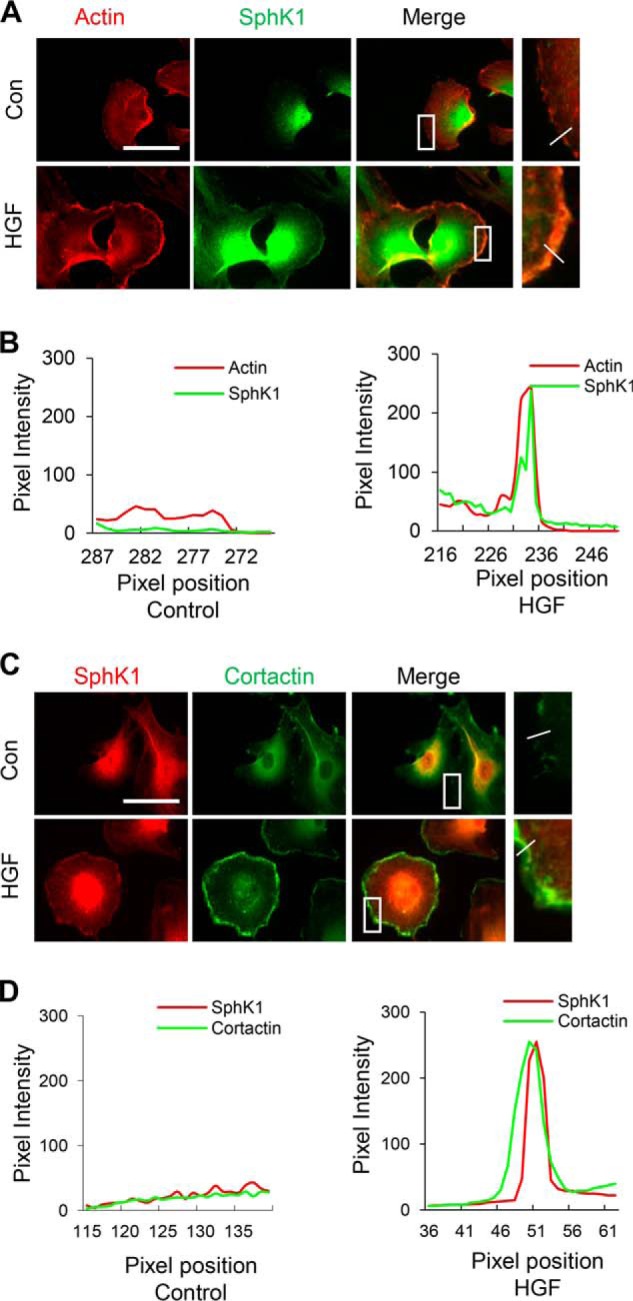
HGF induces SphK1 accumulation in lamellipodia. HLMVECs grown on slide chambers were treated with HGF (20 ng/ml) or PBS for 30 min and probed with anti-actin, anti-SphK1 and anti-cortactin antibodies, and lamellipodia was examined by immunofluorescence microscopy. A and C, co-localization of actin (red) and SphK1 (green) (A) or SphK1 (red) and cortactin (green) (C) to lamellipodia (merge, yellow) was visualized by immunofluorescent staining as described under “Experimental Procedures.” Shown are representative images from three independent experiments. Insets depict enhanced co-localization of actin and SphK1 or cortactin and SphK1 in lamellipodia after HGF treatment. Boxes are enlarged and shown on the right with a line drawn across the cell periphery. A–D, the intensity or distribution of SphK1 with actin (B) or cortactin (D) was quantified from the upper panels (A and C) using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition, and the results are representative of three independent experiments. Con, control.
FIGURE 3.
HGF stimulates SphK1 phosphorylation and accumulation at lamellipodia. HLMVECs grown on slide chambers were treated with HGF (20 ng/ml) or PBS for 30 min and probed with anti-actin, anti-p-SphK1(ser225), and anti-cortactin antibodies, and lamellipodia was examined by immunofluorescence microscopy with 60× oil objective. A and C, co-localization of actin (red) and p-SphK1 (green) (A) or p-SphK1 (red) and cortactin (green) to lamellipodia (merge, yellow) (C) was visualized by immunofluorescent staining as described under “Experimental Procedures.” Shown are representative images from three independent experiments. Insets depict enhanced co-localization of actin and p-SphK1 or cortactin and p-SphK1 in lamellipodia caused by HGF treatment. A–D, the intensity or distribution of p-SphK1 with actin (C) or cortactin (D) was quantified from the upper panels (A and C) using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition, and the results are representative of three independent experiments. Con, control.
Inhibition of c-Met Tyrosine Phosphorylation by SU11274 Attenuates HGF-mediated Phosphorylation of SphK1 and Its Co-localization with Actin in Lamellipodia
Binding of HGF to its receptor, c-Met, induces dimerization and autophosphorylation of specific tyrosine residues, resulting in enhanced kinase activity and activation of diverse intracellular signaling pathways including MAPKs, STAT3, Rac1, and Akt, which promote lamellipodia formation and cell motility. To determine whether phosphorylation of SphK1 by HGF and p-SphK1 co-localization in lamellipodia with actin is mediated by c-Met signaling axis, we used SU11274, an inhibitor of c-Met tyrosine phosphorylation (15). As demonstrated earlier (13), HGF induced c-Met phosphorylation at residues Tyr1234/1235 in HLMVECs (Fig. 3A), and pretreatment of cells with SU11274 (1 μm) for 1 h prior to HGF treatment (20 ng/ml, 15 min) significantly attenuated HGF-induced c-Met phosphorylation at residues Tyr1234/1235, SphK1 phosphorylation at Ser225, and ERK1/2 phosphorylation (Thr202/Tyr204) (Fig. 4, A and B). As a result of inhibition of SphK1, S1P generation induced by HGF was significantly attenuated by SU11274 (Fig. 4C). Furthermore, inhibition of c-Met phosphorylation by SU11274 attenuated phospho-SphK1 (green) co-localization with actin (red) in lamellipodia (merge, yellow) (Fig. 4D). Taken together, these results establish a key role for HGF/c-Met signaling in SphK1 phosphorylation and co-localization with actin in lamellipodia of HLMVECs.
FIGURE 4.

HGF-mediated c-Met activation is essential for SphK1 and ERK1/2 phosphorylation and S1P generation in HLMVECs. A and B, HLMVECs grown to ∼90% confluence on 35-mm dishes were pretreated with c-Met inhibitor SU11274 in DMSO (1 μm, 30 min) or DMSO (1:2000 dilution) followed by HGF (20 ng/ml) treatment for 30 min. Cell lysates (15–30 μg of protein) were subjected to 10% SDS-PAGE, probed with anti-phospho-SphK1 (Ser225), anti-phospho-c-Met (Tyr1234/1235), anti-phospho-ERK1/2 (Thr202/Tyr204), and anti-ERK antibodies as indicated, and fold changes normalized to total ERK were determined from the respective Western blots by ImageJ. Shown are representative blots from three independent experiments. The values are the means ± S.E. *, significantly different from control cells without SU11274 (p < 0.01); #, significantly different from HGF-treated cells without SU11274 (p < 0.01). C, HLMVECs grown to ∼90% confluence on 35-mm dishes were labeled with [32P]orthophosphate (20 μCi/ml) in DMEM-phosphate free medium for 3 h prior to treatment with SU11274 (1 μm, 30 min) followed by stimulation with vehicle or HGF (20 ng/ml) for 30 min. Lipids were extracted and analyzed for [32P]S1P accumulation in cells by thin layer chromatography as described under “Experimental Procedures.” The values are the means ± S.E. of three independent experiments in triplicate, and the S1P formed was normalized to 105 cells. D, HLMVECs grown on slide chambers to ∼95% confluence were pretreated with SU11274 (1 μm, 30 min) prior to stimulation with HGF (20 ng/ml) for 30 min and probed with anti-actin and anti-p-SphK1 (ser225) antibodies, and lamellipodia were examined by immunofluorescence microscopy with 60× oil objective. Co-localization of actin (red) and p-SphK1 (green) in lamellipodia (merge, yellow) was visualized by immunofluorescent staining. Shown are representative images from three independent experiments. Insets depict enhanced actin and p-SphK1 accumulation in lamellipodia that was blocked by SU11274 treatment of cells. E, HGF-induced co-localization of p-SphK1 with actin in the presence or absence of SU11274 was quantified using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition.
Inhibition of SphK1 Activity or Down-regulation of SphK1 Protein Expression Impairs HGF-mediated Lamellipodia Formation in Lung Ecs
Having established that SphK1/p-SphK1 is localized in lamellipodia in response to HGF treatment, we sought to determine whether SphK1 activity and/or protein expression is required for lamellipodia formation. Intrinsic SphK1 activity in HLMVECs was inhibited by PF-543, a specific inhibitor of SphK1, or SphK1 expression was down-regulated by siRNA transfection. As shown in Fig. 5 (A and B), pretreatment of HLMVECs with PF-543 (1 μm) for 1 h significantly attenuated HGF-induced lamellipodia formation as indicated by co-localization of actin (red) and cortactin (green) in the cell periphery (merge, yellow) by immunofluorescent staining. Similarly, knockdown of SphK1, but not SphK2, using specific siRNA significantly attenuated HGF-induced co-localization of actin (red) and cortactin (green) in lamellipodia (Fig. 5, C and D). Knockdown of SphK1 and SphK2 was confirmed by Western blotting (Fig. 5E). Furthermore, intrinsic SphK1 activity was inhibited by transfection of cells with catalytically inactive FLAG-tagged adenoviral SphK1 plasmid. In control cells, there was no difference between transfected and non-transfected cells in terms of cortactin localization; however, cells transfected with SphK1 mutant adenovirus did not show cortactin localization in lamellipodia after stimulation with HGF (data not shown). These results show a potential role for SphK1 activity and protein expression in HGF-induced lamellipodia formation in lung ECs.
FIGURE 5.

Inhibition of SphK1 activity or down-regulation of SphK1, but not SphK2, expression attenuates HGF-induced lamellipodia formation. A, HLMVECs grown on slide chambers to ∼90% confluence were pretreated with PF-543 (1 μm), a specific SphK1 inhibitor, for 1 h prior to stimulation with HGF (20 ng/ml) for 30 min and probed with anti-actin, anti-cortactin antibodies, and lamellipodia were examined by immunofluorescence microscopy with 60× oil objective. Co-localization of actin (red) and cortactin (green) in lamellipodia (merge, yellow) was visualized by immunofluorescent staining. Shown are representative images from three independent experiments. Insets depict enhanced actin and cortactin in lamellipodia that was blocked by PF-543. B, lamellipodia formation was quantified and expressed as a percentage of control. The values are the means ± S.E. of three experiments, and at least 20 cells were analyzed for each treatment. *, significantly different from control cells without HGF (p < 0.01); #, significantly different from HGF treated cells without PF-543 (p < 0.05). C, HLMVECs grown on slide chambers to ∼50% confluence were transfected with scrambled (sc), SphK1, or SphK2 specific siRNA for 48 h prior to challenge with HGF (20 ng/ml) for 30 min. Shown are representative images of three independent experiments. D, lamellipodia formation was quantified and expressed as a percentage of control. The values are the means ± S.E. of three experiments, and at least 20 cells were analyzed for each treatment. *, significantly different from scRNA transfected control cells (p < 0.01); **, significantly different from scRNA transfected cells challenged with HGF (p < 0.05); #, not significantly different from scRNA transfected cells stimulated with HGF (p > 0.05). E, HLMVECs grown on 35-mm dishes to 50% confluence were transfected with scrambled siRNA, SphK1, or SphK2 siRNA for 48 h as indicated in C, and cell lysates were subjected to 10% SDS-PAGE and probed with anti-SphK1, anti-SphK2 and anti-actin antibodies. Shown is a representative blot from three independent experiments. Transfection of HLMVECs with SphK1 and SphK2 siRNA down-regulated the expression of SphK1 and sphK2 proteins ∼75% compared with scRNA transfected cells, and knockdown of protein was specific for the two siRNAs. Con, control; Veh, vehicle.
Role of ERK1/2 and PI3K in HGF-mediated SphK1 Phosphorylation and Co-localization in Lamellipodia of HLMVECs
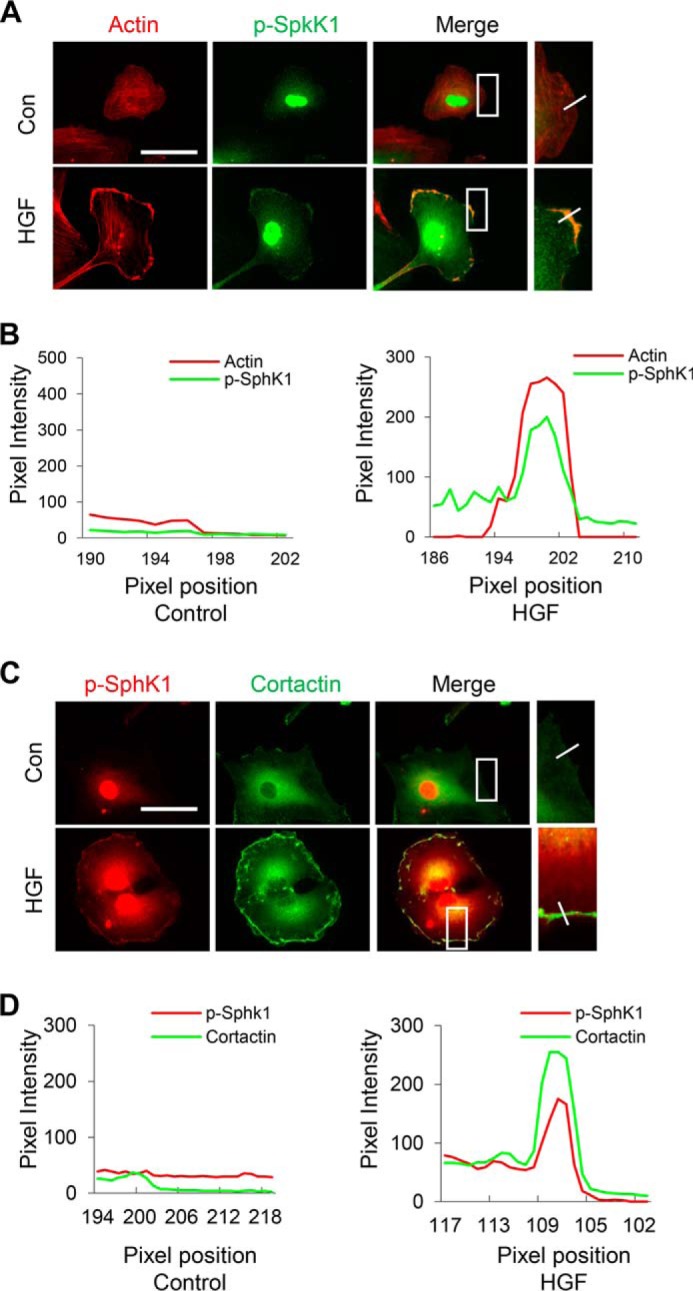
ERK1/2 has been shown to be phosphorylated in response to growth factors and localized to lamellipodial protrusion and adhesions (16). Also, there is evidence that ERK1/2 mediates phosphorylation of SphK1 at Ser225 and other Ser/Thr residues in mammalian cells in response to external stimuli (17). To further investigate the role of ERK1/2 and PI3K in SphK1 phosphorylation and lamellipodia formation, we used PD98059, an ERK1/2 inhibitor, and LY294002, a PI3K specific inhibitor. PD98059 significantly attenuated HGF-induced co-localization of p-SphK1 (green) with cortactin (red) in lamellipodia (merge, yellow) and redistribution of p-SphK1 in cell periphery (Fig. 6, A and B), as well as phosphorylation of ERK1/2 and SphK1 (Fig. 7C). Furthermore, immunofluorescent staining showed enhanced co-localization of phospho-SphK1 with phospho-ERK1/2 at lamellipodia in response to HGF stimulation (Fig. 7, A and B). However, the PI3K inhibitor LY294002 had no effect on HGF-induced SphK1 or ERK1/2 phosphorylation (Fig. 7, E and F). These results indicate that ERK1/2, but not PI3K, mediates phosphorylation of SphK1 and its localization in lamellipodia.
FIGURE 6.
Blocking ERK activation with PD98059 attenuates HGF-induced SphK1 phosphorylation and localization in lamellipodia. A, HLMVECs grown on slide chambers to ∼90% confluence were pretreated with PD98059 (1 μm), a specific inhibitor that blocks Erk phosphorylation, for 1 h prior to stimulation with HGF (20 ng/ml) for 30 min, probed with anti-cortactin and anti-phospho-SphK1 (Ser225) antibodies, and lamellipodia were examined by immunofluorescence microscopy with 60× oil objective. Co-localization of cortactin (green) and p-SphK1 (green) in lamellipodia (merge, yellow) was visualized by immunofluorescent staining. Shown are representative images from three independent experiments. Insets depict enhanced cortactin and p-SphK1 in lamellipodia that was blocked by PD98059. B, the co-localization of cortactin and p-SphK1 in lamellipodia was quantified using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition. Con, control.
FIGURE 7.

HGF stimulates co-localization of p-SphK1 and p-ERK in lamellipodia and SphK1 phosphorylation is mediated by ERK and not PI3K in HLMVECs. A, HLMVECs grown on slide chambers to ∼90% confluence were treated with HGF (20 ng/ml, 30 min) or PBS and probed with anti-p-SphK1 (Ser225) and anti-p-Erk (Thr/Ser204) antibodies, and lamellipodia formation was examined by immunofluorescence microscopy with 60× oil objective. Co-localization of phospho-SphK1 and phospho-ERK was performed as described under “Experimental Procedures.” Co-localization of phospho-SphK1 (red) and phospho-ERK (green) in lamellipodia (merge, yellow) was visualized by immunofluorescent staining. Shown are representative images from three independent experiments. Insets depict enhanced co-localization of p-SphK1 and p-Erk in lamellipodia. B, the co-localization of phospho-SphK1 and phospho-ERK in lamellipodia was quantified from A using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition. C, HLMVECs grown to ∼90% confluence in 35-mm dishes were pretreated with PD98059 (1 μm, 30 min) followed by HGF (20 ng/ml, 30 min) treatment. The cell lysates were subjected to 10% SDS-PAGE and probed with anti-phospho-ERK1/2, anti-phospho-SphK1, anti-ERK1/2, and anti-SphK1 antibodies. Shown are representative blots from three independent experiments. D, phosphorylation of SphK1 and ERK was analyzed from C by ImageJ software. The values are the means ± S.E. *, significantly different compared with cells not stimulated with HGF (p < 0.01); #, significantly different in cells pretreated with PD98059 and exposed to HGF as compared cells exposed to HGF without the inhibitor (p < 0.05). E, HLMVECs grown to ∼90% confluence in 35-mm dishes were pretreated with LY294002 (1 μm), a PI3K inhibitor, for 30 min followed by HGF (20 ng/ml, 30 min) treatment. The cell lysates were subjected to 10% SDS-PAGE and probed with anti-phospho-Akt, anti-phospho-ERK1/2, anti-phospho-SphK1, anti-Akt, anti-ERK1/2, and anti-SphK1 antibodies. Shown is a representative blot from three independent experiments. F, phosphorylation of SphK1, ERK, and Akt was analyzed by image analysis of Western blots from E. The values are the means ± S.E. *, significantly different compared with HGF non-treated control cells (p < 0.01); #, significantly different compared with HGF challenged cells (p < 0.05); **, not significant compared with LY294002 treated cells exposed to HGF (p > 0.05). Con, control.
S1P Transporter, Spns2, Mediation Is Essential for HGF-induced Lamellipodia Formation in HLMVECs
Having established a role for SphK1 in HGF-induced lamellipodia formation, we next investigated whether Spns2, the S1P transporter, plays a role in HGF-induced lamellipodia formation and cell motility. As shown in Fig. 8 (A and B), vehicle-treated HLMVECs revealed a diffused cytosolic distribution of Spns2 (green) and HGF treatment induced redistribution of Spns2 to cell periphery and co-localization with actin (red) in lamellipodia (merge, yellow). To determine the role of Spns2 in lamellipodia formation and cell motility, Spns2 was down-regulated (>80%) by Spns2 specific siRNA for 72 h prior to HGF stimulation (Fig. 8E). Down-regulation of Spns2 with siRNA significantly inhibited HGF-induced lamellipodia formation, as determined by actin (red) and cortactin (green) co-localization in cell periphery (Fig. 8, C and D). We next investigated whether HGF-induced S1P generated inside the cell signals via inside-out mechanism using a specific anti-S1P antibody (18). Exogenous addition of anti-S1P monoclonal antibody (150 μg/ml) to the medium blocked HGF-induced lamellipodia formation compared with control HLMVECs treated with IgG (150 μg/ml) (Fig. 8, F and G).
FIGURE 8.

Spns2 and extracellular S1P are essential for HGF-induced lamellipodia formation in HLMVECs. A, HLMVECs grown to ∼90% confluence in slide chambers were treated with HGF (20 ng/ml) or PBS for 30 min and probed with anti-actin and anti-Spns2 antibodies, and lamellipodia formation was examined by immunofluorescence microscopy. Co-localization of actin (red) and Spns2 (green) in lamellipodia (merge, yellow) was visualized by immunofluorescent staining as described under “Experimental Procedures.” Insets depict enhanced co-localization of actin and Spns2 in lamellipodia after HGF challenge of HLMVECs. B, the co-localization of actin and Spns2 in lamellipodia was quantified from using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition. C, HLMVECs grown to ∼50% confluence on slide chambers were transfected with scrambled (sc) or Spns2 siRNA (100 nm, 48 h) prior to HGF (20 ng/ml) or PBS challenge for 30 min and probed with anti-actin and anti-cortactin antibodies, and lamellipodia formation was examined by immunofluorescence microscopy with 60× oil objective. Co-localization of actin (red) and cortactin (green) in lamellipodia (merge, yellow) was visualized by immunofluorescent staining. Shown are representative images of three independent experiments. D, lamellipodia was quantified using ImageJ software and expressed as a percentage of scRNA control cells. At least 20 cells were analyzed for each condition. The values are the means ± S.E. *, significantly different compared with scRNA control cells (p < 0.01); #, significantly different in Spns2 siRNA transfected cells stimulated with HGF as compared with scRNA cells stimulated with HGF (p < 0.05). E, HLMVECs grown on 35-mm dishes to 50% confluence were transfected with scRNA or Spns2 siRNA for 48 h as indicated in C, and cell lysates were subjected to 10% SDS-PAGE and probed with anti-Spns2 antibody. Shown is a representative blot from three independent experiments. Transfection of HLMVECs with Spns2 siRNA down-regulated ∼85% of native Spns2 protein expression compared with scsiRNA transfected cells. F, HLMVECs grown to ∼90% confluence on slide chambers were preincubated with IgG (150 μg/ml) or S1P neutralizing (150 μg/ml) antibody for 1 h prior to HGF (20 ng/ml) or PBS challenge for 30 min, cells were probed with anti-actin and anti-cortactin antibodies, and lamellipodia formation was examined by immunofluorescence microscopy with 40 × objective. Shown are representative images of three independent experiments. G, lamellipodia was quantified using ImageJ software and expressed as a percentage of IgG control cells. At least 20 cells were analyzed for each condition. The values are the means ± S.E. *, significantly different compared with IgG control cells (p < 0.05); #, significantly different compared with IgG HGF-treated cells (p < 0.01). Con, control.
Intracellular S1P levels in scrambled RNA- and Spns2 siRNA-transfected cells did not show significant difference in the absence of HGF; however, a statistically significant increase in intracellular S1P levels was observed between scrambled RNA-transfected and Spns2 siRNA-transfected cells after HGF treatment (Fig. 9A), indicating that HGF-induced intracellular S1P could not be effectively transported outside. No difference in S1P levels was seen in the medium in cells treated with scrambled or Spns2 siRNA in the absence or presence of HGF (data not shown). To investigate the role of Spns2 in cell motility, wound healing assay was performed with scrambled and Spns2 siRNA-transfected HLMVECs. As shown in Fig. 9 (B and C), HGF enhanced the process of wound closure compared with control cells, and knockdown of Spns2 significantly attenuated HGF-mediated wound healing. These results show an essential role of Spns2 in HGF-induced lamellipodia formation and cell motility in human lung ECs.
FIGURE 9.
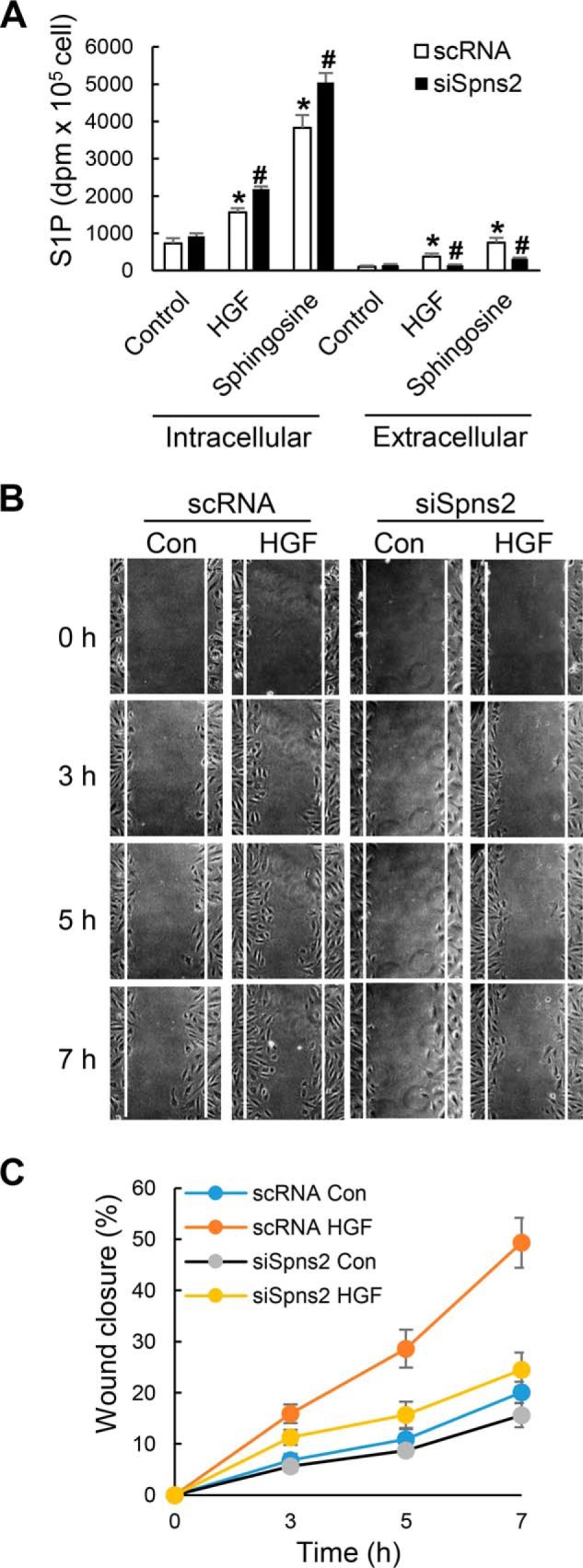
Spns2 siRNA blocks HGF-induced endothelial cell migration. A, HLMVECs grown to ∼50% confluence on 35-mm dishes were transfected with scrambled (sc) or Spns2 siRNA (100 nm, 48 h) as described under “Experimental Procedures.” After 48 h, the medium was aspirated, and cells were labeled with [32P]orthophosphate (20 μCi/ml) in DMEM phosphate-free medium for 3 h prior to stimulation with vehicle or HGF (20 ng/ml) for 30 min in the presence or absence of exogenous sphingosine (1 μm). The lipids were extracted from the media (extracellular) and cells (intracellular) and analyzed for [32P]S1P accumulation by thin layer chromatography as described under “Experimental Procedures.” The values are the means ± S.E. of three independent experiments in triplicate, and the S1P formed was normalized to 105 cells. *, significantly different from control cells not treated with HGF (p < 0.01); #, significantly different from cells transfected with scRNA and stimulated with HGF with or without exogenous sphingosine (p < 0.05). B and C, HLMVECs grown on 35-mm dishes to ∼50% confluence were transfected with sc or Spns2 siRNA as described in A for 48 h. The cells were wounded as described under “Experimental Procedures,” cell migration was viewed and imaged using a confocal microscope. Wound closure was calculated at 3-, 5-, and 7-h time points after the injury. The values are the means ± S.E. of three independent experiments in triplicate. The results show that knockdown of Spns2 with siRNA attenuated HGF-induced wound closure compared with scsiRNA-treated cells challenged with HGF. Con, control.
S1P1, but Not S1P2 or S1P3, Mediates HGF-induced Lamellipodia Formation in HLMVECs
S1P, generated in cells by SphK1/2, can signal intracellularly or extracellularly (inside-out mechanism) by ligation to cell surface G protein-coupled S1P1–5. Human lung ECs exhibit high expression of S1P1 and S1P3 (19). To further characterize the type of S1P receptor(s) involved in HGF-mediated lamellipodia formation, S1P1, S1P2, and S1P3 were down-regulated by specific siRNA, and knockdown of these receptors was confirmed by Western blotting (Fig. 10C). As expected, HGF stimulated redistribution of actin (red) and cortactin (green) to cell periphery and enhanced co-localization of actin and cortactin in lamellipodia (merge, yellow) (Fig. 10A). Furthermore, HGF-induced lamellipodia was inhibited in cells transfected with S1P1, but not S1P2 or S1P3, siRNA (Fig. 10B). These results suggest a role for S1P1 in mediating HGF-induced lamellipodia formation in human lung ECs.
FIGURE 10.

S1P1, but not S1P2 and S1P3, receptor mediates HGF-induced lamellipodia formation. A, HLMVECs grown to ∼50% confluence on 35-mm dishes were transfected with scrambled (sc), S1P1, S1P2, or S1P3 siRNA (50 nm) for 72 h. The cells were then challenged with HGF (20 ng/ml, 30 min) or PBS and probed with anti-actin and anti-cortactin antibodies, and lamellipodia was examined by immunofluorescence microscopy. A, co-localization of actin (red) and cortactin (green) to lamellipodia (merge, yellow) in S1P1 or S1P2 and S1P3 siRNA transfected cells. Shown are representative images from three independent experiments. B, lamellipodia were quantified using ImageJ software and expressed as percentages of scRNA control. At least 20 cells were analyzed for each condition. The values are the means ± S.E. *, significantly different compared with scRNA control cells (p < 0.01); #, significantly different in S1P1 siRNA transfected cells stimulated with HGF as compared with scsiRNA cells stimulated with HGF (p < 0.05). C, knockdown of S1P receptors by siRNA was confirmed by Western blotting of total cell lysates. Shown are representative blots from three independent experiments.
HGF Signaling Enhances Association between SphK1, S1P1, and Spns2 in HLMVECs
Given our results indicating that both Spns2 and SphK1 are localized in lamellipodia in response to HGF, we next investigated potential association between SphK1, S1P1, and Spns2 before and after HGF stimulation. HLMVECs were infected with control adenovirus or FLAG-tagged SphK1 adenovirus prior to HGF stimulation, cell lysates were subjected to immunoprecipitation with anti-FLAG antibody, and the immunoprecipitates were analyzed for co-immunoprecipitation of Spns2, S1P1, or FLAG tag. As shown in Fig. 11A, HGF challenge enhanced SphK1 interaction with S1P1 and Spns2 compared with vehicle treated cells. Similarly, HGF challenge increased co-immunoprecipitation of Spns2 and p-SphK1 in S1P1 immunoprecipitates (Fig. 11C) and S1P1 and p-SphK1 in Spns2 immunoprecipitates (Fig. 11E). A similar increase in co-localization of SphK1 with Spns2 and SphK1 with S1P1 in lamellipodia was observed by immunofluorescence after HGF stimulation of HLMVECs (Fig. 11, G and I). These data suggest that Spns2, SphK1, and S1P1 may be part of a protein platform in lamellipodia of human lung ECs.
FIGURE 11.

HGF enhances association between SphK1, Spns2, and S1P1 in HLMVECs. A, HLMVECs grown to ∼70% confluence on 100-mm dishes were infected with FLAG-tagged SphK1 adenovirus (10 multiplicity of infection) for 48 h, followed by HGF (20 ng/ml) challenge for 30 min. The cells were lysed in lysis buffer, and lysates (1 mg protein) were subjected to immunoprecipitation with anti-FLAG antibody and Western blotted with anti-FLAG, anti-S1P1, or anti-Spns2 antibody. Shown is a representative blot from three independent experiments. B, Western blots from A were subjected to image analysis, immunostaining was quantified, and data were normalized to FLAG pixels. The values are the means ± S.E. *, significantly different from cells not exposed to HGF. The results show enhanced association between SphK1, S1P1, and Spns2 after HGF stimulation (p < 0.05) compared with control cells without HGF treatment. C, HLMVECs grown to ∼90% confluence on 100-mm dishes were exposed to HGF (20 ng/ml) for indicated time points, cell lysates (1 mg of protein) were subjected to immunoprecipitation with anti-S1P1 antibody, and immunoprecipitates were subjected to Western blotting with anti-p-SphK1 (Ser225) and anti-Spns2 antibodies. Shown is a representative blot from three independent experiments. D, Western blots from C were subjected to image analysis, bands were quantified, and data were normalized to total S1P1 immunostaining. The values are the means ± S.E. *, significantly different from cells not exposed to HGF (p < 0.01); #, significantly different compared with control cells not stimulated with HGF (p < 0.05); **, significantly different compared with control cells not exposed to HGF (p < 0.001); ##, significantly different compared with control cells not stimulated with HGF (p < 0.005). The results show enhanced association of p-SphK1 and Spns2 in S1P1 immunoprecipitates after HGF stimulation of HLMVECs. E, HLMVECs grown to ∼90% confluence on 100-mm dishes were exposed to HGF (20 ng/ml) for indicated time points, cell lysates (1 mg protein) were subjected to immunoprecipitation with anti-Spns2 antibody, and immunoprecipitates were subjected to Western blotting with anti-p-SphK1 (Ser225), anti-S1P1, anti-phosphoserine, anti-phosphothreonine, and anti-Spns2 antibodies. Shown is a representative blot from three independent experiments. F, Western blots from E were subjected to image analysis, bands were quantified, and data were normalized to total Spns2 immunostaining. The values are the means ± S.E. *, significantly different from cells not exposed to HGF (p < 0.05); #, significantly different compared with control cells not stimulated with HGF (p < 0.01); **, significantly different compared with control cells not exposed to HGF (p < 0.001). The results show enhanced association of p-SphK1 and S1P1 in Spns2 immunoprecipitates after HGF stimulation of HLMVECs. Further, immunoprecipitation of Spns2 and immunostaining with anti-phosphoserine antibody confirmed that HGF stimulated Spns2 serine phosphorylation and not threonine phosphorylation. G, HLMVECs grown on slide chambers to ∼90% confluence were stimulated with HGF (20 ng/ml) for 30 min and probed with anti-SphK1, and anti-S1P1 antibodies, and lamellipodia was examined by immunofluorescence microscopy. Co-localization of SphK1 (red) and S1P1 (green) to lamellipodia (merge, yellow) is depicted. Insets depict enhanced co-localization of SphK1 with S1P1 in lamellipodia after HGF treatment. H, the co-localization of SphK1 with S1P1 was quantified using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition. I, HLMVECs grown on slide chambers to ∼90% confluence were stimulated with HGF (20 ng/ml) for 30 min and probed with anti-SphK1 and anti-Spns2 antibodies, and lamellipodia was examined by immunofluorescence microscopy. Co-localization of SphK1 (red) and Spns2 (green) to lamellipodia (merge, yellow) is depicted. Insets depict enhanced co-localization of SphK1 with Spns2 in lamellipodia after HGF treatment. J, the co-localization of SphK1 with Spns2 was quantified using ImageJ software and expressed as relative pixel intensity. At least 20 cells were analyzed for each condition. Con, control; IB, immunoblot.
Akt, but Not ERK1/2, Mediates HGF-induced Spns2 Phosphorylation in HLMVECs
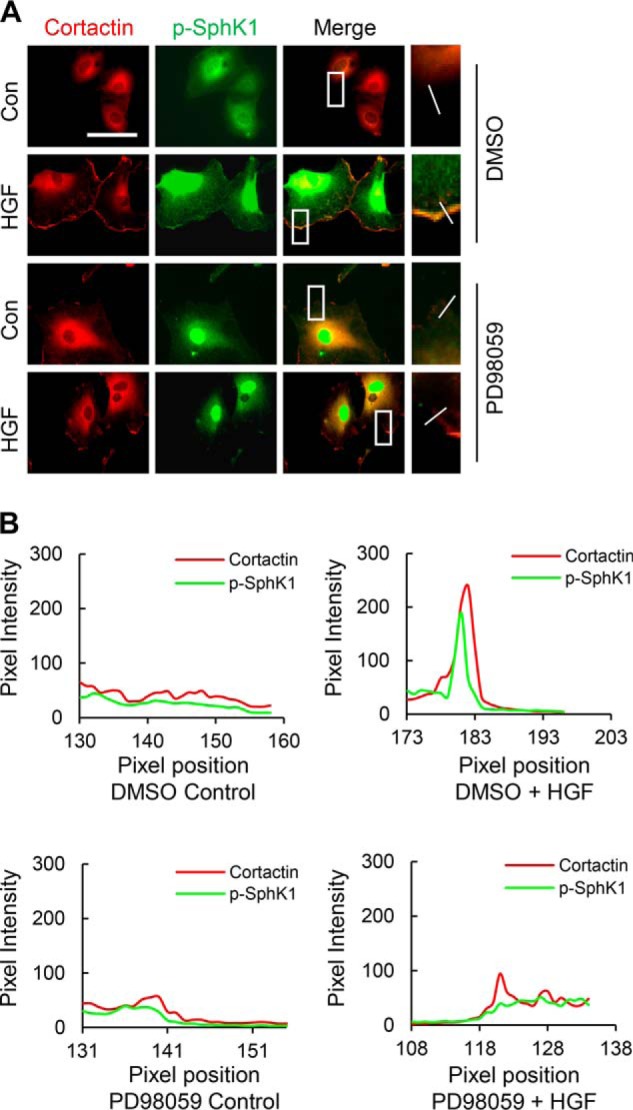
We have previously reported that PI3K/Akt pathway activated by HGF mediated lamellipodia formation in HLMVECs (13); however, the present data did not show involvement of PI3K/Akt in SphK1 phosphorylation. Therefore, we hypothesized that PI3K/Akt pathway may be regulating lamellipodia formation by phosphorylation of Spns2. Exposure of HLMVECs to HGF stimulated phosphorylation of Akt (Fig. 7E) and the S1P transporter Spns2 at the serine residue (Fig. 12A). Interestingly, pretreatment of cells with Akt inhibitor VIII significantly blocked HGF-induced Spns2 phosphorylation (Fig. 12A). However, pretreatment of cells with the ERK inhibitor PD98059 had no effect on Spns2 phosphorylation (Fig. 12C). Additionally, we investigated effect of Akt inhibitor VIII on lamellipodia formation in response to HGF treatment of HLMVECs. As expected, the Akt inhibitor VIII significantly attenuated HGF-induced lamellipodia formation. These results indicate that HGF stimulates SphK1 phosphorylation via ERK and Spns2 phosphorylation via Akt pathway in lung ECs.
FIGURE 12.

Akt, but not ERK, mediates Spns2 phosphorylation. A, HLMVECs grown to ∼90% confluence on 100-mm dishes were pretreated with Akt inhibitor VIII (1 μm, 30 min) or ERK inhibitor PD98059 (1 μm, 30 min), followed by HGF stimulation (20 ng/ml) for 30 min). Cell lysates (1 mg protein) were subjected to immunoprecipitation with anti-Spns2 antibody, and the immunoprecipitates were run on 10% SDS-PAGE and Western blotted with anti-Spns2 and anti-phosphoserine antibodies. Shown are representative blots from three independent experiments. B, Western blots from A were subjected to image analysis, and the bands were quantified. The values are the means ± S.E. *, significantly different from control cells not stimulated with HGF (p < 0.05); #, significantly different in cells pretreated with Akt inhibitor and stimulated with HGF as compared with cells stimulated with HGF in the absence of Akt inhibitor (p < 0.01). C, HLMVECs grown to ∼90% confluence in 100-mm dishes were pretreated with the ERK inhibitor PD98059 (1 μm, 30 min) followed by HGF stimulation (20 ng/ml) for 30 min). The cell lysates (1 mg of protein) were subjected to immunoprecipitation with anti-Spns2 antibody, and the immunoprecipitates were run on 10% SDS-PAGE and Western blotted with anti-Spns2 and anti-phosphoserine antibodies. Shown are representative blots from three independent experiments. D, Western blots from C were subjected to image analysis, and the bands were quantified. The values are the means ± S.E. *, significantly different from control cells not stimulated with HGF (p < 0.05). E, HLMVECs grown to ∼90% confluence in slide chambers were pretreated with Akt inhibitor VII(1 μm, 30 min) prior to HGF (20 ng/ml) or PBS treatment for 30 min and probed with anti-actin and anti-cortactin antibodies, and lamellipodia formation was examined by immunofluorescence was visualized by immunofluorescent staining as described under “Experimental Procedures.” Shown is a representative image from three independent experiments. F, lamellipodia was quantified from E using image analysis software and expressed as a percentage of control. At least 20 cells were analyzed for each condition. The values are the means ± S.E. *, significantly different in cells stimulated with HGF (p < 0.05); #, significantly different in Akt inhibitor treated cells stimulated with HGF as compared with cells without Akt inhibitor (p < 0.01). Con, control; IB, immunoblot; Inh, inhibitor.
Discussion
Disruption of the endothelial barrier integrity in the pulmonary circulation is evidenced in acute lung injury and is related to widespread alveolar edema and consequent life-threatening hypoxemia. Re-establishing endothelial integrity is critical in restoring proper gas exchange. However, the detailed molecular mechanisms regulating resealing of gaps and restoration of barrier integrity are yet to be fully defined. Because lamellipodial protrusions generated at the leading edge of cells are important for closing the gaps, in the current study we extend our previous findings on the role of HGF/c-Met signaling axis in augmenting lamellipodia formation and cell motility in vivo and in vitro (13) and show a role for SphK1/S1P signaling in lamellipodia formation in lung ECs by HGF. Our results demonstrate that HGF/c-Met signaling stimulated SphK1 and Spns2 phosphorylation by ERK1/2 and Akt, respectively, and blocking SphK1 or down-regulation of SphK1 or Spns2 with siRNA attenuated HGF-mediated lamellipodia formation.
The first important and novel aspect of the present study is the activation of SphK1/S1P signaling axis by HGF ligation to c-Met in regulation of lamellipodia formation and EC motility. We have previously reported that HGF-induced c-Met tyrosine phosphorylation stimulated canonical activation of PI3K/Akt pathway required for p47phox/Cortactin/Rac1 co-localization and ROS generation in lamellipodia of lung ECs (13). Our results show that HGF/c-Met signaling also promotes phosphorylation and activation of SphK1 and enhanced S1P generation in lung ECs as treatment of cells with SphK1 siRNA or PF-543, a specific inhibitor of SphK1, decreased S1P levels in the cell and lamellipodia formation. The intracellularly generated S1P is transported out of the cell by ATP binding cassette transporters (20) and/or by the recently identified S1P transporter, Spns2 (21). The functional link between S1P inside-out transport and transactivation was evident because silencing of S1P1 expression with siRNA significantly blunted HGF-induced lamellipodia formation; however, knockdown of S1P2 or S1P3 with siRNA had no significant effect on HGF-mediated lamellipodia formation. These results suggest a transactivation signaling mechanism in operation wherein HGF challenge promotes enhanced S1P generation via SphK1, which signals extracellularly via G protein-coupled S1P1. Activation of SphK1 regulated HGF-induced migration of ECV304 transformed ECs, which was pertussis toxin-insensitive, suggesting non-involvement of Gi-coupled S1P1 signaling in migration (22). This is in contrast to our current observations related to the role of S1P1 in HGF-induced lamellipodia formation. Cross-talk between G protein-coupled receptors and receptor tyrosine kinases is a well established concept in signal transduction (23). Epidermal growth factor receptor, Her2/neu, PDGF-β receptor, insulin-like growth factor-1 receptor, and vascular endothelial growth factor receptors have been shown to be transactivated by lysophosphatidic acid (24–27) and S1P (26, 28–30); however, transactivation of G protein-coupled receptors by receptor tyrosine kinases is under studied. In a recent investigation, HGF/c-Met signaling axis augmented vascular barrier integrity in vitro and in vivo via transactivation of S1P1 and integrin β4, which involved recruitment of c-Met, S1P1, and integrin β4 to caveolin-enriched lipid rafts of lung ECs (8). However, the role of SphK1 in transactivation of S1P1 and integrin β4 was not established (8).
The interdependence between HGF/c-Met signaling and S1P1 is further evidenced by the fact that knockdown of Spns2 abolished lamellipodia formation in HLMVECs (Fig. 8). In many cells including ECs, the intracellularly generated S1P is secreted out via ATP binding cassette transporters (31–33) or S1P specific Spns2 transporter (34–36). The mechanism by which HGF/c-Met signaling modulates Spns2 is unknown. We show here for the first time that stimulation of HLMVECs with HGF enhanced the phosphorylation of Spns2 at serine and threonine but not tyrosine residue(s) (Fig. 11). Further, this phosphorylation was mediated by PI3K and not ERK1/2 as evidenced by studies with Akt and ERK1/2 inhibitors. Inhibition of Akt with the Akt inhibitor not only blocked Spns2 phosphorylation but also reduced HGF-induced lamellipodia formation (Fig. 12). On the contrary, Spns2 knockdown enhanced non-small cell lung carcinoma cell migration, which was reversed by SphK inhibition by SkI-1 (37), thereby suggesting that accumulation of intracellular S1P as a major factor responsible for enhanced cell migration after Spns2 knockdown. Interestingly, knockdown of Spns2 in non-small cell lung carcinoma cells down-regulated S1P1and S1P3 expression and up-regulated PI3K/Akt and JNK/STAT3 pathways, accounting for the increased cell migration. In contrast to A549 cells, migration of lung ECs required Spns2-mediated transport of intracellular S1P to extracellular milieu for signaling via S1PRs, especially S1P1. It is unclear whether Spns2 knockdown in lung ECs also modulate expression of S1PRs. Further, in this study we have not demonstrated a direct link between the Spns2 phosphorylation to lamellipodia formation, and mapping of the Akt dependent serine/threonine phosphorylation sites on Spns2 will be necessary to generate site-specific mutants to link specific serine/threonine phosphorylation to lamellipodia formation. Further, using anti-S1P antibody to neutralize S1P, we have demonstrated that extracellular S1P released from HLMVECs via Spns2 was critical to HGF-mediated lamellipodia formation. In addition, our results suggest potential interactions between SphK1, Spns2, and S1P1 that facilitate the inside-out signaling of secreted S1P in an autocrine or paracrine fashion. However, the co-immunoprecipitation and pulldown data do not conclusively prove that these proteins bind to each other. In fact, it is possible that these proteins are somehow interconnected by binding to cytoskeletal elements such as actin and/or cortactin. Further studies using recombinant proteins and FRET analysis are necessary to prove potential direct interaction between SphK1, Spns2, and S1P1 in the lamellipodia after stimulation with HGF.
Another interesting finding of this study is involvement of two independent kinases mediating HGF-induced phosphorylation of SphK1 and Spns2 in HLMVECs. ERK plays an important role in SphK1 activation. The ERK pathway is one of the principle signaling cascades by which cells respond to extracellular and intracellular cues. ERK is known to be required for membrane ruffling and extension (16, 38). There is evidence for localization of ERK in lamellipodia in response to growth factors. ERK regulates protrusion via its phosphorylation and activation of focal adhesion proteins and WAVE2 regulatory complex (16). We found that HGF activates ERK as evidenced by ERK phosphorylation. Inhibition of ERK by PD98059 significantly inhibited SphK1 phosphorylation at Ser225. In addition to ERK, SphK1 is also phosphorylated by PKC. Interestingly, in a recent study mouse SphK1 was negatively regulated by PKCα-dependent phosphorylation of Ser373 residue (39). Thus, phosphorylation of SphK1 at different residues may have opposing effects on its activity, which needs further investigation. Very little is known regarding the role of Spns2 phosphorylation in cell migration. Unlike SphK1 phosphorylation, Spns2 phosphorylation is regulated by PI3K/Akt pathway. We found that Akt inhibitor significantly reduced Spns2 phosphorylation, but not SphK1 phosphorylation. Further, inhibition of PI3K activity by LY294002 blocked Akt phosphorylation, but not ERK. Nonetheless, inhibition of c-Met by SU11274 inhibited both ERK and SphK1 phosphorylation and lamellipodia formation.
In summary, our findings support the transactivation of SphK1/S1P/S1P1 pathway by HGF/c-Met signaling axis in lamellipodia formation and motility of lung ECs (35). These findings also support a critical role of the S1P transporter, Spns2, and potential interaction and functional link between SphK1, Spns2, and S1P1 in HGF-mediated lamellipodia formation (Fig. 13). The importance of our findings on SphK1, Spns2, and S1P1 attests to the significance of sphingolipids and sphingolipid metabolizing enzymes in lung inflammation and injury in respiratory diseases and development of drugs against sphingolipid metabolizing enzyme(s) for intervention.
FIGURE 13.
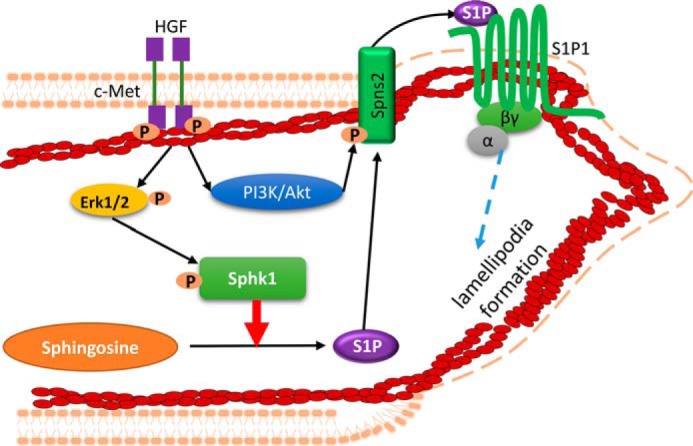
Proposed model on the cross-talk between HGF/c-Met and SphK1/S1P/Spns2/S1P1 signaling axis in HGF-mediated lamellipodia formation. In this model, HGF initiates activation of its receptor c-Met, which initiates phosphorylation of ERK and Akt. Activation of ERK by HGF stimulates SphK1 phosphorylation, which in turn converts sphingosine into S1P. HGF-mediated activation of PI3K/Akt pathway phosphorylates S1P transporter, Spns2, which facilitates efflux of intracellular S1P to extracellular milieu. The secreted S1P binds to its receptor S1P1 to initiate downstream pathways mediating lamellipodia formation of lung ECs. This proposed schema is a static model and does not convey the possibility that in a dynamic model the interacting partners might function differently over time.
Experimental Procedures
Materials
HLMVECs (catalog no. cc-2583, lot number 441097) and endothelial basal medium (EBM-2) (catalog no. cc-3162) were obtained from Lonza (San Diego, CA). Gold antifade mounting medium (catalog no. P36935), Hoechst and precast Tris-glycine PAGE, Alexa Fluor 488, Alexa Fluor 568, Alexa Fluor Phalloidin 568, were procured from Life Technologies Inc. HGF was from PeproTech (Rocky Hill, NJ). Antibodies of phospho-SphK1 (Ser225) (catalog no. SP1641) and phospho-SphK2 (Thr578) (catalog no. SP4631) were from ECM Biosciences. SphK1 (catalog no. ab16491) and SphK2 (ab37977) antibodies were from Abcam. Spns2 (catalog no. SAB1304232), phospho-Ser antibodies (catalog no. T1325), ERK inhibitor PD98059 (catalog no. P215), and PI3K inhibitor LY294002 (catalog no. L9908) were from Sigma. Cortactin antibody (catalog no. 05-180) was from Millipore (Bedford, MA). Phospho-ERK (Thr202/Tyr204) (catalog no. 4370S), phospho-c-Met (Tyr1234/1235) (catalog no. 3126) antibodies, and cell lysis buffer (catalog no. 9803) were from Cell Signaling Technology (Danvers, MA). Antibodies for S1P1 (catalog no. LS-C118016-100), S1P2 (catalog no. LS-C47405-100), and S1P3 (catalog no. LS-C146684-1) were from Life Span Biosciences (Seattle, WA). Anti-S1P antibody was provided by Dr. Roger A. Sabbadini of Lpath Inc. (San Diego, CA). Scrambled siRNA, siRNA for SphK1 (catalog no. sc-156038), SphK2 (catalog no. sc-39225), and Spns2 (catalog no. sc-106749) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). RNA transfection reagent Gene Silencer (T500020) was from Genlantis (San Diego, CA). SU11274 (catalog no. S1080) was purchased from Selleck Chemicals (Houston, TX). Bovine serum albumin (sc-2323) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Akt inhibitor VIII (catalog no. ENZ-CHM125–0001) was from Enzo Life Sciences Inc. (Farmingdale, NY).
Cell Culture
HLMVECs were cultured in complete growth medium containing endothelial growth medium-2 (EGM-2) with 10% fetal bovine serum and growth factors provided as a kit by the supplier (Lonza, San Diego, CA). The cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air as previously described (13). Endothelial cells were used at passages 5–7, and on the day prior to experimentation, the cells were incubated in EGM-2 containing 2% FBS.
Analysis of Cellular S1P by LC-MS/MS
Total lipids from HLMVECs were extracted under acidic conditions using 0.1 m HCl and C17-S1P, which was added as an internal standard during the lipid extraction step (18, 40). The lipid extracts were dissolved in ethanol (200 μl), and aliquots were analyzed for total lipid phosphorus and then subjected to LC-MS/MS for quantification of S1P as described previously (18, 40).
Intracellular and Extracellular Levels of [32P]S1P Formation in HLMVECs Stimulated by HGF
HLMVECs in 35-mm dishes, transfected with either scrambled or Spns2 siRNA, were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h. The radioactive medium was aspirated, 1 ml of EGM-2 containing 0.1% fatty acid-free BSA was added, and cells were exposed either to vehicle or HGF (20 ng/ml) in the presence or absence of sphingosine for 30 min. Lipid labeling was terminated by the addition of 100 μl of 1 m HCl, followed by 2 ml of methanol. The cells were harvested with a cell scraper, and the lipids were partitioned after vortexing into the chloroform phase by the addition of 2 ml of chloroform and 700 μl of 1 m HCl to get a final ratio of 1:1:0.9 (v/v/v) of chloroform:methanol:acidic aqueous phase. The total lipid extracts were dried under N2 and subjected to TLC and autoradiography; the area corresponding S1P was excised, and radioactivity was determined by liquid scintillation counting (41). The data were normalized to total radioactivity in 105 cells.
SphK Activity Assay
HLMVECs grown to ∼90% confluence in 35-mm dishes were challenged with EBM-2 medium alone or medium containing HGF (20 ng/ml) in the presence of 0.1% fatty acid free BSA for 15 min. The cells were rinsed in ice-cold PBS, scraped, and centrifuged at 1000 × g for 10 min. The pellet was resuspended in 1 ml of 5 mm MOPS buffer (pH 7.4) containing 1 mm EDTA, 0.25 m sucrose, 1 mm PMSF, and 10% glycerol (v/v) sonicated with a probe sonicator (3 × 20 s), centrifuged at 5000 × g at 4 °C for 5 min, and the supernatants were used for SphK activity in vitro assay. The SphK activity was carried out in 10 mm HEPES buffer (pH7.4) containing 10 mm MgCl2, 5 mm β-glycerophosphate, 2 mm dithiothreitol, 0.1 mm Na3VO4, 100 μm Triton X-100, and 25 μg of total cell lysate protein. The reaction was initiated by the addition of the substrate, sphingosine (5 μm) in 0.1% fatty acid-free BSA and [γ-32P]ATP (200 μm; specific activity 2200 dpm/pmol) in a final volume of 0.1 ml. The reaction was terminated by the addition of 1 ml of methanol:HCl (100:1, v/v), and lipids were extracted by the addition of 1 ml of chloroform and 0.8 ml of 0.1 n HCl (41). The tubes were thoroughly mixed and centrifuged, and the lower chloroform phase was analyzed for [32P]S1P formed by thin layer chromatography (41). The plates were exposed to X-ray film, and radioactivity associated with S1P was quantified by scintillation counting.
Transfection of HLMVECs with Small Interfering RNA
Depletion of endogenous SphK1, SphK2, S1P1, S1P2, S1P2, and Spns2 proteins in HLMVECs was carried out using gene-specific siRNA as described previously (13, 42). In brief, predesigned siRNA of human SphK1, SphK2, S1P1, S1P2, S1P3, Spns2, or nonspecific/non-targeting siRNA were used to transfect HLMVECs (passage of 5–7) for 72 h. Each siRNA contained at least three different sequences targeting the mRNA of each gene. Prior to transfection, the cells were starved in EBM-2 basal medium containing 2% BSA for 24 h. The next day, 50 nm scrambled or gene specific siRNA complexes were prepared in Gene Silencer transfection reagent according to the manufacturer's recommendation, and the cells were transfected in serum-free medium for 4 h, and the medium was replaced with fresh complete EBM-2 medium supplemented with 10% FBS and growth factors, and the cells were stimulated with HGF 72 h post-transfection.
Immunoblotting and Immunoprecipitation
Immunoblotting and immunoprecipitation (IP) of cell lysates were performed as described previously (43). In brief, after appropriate treatments, the cells were pelleted in ice-cold PBS, lysed in standard lysis buffer (Cell Signaling), and sonicated. The lysates were then centrifuged at 1000 × g for 10 min at 4 °C. Supernatants were collected and protein assayed using BCA protein assay kit. For IP, equivalent amounts of protein (1 mg) from each sample were precleared with control IgG conjugated to A/G-agarose beads at 4 °C for 1 h and centrifuged at 1000 × g for 10 min in a microcentrifuge, and supernatants were collected and incubated overnight with primary antibody conjugated to A/G-agarose beads at 4 °C. After 18–24 h, the samples were centrifuged at 1000 × g for 5 min in a microcentrifuge centrifuge, and the pellets containing the agarose beads were washed three times with lysis buffer at room temperature. After brief centrifugation at 1,000 × g for 5 min, the beads were collected by removing supernatant buffer, and 40 μl of SDS sample buffer (100 mm Tris-HCl, pH 6.8, 4% SDS, 0.1% bromphenol blue, 20% glycerol, 200 mm DTT) were added to the beads and boiled. The lysates were then subjected to 10% SDS-PAGE followed by Western blotting. Proteins were detected by immunoblotting using appropriate primary antibodies and HRP-conjugated anti-rabbit or anti-mouse secondary antibodies. Band intensities were quantified by densitometry using ImageJ software.
Immunofluorescence Staining
Human lung HLMVECs were fixed with 3.7% formaldehyde at room temperature for 10 min and permeabilized with 0.1% Triton X-100 at room temperature for 10 min. After washing with PBS three times, the cells were incubated with cortactin, Spns2, SphK1, p-SphK1, or p-Erk antibody (1:100 in 2% BSA/PBS) at room temperature for 1 h. The cells were then rinsed with PBS three times and subsequently incubated with respective secondary antibody conjugated with Alex Fluor 488/568 (1:2000 in 2% BSA/PBS) and Phalloidin 568 at room temperature for 1 h. The cells were then rinsed with PBS three times, coverslips were mounted with gold antifade mounting medium containing Hoechst for DAPI staining, and the cells were examined under Nikon Microscope with 60× oil immersion objective and Meta Vue software.
Quantification of Co-localization of Actin, Cortactin, and SphK1 in Lamellipodia
Co-localization of proteins in lamellipodia was quantified as described previously (13). Briefly, for each image, background signal was subtracted by drawing a region of interest around the cell periphery of individual cells. All areas outside the cell were cleared to best visualize the leading edges including cell periphery, and the fluorescence intensity within the entire cell was summed by MBF ImageJ bundle (Tony Collins, McMaster University and Wayne Rasband, National Institutes of Health). Pearson's correlation coefficient (PCC) was used as a statistic for quantifying co-localization. PCC was defined by the following formula,
| (Eq. 1) |
where Ri and Gi refer to the intensity values of the red and green channels, respectively, of pixel i, and [macron]R and [macron]G refer to the mean intensities of the red and green channels, respectively, across the entire image. Values near 1 reflect the fluorescence intensities of two images that are perfectly and linearly related to one another. Values near 0 reflect distribution of probes that are uncorrelated to one another.
Wound Healing Assay
Wound healing assay was performed as described previously (13, 18) to address effect of siSpns2 on tissue repair. In brief, scramble siRNA-transfected and Spns2 siRNA-transfected HLMVECs were inoculated and grown in a 6-well plate. After confluence, the cell monolayers were scratched along center of the culture well diameter by using a 200-μl pipette tip to create a cell-free area (13, 18). The cells were then treated with PBS or HGF (20 ng/ml) and maintained in growth medium that was supplemented with 2% FBS containing mitomycin C (1 μg/ml)) at 37 °C with 5% CO2. At the indicated time points (0, 3, 5, and 7 h) cells were scratched), and the scratched wound was visualized under a phase contrast microscope (Nikon Eclips TS2000-S), and imaged by a digital camera. Cell-free areas (at least 5–10 fields) were measured by using ImageJ software.
Statistical Analysis
The data are presented as group means ± S.E. Statistical significance was determined with the non-parametric Wold-Wolfowitz runs test. This statistical test was selected because it allows for the evaluation of two different kinds of statistical assumptions, binomial to determine whether co-localization is present or not and Poisson distribution to assess for the randomness of this co-localization. In all cases, statistical significance was defined at p < 0.05.
Author Contributions
P. F. conducted most of the experiments, analyzed the results, and wrote the first draft of the manuscript. I. A. B. analyzed the data of lamellipodia formation. E. V. B. conducted measurements of S1P generation after HGF treatment by LC-MS/MS. D. L. E. conducted experiment of interactions between SphK1 and Spns2. M. S. and A. H. analyzed Western blots and IP data. V. N. conceived the idea, discussed the experiments, conducted the [32P]S1P assay, and wrote and edited the manuscript.
Acknowledgments
We thank Dr. P. V. Usatyuk in helping quantification of lamellipodia and Dr. Prasad Kanteti for valuable suggestions and editing the manuscript. We thank Dr. Roger A Sabbadini of Lpath Inc. (San Diego, CA) for providing mouse anti-S1P monoclonal antibody.
This work was supported in part by National Institutes of Health Grant P01 HL060678 Project 4 (to V. N.) and Grant P01 HL098050 (to V. N.) and by the College of Medicine, University of Illinois. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EC
- endothelial cell
- HGF
- hepatocyte growth factor
- HLMVEC
- human lung microvascular endothelial cell
- S1P
- sphingosine-1-phosphate
- SphK1
- sphingosine kinase 1
- SphK2
- sphingosine kinase 2
- Spns2
- spinster homolog 2
- S1PR
- sphingosine-1-phosphate receptor
- PI3K
- phosphatidylinositol-3-kinase
- EBM
- endothelial basal medium
- IP
- immunoprecipitation
- ROS
- reactive oxygen species
- EGM-2
- endothelial growth medium 2
- PCC
- Pearson's correlation coefficient.
References
- 1. Lamalice L., Le Boeuf F., and Huot J. (2007) Endothelial cell migration during angiogenesis. Circ. Res. 100, 782–794 [DOI] [PubMed] [Google Scholar]
- 2. Mudau M., Genis A., Lochner A., and Strijdom H. (2012) Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc. J. Afr.. 23, 222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krause M., and Gautreau A. (2014) Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 15, 577–590 [DOI] [PubMed] [Google Scholar]
- 4. Fletcher D. A., and Mullins R. D. (2010) Cell mechanics and the cytoskeleton. Nature 463, 485–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nobes C. D., and Hall A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 6. Bussolino F., Di Renzo M. F., Ziche M., Bocchietto E., Olivero M., Naldini L., Gaudino G., Tamagnone L., Coffer A., and Comoglio P. M. (1992) Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol. 119, 629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cai W., Rook S. L., Jiang Z. Y., Takahara N., and Aiello L. P. (2000) Mechanisms of hepatocyte growth factor-induced retinal endothelial cell migration and growth. Invest. Ophthalmol. Vis. Sci. 41, 1885–1893 [PubMed] [Google Scholar]
- 8. Ephstein Y., Singleton P. A., Chen W., Wang L., Salgia R., Kanteti P., Dudek S. M., Garcia J. G., and Jacobson J. R. (2013) Critical role of S1PR1 and integrin β4 in HGF/c-Met-mediated increases in vascular integrity. J. Biol. Chem. 288, 2191–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu F., Schaphorst K. L., Verin A. D., Jacobs K., Birukova A., Day R. M., Bogatcheva N., Bottaro D. P., and Garcia J. G. (2002) Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3β. FASEB J. 16, 950–962 [DOI] [PubMed] [Google Scholar]
- 10. Webb C. P., Taylor G. A., Jeffers M., Fiscella M., Oskarsson M., Resau J. H., and Vande Woude G. F. (1998) Evidence for a role of Met-HGF/SF during Ras-mediated tumorigenesis/metastasis. Oncogene 17, 2019–2025 [DOI] [PubMed] [Google Scholar]
- 11. Organ S. L., and Tsao M. S. (2011) An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 3, S7–S19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Purdie K. J., Whitley G. S., Johnstone A. P., and Cartwright J. E. (2002) Hepatocyte growth factor-induced endothelial cell motility is mediated by the upregulation of inducible nitric oxide synthase expression. Cardiovasc. Res. 54, 659–668 [DOI] [PubMed] [Google Scholar]
- 13. Usatyuk P. V., Fu P., Mohan V., Epshtein Y., Jacobson J. R., Gomez-Cambronero J., Wary K. K., Bindokas V., Dudek S. M., Salgia R., Garcia J. G., and Natarajan V. (2014) Role of c-Met/phosphatidylinositol 3-kinase (PI3K)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation, and motility of lung endothelial cells. J. Biol. Chem. 289, 13476–13491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schnute M. E., McReynolds M. D., Kasten T., Yates M., Jerome G., Rains J. W., Hall T., Chrencik J., Kraus M., Cronin C. N., Saabye M., Highkin M. K., Broadus R., Ogawa S., Cukyne K., Zawadzke L. E., Peterkin V., Iyanar K., Scholten J. A., Wendling J., Fujiwara H., Nemirovskiy O., Wittwer A. J., and Nagiec M. M. (2012) Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 444, 79–88 [DOI] [PubMed] [Google Scholar]
- 15. Wang X., Le P., Liang C., Chan J., Kiewlich D., Miller T., Harris D., Sun L., Rice A., Vasile S., Blake R. A., Howlett A. R., Patel N., McMahon G., and Lipson K. E. (2003) Potent and selective inhibitors of the Met [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced tumor cell growth and invasion. Mol. Cancer Ther. 2, 1085–1092 [PubMed] [Google Scholar]
- 16. Mendoza M. C., Er E. E., Zhang W., Ballif B. A., Elliott H. L., Danuser G., and Blenis J. (2011) ERK-MAPK drives lamellipodia protrusion by activating the WAVE2 regulatory complex. Mol. Cell 41, 661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pitson S. M., Moretti P. A., Zebol J. R., Lynn H. E., Xia P., Vadas M. A., and Wattenberg B. W. (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 22, 5491–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berdyshev E. V., Gorshkova I., Usatyuk P., Kalari S., Zhao Y., Pyne N. J., Pyne S., Sabbadini R. A., Garcia J. G., and Natarajan V. (2011) Intracellular S1P generation is essential for S1P-induced motility of human lung endothelial cells: role of sphingosine kinase 1 and S1P lyase. PLoS One 6, e16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waeber C., Blondeau N., and Salomone S. (2004) Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect. 17, 365–382 [DOI] [PubMed] [Google Scholar]
- 20. Nishi T., Kobayashi N., Hisano Y., Kawahara A., and Yamaguchi A. (2014) Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochim. Biophys. Acta 1841, 759–765 [DOI] [PubMed] [Google Scholar]
- 21. Kawahara A., Nishi T., Hisano Y., Fukui H., Yamaguchi A., and Mochizuki N. (2009) The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 323, 524–527 [DOI] [PubMed] [Google Scholar]
- 22. Duan H. F., Wu C. T., Lu Y., Wang H., Liu H. J., Zhang Q. W., Jia X. X., Lu Z. Z., and Wang L. S. (2004) Sphingosine kinase activation regulates hepatocyte growth factor induced migration of endothelial cells. Exp. Cell Res. 298, 593–601 [DOI] [PubMed] [Google Scholar]
- 23. Liebmann C., and Böhmer F. D. (2000) Signal transduction pathways of G protein-coupled receptors and their cross-talk with receptor tyrosine kinases: lessons from bradykinin signaling. Curr. Med. Chem. 7, 911–943 [DOI] [PubMed] [Google Scholar]
- 24. Cerutis D. R., Dreyer A., Cordini F., McVaney T. P., Mattson J. S., Parrish L. C., Romito L., Huebner G. R., and Jabro M. (2004) Lysophosphatidic acid modulates the regenerative responses of human gingival fibroblasts and enhances the actions of platelet-derived growth factor. J. Periodontol. 75, 297–305 [DOI] [PubMed] [Google Scholar]
- 25. Gschwind A., Prenzel N., and Ullrich A. (2002) Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 62, 6329–6336 [PubMed] [Google Scholar]
- 26. Shida D., Kitayama J., Yamaguchi H., Yamashita H., Mori K., Watanabe T., and Nagawa H. (2005) Lysophospholipids transactivate HER2/neu (erbB-2) in human gastric cancer cells. Biochem. Biophys. Res. Commun. 327, 907–914 [DOI] [PubMed] [Google Scholar]
- 27. So J., Wang F. Q., Navari J., Schreher J., and Fishman D. A. (2005) LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2). Gynecol. Oncol. 97, 870–878 [DOI] [PubMed] [Google Scholar]
- 28. Endo A., Nagashima K., Kurose H., Mochizuki S., Matsuda M., and Mochizuki N. (2002) Sphingosine 1-phosphate induces membrane ruffling and increases motility of human umbilical vein endothelial cells via vascular endothelial growth factor receptor and CrkII. J. Biol. Chem. 277, 23747–23754 [DOI] [PubMed] [Google Scholar]
- 29. Sukocheva O., Wadham C., and Xia P. (2013) Estrogen defines the dynamics and destination of transactivated EGF receptor in breast cancer cells: role of S1P3 receptor and Cdc42. Exp. Cell Res. 319, 455–465 [DOI] [PubMed] [Google Scholar]
- 30. Tanimoto T., Lungu A. O., and Berk B. C. (2004) Sphingosine 1-phosphate transactivates the platelet-derived growth factor β receptor and epidermal growth factor receptor in vascular smooth muscle cells. Circ. Res. 94, 1050–1058 [DOI] [PubMed] [Google Scholar]
- 31. Nieuwenhuis B., Lüth A., Chun J., Huwiler A., Pfeilschifter J., Schäfer-Korting M., and Kleuser B. (2009) Involvement of the ABC-transporter ABCC1 and the sphingosine 1-phosphate receptor subtype S1P3 in the cytoprotection of human fibroblasts by the glucocorticoid dexamethasone. J. Mol. Med. 87, 645–657 [DOI] [PubMed] [Google Scholar]
- 32. Sato K., Malchinkhuu E., Horiuchi Y., Mogi C., Tomura H., Tosaka M., Yoshimoto Y., Kuwabara A., and Okajima F. (2007) Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J. Neurochem. 103, 2610–2619 [DOI] [PubMed] [Google Scholar]
- 33. Takabe K., Kim R. H., Allegood J. C., Mitra P., Ramachandran S., Nagahashi M., Harikumar K. B., Hait N. C., Milstien S., and Spiegel S. (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 285, 10477–10486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fukuhara S., Simmons S., Kawamura S., Inoue A., Orba Y., Tokudome T., Sunden Y., Arai Y., Moriwaki K., Ishida J., Uemura A., Kiyonari H., Abe T., Fukamizu A., Hirashima M., Sawa H., Aoki J., Ishii M., and Mochizuki N. (2012) The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest. 122, 1416–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mendoza A., Bréart B., Ramos-Perez W. D., Pitt L. A., Gobert M., Sunkara M., Lafaille J. J., Morris A. J., and Schwab S. R. (2012) The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2, 1104–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nijnik A., Clare S., Hale C., Chen J., Raisen C., Mottram L., Lucas M., Estabel J., Ryder E., Adissu H., Adams N. C., Ramirez-Solis R., White J. K., Steel K. P., Dougan G., and Hancock R. E. (2012) The role of sphingosine-1-phosphate transporter Spns2 in immune system function. J. Immunol. 189, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bradley E., Dasgupta S., Jiang X., Zhao X., Zhu G., He Q., Dinkins M., Bieberich E., and Wang G. (2014) Critical role of Spns2, a sphingosine-1-phosphate transporter, in lung cancer cell survival and migration. PLoS One 9, e110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mendoza M. C., Vilela M., Juarez J. E., Blenis J., and Danuser G. (2015) ERK reinforces actin polymerization to power persistent edge protrusion during motility. Sci. Signal. 8, ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oh Y. S., Bae S. S., Park J. B., Ha S. H., Ryu S. H., and Suh P. G. (2015) Mouse sphingosine kinase 1a is negatively regulated through conventional PKC-dependent phosphorylation at S373 residue. PLoS One 10, e0143695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berdyshev E. V., Goya J., Gorshkova I., Prestwich G. D., Byun H. S., Bittman R., and Natarajan V. (2011) Characterization of sphingosine-1-phosphate lyase activity by electrospray ionization-liquid chromatography/tandem mass spectrometry quantitation of (2E)-hexadecenal. Anal. Biochem. 408, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao Y., Kalari S. K., Usatyuk P. V., Gorshkova I., He D., Watkins T., Brindley D. N., Sun C., Bittman R., Garcia J. G., Berdyshev E. V., and Natarajan V. (2007) Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J. Biol. Chem. 282, 14165–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fu P., Mohan V., Mansoor S., Tiruppathi C., Sadikot R. T., and Natarajan V. (2013) Role of nicotinamide adenine dinucleotide phosphate-reduced oxidase proteins in Pseudomonas aeruginosa-induced lung inflammation and permeability. Am. J. Respir. Cell Mol. Biol. 48, 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Usatyuk P. V., Burns M., Mohan V., Pendyala S., He D., Ebenezer D. L., Harijith A., Fu P., Huang L. S., Bear J. E., Garcia J. G., and Natarajan V. (2013) Coronin 1B regulates S1P-induced human lung endothelial cell chemotaxis: role of PLD2, protein kinase C and Rac1 signal transduction. PLoS One 8, e63007. [DOI] [PMC free article] [PubMed] [Google Scholar]