

Abstract
Hypothiocyanite (OSCN−) serves as a potent innate defense system against microbes in the lungs. OSCN− is generated by the catalysis of peroxidases using thiocyanate transported via several anion transporters, including pendrin/SLC26A4 and hydrogen peroxide (H2O2) generated by Duox1 and Duox2. We previously demonstrated that expression of pendrin, peroxidases, and Duox1/Duox2 is up-regulated in bronchial asthma patients and/or asthma model mice and that these molecules are important in accelerating airway inflammation. However, it remained unclear how activating these molecules would lead to airway inflammation. In this study, we examined whether OSCN− produced via the pendrin/peroxidase/Duox pathway causes inflammation via airway epithelial cells. In an in vitro OSCN− production system, OSCN−, but not H2O2, activated NF-κB, a transcription factor critical for inflammatory responses, in the airway epithelial cells. OSCN− was sensed by protein kinase A (PKA) followed by formation of the dimerization of PKA. The dimerized PKA, the active form, was critical in activating NF-κB. Detoxifying H2O2, mainly by catalase, enabled the dominant abilities of OSCN− to dimerize PKA and activate NF-κB, compared with untreated H2O2. Furthermore, OSCN− in high doses caused necrosis of the cells, inducing release of IL-33, a trigger to initiate type 2 inflammation. These results demonstrate that OSCN− in low doses activates NF-κB via PKA in airway epithelial cells, whereas OSCN− in high doses causes necrosis, suggesting an important role in airway allergic inflammation for the production of OSCN− via the pendrin/peroxidase/Duox pathway.
Keywords: anion transport, asthma, epithelial cell, hydrogen peroxide, inflammation, NF-κB, peroxidase, protein kinase A (PKA)
Introduction
Asthma is a common disease affecting up to 10% of adults and 30% of children in the Western world (1). Type 2 immunity is dominant in the pathogenesis of asthma, and it is estimated that 50–70% of adult asthma patients have type 2 immunity (2, 3). Among type 2 cytokines, IL-13 plays a central role in the pathogenesis of asthma; inhalation of IL-13 alone causes asthma-like phenotypes in mice, whereas blocking the IL-13 signals diminishes asthma-like phenotypes in ovalbumin-induced asthma model mice (4, 5). Based on such an immunological background, several anti-asthma drugs targeting IL-13 are now under development (6).
IL-13 is a multifunctional cytokine; it acts on both immune cells (e.g. eosinophils and mast cells) and resident cells (e.g. epithelial cells, fibroblasts, and smooth muscle cells) (7). Analyses of the gene-manipulated mice showed the importance of direct effects of IL-13 on airway epithelial cells among various IL-13-targeted cells; ectopic expression of IL-13 in airway epithelial cells led to asthma-like phenotypes (8). Moreover, selective expression of STAT6, a transcriptional factor critical for the IL-13 signals, to airway epithelial cells in STAT6-deficient mice recovered IL-13-inducing asthma-like phenotypes (9). However, it is not fully understood how IL-13's actions on epithelial cells would lead to airway allergic inflammation.
Hypothiocyanite (OSCN−) serves as a potent innate defense system against microbes in lungs as do lysozyme, defensin, cathelicidin peptides, lactoferrin, and leukocyte protease inhibitors (10, 11). In the OSCN− production system, thiocyanate (SCN−), a pseudohalide, is first incorporated from the basal side via the Na+I− symporter (NIS)2/the solute carrier family (SLC)5A5 into airway epithelial cells. SCN− is then actively transported into pulmonary lumens at the apical side via several anion transporters, cystic fibrosis transmembrane conductance regulator (CFTR) and pendrin/SLC26A4. SCN− together with hydrogen peroxide (H2O2) generated by Duox1 and Duox2, members of the Nox/Duox family, are catalyzed by peroxidases into OSCN−. Three peroxidases, myeloperoxidase (MPO), eosinophil peroxidase (EPX), and lactoperoxidase (LPO), are involved in this reaction in lung tissues. OSCN− is thought to be the major product of these peroxidases because SCN− is abundant in the airway surface liquid and is the most preferred substrate (10–13). OSCN− has potent antimicrobial properties against bacteria, viruses, and fungi. The importance of the OSCN− production system in pulmonary defense is shown in cystic fibrosis patients in whom susceptibility to chronic respiratory infections increases in proportion to impaired CFTR function (14). In contrast to the powerful effects of OSCN− on microbes, it is not clear how the OSCN− production system disturbs homeostasis of hosts leading to pathological conditions.
We previously searched for IL-13-inducible genes in airway epithelial cells by a comprehensive method using DNA microarray, finding that pendrin is involved in those cells (15). We and others then showed the significance of pendrin in airway allergic inflammation using model mice as follows: overexpression of pendrin in bronchial tissues causes mucus hyper-production, enhanced airway reactivity, infiltration of inflammatory cells, and up-regulated expression of inflammatory mediators (15). Reciprocally, pendrin deficiency decreases airway reactivity and infiltration of inflammatory cells in bronchoalveolar lavage fluid (16). These findings suggest to us that anions transported by pendrin, or their derivatives, play an important role in airway allergic inflammation. Furthermore, we recently found that expression of peroxidases and Duox1/Duox2 is up-regulated in both asthma model mice and asthma patients and that the blockage of peroxidases by its inhibitors, widely used as thyroid pharmaceuticals, improves airway inflammation (17). Given that much of the machinery of the OSCN− production system is induced by the asthmatic conditions and shows important roles in airway inflammation, we hypothesized that the OSCN− produced via the pendrin/peroxidase/Duox pathway would be involved in airway inflammation.
In this study, we examined the molecular mechanism of how OSCN− activates airway epithelial cells using an in vitro OSCN− production system. We found that OSCN− in low doses is sensed by protein kinase A (PKA) followed by activation of NF-κB, a transcription factor critical for inflammatory responses, in the cells, whereas OSCN− in high doses causes necrosis. These results suggest that OSCN− has deleterious effects on airway epithelial effects leading to airway inflammation.
Results
Activation of NF-κB by OSCN−, but Not by H2O2, in Airway Epithelial Cells
We first established an in vitro OSCN− production system to investigate the underlying molecular mechanism of how OSCN− causes inflammation by acting on airway epithelial cells (Fig. 1A). In this system, H2O2 was first generated by glucose oxidase (GOX). Then SCN− and H2O2 were catalyzed in the presence of LPO into OSCN−. H2O2 was generated from β-d-glucose in a dose-dependent manner with up to 16 milliunits/ml of GOX in the absence of LPO (Fig. 1B). H2O2 production continuously increased up to 9 h. When we added 10 μg/ml LPO in this system, OSCN− production also behaved in a dose-dependent manner with GOX, reaching a peak at 3 h (Fig. 1C). Therefore, we performed the following experiments with 16 milliunits/ml of GOX in the presence or absence of 10 μg/ml LPO.
FIGURE 1.
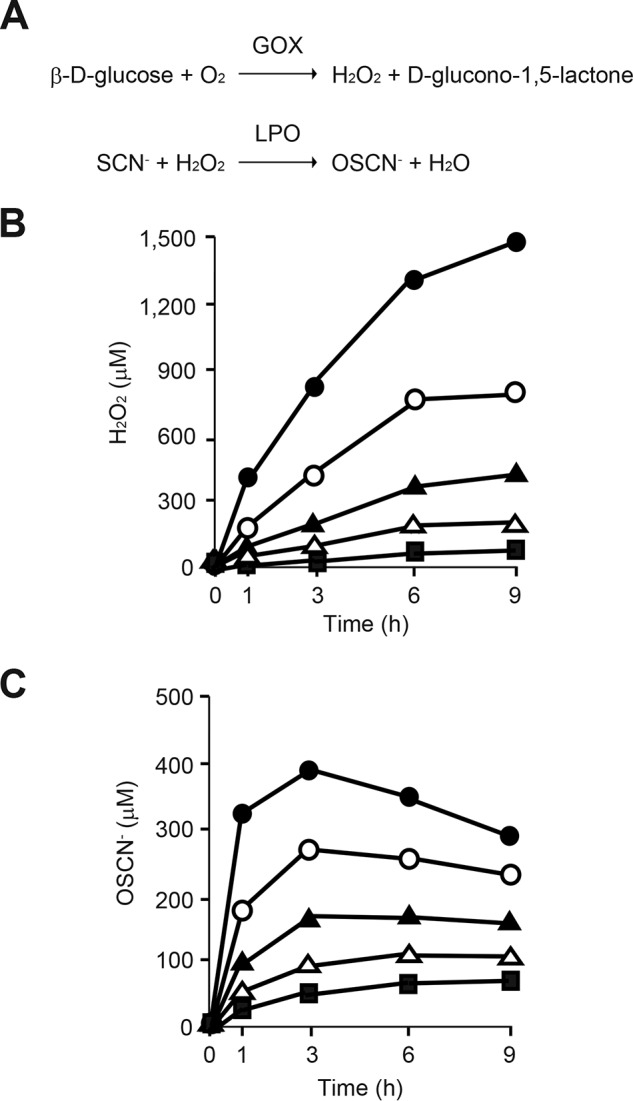
Establishment of in vitro OSCN-system. A, chemical reactions of in vitro H2O2 and OSCN− production. B and C, medium containing 11 mm β-d-glucose, 3 mm HSCN, and GOX (closed square, 1 milliunit/ml; open triangle, 2 milliunits/ml; closed triangle, 4 milliunits/ml; open circle, 8 milliunits/ml, and closed circle, 16 milliunits/ml) was incubated without (B) or with (C) 10 μg/ml LPO for the indicated times. Produced H2O2 (B) or OSCN− (C) is depicted. All experiments were performed at least more than two times.
We next examined whether H2O2 or OSCN− activates NF-κB, a transcription factor critical for inflammatory responses, in the human airway epithelial cell line H292 using the in vitro OSCN− production system. When we incubated the cells with GOX or LPO alone for 5 h in the system, no activation of NF-κB was observed (Fig. 2, A and B). In the former condition, ∼1 mm H2O2 was generated (Fig. 1B). In contrast, when the cells were incubated in the presence of both GOX and LPO, the condition in which ∼350 μm OSCN− was generated (Fig. 1C), activation of NF-κB was clearly observed. The supershift assay showed that NF-κB activated by OSCN− in H292 cells contained p50, but not p52, p65, Rel B, or c-Rel (Fig. 2B). These results suggest that OSCN−, but not H2O2, activates the formation of the p50 homodimer in H292 cells.
FIGURE 2.
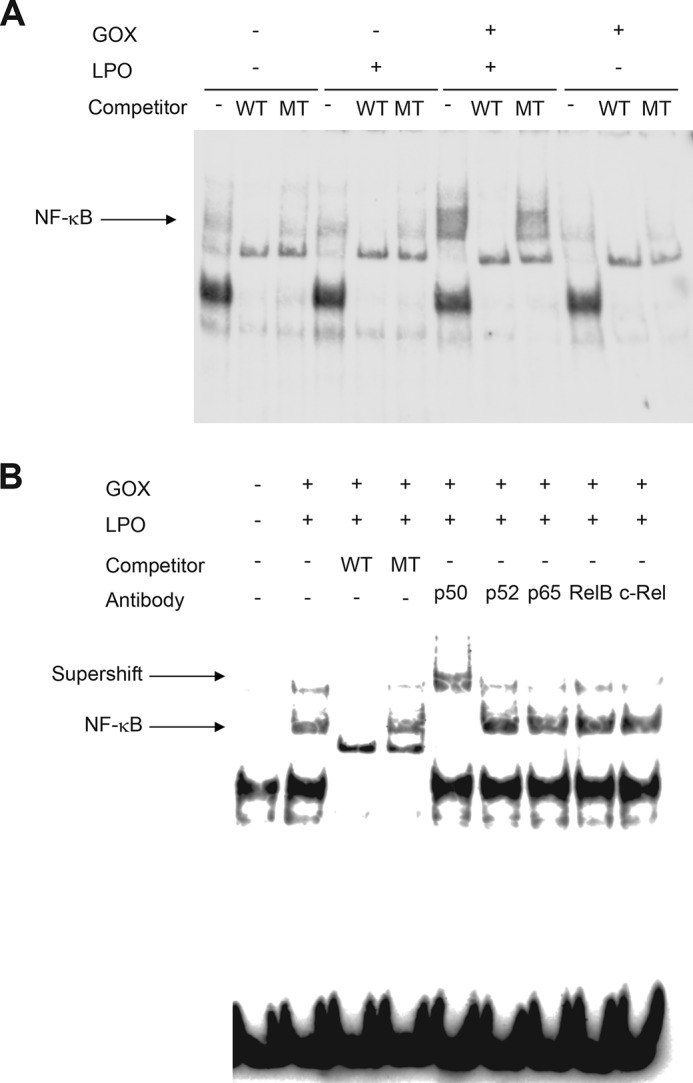
Activation of NF-κB by OSCN−, but not by H2O2, in airway epithelial cells. A, H292 cells were stimulated for 5 h with medium containing 11 mm β-d-glucose, 3 mm HSCN, and in the presence or absence of 16 milliunits/ml GOX or 10 μg/ml LPO. The nuclear extracts were applied to the EMSA. Excess amounts of cold wild competitors (WT) or mutated competitors (MT) were added. B, H292 cells were incubated as in A in the presence or absence of 16 milliunits/ml GOX or 10 μg/ml LPO. The nuclear extracts were applied to the EMSA. Excess amounts of cold WT competitors, cold mutated competitors, or the antibodies were added to the nuclear extracts from the cells stimulated in the presence of GOX or LPO. The antibodies against p50, p52, p65, Rel B, or c-Rel were used. The arrows indicate the probe·NF-κB complex or the super-shifted band. All experiments were performed at least more than two times.
PKA Senses OSCN− Leading to NF-κB Activation in Airway Epithelial Cells
We next investigated the molecular mechanism of NF-κB activation by OSCN− in airway epithelial cells. For that purpose, we examined the effects of inhibitors against PKA (H-89), p38 (SB203580), MEK1/2 (PD98059), and JNK (SP600125), signal-transducing molecules potentially located in the upstream pathway of NF-κB, on NF-κB activation by OSCN−. H-89 diminished NF-κB activation in a dose-dependent manner, whereas SB203580 and PD98059 did not change NF-κB activation (Fig. 3A). SP600125 inhibited NF-κB activation (Fig. 3A); however, we could detect phosphorylation of p38, but not JNK, by OSCN− (Fig. 3, B and C). These results demonstrate that PKA plays an important role in NF-κB activation by OSCN−.
FIGURE 3.

PKA causes NF-κB activation by OSCN− in airway epithelial cells. A, H292 cells were stimulated for 5 h with medium containing 11 mm β-d-glucose, 3 mm HSCN, 16 milliunits/ml GOX, and the indicated concentrations of H-89, SB203580, PD98059, or SP600126 in the presence or the absence of 10 μg/ml LPO. The nuclear extracts were applied to the EMSA. B and C, H292 cells were stimulated for 5 h with medium containing 11 mm β-d-glucose, and 3 mm HSCN with the indicated concentrations of GOX, in the presence or the absence of 10 μg/ml LPO. The cell lysates were applied to Western blotting using anti-phospho-JNK antibodies (B) or anti-phospho-p38 antibodies (C). D and E, cell lysates prepared in B or C were applied to Western blotting under non-reducing conditions using anti-PKA antibodies (D) or antibodies against phosphorylated PKA substrates (pPKAS) or GAPDH (E). E, 10 μΜ H-89 was added 10 min before stimulation. As positive control, cells were stimulated with 100 nm isoprenaline for 5 min. Asterisks indicate the phosphorylated PKA substrates. All experiments were performed at least more than two times.
It has been reported that PKA is a sensor of oxidants in the cells and that upon stimulation an oxidant generates a dimer, the active form, by forming an intermolecular disulfide bond independently of cAMP (18, 19). We then examined whether OSCN− is sensed by PKA followed by activation of PKA in airway epithelial cells. When we exposed H292 cells with OSCN− in the presence of LPO, PKA was dimerized in a dose-dependent manner (Fig. 3D). In contrast, when H292 cells were exposed to H2O2 in the absence of LPO, no dimerization of PKA was observed. To confirm whether dimerized PKA by OSCN− is the active form, we then examined phosphorylation of PKA substrates by OSCN−. Phosphorylation of PKA substrates was up-regulated in the presence of OSCN− as well as isoprenaline, a β-agonist, and phosphorylation of PKA substrates by both OSCN− and isoprenaline was inhibited by H-89 (Fig. 3E). Although isoprenaline caused a 100-fold elevation of cAMP, OSCN− did not cause any changes of it throughout the time course of the culture, excluding the possibility that OSCN− activates PKA by up-regulating the cAMP concentrations (data not shown). These results demonstrate that OSCN− is sensed by PKA in the airway epithelial cells, causing PKA dimerization followed by activation of NF-κB independently of cAMP.
H2O2 Is Detoxified Mainly by Catalase in Airway Epithelial Cells
H2O2 has a potent oxidation activity so that it has the potential to dimerize PKA as well as OSCN− (19). However, our present finding showed that H2O2 has much weaker oxidation activity than OSCN− in airway epithelial cells. We hypothesized that the different abilities of OSCN− and H2O2 for dimer formation of PKA in airway epithelial cells would be due to the difference in their clearance from these cells. To address this question, we first analyzed the kinetics of the OSCN− and H2O2 concentrations in the medium in the presence of H292 cells. H2O2 was promptly removed from the medium in the presence of H292 cells, and it mostly disappeared by 1 h (Fig. 4A). In contrast, clearance of OSCN− was slow even in the presence of H292 cells, with almost the half of the initial activity remaining after 1 h. These results suggest that H2O2, but not OSCN−, would be rapidly incorporated into H292 cells or detoxified in H292 cells.
FIGURE 4.

H2O2 is detoxified mainly by catalase in airway epithelial cells. A, medium was prepared with 11 mm β-d-glucose, 3 mm HSCN, 16 milliunits/ml GOX in the presence (left) or absence (right) of 10 μg/ml LPO. The medium was incubated with H292 cells (open circle) or without H292 cells (closed circle) for 5 h. Produced OSCN− (left) and H2O2 (right) are depicted. The consumed amounts (closed triangle) were calculated by subtracting the concentrations in the presence of H292 cells from the concentrations in the absence of H292 cells. B, chemical reactions of three H2O2 detoxification systems. C, H292 cells were incubated for 5 h with 100 mm aminotriazole (closed triangle) or 1 mm BCNU (open triangle) or 1 μm auranofin (closed square) or no inhibitor (open circle) using the medium prepared in A. The medium alone, without the cells, is depicted as a closed circle. The residual H2O2 ratios compared with the initial amounts at the indicated times are depicted. D, H292 cells were stimulated for 5 h with medium containing 11 mm β-d-glucose and 3 mm HSCN with the indicated concentrations of GOX in the presence or the absence of 10 μg/ml LPO and/or 100 mm aminotriazole. E, H292 cells were stimulated with medium containing the indicated concentrations of H2O2 alone or 11 mm β-d-glucose, 3 mm HSCN, and 10 μg/ml LPO with the indicated concentrations of GOX for 5 h. D and E, cell lysates were applied to Western blotting under non-reducing conditions using anti-PKA antibodies. All experiments were performed at least more than two times.
We next examined the effects of the detoxification system for H2O2 on dimerization of PKA in H292 cells. Three detoxification systems (catalase, the glutathione peroxidase/reductase system, and the peroxiredoxin/thioredoxin/thioredoxin reductase system) were expected to function in the cells (Fig. 4B). We first examined which detoxification system was dominant for H2O2 in H292 cells using inhibitors against catalase (aminotriazole), glutathione reductase (carmustine; 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)), and thioredoxin reductase (auranofin). Adding aminotriazole recovered the H2O2 concentrations in the medium, whereas BCNU or auranofin did not (Fig. 4C). Accordingly, adding aminotriazole to the in vitro H2O2 production system significantly enhanced the formation of PKA dimer to the same level of OSCN− (Fig. 4D). Moreover, when we added more than 10 mm H2O2 into the system, PKA was dimerized at the same level of OSCN− (Fig. 4E), suggesting that excess amounts of H2O2 overcame the detoxification system in H292 cells. These results demonstrate that catalase is dominantly involved in the detoxification system for H2O2 in H292 cells and that in airway epithelial cells, the detoxification of H2O2 contributes to the dominant abilities of OSCN− for dimerization of PKA compared with H2O2.
Induction of Necrosis in Airway Epithelial Cells by High Concentrations of OSCN−
Epithelial desquamation is a typical histological feature of asthma reflecting damage and injury in airway epithelial cells (20, 21). Furthermore, necrosis of epithelial cells causes release of IL-33, which triggers type 2 immune responses targeting mainly the innate lymphoid cell type 2 (ILC2), from their nuclei (22, 23). Moreover, it has been reported that OSCN− induces both apoptosis and necrosis in macrophage cell lines but apoptosis in endothelial cells (24, 25). Finally, we evaluated the effects of high dose OSCN− on airway epithelial cells to determine whether the observed effects were necrotic and apoptotic in character. This evaluation was carried out in the following two ways: fractions estimated by annexin V/propidium iodide (PI) and quantitation of DNA content. H292 cells treated with cycloheximide and TNF-α caused apoptosis in a time-dependent manner, estimated by the increases of the annexin V+PI− fraction and the low DNA content fraction (Fig. 5, A and B, lower panels). In contrast, high dose OSCN− (≥32 milliunits/ml GOX) caused necrosis (increase of the annexin V+PI+ fraction and no change in the low DNA content fraction) in H292 cells (Fig. 5, A and B, upper panels). A trace amount of IL-33 (0.063 ± 0.022 pg/ml) was detected in the medium of H292 cells treated with high dose OSCN−. In low dose OSCN− (≤16 milliunits/ml GOX) in which PKA dimerization occurred, the cells were still intact. These results demonstrate that high dose OSCN− causes necrosis, whereas low dose OSCN− activates NF-κB via PKA in airway epithelial cells.
FIGURE 5.

High concentrations of OSCN− cause necrosis in human pulmonary epithelial cells. H292 cells were treated for 5 h with 11 mm β-d-glucose, 3 mm HSCN, the indicated concentrations of GOX, and 10 μg/ml LPO or with 50 μg/ml cycloheximide and 50 ng/ml TNF-α for the indicated times. The populations of the annexin V+PI+ fraction (necrosis) and the annexin V+PI− fraction (apoptosis) are shown in A, and the populations of the low DNA content fraction (apoptosis) are shown in B. All experiments were performed at least more than two times.
Discussion
In this study, we demonstrated that OSCN− produced via the pendrin/peroxidase/Duox pathway activates NF-κB via PKA in epithelial cells, which would lead to the onset of airway inflammation (Fig. 6). In this pathway, SCN− is first actively transported into pulmonary lumens via NIS/SLC5A5 at the basal side and via anion transporters, including CFTR and pendrin/SLC26A4, at the apical side in airway epithelial cells (10, 11). OSCN− is then generated by the catalysis of peroxidases composed of MPO, EPX, and LPO using SCN− and H2O2 generated by Duox1/Duox2. It has been known that many extrinsic or intrinsic factors activate NF-κB via pattern recognition receptors (PRRs) on epithelial cells (26); however, to our knowledge, OSCN− is the first anion to activate NF-κB in epithelial cells.
FIGURE 6.
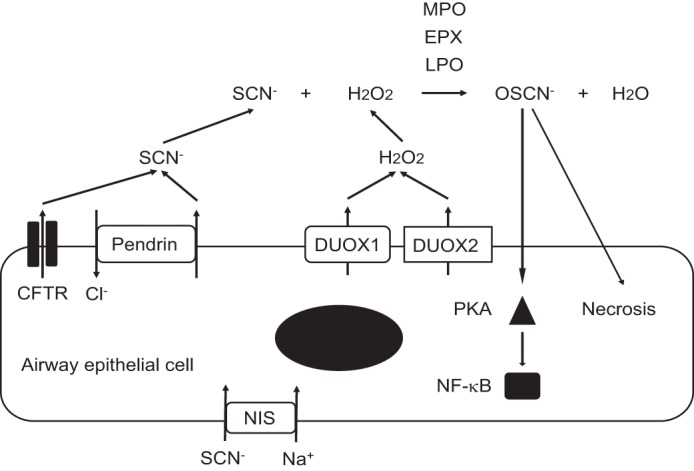
Schematic model of the OSCN− production and its activation of NF-κB via PKA in airway epithelial cells. SCN− is actively transported into pulmonary lumens via NIS/SLC5A5 at the basal side and via several anion transporters, including CFTR and pendrin/SLC26A4 at the apical side in airway epithelial cells. SCN− together with H2O2 generated by Duox1 and Duox2 is catalyzed by peroxidases into OSCN−. Three peroxidases, including MPO, EPX, and LPO, are involved in this reaction. The produced OSCN− activates NF-κB via PKA or causes necrosis in airway epithelial cells. All experiments were performed at least more than two times.
We have recently demonstrated that expression of pendrin, peroxidases, and Duox1/Duox2 is enhanced, resulting in that the OSCN− production would be up-regulated in the airways of allergic inflammation in asthma model mice and asthma patients (17). These data demonstrate that NF-κB activation by OSCN− in airway epithelial cells would be one underlying mechanism of airway allergic inflammation. Given that OSCN− has harmful effects on various microbes (10, 11) and that type 2 immune responses are equipped to be a defense mechanism against parasites (27, 28), we hypothesize that whereas the OSCN− production system may be an innate host defense mechanism in the lung, this misplaced production of OSCN− is a likely contributor to pulmonary inflammation, causing deleterious effects in response to airway allergen provocation.
Various extrinsic or intrinsic danger signals, including allergens, activate NF-κB via PRRs or cause necrosis followed by release of IL-33 in epithelial cells (29, 30). Activation of NF-κB in airway epithelial cells is important for production of chemokines and inflammatory cytokines and expression of adhesion molecules accelerating type 2 immunity (29). IL-33 acts on several immune cells as follows: ILC2; mast cells; basophils; eosinophils; Th2 cells; NKT; and NK cells (30). IL-33 acts on ILC2 inducing production of IL-5 and IL-13. Thus, activation of NF-κB in epithelial cells and release of IL-33 from epithelial cells play critical roles in the transition from innate immunity to acquired immunity in the pathogenesis of asthma. However, this study has shown that OSCN−, a downstream molecule of the IL-13 signals, activates NF-κB in epithelial cells or induces release of IL-33 from epithelial cells, suggesting that IL-13, OSCN−, and IL-33 may make a vicious circle. These results suggest the potential that type 2 inflammation is exaggerated or prolonged by this vicious circle involving the OSCN− production pathway in airway epithelial cells.
Our data showed that OSCN− is sensed by PKA followed by activation of NF-κB in airway epithelial cells. It has also been shown that oxidative stress affects mobilization of several signaling molecules, PKA, nucleotide diphosphate kinase B, serine/threonine protein phosphatase, and PKC δ/ϵ, in myocytes (18). Among these molecules, upon stimulation of oxidants, the regulatory subunit of type I PKA is dimerized through a disulfide bond followed by increased affinity for the substrates, demonstrating that oxidative stress is changed to intracellular signaling through PKA, independently of cAMP (19). This study shows PKA acting as a dominant sensor for OSCN−, leading to activation of NF-κB in airway epithelial cells (Fig. 3). A previous report that inhibition of PKA by H-89 abolished airway allergy inflammation in model mice supports our suggested importance of the PKA-mediated pathway (31).
The present finding showed that the detoxification system of H2O2 mainly by catalase causes the more potent abilities of OSCN− for dimerization of PKA and activation of NF-κB, compared with H2O2 (Fig. 4). H2O2, abundant in the environmental milieu, is easily incorporated into the cells and is toxic to living organisms, causing DNA damage (32). Therefore, the detoxification system of H2O2, including catalase, is found widely among microbes, plants, and animals. Although it has been reported that OSCN− is detoxified by thioredoxin reductase (33), it is unclear that the detoxification system of OSCN− is as ubiquitous as that of H2O2. Actually, it is likely that H292 cells are susceptible to OSCN−, which is at least partially explained by the lack of a detoxification system for OSCN− (Fig. 4A). In microbes, this lack becomes advantageous in that animals can use OSCN− as an effector of their innate defense system against microbes. In turn, it becomes disadvantageous for hosts because of the high susceptibility to insults by OSCN−.
In addition to its application to the pathogenesis of type 2 immunity, the present finding can be also applied to airway inflammation in smokers. Plasma SCN− levels in smokers are almost three times higher than in non-smokers (130–140 μm versus 40–50 μm) (34, 35). It is well known that in asthma, smoking is associated with poor control, decrease of lung function, and enhanced corticosteroid resistance (36, 37). However, no report has yet shown any deleterious effects of SCN− or OSCN− derived from tobacco on lungs. The present results suggest the possible involvement of the OSCN− production system in how smoking affects asthma or other smoking-related pulmonary diseases.
The present findings indicate that all of the machineries of the OSCN− production system can be listed as novel therapeutic targets for bronchial asthma downstream of type 2 immunity. We have recently shown that among them peroxidase can be a novel therapeutic target for bronchial asthma using antagonists against peroxidases (17). Elucidation of the molecular mechanisms underlying OSCN− production and the activation of epithelial cells by OSCN− have promising clinical significance for developing novel treatments for bronchial asthma.
Experimental Procedures
In Vitro Production of H2O2 and OSCN−
A human lung carcinoma cell line, H292, was purchased from the ATCC. H292 cells (1.8 × 106 cells/well) were stimulated for 5 h with phenol red-free RPMI 1640 medium (1.5 ml/well) containing 11 mm β-d-glucose (Life Technologies, Inc.), 3 mm NaSCN (Wako, Japan), and the indicated concentrations of GOX (Wako) in the presence or absence of 10 μg/ml LPO (Calzyme Laboratories). For the experiments to inhibit signal-transducing molecules, the indicated concentrations of inhibitors against PKA (H89, Merck Millipore), p38 (SB203580, Wako), MEK1/2 (PD98059, Cell Signaling Technology), and JNK (SP600126, Wako) were added. H2O2 was measured by BIOXYTECH H2O2–560TM (Percipio Biosciences), and OSCN− was measured by reacting with 5-thio-2-nitrobenzoic acid and by estimating the decrease of the absorbance at 412 nm as described previously (38). For the experiments to inhibit H2O2 detoxification systems, inhibitors against catalase (100 mm aminotriazole, Sigma), glutathione reductase (1 mm BCNU, Sigma), and thioredoxin reductase (1 μm auranofin, Sigma) were added.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extraction and the EMSA were prepared as described previously (39, 40). The sequence of the used probe was 5′-AGTTGAGGGGACTTTCCCAGGC-3′, and biotin was conjugated with the 5′ end of the sense probe. To detect supershifted bands, antibodies against p50, p52, p65, Rel B, and c-Rel (Santa Cruz Biotechnology) were added to the assay mixtures.
Western Blotting
The procedure was performed as described previously (41) with modification. For blotting of PKA RI subunit dimer, cells were lysed by non-reducing RIPA buffer containing 100 mm maleimide (Sigma). The proteins were blotted by antibodies against PKA RI (Cell Signaling Technology), phospho-p38 (Cell Signaling Technology), and phospho-JNK (Cell Signaling Technology). In some experiments, the cells were incubated with H2O2 (Wako). For blotting of phosphorylation of PKA substrates, cells were treated with 10% (w/v) trichloroacetic acid (Wako) in 0.15 m NaCl (Wako) for 15 min on ice and then lysed as described previously (42).
In some experiments, 100 nm isoprenaline (Sigma) and/or 10 μm H-89 (Calbiochem) were added. The proteins were blotted by antibodies against phospho-(Ser/Thr) PKA substrate (Cell Signaling Technology) or GAPDH (Cell Signaling Technology). The proteins were visualized by SuperSignal West Pico Substrate (Life Technologies, Inc.).
Cell Death Assay
H292 cells treated with phenol red-free RPMI 1640 medium (1.5 ml/well) containing 11 mm β-d-glucose, 3 mm NaSCN, 10 μg/ml LPO, and the indicated concentrations of GOX or with 50 μg/ml cycloheximide (Wako) and 50 ng/ml TNF-α (PeproTech) for the indicated times were used. Cell death was quantified in two ways. One was double staining of anti-annexin V antibodies (Apoptosis Detection Kit I, BD Biosciences) and PI. The other was quantitation of nuclear DNA (43), performed using FACSCalibur and Cell Quest (BD Biosciences). IL-33 was measured with a commercial ELISA kit (Bio-Techne).
cAMP Assay
Intracellular cAMP levels were measured using the DetectX direct cyclic AMP enzyme immunoassay kit (Arbor Assays) according to the manufacturer's protocol.
Author Contributions
S. S., M. O., S. O., Y. N., H. S., Y. M., and T. Y. performed the experiments. S. N. also contributed with performing the experiments. S. S. and K. I. contributed to the experimental design. J. J. L. and K. I. wrote the manuscript.
Acknowledgment
We thank Dr. Dovie R. Wylie for the critical review of this manuscript.
This work was supported in part by grants-in-aid for scientific research from the Japan Society for the Promotion of Science. The authors declare that they have no conflicts of interest with the contents of this article.
- NIS
- Na+I− symporter
- CFTR
- cystic fibrosis transmembrane conductance regulator
- EPX
- eosinophil peroxidase
- LPO
- lactoperoxidase
- GOX
- glucose oxidase
- BCNU
- 1,3-bis(2-chloroethyl)-1-nitrosourea
- PI
- propidium iodide
- PRR
- pattern recognition receptor.
References
- 1. Jackson D. J., Sykes A., Mallia P., and Johnston S. L. (2011) Asthma exacerbations: origin, effect, and prevention. J. Allergy Clin. Immunol. 128, 1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woodruff P. G., Modrek B., Choy D. F., Jia G., Abbas A. R., Ellwanger A., Koth L. L., Arron J. R., and Fahy J. V. (2009) T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 180, 388–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peters M. C., Mekonnen Z. K., Yuan S., Bhakta N. R., Woodruff P. G., and Fahy J. V. (2014) Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J. Allergy Clin. Immunol. 133, 388–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grünig G., Warnock M., Wakil A. E., Venkayya R., Brombacher F., Rennick D. M., Sheppard D., Mohrs M., Donaldson D. D., Locksley R. M., and Corry D. B. (1998) Requirement for IL-13 independently of IL-4 in experimental asthma. Science 282, 2261–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T. Y., Karp C. L., and Donaldson D. D. (1998) Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261 [DOI] [PubMed] [Google Scholar]
- 6. Izuhara K., Matsumoto H., Ohta S., Ono J., Arima K., and Ogawa M. (2015) Recent developments regarding periostin in bronchial asthma. Allergol. Int. 64, S3–S10 [DOI] [PubMed] [Google Scholar]
- 7. Izuhara K. (2003) The role of interleukin-4 and interleukin-13 in the non-immunologic aspects of asthma pathogenesis. Clin. Chem. Lab. Med. 41, 860–864 [DOI] [PubMed] [Google Scholar]
- 8. Zhu Z., Homer R. J., Wang Z., Chen Q., Geba G. P., Wang J., Zhang Y., and Elias J. A. (1999) Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103, 779–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuperman D. A., Huang X., Koth L. L., Chang G. H., Dolganov G. M., Zhu Z., Elias J. A., Sheppard D., and Erle D. J. (2002) Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8, 885–889 [DOI] [PubMed] [Google Scholar]
- 10. Hawkins C. L. (2009) The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radic. Res. 43, 1147–1158 [DOI] [PubMed] [Google Scholar]
- 11. Barrett T. J., and Hawkins C. L. (2012) Hypothiocyanous acid: benign or deadly? Chem. Res. Toxicol. 25, 263–273 [DOI] [PubMed] [Google Scholar]
- 12. Lorentzen D., Durairaj L., Pezzulo A. A., Nakano Y., Launspach J., Stoltz D. A., Zamba G., McCray P. B. Jr, Zabner J., Welsh M. J., Nauseef W. M., and Bánfi B. (2011) Concentration of the antibacterial precursor thiocyanate in cystic fibrosis airway secretions. Free Radic Biol. Med. 50, 1144–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wijkstrom-Frei C., El-Chemaly S., Ali-Rachedi R., Gerson C., Cobas M. A., Forteza R., Salathe M., and Conner G. E. (2003) Lactoperoxidase and human airway host defense. Am. J. Respir. Cell Mol. Biol. 29, 206–212 [DOI] [PubMed] [Google Scholar]
- 14. Moskwa P., Lorentzen D., Excoffon K. J., Zabner J., McCray P. B. Jr, Nauseef W. M., Dupuy C., and Bánfi B. (2007) A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir Crit. Care Med. 175, 174–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakao I., Kanaji S., Ohta S., Matsushita H., Arima K., Yuyama N., Yamaya M., Nakayama K., Kubo H., Watanabe M., Sagara H., Sugiyama K., Tanaka H., Toda S., Hayashi H., et al. (2008) Identification of pendrin as a common mediator for mucus production in bronchial asthma and chronic obstructive pulmonary disease. J. Immunol. 180, 6262–6269 [DOI] [PubMed] [Google Scholar]
- 16. Nakagami Y., Favoreto S. Jr, Zhen G., Park S. W., Nguyenvu L. T., Kuperman D. A., Dolganov G. M., Huang X., Boushey H. A., Avila P. C., and Erle D. J. (2008) The epithelial anion transporter pendrin is induced by allergy and rhinovirus infection, regulates airway surface liquid, and increases airway reactivity and inflammation in an asthma model. J. Immunol. 181, 2203–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki S., Ogawa M., Ohta S., Arima K., Nunomura S., Nanri Y., Mitamura Y., Yoshihara T., Nakamura Y., Yamauchi K., Chibana K., Ishii Y., Lee J. J., Aratani Y., Kakuta S., et al. (2016) The potential for repositioning anti-thyroid agents as anti-asthma drugs. J. Allergy Clin. Immunol. 138, 1458–1461 [DOI] [PubMed] [Google Scholar]
- 18. Brennan J. P., Wait R., Begum S., Bell J. R., Dunn M. J., and Eaton P. (2004) Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J. Biol. Chem. 279, 41352–41360 [DOI] [PubMed] [Google Scholar]
- 19. Brennan J. P., Bardswell S. C., Burgoyne J. R., Fuller W., Schröder E., Wait R., Begum S., Kentish J. C., and Eaton P. (2006) Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 281, 21827–21836 [DOI] [PubMed] [Google Scholar]
- 20. Gras D., Chanez P., Vachier I., Petit A., and Bourdin A. (2013) Bronchial epithelium as a target for innovative treatments in asthma. Pharmacol. Ther. 140, 290–305 [DOI] [PubMed] [Google Scholar]
- 21. Holgate S. T. (2008) The airway epithelium is central to the pathogenesis of asthma. Allergol. Int. 57, 1–10 [DOI] [PubMed] [Google Scholar]
- 22. Lefrançais E., and Cayrol C. (2012) Mechanisms of IL-33 processing and secretion: differences and similarities between IL-1 family members. Eur. Cytokine Netw. 23, 120–127 [DOI] [PubMed] [Google Scholar]
- 23. Lambrecht B. N., and Hammad H. (2013) Asthma: the importance of dysregulated barrier immunity. Eur. J. Immunol. 43, 3125–3137 [DOI] [PubMed] [Google Scholar]
- 24. Lloyd M. M., van Reyk D. M., Davies M. J., and Hawkins C. L. (2008) Hypothiocyanous acid is a more potent inducer of apoptosis and protein thiol depletion in murine macrophage cells than hypochlorous acid or hypobromous acid. Biochem. J. 414, 271–280 [DOI] [PubMed] [Google Scholar]
- 25. Lloyd M. M., Grima M. A., Rayner B. S., Hadfield K. A., Davies M. J., and Hawkins C. L. (2013) Comparative reactivity of the myeloperoxidase-derived oxidants hypochlorous acid and hypothiocyanous acid with human coronary artery endothelial cells. Free Radic. Biol. Med. 65, 1352–1362 [DOI] [PubMed] [Google Scholar]
- 26. Lambrecht B. N., and Hammad H. (2012) The airway epithelium in asthma. Nat. Med. 18, 684–692 [DOI] [PubMed] [Google Scholar]
- 27. Liu Z., Liu Q., Pesce J., Anthony R. M., Lamb E., Whitmire J., Hamed H., Morimoto M., Urban J. F. Jr, and Gause W. C. (2004) Requirements for the development of IL-4-producing T cells during intestinal nematode infections: what it takes to make a Th2 cell in vivo. Immunol. Rev. 201, 57–74 [DOI] [PubMed] [Google Scholar]
- 28. Ramalingam T. R., Reiman R. M., and Wynn T. A. (2005) Exploiting worm and allergy models to understand Th2 cytokine biology. Curr. Opin. Allergy Clin. Immunol. 5, 392–398 [DOI] [PubMed] [Google Scholar]
- 29. Schuliga M. (2015) NF-κB signaling in chronic inflammatory airway disease. Biomolecules 5, 1266–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cayrol C., and Girard J. P. (2014) IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 31, 31–37 [DOI] [PubMed] [Google Scholar]
- 31. Reber L. L., Daubeuf F., Nemska S., and Frossard N. (2012) The AGC kinase inhibitor H89 attenuates airway inflammation in mouse models of asthma. PLoS ONE 7, e49512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mishra S., and Imlay J. (2012) Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch. Biochem. Biophys. 525, 145–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chandler J. D., Nichols D. P., Nick J. A., Hondal R. J., and Day B. J. (2013) Selective metabolism of hypothiocyanous acid by mammalian thioredoxin reductase promotes lung innate immunity and antioxidant defense. J. Biol. Chem. 288, 18421–18428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Husgafvel-Pursiainen K., Sorsa M., Engström K., and Einistö P. (1987) Passive smoking at work: biochemical and biological measures of exposure to environmental tobacco smoke. Int. Arch. Occup. Environ. Health 59, 337–345 [DOI] [PubMed] [Google Scholar]
- 35. Saloojee Y., Vesey C. J., Cole P. V., and Russell M. A. (1982) Carboxyhaemoglobin and plasma thiocyanate: complementary indicators of smoking behaviour? Thorax 37, 521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tamimi A., Serdarevic D., and Hanania N. A. (2012) The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir. Med. 106, 319–328 [DOI] [PubMed] [Google Scholar]
- 37. Polosa R., and Thomson N. C. (2013) Smoking and asthma: dangerous liaisons. Eur. Respir. J. 41, 716–726 [DOI] [PubMed] [Google Scholar]
- 38. Bozonet S. M., Scott-Thomas A. P., Nagy P., and Vissers M. C. (2010) Hypothiocyanous acid is a potent inhibitor of apoptosis and caspase 3 activation in endothelial cells. Free Radic. Biol. Med. 49, 1054–1063 [DOI] [PubMed] [Google Scholar]
- 39. Schreiber E., Matthias P., Müller M. M., and Schaffner W. (1989) Rapid detection of octamer binding proteins with 'mini-extracts', prepared from a small number of cells. Nucleic Acids Res. 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Midwinter R. G., Cheah F. C., Moskovitz J., Vissers M. C., and Winterbourn C. C. (2006) IκB is a sensitive target for oxidation by cell-permeable chloramines: inhibition of NF-κB activity by glycine chloramine through methionine oxidation. Biochem. J. 396, 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taniguchi K., Arima K., Masuoka M., Ohta S., Shiraishi H., Ontsuka K., Suzuki S., Inamitsu M., Yamamoto K., Simmons O., Toda S., Conway S. J., Hamasaki Y., and Izuhara K. (2014) Periostin controls keratinocyte proliferation and differentiation by interacting with the paracrine IL-1α/IL-6 loop. J. Invest. Dermatol. 134, 1295–1304 [DOI] [PubMed] [Google Scholar]
- 42. Niikura Y., Nonaka T., and Imajoh-Ohmi S. (2002) Monitoring of caspase-8/FLICE processing and activation upon Fas stimulation with novel antibodies directed against a cleavage site for caspase-8 and its substrate, FLICE-like inhibitory protein (FLIP). J. Biochem. 132, 53–62 [DOI] [PubMed] [Google Scholar]
- 43. Riccardi C., and Nicoletti I. (2006) Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 1, 1458–1461 [DOI] [PubMed] [Google Scholar]