

Abstract
Previously, we showed that levels of sphingosine-1 phosphate receptor 3 (S1PR3) are increased in a panel of cultured human lung adenocarcinoma cell lines, and that S1PR3-mediated signaling pathways regulate proliferation, soft agar growth, and invasion of human lung adenocarcinoma cells in vitro. In the present study, we examine S1PR3 levels in human lung adenocarcinoma specimens. cDNA array and tumor microarray analysis shows that mRNA and protein levels of S1PR3 are significantly increased in human lung adenocarcinomas when compared with normal lung epithelial cells. Promoter analysis shows 16 candidate SMAD3 binding sites in the promoter region of S1PR3. ChIP indicates that TGF-β treatment stimulates the binding of SMAD3 to the promoter region of S1PR3. Luciferase reporter assay demonstrates that SMAD3 transactivates S1PR3 promoter. TGF-β stimulation or ectopic expression of TGF-β up-regulates S1PR3 levels in vitro and ex vivo. Pharmacologic inhibition of TGF-β receptor or SMAD3 abrogates the TGF-β-stimulated S1PR3 up-regulation. Moreover, S1PR3 knockdown dramatically inhibits tumor growth and lung metastasis, whereas ectopic expression of S1PR3 promotes the growth of human lung adenocarcinoma cells in animals. Pharmacological inhibition of S1PR3 profoundly inhibits the growth of lung carcinoma in mice. Our studies suggest that levels of S1PR3 are up-regulated in human lung adenocarcinomas, at least in part due to the TGF-β/SMAD3 signaling axis. Furthermore, S1PR3 activity promotes the progression of human lung adenocarcinomas. Therefore, S1PR3 may represent a novel therapeutic target for the treatment of deadly lung adenocarcinomas.
Keywords: lung cancer, SMAD transcription factor, sphingolipid, sphingosine-1 phosphate (S1P), transforming growth factor β (TGF-B), S1PR3, TGF-β
Introduction
Sphingosine-1-phospahte (S1P)3 is a serum-borne bioactive lipid mediator, which is generated by two sphingosine kinase isozymes, SphK1 and SphK2, using sphingosine as the substrate (1). S1P functions as an extracellular ligand or intracellular lipid mediator (2–5), and regulates various physiological and pathophysiological functions (5–8). When S1P is functioning as an extracellular ligand, its activities are mediated by the S1P family of G protein-coupled receptors (S1PR1–S1PR5) (2, 9–11). Several lines of evidence suggest that S1P-mediated signaling pathways are closely linked to the tumorigenesis of various human cancers (12–16). However, the pathological link between the S1P-mediated signaling pathways and human lung adenocarcinoma is poorly understood. Previously, we showed that levels of sphingosine-1 phosphate receptor 3 (S1PR3) are significantly increased in cultured human lung adenocarcinoma cell lines (16). Moreover, we demonstrated that the S1PR3-activated signaling pathways play an important role in promoting the progression and invasiveness of human lung adenocarcinoma cells (11, 16).
TGF-β activates multiple signaling pathways to regulate various tumorigenic processes. For example, TGF-β regulates epithelial-mesenchymal transition, which is a critical process in cancer initiation and progression (17–20). Also, TGF-β stimulates the production of inflammatory cytokines in tumor microenvironments (21), and promotes tumor progression through extracellular matrix remodeling, cell adhesion, migration, and immune tolerance (17, 22, 23). Upon TGF-β ligation, TGF-β receptors phosphorylate SMAD (homolog of mothers against decapentaplegic) signaling molecules, leading to the nuclear translocation of SMADs. The nucleus-localized SMADs interact with specific transcriptional activators and repressors and regulate the expression of tumorigenic genes (24). In addition, TGF-β activates SMAD-independent pathways such as MAPK, JNK, NFκB, Ras/Raf/ERK, and Rho kinase pathways in a cell type-dependent manner (24, 25). Although both SMAD and non-SMAD pathways were reported to be involved in tumorigenic process, the mechanistic details remain to be elucidated.
Previous studies have suggested the cross-talk between TGF-β and S1P signaling pathways. TGF-β was shown to activate SphK1 and stimulate the production of S1P (26), which may be involved in extracellular matrix deposition and fibrosis. On the other hand, S1P transactivates the TGF-β pathway and regulates several TGF-β-mediated physiological and pathological functions (27, 28). Thus, a better understanding of the cross-talk between the S1P- and TGF-β mediated signaling pathways is expected to open new perspectives for the treatment of TGF-β-triggered pathologies such as inflammation, fibrosis, and cancer.
In the present study, we show that levels of S1PR3 are significantly increased in human lung adenocarcinoma specimens. Mechanistically, our data suggest that the TGF-β/SMAD 3 signaling pathway contributes to S1PR3 up-regulation in lung adenocarcinomas. Moreover, our study suggests that S1PR3 represents a novel therapeutic target for the treatment of human lung cancers.
Results
Up-regulation of S1PR3 in Human Lung Adenocarcinomas
Previously, we showed that levels of S1PR3 are significantly increased in a panel of cultured human lung adenocarcinoma cell lines when compared with normal lung epithelial cells (16). The pathological relevance of this in vitro observation was investigated by measuring mRNA levels of S1PR3 in cDNA microarrays of human lung adenocarcinoma specimens (OriGene, HLRT). Quantitative PCR analysis showed that mRNA levels of S1PR3 are significantly increased in human lung adenocarcinoma specimens when compared with normal lung tissues (Fig. 1A). We previously showed that S1PR2 levels are increased in endothelial senescence and inflammation (10, 29). However, we observed that levels of S1PR2 are decreased in human lung cancers (Fig. 1B).
FIGURE 1.

Up-regulation of S1PR3 in human lung adenocarcinomas. A, qPCR quantitation of S1PR3 mRNA in cDNA arrays of human lung adenocarcinoma specimens (OriGene, HLRT101 and HLRT105). **, p < 0.01, Student's t test. B, qPCR quantitation of S1PR2 mRNA in a cDNA array of human lung cancers (OriGene, HLRT105). **, p < 0.01, Student's t test. C, HEK293 cells were transfected with S1PR3 or pcDNA vector. Transfected cells were immunostained with anti-S1PR3 (Cayman Chemical) (IMF, left panels). Arrows, nonspecific fluorescent precipitates used for image orientation. Scale bar = 33 μm. D, anti-S1PR3 staining of human lung adenocarcinoma tumor microarray (Accumax 306). AdC, adenocarcinoma; N, adjacent normal lung tissue. E, immunostaining intensity was quantitated with the National Institutes of Health ImageJ software. Data, analyzed with GraphPad Prism 5 software, are shown as mean ± S.E. Statistical significance was analyzed by Student's t test. F, representative images of anti-S1PR3 staining of human lung adenocarcinoma and the respective adjacent normal lung epithelial tissue. G, quantitation of anti-S1PR3 staining of human lung squamous carcinoma microarray (Accumax 306). Data are mean ± S.E. Statistical significance was analyzed by Student's t test. H, representative images of anti-S1PR3 staining of human lung squamous carcinoma and the respective adjacent normal lung epithelial tissue.
Next, we utilized immunohistochemical staining to examine protein levels of S1PR3 in a paraffin-embedded tumor microarray of human lung adenocarcinoma specimens (Accumax). Anit-S1PR3 (Cayman) immunoreacted with plasma membrane-localized S1PR3 in HEK293 cells transiently transfected with S1PR3 vector. No immunoreactivity was observed in HEK293 transfected with pcDNA control vector (Fig. 1C). Immunohistochemical staining analysis showed that the intensity of anti-S1PR3 immunostaining is significantly increased in human lung adenocarcinomas when compared with their respective adjacent normal lung epithelial cells (Fig. 1, D–F). Moreover, levels of S1PR3 are increased in human lung squamous carcinoma specimens (Fig. 1, G and H).
Oncogenic K-Ras mutation is found in more than 25% of non-small cell lung carcinomas and represents one of the most prevalent oncogenic drivers in non-small cell lung carcinomas (30, 31). We utilized a conditionally inducible knock-in K-RasG12D (Lox-Stop-Lox-K-RasG12D, LSL-K-RasG12D) mouse model (32, 33) to measure S1PR3 levels in lung adenocarcinomas and normal lung tissues. As shown in Fig. 2A, lung tumors were readily observed in heterozygous LSL-K-RasG12D mice following intratracheal injection of adenoviral particles carrying Cre recombinase (Ad-Cre). S1PR3 levels were increased ∼20-fold in lungs of K-RasG12D-expressing mice when compared with that in mice treated with empty adenoviral particles (Ad-Ctrl) (Fig. 2B). A minimal increase of S1PR4 was observed in lungs of K-RasG12D-expressing mice. There were no significant changes of S1PR1 and S1PR2, and S1PR5 was not detected in lungs of Ad-Cre-injected mice (Fig. 2B). In addition, immunohistochemical staining showed that protein levels of S1PR3 were markedly increased in lung carcinoma specimens of K-RasG12D transgenic mice (Fig. 2C) when compared with normal lung tissues of wild-type mice. In a control, no staining was detected in lung adenocarcinoma specimens of K-RasG12D transgenic mice when immunohistochemical staining was performed without S1PR3 antibody (data not shown). These data suggest that S1PR3 levels are increased in lung adenocarcinomas.
FIGURE 2.
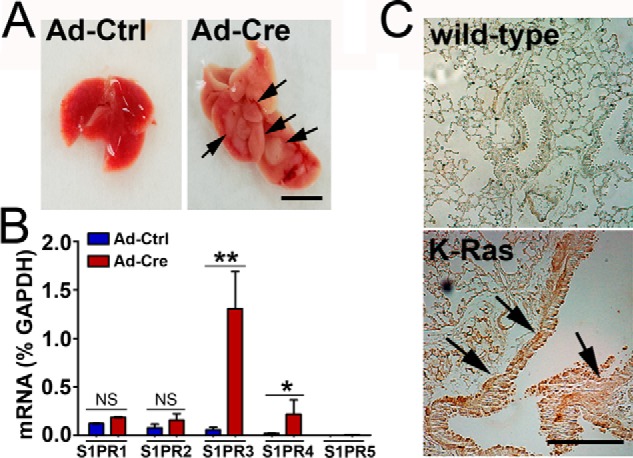
Oncogenic K-Ras mutant stimulates S1PR3 expression. A, LSL-K-RasG12D mice were intratracheally injected with empty adenoviral (Ad-Ctrl) or Ad-Cre particles (1 × 108 pfu). The development of lung adenocarcinomas (arrows) was analyzed 2 months later. Scale bar = 0.5 cm. B, K-RasG12D mice were injected with Ad-Ctrl or Ad-Cre particles. 2 months later, levels of S1PRs in lungs were measured by qPCR analysis. ** and *, p < 0.01 and 0.05, respectively. NS, non-statistically significant. n = 5, Student's t test. C, immunohistochemical staining of S1PR3 in lung specimens from wild-type or K-Ras transgenic mice. Note that levels of S1PR3 are profoundly increased in lung adenocarcinoma of K-Ras transgenic mice (arrows). Scale bar = 200 μm.
TGF-β/SMAD3 Signaling Pathway Stimulates S1PR3 Expression
Promoter analysis suggested that the promoter region of S1PR3 contains 16 potential binding elements for the SMAD3 molecule (Fig. 3A, Table 1), a critical signal transducer downstream of TGF-β/TGF-β receptor signaling. Also, it was shown that K-Ras mutant up-regulated TGF-β, which is required for tumor angiogenesis (34). Therefore, we examined whether the TGF-β/SMAD3 signaling contributes to oncogenic K-Ras mutant-stimulated S1PR3 up-regulation. Ectopic expression of oncogenic K-RasG12V mutant significantly increased S1PR3 (Fig. 3, B and C). Expression of K-RasG12V did not alter levels of other S1P receptor subtypes. In agreement with a previous study (34), levels of TGF-β were increased in K-RasG12V-expressing cells (Fig. 3D). Treatments with TGF-β antibody (Fig. 3E) and inhibition of TGF-β receptor I and SMAD3 using compound SB-431542 and SIS3 (Fig. 3F), respectively, abrogated the S1PR3 up-regulation in K-RasG12V-expressing cells.
FIGURE 3.
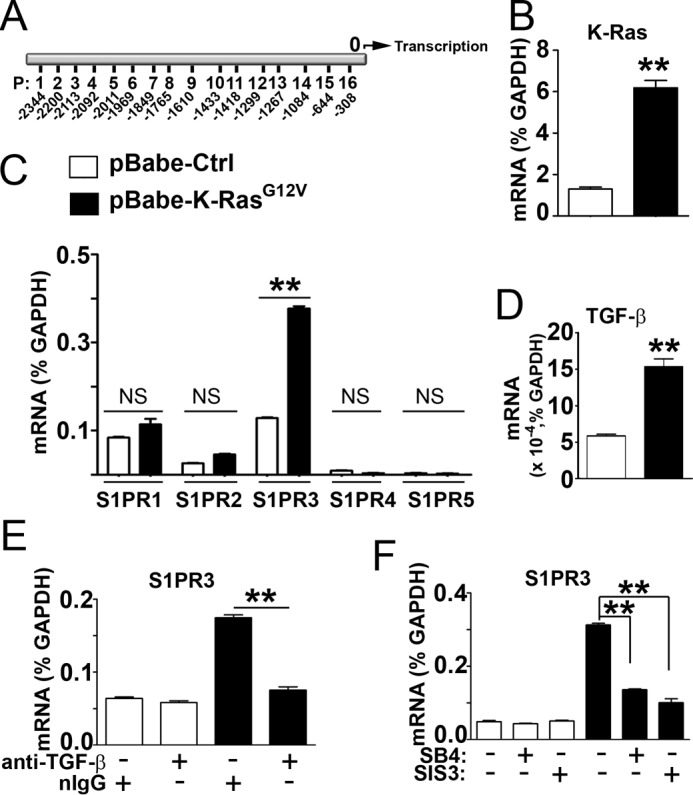
TGF-β/SMAD3 signaling contributes to oncogenic K-Ras mutant-stimulated S1PR3 up-regulation. A, P, potential SMAD3 binding sites in S1PR3 promoter. 0, transcription initiation site. B–D, HEK293 cells were stably transfected with pBabe-K-RasG12V or pBabe control vector. Levels of total cellular K-Ras (B), S1PRs (C), and TGF-β (D) were measured by qPCR analysis. E, HEK293 cells transfected with pBabe-K-RasG12V or pBabe control vector were incubated with anti-TGF-β (Cell Signaling, antibody number 3711, 10 μg/ml) or irrelevant normal rabbit IgG (10 μg/ml) at 37 °C for 24 h. Levels of S1PR3 were quantitated by qPCR. F, HEK293 cells transfected with pBabe-K-RasG12V or pBabe control vector were treated with or without SB-431542 (SB4) (inhibitor of TGF-β receptor I, 10 μm) or SIS3 (inhibitor of SMAD3, 2 μm) at 37 °C for 24 h. Levels of S1PR3 were quantitated by qPCR. **, p < 0.01, n = 3, Student's t test.
TABLE 1.
Candidate SMAD3 binding elements (SBEs) on the promoter region of S1PR3 gene
| SBE | Sequence | Position | Primer name | Primer sequence |
|---|---|---|---|---|
| 1 | GCCAGA | (−2344) to (−2399) | SBE1 F | CCAGGTTGAGCCAGTATTAG |
| SBE1 R | CAGGCGAACGGGTGCTAAT | |||
| 2 | TACAGA | (−2200) to (−2195) | SBE2 F | GACACCCACTAGTGCACAC |
| SBE2 R | GAATGCCAGCTCATAAACG | |||
| 3 | TGCAGA | (−2113) to (−2108) | SBE3/4 F | CGTTTATGAGCTGGCATTC |
| SBE3/4 R | CCATCTTCTCTCACCACTCC | |||
| 4 | AACAGA | (−2092) to (−2087) | SBE3/4 F | CGTTTATGAGCTGGCATTC |
| SBE3/4 R | CCATCTTCTCTCACCACTCC | |||
| 5 | GTCAGA | (−2011) to (−2006) | SBE5/6 F | GGAGTGGTGAGAGAAGATG |
| SBE5/6 R | TCCCCACACACGGGCCTCT | |||
| 6 | AGCAGA | (−1969) to (−1964) | SBE5/6 F | GGAGTGGTGAGAGAAGATG |
| SBE5/6 R | TCCCCACACACGGGCCTCT | |||
| 7 | TCAGA | (−1849) to (−1845) | SBE7 F | CCCACTGAGGACAGGGATC |
| SBE7 R | GCCCAGAAGCCCTGAGGAG | |||
| 8 | CAGACT | (−1765) to (−1760) | SBE8 F | GCCATAAAGCATGAGAGCC |
| SBE8 R | GAGAAAGTCAGTTACCCTG | |||
| 9 | ACAGA | (−1610) to (−1606) | SBE9 F | CAACCCTCAGCCAGTATCC |
| SBE9 R | GCATACTGTGGCCACTGTGTC | |||
| 10 | ACAGA | (−1433) to (−1429) | SBE10/11 F | GTAGGAGTCCAACAAAGGG |
| SBE10/11 R | CACTTCGCCTGTGCTACTGTG | |||
| 11 | CAGA | (−1418) to (−1415) | SBE10/11 F | GTAGGAGTCCAACAAAGGG |
| SBE10/11 R | CACTTCGCCTGTGCTACTGTG | |||
| 12 | CCAGA | (−1299) to (−1295) | SBE12/13 F | CACAGTAGCACAGGCGAAG |
| SBE12/13 R | GCCCTCAGCAATTGAGCTG | |||
| 13 | CAGA | (−1267) to (−1264) | SBE12/13 F | CACAGTAGCACAGGCGAAG |
| SBE12/13 R | GCCCTCAGCAATTGAGCTG | |||
| 14 | AGACAGA | (−1084) to (−1078) | SBE14 F | GCACCCTGCTCCAAGAGAAG |
| SBE14 R | GGCCGGCAGGATTGGGCCGC | |||
| 15 | CAGA | (−644) to (−641) | SBE15 F | CTGGCTGCCGGTCTAGGAGG |
| SBE15 R | CCAGGCACGCAGAGACTTGG | |||
| 16 | CCAGAC | (−350) to (−345) | SBE16 F | GAATCGGCCCAAACAAACCC |
| SBE16 R | GAGCTCCAGGTGCAGAAAC |
Next, we investigated whether TGF-β treatment stimulates S1PR3 expression in lung epithelial cells. HBEC2-KT cells, an immortalized normal human lung epithelial cell line (16), were treated with TGF-β for various times. Quantitative analysis of the expression of S1P receptor subtypes by qPCR analysis showed that TGF-β treatment increased mRNA levels of S1PR3 in a time-dependent manner (Fig. 4A). TGF-β treatment did not affect levels of other subtypes of S1PRs such as S1PR1, S1PR2, and S1PR4. S1PR5 was not detected in HBEC2-KT cells. Also, TGF-β treatment increased protein levels of S1PR3 in HBEC2-KT cells (Fig. 4B). Validation of the specificity of anti-S1PR3 for Western blotting analysis showed that anti-S1PR3 specifically immunoreacts with S1PR3 (Fig. 4C).
FIGURE 4.

TGF-β/SMAD3 signaling axis up-regulates S1PR3. A, HBEC2-KT cells were treated with TGF-β (1 ng/ml) for various times. mRNA levels of S1P receptors were measured by qPCR analysis. Data are mean ± S.D. of triplicate determinations. *, p < 0.05, Student's t test. B, protein levels of S1PR3 in TGF-β (1 ng/ml)-treated HBEC2-KT cells. Lower panel, Western blot intensity was quantitated by National Institutes of Health ImageJ. Data (normalized to actin) are mean ± S.D. of triplicate determinations. * and **, p < 0.05 and 0.01, respectively, Student's t test. C, CHO cells were transduced with adenoviral particles (multiplicity of infection of 200) carrying S1PR1, S1PR2, or S1PR3 vector for 20 h as we described (8). Extracts were blotted with antibody against S1PR3 (Cayman), S1PR2 (Cayman), or S1PR1 (E49) (8). D, mRNAs of S1PR3 and TGF-β in minced C57BL/6 mouse lungs (∼1–2 mm3) infected with adenoviral active TGF-β (Ad-TGF-β, 1 × 108 pfu/ml) or empty vector (Ad-Ctrl) (37 °C, 24 h). **, p < 0.01, n = 5, Student's t test. E, mRNA levels of SphK1 and SphK2 in TGF-β-treated HBEC2-KT cells. F, HBEC2-KT cells (2 × 106 cells in 100-mm dish, 10 ml of cultural medium) were treated with TGF-β (1 ng/ml) for 24 h. Medium was quantitated for S1P, ceramide (Cer), and sphingomyelin (SPM) by LC-MS/MS (29, 46). G, HBEC2-KT were pretreated for 30 min with inhibitors. S1PR3 levels were measured by qPCR, following TGF-β treatment (4 h). The following inhibitors were used: SB4, TGF-β receptor I (SB-431542, 10 μm); SIS3, SMAD3 (2 μm); SB2, p38 kinase (SB-203580, 50 nm); BAY, NFκB (BAY11-7085, 10 μm); JII, JNK (JNK inhibitor II, 10 μm). *, p < 0.05; dashed line, non-statistical significance; n = 3, ANOVA. Each experiment was repeated 2–3 times with similar results. H, cells were pretreated for 30 min with inhibitors, followed by stimulation with TGF-β (1 ng/ml). Activation of p38, JNK, and NFκB was measured by Western blotting with phospho-p38 (P-p38), phospho-JNK (P-p54JNK and P-p46JNK), and phospho-IκBα (p-IκBα). Inhibitors used are: SB-203580 (50 nm) for p38 kinase, JNK inhibitor II (10 μm) for JNK, and BAY11-7085 (10 μm) for NFκB.
Moreover, transduction with adenoviral particles carrying an active form of the TGF-β vector (35–37) effectively increased S1PR3 when compared with transduction with control adenoviral particles, in ex vivo mouse lung minces (Fig. 4D). Furthermore, TGF-β treatment time-dependently increased levels of SphK1 (Fig. 4E) and S1P production (Fig. 4F) in HBEC2-KT normal lung epithelial cells. TGF-β treatment did not alter levels of SphK2 in HBEC2-KT cells.
Next, we used selective pharmacological inhibitor to investigate the role of SMAD3 in TGF-β-stimulated S1PR3 up-regulation. Inhibition of TGF-β receptor I and SMAD3 using compound SB-431542 and SIS3, respectively, abrogated the TGF-β-stimulated S1PR3 up-regulation (Fig. 4G). In contrast, inhibition of other signaling molecules downstream of TGF-β signaling (e.g. NFκB, JNK, and p38 kinase) did not significantly diminish the TGF-β-stimulated S1PR3 up-regulation. In a parallel control experiment, treatment with inhibitor effectively diminished the activation of their respective target following TGF-β stimulation (Fig. 4H). These data suggest that the TGF-β receptor I/Samd3 signaling pathway contributes to the TGF-β-stimulated S1PR3 expression in lung epithelial cells.
Treatment of HBEC2-KT cells with TGF-β markedly stimulated the nuclear accumulation of phosphorylated SMAD3 (Fig. 5A, arrows), indicating that TGF-β treatment activates SMAD3 in HBEC2-KT lung epithelial cells. Subsequently, we designed 16 pairs of primers (Table 1) that amplify these candidate SMAD3 binding elements in the promoter region of S1PR3. ChIP assay showed that TGF-β treatment significantly increased the binding of phospho-SMAD3 to P13, P14, and P15 sites in the promoter region of S1PR3 (Fig. 5B). No specific binding was observed when ChIP assays were performed using irrelevant normal IgG as a control, suggesting that bindings of phospho-SMAD3 are specific.
FIGURE 5.
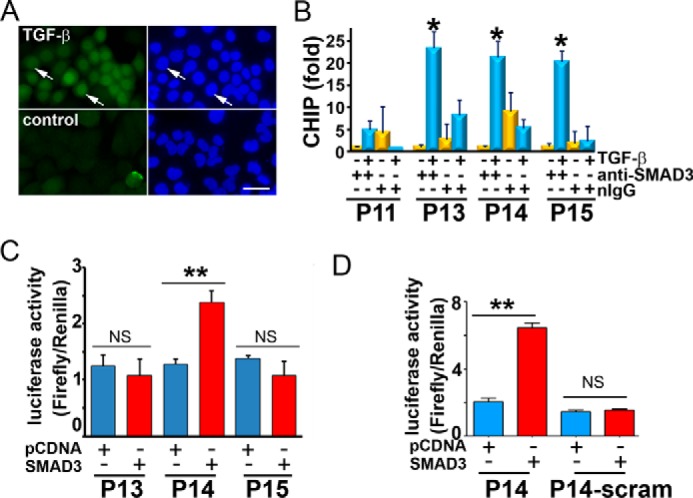
SMAD3 transactivates S1PR3 promoter. A, immunostaining with anti-phospho-SMAD3 in HBEC2-KT treated with or without TGF-β (1 ng/ml, 15 min). Left, fluorescence; right, DAPI nuclear staining. Scale bar = 15.2 μm. B, ChIP was performed with anti-phopho-SMAD3 or normal IgG in HBEC2-KT treated with or without TGF-β (1 h). *, p < 0.05, TGF-β (+)/anti-phospho-SMAD3 versus TGF-β (−)/anti-phospho-SMAD3 (n = 3, Student's t test). C, HEK293 cells were co-transfected with pGL3 luciferase vector carrying double-stranded P13, P14, or P15 oligonucleotides, pcDNA-SMAD3 or empty pcDNA plasmids, and Renilla luciferase vector (5:5:1). 24 h later, both firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activities were normalized to Renilla luciferase activities. D, HEK293 cells were co-transfected with pGL3 luciferase vector carrying P14 or scrambled P14 oligonucleotides, pcDNA-SMAD3 or empty pcDNA plasmids, and Renilla luciferase vector (5:5:1). 24 h later, luciferase activities (firefly/Renilla luciferase activity) were measured. **, p < 0.01; NS, non-statistical significance; n = 3, Student's t test.
Next, we used a luciferase reporter assay to examine whether SMAD3 transactivates those candidate SMAD3 binding sites present in the S1PR3 promoter region. As shown in Fig. 5C, SMAD3 activates PGL3-promoter luciferase vector carrying P14, whereas SMAD3 did not activate PGL3-promoter luciferase vector carrying P13 and P15. The luciferase reporter assay is specific, because SMAD3 was unable to activate scrambled P14 (Fig. 5D).
S1PR3 Promotes Lung Adenocarcinoma Progression
We previously showed that S1PR3 activation promotes proliferation, soft agar growth, and invasion of human lung adenocarcinoma cells in vitro (11, 16). Therefore, we utilized animal models to examine the role of S1PR3 in human lung adenocarcinoma progression. Human H1793 lung adenocarcinoma cells, abundantly expressing S1PR3 (16), were stably transfected with sh-S1PR3 or sh-control vectors. Expression of sh-S1PR3 effectively knocked down ∼67% of S1PR3 in H1793 cells (Fig. 6A). Moreover, S1PR3 knockdown significantly inhibited tumor growth in a subcutaneous xenograft mouse model (Fig. 6, B and C). Similarly, S1PR3 knockdown diminished lung colonization of H1793 cells, which were injected via the tail vein route (Fig. 6, D and E). In contrast, H1299 human lung adenocarcinoma cells express very low levels of S1PR3 among human lung adenocarcinoma cell lines (16) and are poorly tumorigenic in athymic mice. Ectopic expression of S1PR3 profoundly promoted tumor growth in athymic mice (Fig. 6F). These results suggest that S1PR3 activity promotes tumorigenesis of human lung adenocarcinomas.
FIGURE 6.
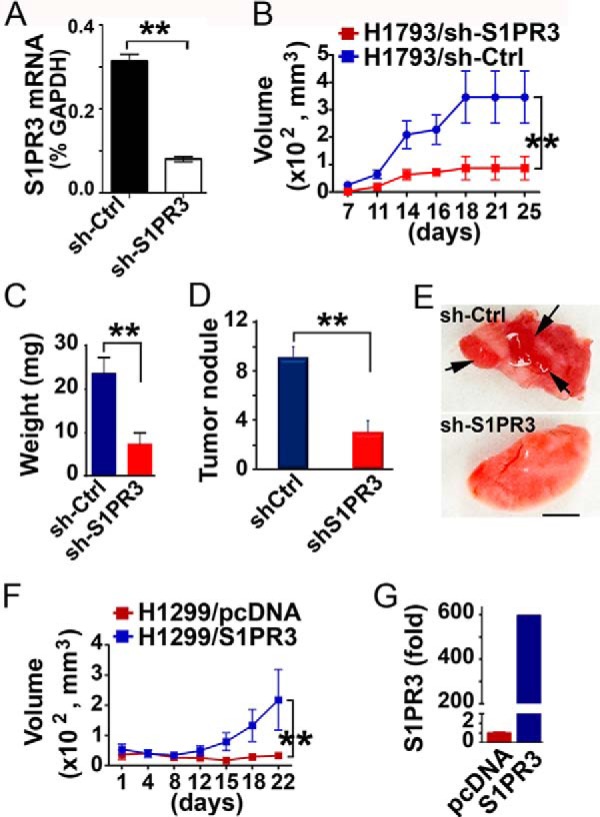
S1PR3 regulates growth and lung colonization of lung adenocarcinoma cells. A, H1793 cells were stably transfected with sh-S1PR3 or pRS (sh-Ctrl) vector (11, 16). mRNA levels of S1PR3 were quantitated with qPCR analysis. B, H1793 cells (1 × 106 cells), stably transfected with sh-S1PR3 or sh-Ctrl vector, were subcutaneously inoculated in Scid mice. Tumor volume was measured in two dimensions using calipers, and volume was determined using the formula width2 × length × 0.52 (49). C, 4 weeks after inoculation, tumors were removed and weighed. D, Scid mice were injected with H1793 cells transfected with sh-S1PR3 or sh-Ctrl vector (1 × 106 cells) via tail vein route. 28 days later, tumor nodules on lung surface were scored. E, representative images of lung injected with H1793-sh-Ctrl and H1793-sh-S1PR3 cells. Arrows, tumor nodules. Scale bar = 0.5 cm. F, volume of xenograft tumors in athymic nude mice subcutaneously implanted with H1299 cells stably transfected with S1PR3 or control pcDNA vector (1 × 106 cells) (11, 16). G, qPCR quantitation of S1PR3 levels in H1299/pcDNA and H1299/S1PR3 cells. **, p < 0.01, n = 6, ANOVA.
Pharmacological Inhibition of S1PR3 Diminishes Lung Adenocarcinoma Growth
Next, we investigated whether treatment with S1PR3 antagonist diminishes the growth of human lung adenocarcinoma cells. C57BL/6 mice were subcutaneously implanted with murine Lewis lung carcinoma (LLC) cells. 1 week after tumor implantation, mice were intraperitoneally injected every 3 days with VPC23019, an antagonist of S1PR1 and S1PR3 receptors (38). Administration of VPC23019 significantly inhibited tumor growth (Fig. 7A). Lewis lung carcinoma cells predominantly express S1PR3, and S1PR1 is barely detected (Fig. 7B). Thus, the effect of VPC23019 on inhibition of tumor growth is most likely due to its antagonistic activity on S1PR3 present in LLC cells. Indeed, treatment with TY-52156, a highly selective antagonist of S1PR3 (39–41) (Fig. 7C), significantly suppressed the growth of Lewis lung carcinoma cells (Fig. 7, D and E). These results suggest that S1PR3 represents a novel therapeutic target for the treatment of lung carcinomas.
FIGURE 7.
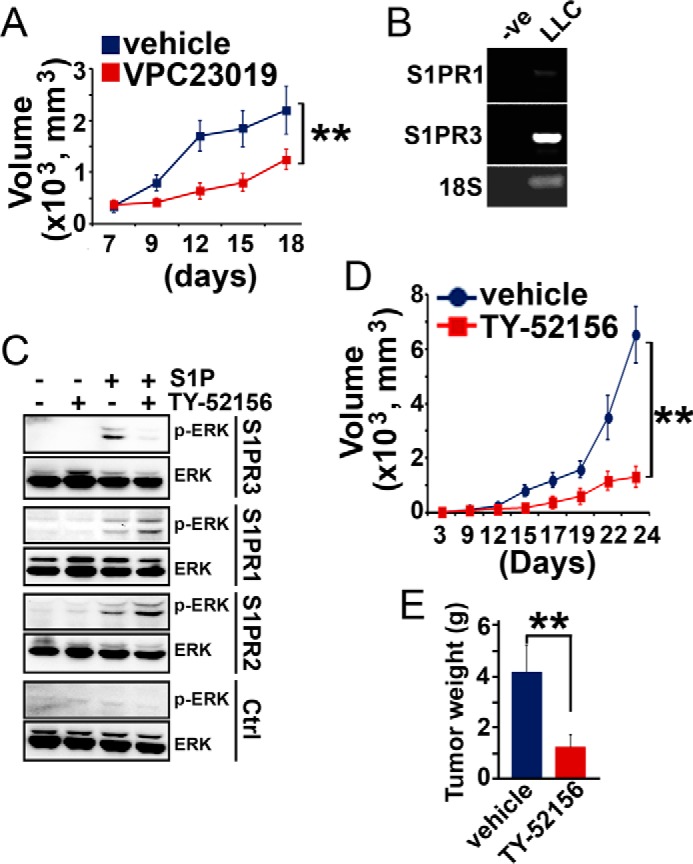
Inhibition of S1PR3 diminishes lung carcinoma growth. A, C57BL/6 mice were subcutaneously inoculated with LLC cells (1 × 106 cells). 1 week later, mice were intraperitoneally administered with VPC23019 (1.5 mg/kg of body weight) or control vehicle every 3 days. B, S1PR1 and S1PR3 levels in Lewis lung carcinoma cells. −ve, PCR reactions were performed without cDNA. **, p < 0.01, n = 6, ANOVA. C, CHO cells were transduced with adenoviral particles carrying S1PR1, S1PR2, S1PR3, or pcDNA control vector. Cells were serum-starved for 24 h. Subsequently, cells were treated with TY-52156 (10 μm) for 10 min, followed by stimulating with S1P (200 nm, 10 min). ERK1/2 activation (p-ERK) was measured by Western blotting analysis. D, C57BL/6 mice were subcutaneously inoculated with LLC cells (1 × 106 cells). 1 week later, mice were intraperitoneally administered with TY-52156 (10 mg/kg of body weight) or control vehicle every 2 days. **, p < 0.01, n = 6, ANOVA. E, tumor weights were measured 24 days after implantation. **, p < 0.01, n = 6, ANOVA.
Discussion
We previously showed that levels of S1PR3 are increased in a panel of cultured human lung adenocarcinoma cell lines when compared with normal lung epithelial cells (16). In this report, we observed that mRNA and protein levels of S1PR3 are significantly up-regulated in human lung adenocarcinoma specimens. Our observation is supported by the analysis of Oncomine data sets (42–45) showing that S1PR3 expression correlates with clinical stages (42, 44), EML4-ALK gene fusion (42), lymphatic and perineural invasion (44), metastasis to bone (44), vascular invasion (44), BCL amplification (45), and APC deletion and family history (43) of human lung adenocarcinomas. These data suggest that S1PR3 is up-regulated in human lung adenocarcinomas, and S1PR3 expression correlates with the aggressiveness of lung adenocarcinomas.
Oncogenic K-Ras mutation is found in more than 25% of non-small cell lung cancers (30, 31). In the LSL-K-RasG12D transgenic mouse model, we found that the expression of K-RasG12D mutant triggered the development of lung cancers and concurrently stimulated the expression of S1PR3. In agreement with our study, Oncomine data sets analysis showed that S1PR3 up-regulation correlates with K-Ras mutation status in human lung cancers (42, 43, 48, 50, 51) (see Genomic Data Commons (https://gdc.cancer.gov)). Mechanistically, our data suggest that the oncogenic K-Ras mutant-stimulated S1PR3 expression is mediated by an autocrine TGF-β/SMAD3 axis in lung epithelial cells. In supporting our observations, it was shown that oncogenic K-Ras mutant stimulated the expression of TGF-β, which plays a critical role in tumor angiogenesis in K-Ras mutant-driven cancers (34). It should be noted that lung cancers driven by K-Ras mutant are generally refractory to chemotherapy as well as targeted agents (31, 52). To date, the identification of drugs to therapeutically inhibit K-Ras mutant has been unsuccessful, suggesting that other approaches are required. We showed that oncogenic K-Ras mutant stimulates S1PR3 expression, suggesting that S1PR3 represents a novel therapeutic target for the treatment of K-Ras mutant-driven lung cancers.
Previously, we showed that S1PR3 regulates the proliferation, colony formation, and invasiveness of human lung adenocarcinoma cells in vitro (11, 16). In the present study, we utilized animal models to examine the role of S1PR3 in the progression of human lung adenocarcinomas. H1793 human lung adenocarcinoma cells abundantly express S1PR3, and S1PR3 knockdown profoundly abrogated proliferation, colony formation in soft agar, and invasion of tumor cells in vitro (11, 16). Similarly, S1PR3 knockdown significantly inhibited tumor growth in a xenograft model, as well as lung colonization of adenocarcinoma cells in a tail vein implantation model. In contrast, H1299 human lung adenocarcinoma cells express very low levels of S1PR3 among lung adenocarcinoma cell lines (16). Expression of S1PR3 significantly promoted growth of tumor xenograft. These results suggest that the S1PR3-mediated signaling pathways play an important role in promoting the progression of lung adenocarcinoma cells. We previously characterized two S1PR3-mediated signaling pathways that may have functional implications in promoting lung adenocarcinoma progression. We found that S1PR3 activation transcriptionally up-regulates EGFR levels and greatly potentiates the effect of EGF on the proliferation of lung adenocarcinoma cells (16). Moreover, we characterized a novel signaling pathway, namely S1PR3/JNK/AP-1/ETS-1/CD44 axis, which critically regulates the invasiveness of human lung adenocarcinoma cell in vitro (11). Collectively, our studies suggest that S1PR3 represents a potential therapeutic target for the treatment of human lung adenocarcinomas. Indeed, our study using pharmacological inhibitors supports this notion. We found that administration of VPC23019 (an antagonist of S1PR1 and S1PR3 receptors (38)) and TY-52156 (a selective inhibitor of S1PR3 (39–41)) significantly diminished lung tumor growth in xenograft mouse model.
Mechanistically, we showed that TGF-β/SMAD3 signaling pathway transactivates S1P/S1PR3 axis in lung epithelial cells. A previous study showed that TGF-β activates sphingosine kinase via a non-SMAD signaling pathway and that the TGF-β/sphingosine kinase axis is important for the migration and invasion of esophageal cancer cells in vitro (53). However, the role of the TGF-β signaling axis on the regulation of S1PRs was not investigated in that study. Moreover, in agreement with our observation, Cencetti et al. (54) showed that TGF-β stimulated S1PR3 expression in C2C12 myoblasts. In contrast to their study, we precisely defined the SMAD3 binding sites on the promoter region of S1PR3 and demonstrated that the TGF-β-stimulated S1PR3 up-regulation is dependent on the SMAD3 signaling molecule. Furthermore, we found that TGF-β concomitantly stimulated SphK1 expression and increased S1P production in lung epithelial cells. Collectively, our results suggest that TGF-β activates an autocrine S1P/S1PR3 signaling in lung epithelial cells, which may contribute to lung adenocarcinoma progression.
Several tumors, including lung cancers, express high levels of TGF-β (55–57), which correlates with tumor progression and clinical prognosis (58–63). Thus, our observation of the TGF-β-mediated S1PR3 up-regulation in lung cancers is pathologically relevant. In addition, TGF-β plays an important role in regulating the tumorigenic processes including epithelial-mesenchymal transition (17, 20, 64–66) and tumor inflammation (67–72). For example, TGF-β stimulates the expression of pro-inflammatory and pro-tumorigenic cytokine IL-6 (71, 72). Elevated systemic and pulmonary productions of IL-6 are commonly observed in lung adenocarcinoma patients and correlate with poor patient survival (73, 74). Moreover, the TGF-β/IL-6 axis was recently shown to mediate the chemo-resistance in lung cancer (71). Our results show that TGF-β activates the autocrine S1P/S1PR3 signaling axis in lung epithelial cells. Thus, it is of significant interest, both clinical and scientific, to investigate the role of S1P/S1PR3 signaling axis in regulating the TGF-β-mediated lung pathologies in the future.
Experimental Procedures
Reagents
Sphingosine-1 phosphate (Biomol) and VPC23019 (Cayman Chemical) were prepared as micelles by sonicating in aqueous solution of fatty acid-free bovine serum albumin (0.4 mg/ml, Sigma). TY-52156 was chemically synthesized as described (39). TGF-β was from R&D Systems. Anti-S1PR3 and anti-phospho-SMAD3 were from Cayman and Abcam, respectively. SB-431542 and SIS3 were purchased from Sigma. Unless specified, other reagents are from Sigma.
Cell Cultures
Immortalized normal human lung epithelial cells (HBEC2-KT and HBEC3-KT) were cultured using keratinocyte-serum free medium (Invitrogen) (75). H1793 human lung adenocarcinoma cells were cultured using HITES medium (RPMI 1640 medium supplemented with hydrocortisone (10 nm), insulin (5 μg/ml), transferrin (100 μg/ml), 17 β-estradiol (10 nm), sodium selenite (30 nm), and 5% fetal bovine serum) (75). H1299 and mouse Lewis lung carcinoma cells were cultured essentially as we described previously (16). Cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C.
Real-time PCR Analysis
Total RNA was isolated using TRIzol reagent (Invitrogen) and was reverse-transcribed with an oligo(dT) primer (Promega) by Moloney Murine Leukemia Virus (M-MLV) Reverse Transcriptase (Promega) for first-strand cDNA synthesis. For real-time PCR quantitation, 50 ng of reversely transcribed cDNAs were amplified with the ABI 7500 system (Applied Biosystems) in the presence of TaqMan DNA polymerase. The qPCR reaction was performed by using a universal PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. The sense and antisense primers used for qPCR analysis are: human and mouse S1PR1, sense, 5′-ATC ATG GGC TGG AAC TGC ATC A-3′, antisense, 5′-CGA GTC CTG ACC AAG GAG TAG AT-3; human and mouse S1PR2, sense, 5′-CAG ACG CTA GCC CTG CTC AAG A-3′, antisense, 5′-TAG TGG GCT TTG TAG AGG A-3′; human and mouse S1PR3, sense, 5′-ACA ACC GCA TGT ACT TTT TCA T-3′, antisense, 5′-TAC TGC CCT CCC TGA GGA ACC A-3′; human S1PR4, sense, 5′-GGG CCA TCT TCC GCC TGG TG-3′, antisense, 5′-TGC CCC GCA GGT ACT CCT GG-3′; human S1PR5, sense, 5′-GGC GCG CAC CTG TCC TGT AC-3′, antisense, 5′-TCG GGT CTC TGC CGC AGG AG-3′; human and mouse SphK1, sense, 5′-AAA CCC CTG TGT AGC CTC CC-3′, antisense, 5′-AGC AGG TTC ATG GGT GAC AG-3′; human SphK2, sense, 5′-GCA CAG CAA CAG TGA GCA-3′, antisense, 5′-GAG CCT GAG TGA GTG GGA-3′; porcine TGF-β, sense, 5′-GCA CGT GGA GCT ATA CCA GAA-3′, antisense, 5′-CAT CAA AGG ACA GCC ACT CC-3′; human GAPDH, sense, 5′-GAA GGT GAA GGT CGG AGT-3′, antisense, 5′-GAA GAT GGT GAT GGG TTT C-3′; and mouse GAPDH, sense, 5′-CAC CTT CGA TGC CGG GGC TG-3′, antisense, 5′-GGC CAT GAG GTC CAC CAC CC-3′. cDNA array analysis of mRNA levels of S1PR3 was performed using TissueScan qPCR arrays (HLRT101 and HLRT105, OriGene) following the manufacturer's instructions. The results of adenocarcinomas were extracted, and then analyzed by Student's t test.
Immunofluorescence Microscopy
Cells were fixed with 4% paraformaldehyde for 30 min, followed by permeabilization with PBS containing 0.05% Triton X-100. After washing three times with PBS, cells were incubated with primary antibody at room temperature overnight. Cells were then washed three times with PBS, and incubated with FITC-conjugated secondary antibody for 1 h. Fluorescence images were captured by the Leica TCS SP5 confocal system (Leica, Wetzlar, Germany).
Immunohistochemical Staining
Human lung carcinoma tumor microarray (TMA) was purchased from Accumax (Accumax 306). Immunohistochemical staining was performed using VECTASTAIN ABC kit (Vector Laboratories, catalog number PK-6200) following the manufacturer's instructions. Briefly, TMA sections were deparaffinized and dehydrated. Antigen retrieval was performed by microwave irradiation (two cycles of 5 min each) in 10 mm citrate buffer (pH 6.0). TMA was incubated with rabbit polyclonal S1P3 antibody (1:200, Cayman) for 60 min, and then with biotinylated secondary antibody solution for 30 min and VECTASTAIN ABC Reagent for 30 min at room temperature. Subsequently, sections were incubated with peroxidase substrate (ImmPACT DAB (3,3′-diaminobenzidine), Vector Laboratories, catalog number SK-4105) until the desired stain intensity develops. Levels of S1PR3 were visualized by light microscopy (Leica DMI3000B).
Western Blotting Analysis
Protein extraction and Western blotting were performed as described (11). Briefly, cells were collected in ice-cold PBS using cell scrapers followed by centrifugation (500 × g, 5 min). Cell extracts were prepared with radioimmunoprecipitation assay buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitors (Calbiochem) with constant agitation at 4 °C for 30 min. After centrifugation at 15,000 × g for 20 min, supernatant was collected and protein concentration was measured using a bicinchoninic acid protein assay kit with BSA as standard. 50 μg of protein extracts were dissolved in 2× Laemmli sample buffer, heated at 95 °C for 5 min, and resolved on a 10% SDS-PAGE gel. After electrophoresis, gels were transferred to nitrocellulose membranes. Subsequently, membranes were blocked in 5% nonfat dry milk (Lab Scientific) in TBST buffer (20 mm Tris-HCl, pH 7.4, 500 mm NaCl, and 0.05% Tween 20). Membranes were washed and incubated with indicated primary antibodies (1:1000 dilution) on a rotary shaker at 4 °C overnight. The blots were then incubated with peroxidase-conjugated secondary antibody for 1 h at room temperature and developed with enhanced chemiluminescent reagent (Thermo Scientific).
ChIP Analysis
The ChIP assay was performed using Pierce Agarose ChIP Kit, following the manufacturer's instructions. Briefly, 1 × 107 cells were cross-linked with 1% formaldehyde for 10 min. Following the addition of glycine quenching solution, cells were scraped and resuspended in 1× PBS with protease inhibitor cocktails (Calbiochem). Cells were then lysed in lysis buffer, and nuclear lysates were treated with micrococcal nuclease. Lysates were immunoprecipitated with anti-phospho-SMAD3 (Thermo Scientific) at 4 °C overnight. Immunoprecipitation with irrelevant normal IgG was used as a control. Immune complexes were isolated with protein A/G-Sepharose beads at 4 °C for 1 h. After washings, DNA fragments contained in immune complexes were purified, and then amplified by qPCR reactions. Sequences of primer pairs used for ChIP assay of SMAD3 binding to S1PR3 promoter are shown in Table 1.
Luciferase Reporter Assay
Oligonucleotides of candidate SMAD3 binding sites in the S1PR3 promoter region were synthesized, with an overhanging NheI and SacI restriction site sequence at the 5′-end and 3′-end, respectively, of the antisense strand. Synthesized oligonucleotides are: P13 sense, 5′-GTC AGC AGG CAG AGT CAC TTG C-3′; P13 antisense, 5′-CTA GGC AAG TGA CTC TGC CTG CTG ACA GCT-3′; P14 sense, 5′-GGG CAA AAG ACA GAA AGT AAC C-3′; P14 antisense, 5′-CTA GGG TTA CTT TCT GTC TTT TGC CCA GCT-3′; P15 sense, 5′-GTG CAC CAG CAG AGG CTG GGG C-3′; P15 antisense, 5′-CTA GGC CCC AGC CTC TGC TGG TGC ACA GCT-3′; Scramble P14 sense, 5′-GGG CAA ATG GCG AAA AGT AAC C-3′; Scramble P14 antisense, 5′-CTA GGG TTA CTT TTC GCC ATT TGC CCA GCT-3′. Equimolar amounts of sense and antisense oligonucleotides were mixed at 95 °C for 5 min, followed by cooling to room temperature. Annealed double-strand oligonucleotides were ligated with NheI- and SacI-digested pGL3-promoter luciferase reporter vector (Promega). Recombinant luciferase vectors were verified by DNA sequencing.
HEK293 cells were co-transfected with recombinant pGL3 luciferase vector, pcDNA-SMAD3 (47) or empty pcDNA plasmids, and pRL-null vector (Promega) carrying the Renilla luciferase gene (5:5:1) by using Lipofectamine 2000 reagent (Life Technologies). 24 h after transfection, both firefly and Renilla luciferase activities were measured with the Dual-Luciferase Reporter Assay System (Promega) using a SpectraMax M3 Multi-mode Microplate Reader (Molecular Devices). Firefly luciferase activities (M1) were normalized to Renilla luciferase activities (M2).
Sphingolipid Measurement by LC-MS/MS
Sphingolipids were extracted from culture medium as we described previously (29, 46). Samples were filtered through 0.45-μm nylon filters directly into auto sampler vials for LC-MS/MS analysis. Reverse phase HPLC was performed using BDS HYPERSIL C8 columns (100 × 2.1 mm, 2.4 μm, Thermo Scientific) and gradient elution on Waters Alliance 2695 system (Waters Corp.). The mobile phase consisted of methanol, water, and ammonium formate. Solvent A was 2 mm ammonium formate in methanol with 0.2% formic acid. The column was equilibrated with solvent A for 5 min. Samples were injected using the autosampler (an integral part of the Waters Alliance 2695 system) maintained at 10 ± 2 °C. The injection volumes were 80 μl for each sample. A complete injection of each sample took 7 min including column equilibration. The flow rate was 0.3 ml/min. The HPLC eluent was directly introduced to Quattro LC mass spectrometer (Micromass, Waters), equipped with an electrospray ion source that was used for ESI-MS/MS. The ESI-MS/MS experiments for the quantitation of sphingolipids were carried out in the positive ion mode with ESI needle voltage, 2.8 kV; source block temperature, 120 °C; desolvation temperature, 350 °C; desolvation gas flow, 540 liters/h; nebulizer gas flow, 80 liters/h; and collision gas pressure, 3.2 × 10−4 bars. Cone voltage and collision energy for each multiple reaction monitoring transition were optimized. Chromatographic data were analyzed by the QuanLynx module of the MassLynx software (Waters) to integrate the chromatograms for each multiple reaction monitoring transition.
Tumor Growth and Lung Colonization in Mice
All animal procedures were performed according to the National Institutes of Health and institutional guidelines, and were approved by the Wayne State University Animal Use and Care Committee. For subcutaneous implantation, lung carcinoma cells were adjusted to 1 × 107 cells/ml. Mice were injected with 0.1 ml of cell suspension into the subcutaneous dorsa in the proximal midline. Alternatively, 1 × 106 cells (in 50 μl) were injected via the tail vein route. NOD-Scid mice (8 weeks old, female, Taconic) were used for H1793, athymic nude mice (8 weeks old, female, Harlan) were used for H1299 cells, and C57BL/6 mice (8 weeks old, female, The Jackson Laboratory) were used for Lewis lung carcinoma cells. Tumor volume was measured in two dimensions using calipers, and volume was determined using the formula width2 × length × 0.52 (49). For VPC23019 treatment, mice were randomized into two groups (six animals per group) 1 week after inoculation of tumor cells. One group of mice was intraperitoneally injected with VPC23019 (1.5 mg/kg of body weight), and the other was injected with 100 μl of 0.4% BSA (vehicle control) every 3 days. For TY-52156 treatment, mice (six mice) were intraperitoneally injected with TY-52156 (10 mg/kg of body weight) or DMSO control vehicle every 2 days.
Statistical Analysis
Results are shown as mean ± S.D. Differences between paired samples were analyzed by Student's t test. ANOVA analysis was performed to analyze tumor progression in mouse experiments. p value < 0.01 is considered highly significant, and p < 0.05 is considered statistically significant.
Author Contributions
J. Z., J. L., J. F. L., W. Z., and M. K. designed the study and conducted experiments. G. C. V. and Y. H. A. chemically synthesized TY-52156. S. C. prepared research reagents, F. L. pathological evaluation of lung adenocarcinoma. J. Z., J. L., and M. J. L. designed experiments and prepared manuscript.
Acknowledgments
We are grateful for the generous gift of adenoviral particles carrying active TGF-β from Dr. Jack Gauldie (Department of Pathology and Molecular Medicine, McMaster University, Hamilton, Ontario, Canada).
This work was supported by Department of Defense Grant W81XWH-14-1-0346, a funding support of Wayne State University, a Strategic Research Initiative Grant (SRIG) award of the Karmanos Cancer Institute (to M. L.), and National Institutes of Health Grants HL123302 and HL119053 (to S. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- S1P
- sphingosine-1-phosphate
- TGF-β
- transforming growth factor beta
- SBE
- SMAD3 binding element
- LLC
- Lewis lung carcinoma
- Ad
- adenovirus
- Ctrl
- control
- qPCR
- quantitative PCR
- TMA
- tumor microarray
- ESI
- electrospray ionization
- ANOVA
- analysis of variance.
References
- 1. Siow D. L., Anderson C. D., Berdyshev E. V., Skobeleva A., Natarajan V., Pitson S. M., and Wattenberg B. W. (2011) Sphingosine kinase localization in the control of sphingolipid metabolism. Adv. Enzyme Regul. 51, 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee M. J., Van Brocklyn J. R., Thangada S., Liu C. H., Hand A. R., Menzeleev R., Spiegel S., and Hla T. (1998) Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 279, 1552–1555 [DOI] [PubMed] [Google Scholar]
- 3. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., and Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alvarez S. E., Harikumar K. B., Hait N. C., Allegood J., Strub G. M., Kim E. Y., Maceyka M., Jiang H., Luo C., Kordula T., Milstien S., and Spiegel S. (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee M. J., Thangada S., Claffey K. P., Ancellin N., Liu C. H., Kluk M., Volpi M., Sha'afi R. I., and Hla T. (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99, 301–312 [DOI] [PubMed] [Google Scholar]
- 6. Green J. A., Suzuki K., Cho B., Willison L. D., Palmer D., Allen C. D., Schmidt T. H., Xu Y., Proia R. L., Coughlin S. R., and Cyster J. G. (2011) The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 12, 672–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jenne C. N., Enders A., Rivera R., Watson S. R., Bankovich A. J., Pereira J. P., Xu Y., Roots C. M., Beilke J. N., Banerjee A., Reiner S. L., Miller S. A., Weinmann A. S., Goodnow C. C., Lanier L. L., Cyster J. G., and Chun J. (2009) T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J. Exp. Med. 206, 2469–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee M. J., Thangada S., Paik J. H., Sapkota G. P., Ancellin N., Chae S. S., Wu M., Morales-Ruiz M., Sessa W. C., Alessi D. R., and Hla T. (2001) Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol. Cell 8, 693–704 [DOI] [PubMed] [Google Scholar]
- 9. An S., Goetzl E. J., and Lee H. (1998) Signaling mechanisms and molecular characteristics of G protein-coupled receptors for lysophosphatidic acid and sphingosine 1-phosphate. J. Cell. Biochem. Suppl. 30–31, 147–157 [PubMed] [Google Scholar]
- 10. Estrada R., Zeng Q., Lu H., Sarojini H., Lee J. F., Mathis S. P., Sanchez T., Wang E., Kontos C. D., Lin C. Y., Hla T., Haribabu B., and Lee M. J. (2008) Up-regulating sphingosine 1-phosphate receptor-2 signaling impairs chemotactic, wound-healing, and morphogenetic responses in senescent endothelial cells. J. Biol. Chem. 283, 30363–30375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang W., Zhao J., Lee J. F., Gartung A., Jawadi H., Lambiv W. L., Honn K. V., and Lee M. J. (2013) ETS-1-mediated transcriptional up-regulation of CD44 is required for sphingosine-1-phosphate receptor subtype 3-stimulated chemotaxis. J. Biol. Chem. 288, 32126–32137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yester J. W., Tizazu E., Harikumar K. B., and Kordula T. (2011) Extracellular and intracellular sphingosine-1-phosphate in cancer. Cancer Metastasis Rev. 30, 577–597 [DOI] [PubMed] [Google Scholar]
- 13. Pyne N. J., Tonelli F., Lim K. G., Long J. S., Edwards J., and Pyne S. (2012) Sphingosine 1-phosphate signalling in cancer. Biochem. Soc. Trans. 40, 94–100 [DOI] [PubMed] [Google Scholar]
- 14. Furuya H., Shimizu Y., and Kawamori T. (2011) Sphingolipids in cancer. Cancer Metastasis Rev. 30, 567–576 [DOI] [PubMed] [Google Scholar]
- 15. Aoyagi T., Nagahashi M., Yamada A., and Takabe K. (2012) The role of sphingosine-1-phosphate in breast cancer tumor-induced lymphangiogenesis. Lymphat. Res. Biol. 10, 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu A., Zhang W., Lee J. F., An J., Ekambaram P., Liu J., Honn K. V., Klinge C. M., and Lee M. J. (2012) Sphingosine-1-phosphate receptor-3 signaling up-regulates epidermal growth factor receptor and enhances epidermal growth factor receptor-mediated carcinogenic activities in cultured lung adenocarcinoma cells. Int. J. Oncol. 40, 1619–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papageorgis P. (2015) TGFβ signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J. Oncol. 2015, 587193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nowrin K., Sohal S. S., Peterson G., Patel R., and Walters E. H. (2014) Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert Rev. Respir. Med. 8, 547–559 [DOI] [PubMed] [Google Scholar]
- 19. Giannelli G., Villa E., and Lahn M. (2014) Transforming growth factor-β as a therapeutic target in hepatocellular carcinoma. Cancer Res. 74, 1890–1894 [DOI] [PubMed] [Google Scholar]
- 20. Derynck R., Muthusamy B. P., and Saeteurn K. Y. (2014) Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr. Opin Cell Biol. 31, 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fuxe J., and Karlsson M. C. (2012) TGF-β-induced epithelial-mesenchymal transition: a link between cancer and inflammation. Semin. Cancer Biol. 22, 455–461 [DOI] [PubMed] [Google Scholar]
- 22. Nalluri S. M., O'Connor J. W., and Gomez E. W. (2015) Cytoskeletal signaling in TGFβ-induced epithelial-mesenchymal transition. Cytoskeleton (Hoboken) 72, 557–569 [DOI] [PubMed] [Google Scholar]
- 23. Mantel P. Y., and Schmidt-Weber C. B. (2011) Transforming growth factor-β: recent advances on its role in immune tolerance. Methods Mol. Biol. 677, 303–338 [DOI] [PubMed] [Google Scholar]
- 24. Nagaraj N. S., and Datta P. K. (2010) Targeting the transforming growth factor-β signaling pathway in human cancer. Expert Opin. Investig. Drugs 19, 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Derynck R., Akhurst R. J., and Balmain A. (2001) TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129 [DOI] [PubMed] [Google Scholar]
- 26. Yamanaka M., Shegogue D., Pei H., Bu S., Bielawska A., Bielawski J., Pettus B., Hannun Y. A., Obeid L., and Trojanowska M. (2004) Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-β and mediates TIMP-1 up-regulation. J. Biol. Chem. 279, 53994–54001 [DOI] [PubMed] [Google Scholar]
- 27. Xin C., Ren S., Kleuser B., Shabahang S., Eberhardt W., Radeke H., Schäfer-Korting M., Pfeilschifter J., and Huwiler A. (2004) Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-β-induced cell responses. J. Biol. Chem. 279, 35255–35262 [DOI] [PubMed] [Google Scholar]
- 28. Bu S., Kapanadze B., Hsu T., and Trojanowska M. (2008) Opposite effects of dihydrosphingosine 1-phosphate and sphingosine 1-phosphate on transforming growth factor-β/Smad signaling are mediated through the PTEN/PPM1A-dependent pathway. J. Biol. Chem. 283, 19593–19602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang W., An J., Jawadi H., Siow D. L., Lee J. F., Zhao J., Gartung A., Maddipati K. R., Honn K. V., Wattenberg B. W., and Lee M. J. (2013) Sphingosine-1-phosphate receptor-2 mediated NFκB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 106, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sholl L. M., Aisner D. L., Varella-Garcia M., Berry L. D., Dias-Santagata D., Wistuba I. I., Chen H., Fujimoto J., Kugler K., Franklin W. A., Iafrate A. J., Ladanyi M., Kris M. G., Johnson B. E., Bunn P. A., et al. (2015) Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 10, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karachaliou N., Mayo C., Costa C., Magrí I., Gimenez-Capitan A., Molina-Vila M. A., and Rosell R. (2013) KRAS mutations in lung cancer. Clin. Lung Cancer 14, 205–214 [DOI] [PubMed] [Google Scholar]
- 32. Jackson E. L., Willis N., Mercer K., Bronson R. T., Crowley D., Montoya R., Jacks T., and Tuveson D. A. (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao P., Damerow M. S., Stern P., Liu A. H., Sweet-Cordero A., Siziopikou K., Neilson J. R., Sharp P. A., and Cheng C. (2013) CD44 promotes Kras-dependent lung adenocarcinoma. Oncogene 32, 5186–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rak J., Filmus J., Finkenzeller G., Grugel S., Marmé D., and Kerbel R. S. (1995) Oncogenes as inducers of tumor angiogenesis. Cancer Metastasis Rev. 14, 263–277 [DOI] [PubMed] [Google Scholar]
- 35. Gauldie J., and Kolb M. (2008) Animal models of pulmonary fibrosis: how far from effective reality? Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L151. [DOI] [PubMed] [Google Scholar]
- 36. Decologne N., Kolb M., Margetts P. J., Menetrier F., Artur Y., Garrido C., Gauldie J., Camus P., and Bonniaud P. (2007) TGF-β1 induces progressive pleural scarring and subpleural fibrosis. J. Immunol. 179, 6043–6051 [DOI] [PubMed] [Google Scholar]
- 37. Cui Y., Robertson J., Maharaj S., Waldhauser L., Niu J., Wang J., Farkas L., Kolb M., and Gauldie J. (2011) Oxidative stress contributes to the induction and persistence of TGF-β1 induced pulmonary fibrosis. Int. J. Biochem. Cell Biol. 43, 1122–1133 [DOI] [PubMed] [Google Scholar]
- 38. Davis M. D., Clemens J. J., Macdonald T. L., and Lynch K. R. (2005) Sphingosine 1-phosphate analogs as receptor antagonists. J. Biol. Chem. 280, 9833–9841 [DOI] [PubMed] [Google Scholar]
- 39. Murakami A., Takasugi H., Ohnuma S., Koide Y., Sakurai A., Takeda S., Hasegawa T., Sasamori J., Konno T., Hayashi K., Watanabe Y., Mori K., Sato Y., Takahashi A., Mochizuki N., and Takakura N. (2010) Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 77, 704–713 [DOI] [PubMed] [Google Scholar]
- 40. Nussbaum C., Bannenberg S., Keul P., Gräler M. H., Gonçalves-de-Albuquerque C. F., Korhonen H., von Wnuck Lipinski K., Heusch G., de Castro Faria Neto H. C., Rohwedder I., Göthert J. R., Prasad V. P., Haufe G., Lange-Sperandio B., Offermanns S., et al. (2015) Sphingosine-1-phosphate receptor 3 promotes leukocyte rolling by mobilizing endothelial P-selectin. Nat. Commun. 6, 6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hirata N., Yamada S., Shoda T., Kurihara M., Sekino Y., and Kanda Y. (2014) Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat. Commun 5, 4806. [DOI] [PubMed] [Google Scholar]
- 42. Bild A. H., Yao G., Chang J. T., Wang Q., Potti A., Chasse D., Joshi M.-B., Harpole D., Lancaster J. M., Berchuck A., Olson J. A. Jr., Marks J. R., Dressman H. K., West M., and Nevins J. R. (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439, 353–357 [DOI] [PubMed] [Google Scholar]
- 43. Ding L., Getz G., Wheeler D. A., Mardis E. R., McLellan M. D., Cibulskis K., Sougnez C., Greulich H., Muzny D. M., Morgan M. B., Fulton L., Fulton R. S., Zhang Q., Wendl M. C., Lawrence M. S., et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larsen J. E., Pavey S. J., Passmore L. H., Bowman R., Clarke B. E., Hayward N. K., and Fong K. M. (2007) Expression profiling defines a recurrence signature in lung squamous cell carcinoma. Carcinogenesis 28, 760–766 [DOI] [PubMed] [Google Scholar]
- 45. Olejniczak E. T., Van Sant C., Anderson M. G., Wang G., Tahir S. K., Sauter G., Lesniewski R., and Semizarov D. (2007) Integrative genomic analysis of small-cell lung carcinoma reveals correlates of sensitivity to Bcl-2 antagonists and uncovers novel chromosomal gains. Mol. Cancer Res. 5, 331–339 [DOI] [PubMed] [Google Scholar]
- 46. Zhang W., Mottillo E. P., Zhao J., Gartung A., VanHecke G. C., Lee J. F., Maddipati K. R., Xu H., Ahn Y. H., Proia R. L., Granneman J. G., and Lee M. J. (2014) Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J. Biol. Chem. 289, 32178–32185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie W. B., Li Z., Miano J. M., Long X., and Chen S. Y. (2011) Smad3-mediated myocardin silencing: a novel mechanism governing the initiation of smooth muscle differentiation. J. Biol. Chem. 286, 15050–15057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Okayama H., Kohno T., Ishii Y., Shimada Y., Shiraishi K., Iwakawa R., Furuta K., Tsuta K., Shibata T., Yamamoto S., Watanabe S., Sakamoto H., Kumamoto K., Takenoshita S., Gotoh N., et al. (2012) Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 72, 100–111 [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto H., Toyooka S., and Mitsudomi T. (2009) Impact of EGFR mutation analysis in non-small cell lung cancer. Lung Cancer 63, 315–321 [DOI] [PubMed] [Google Scholar]
- 50. Selamat S. A., Chung B. S., Girard L., Zhang W., Zhang Y., Campan M., Siegmund K. D., Koss M. N., Hagen J. A., Lam W. L., Lam S., Gazdar A. F., and Laird-Offringa I. A. (2012) Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 22, 1197–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yauch R. L., Januario T., Eberhard D. A., Cavet G., Zhu W., Fu L., Pham T. Q., Soriano R., Stinson J., Seshagiri S., Modrusan Z., Lin C. Y., O'Neill V., and Amler L. C. (2005) Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 11, 8686–8698 [DOI] [PubMed] [Google Scholar]
- 52. Riely G. J., Marks J., and Pao W. (2009) KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 6, 201–205 [DOI] [PubMed] [Google Scholar]
- 53. Miller A. V., Alvarez S. E., Spiegel S., and Lebman D. A. (2008) Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor β-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell. Biol. 28, 4142–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cencetti F., Bernacchioni C., Nincheri P., Donati C., and Bruni P. (2010) Transforming growth factor-β1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 21, 1111–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barthelemy-Brichant N., David J. L., Bosquée L., Bury T., Seidel L., Albert A., Bartsch P., Baugnet-Mahieu L., and Deneufbourg J. M. (2002) Increased TGFβ1 plasma level in patients with lung cancer: potential mechanisms. Eur. J. Clin. Invest. 32, 193–198 [DOI] [PubMed] [Google Scholar]
- 56. Domagała-Kulawik J., Hoser G., Safianowska A., Grubek-Jaworska H., and Chazan R. (2006) Elevated TGF-β1 concentration in bronchoalveolar lavage fluid from patients with primary lung cancer. Arch. Immunol. Ther. Exp. (Warsz.) 54, 143–147 [DOI] [PubMed] [Google Scholar]
- 57. Lee J. C., Lee K. M., Kim D. W., and Heo D. S. (2004) Elevated TGF-β1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 172, 7335–7340 [DOI] [PubMed] [Google Scholar]
- 58. Hasegawa Y., Takanashi S., Kanehira Y., Tsushima T., Imai T., and Okumura K. (2001) Transforming growth factor-β1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer 91, 964–971 [PubMed] [Google Scholar]
- 59. Bruna A., Darken R. S., Rojo F., Ocaña A., Peñuelas S., Arias A., Paris R., Tortosa A., Mora J., Baselga J., and Seoane J. (2007) High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160 [DOI] [PubMed] [Google Scholar]
- 60. Saito H., Tsujitani S., Oka S., Kondo A., Ikeguchi M., Maeta M., and Kaibara N. (1999) The expression of transforming growth factor-β1 is significantly correlated with the expression of vascular endothelial growth factor and poor prognosis of patients with advanced gastric carcinoma. Cancer 86, 1455–1462 [DOI] [PubMed] [Google Scholar]
- 61. Tsushima H., Kawata S., Tamura S., Ito N., Shirai Y., Kiso S., Imai Y., Shimomukai H., Nomura Y., Matsuda Y., and Matsuzawa Y. (1996) High levels of transforming growth factor β1 in patients with colorectal cancer: association with disease progression. Gastroenterology 110, 375–382 [DOI] [PubMed] [Google Scholar]
- 62. Wikström P., Stattin P., Franck-Lissbrant I., Damber J. E., and Bergh A. (1998) Transforming growth factor β1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 37, 19–29 [DOI] [PubMed] [Google Scholar]
- 63. Levy L., and Hill C. S. (2006) Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 17, 41–58 [DOI] [PubMed] [Google Scholar]
- 64. Yu H., Shen Y., Hong J., Xia Q., Zhou F., and Liu X. (2015) The contribution of TGF-β in epithelial-mesenchymal transition (EMT): down-regulation of E-cadherin via snail. Neoplasma 62, 1–15 [DOI] [PubMed] [Google Scholar]
- 65. Wang Y., Shi J., Chai K., Ying X., and Zhou B. P. (2013) The role of Snail in EMT and tumorigenesis. Curr. Cancer Drug Targets 13, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Saitoh M. (2015) Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-β signaling during tumor progression. Cancer Sci. 106, 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang L. (2010) TGFβ and cancer metastasis: an inflammation link. Cancer Metastasis Rev. 29, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tian M., Neil J. R., and Schiemann W. P. (2011) Transforming growth factor-β and the hallmarks of cancer. Cell. Signal. 23, 951–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Naber H. P., ten Dijke P., and Pardali E. (2008) Role of TGF-β in the tumor stroma. Curr. Cancer Drug Targets 8, 466–472 [DOI] [PubMed] [Google Scholar]
- 70. Hong S., Lee H. J., Kim S. J., and Hahm K. B. (2010) Connection between inflammation and carcinogenesis in gastrointestinal tract: focus on TGF-β signaling. World J. Gastroenterol. 16, 2080–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yao Z., Fenoglio S., Gao D. C., Camiolo M., Stiles B., Lindsted T., Schlederer M., Johns C., Altorki N., Mittal V., Kenner L., and Sordella R. (2010) TGF-β IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. U.S.A. 107, 15535–15540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen M. F., Wang W. H., Lin P. Y., Lee K. D., and Chen W. C. (2012) Significance of the TGF-β1/IL-6 axis in oral cancer. Clin. Sci. (Lond.) 122, 459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yanagawa H., Sone S., Takahashi Y., Haku T., Yano S., Shinohara T., and Ogura T. (1995) Serum levels of interleukin 6 in patients with lung cancer. Br. J. Cancer 71, 1095–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haura E. B., Livingston S., and Coppola D. (2006) Autocrine interleukin-6/interleukin-6 receptor stimulation in non-small-cell lung cancer. Clin. Lung Cancer 7, 273–275 [DOI] [PubMed] [Google Scholar]
- 75. Ivanova M. M., Mazhawidza W., Dougherty S. M., Minna J. D., and Klinge C. M. (2009) Activity and intracellular location of estrogen receptors α and β in human bronchial epithelial cells. Mol. Cell. Endocrinol. 305, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]