

Abstract
Mitsugumin 53 (MG53) is an E3 ligase that interacts with and ubiquitinates insulin receptor substrate-1 (IRS-1) in skeletal muscle; thus, an MG53-IRS-1 interaction disruptor (MID), which potentially sensitizes insulin signaling with an elevated level of IRS-1 in skeletal muscle, is an excellent candidate for treating insulin resistance. To screen for an MID, we developed a bimolecular luminescence complementation system using an N-terminal luciferase fragment fused with IRS-1 and a C-terminal luciferase fragment fused with an MG53 C14A mutant that binds to IRS-1 but does not have E3 ligase activity. An MID, which was discovered using the bimolecular luminescence complementation system, disrupted the molecular association of MG53 with IRS-1, thus abolishing MG53-mediated IRS-1 ubiquitination and degradation. Thus, the MID sensitized insulin signaling and increased insulin-elicited glucose uptake with an elevated level of IRS-1 in C2C12 myotubes. These data indicate that this MID holds promise as a drug candidate for treating insulin resistance.
Keywords: diabetes, drug discovery, insulin receptor substrate 1 (IRS-1), insulin resistance, skeletal muscle
Introduction
Noninsulin-dependent diabetes mellitus (type 2 diabetes) has become a worldwide epidemic disease due to the increased incidence of obesity. Insulin receptor (IR) and insulin receptor substrate (IRS)4 are inactivated by elevated serum levels of free fatty acids, leading to insulin resistance and noninsulin-dependent diabetes mellitus (1–3). Because skeletal muscle is the largest organ participating in glucose uptake, exercise-induced skeletal muscle development is an excellent treatment for insulin resistance. However, no current candidates target skeletal muscle to treat insulin resistance.
Mitsugumin 53 (MG53), which is also called tripartite motif-containing protein 72 (TRIM72) and which is largely expressed in skeletal muscle, was independently identified by proteomic analysis of lipid rafts and triad-rich membranes (4). MG53 contains a tripartite domain (an E3 ligase RING domain, a B-box, and two coiled-coil domains) and a SPRY domain. Insulin-like growth factor-1 (IGF-1) initiates MyoD activation via an IGF-1 receptor-PI3K-Akt pathway during skeletal myogenesis (5, 6). MyoD and myocyte enhancer factor 2 (MEF2) binds to two proximal E-boxes and an MEF2 site in MG53 promoter, activating MG53 gene transcription (7). The MG53 protein interacts with IRS-1 and focal adhesion kinase (FAK), inducing IRS-1 and FAK ubiquitination and degradation in skeletal muscle with the help of E2 ligase UBE2H (8–10). Moreover, RING domain-disrupted MG53 mutants (ΔR and C14A) abolish IRS-1 and FAK ubiquitination and degradation in skeletal muscle, indicating that MG53 is an E3 ligase that targets IRS-1 and FAK.
Systemic MG53 ablation abrogates IRS-1 ubiquitination and degradation in skeletal and cardiac muscle, leading to elevated IRS-1 expression level and increased insulin signaling (8, 9). Thus, MG53 knock-out mice do not develop diet-induced insulin resistance. In contrast, skeletal muscle-specific MG53 transgenic mice exhibit metabolic disorders such as obesity, high blood pressure, and insulin resistance with decreased IRS-1 expression and decreased insulin signaling in skeletal muscle (9). Moreover, the MG53 expression level is 3–4-fold higher in the skeletal and cardiac muscle obtained from animal models with metabolic syndromes such as high blood pressure, obesity, and diabetes compared with that from control animals. These data demonstrate that MG53 is a therapeutic target protein for treating insulin resistance.
MG53 functions as a critical component for cell membrane repair machinery in muscle and epithelial cells by interacting with dysferlin-1 and caveolin-3 (5, 11, 12). Acute membrane injury induces the fusion of MG53-containing intracellular vesicles with sarcolemma to seal wounds. MG53 ablation impairs membrane repair capacity in skeletal muscle, heart, lung, and kidney (5, 13–17). In addition, the extracellular administration of recombinant MG53 or gene delivery of MG53 improves membrane repair in skeletal muscle obtained from mice with muscular dystrophy (18, 19).
Both functions of MG 53, as an E3 ligase for IRS-1 ubiquitination and a component for membrane repair, should be considered two sides of the same coin when developing therapies targeting MG53. For example, simple MG53 inhibition may result in impaired membrane repair in cardiac and skeletal muscle cells and in lung alveolar and kidney epithelial cells after acute injury. To treat insulin resistance, a smart strategy is necessary for developing an MG53-targeting drug that disrupts only the molecular interaction between MG53 and IRS-1 without affecting the function of MG53 in cell membrane repair. Here, we developed a novel method for screening for an MG53-IRS-1 interaction disruptor (MID) using bimolecular luciferase complement (BiLC) assay. We demonstrated that one MID compound disrupted molecular association of MG53 with IRS-1 and abolished MG53-induced IRS-1 ubiquitination and degradation in skeletal muscle, leading to elevated IRS-1 expression level and increased insulin signaling and glucose uptake.
Results
Development of the BiLC System to Monitor the Molecular Association of MG53 with IRS-1
To screen for chemicals that disrupt the molecular association of MG53 with IRS-1, we prepared two fusion genes (Fig. 1A). N-terminal luciferase fragment was fused with IRS-1 in one fusion gene (NLUC-IRS-1), whereas the C-terminal luciferase fragment was fused with MG53 C14A in the other fusion gene (CLUC-C14A). Note that the C14A mutant loses its unique E3 ligase activity despite its molecular association with IRS-1 (8). The molecular association of C14A with IRS-1 was demonstrated by reciprocal exogenous co-immunoprecipitation and luciferase activity assay after transient co-expression of NLUC-IRS-1 and CLUC-C14A in HEK 293 cells (Fig. 1, B and C). Next, we prepared two stable HEK 293 cell lines expressing NLUC-IRS-1 and CLUC-C14A. Reciprocal co-immunoprecipitation and luciferase activity assay results (Fig. 1, D and E) confirmed that the stable HEK 293 clones could be used for monitoring the molecular association of C14A with IRS-1.
FIGURE 1.
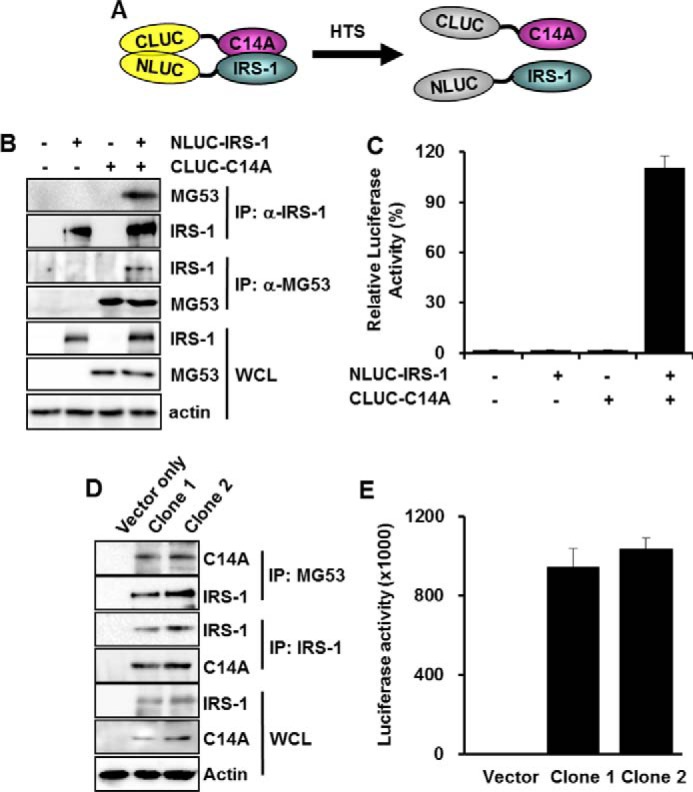
The development of the BiLC system to screen for MID chemicals. The BiLC system uses two fusion genes, the C-terminal luciferase fragment fused with MG53 C14A (CLUC-C14A) and the N-terminal luciferase fragment fused with IRS-1 (NLUC-IRS-1). HTS, high throughput screening (A). Different combinations of CLUC-C14A (5 μg) and NLUC-IRS-1 (5 μg) were transiently expressed in HEK 293 cells for 48 h. The molecular association of CLUC-C14A with NLUC-IRS-1 and luciferase activity was determined by immunoprecipitation (B) and luciferase assay (C), respectively. Two HEK 293 clones were selected after transfection of CLUC-C14A and NLUC-IRS-1. The molecular association of CLUC-C14A with NLUC-IRS-1 and luciferase activity was determined by immunoprecipitation (D) and luciferase assay (E), respectively. Relative luciferase activity was presented as a percentage of the control, which did not express CLUC-C14A and NLUC-IRS-1.
Finding Small Chemicals That Disrupt the MG53-IRS-1 Interaction Using the BiLC System
To screen for small chemicals that disrupt the molecular association of MG53 with IRS-1, luciferase activity was measured in the presence of 10 μm small chemicals from whole-cell lysates (WCL) of stable HEK 293 cell line (clone 1) that expressed NLUC-IRS-1 and CLUC-C14A. Among 20,000 small chemicals, only 23 chemicals reduced the luciferase activity by >50% in the WCL-based BiLC assay, compared with carrier (DMSO)-treated WCL (Table 1). Luciferase activity was also measured from the stable HEK 293 cell line (clone 1) expressing NLUC-IRS-1 and CLUC-C14A after treatment with these 23 chemicals for 12 h. In the cell-based BiLC assay, four chemicals decreased the luciferase activity by >35% compared with carrier (DMSO)-treated cells (Table 1). We named them MG53-IRS-1 interaction disruptors (MIDs).
TABLE 1.
The list of MIDs that decreased luciferase activity in the BiLC system with CLUC-C14A and NLUC-IRS-1
| Compound ID | Lysate-based complimentary luciferase assay | Cell-based complimentary luciferase assay |
|---|---|---|
| % | % | |
| DMSO | 100 | 100 |
| ChemBridge5655896 (MID-1) | 16 | 24 |
| ChemBridge5714547 | 17 | 54 |
| ChemBridge5857986 | 46 | 78 |
| ChemBridge5890847 (MID-2) | 38 | 25 |
| ChemBridge5897483 | 42 | 37 |
| ChemBridge6005705 | 44 | 71 |
| ChemBridge6142797 | 46 | 69 |
| Ambiter2641019 | 42 | 67 |
| ChemBridge6445534 | 23 | 45 |
| ChemBridge6623100 (MID-3) | 37 | 24 |
| AKOS001439160 | 26 | 40 |
| ChemBridge7543255 | 22 | 90 |
| ChemBridge7727116 | 20 | 68 |
| ChemBridge7747515 | 34 | 70 |
| ChemBridge7757756 | 35 | 51 |
| ChemBridge7767668 | 37 | 50 |
| AKOS00339207 | 47 | 34 |
| ChemBridge9041500 | 46 | 55 |
| ChemBridge9076403 | 39 | 55 |
| ChemBridge9092435 | 49 | 54 |
| ChemBridge9127124 | 47 | 58 |
| ChemBridge9151854 (MID-4) | 30 | 35 |
| ChemBridge9192560 | 24 | 59 |
MID-1 Disrupts MG53-IRS-1 Interaction
To monitor whether MIDs disrupt the MG53-IRS-1 interaction, IRS-1 expression levels in C2C12 myotubes were determined by immunofluorescence. Note that IRS-1 expression level is low in C2C12 myotubes because of MG53-induced IRS-1 ubiquitination and degradation (8). Thus, MIDs might increase IRS-1 protein level if they disrupt MG53-IRS-1 interaction, abolishing MG53-induced ubiquitination and degradation. To determine which MID increases IRS-1 protein level, we monitored IRS-1 protein levels by immunofluorescence and immunoblotting after treating 4-day-differentiated C2C12 myotubes with MID-1, -2, -3, and -4. As shown in Fig. 2, A and B, the IRS-1 protein level was the highest after MID-1 treatment. These data suggest that MID-1 could increase the IRS-1 expression level in skeletal muscle by disrupting the MG53-IRS-1 interaction.
FIGURE 2.

MID-1 increased the IRS-1 protein level in C2C12 myotubes. 4-Day-differentiated C2C12 myotubes were incubated with 5 μm MID-1, -2, -3, or -4 for 24 h. The IRS-1 and MG53 protein level was determined by immunofluorescence (A) and immunoblotting (B). GAPDH was used as a loading control in immunoblotting.
Fig. 3A shows the chemical structural formula of MID-1: 4-ethoxy-N-(5-nitrothiazol-2-yl)benzamide. To confirm the effect of MID-1 on the MG53-IRS-1 interaction, we used an in situ proximity ligation assay that detects protein-protein interactions by immunoassay (20). As shown in Fig. 3B, the fluorescent interaction signal between MG53 and IRS-1 (or FAK) appeared only in the presence of proteasome inhibitor MG132 because MG53 ubiquitinates and degrades both IRS-1 and FAK in C2C12 myotubes. Indeed, the MG53-IRS-1 and MG53-FAK interaction signals increased in the presence of MG132 (Fig. 3, B and D). As expected, the MG132-induced MG53-IRS-1 interaction signal disappeared with inclusion of MID-1, but the MG132-induced MG53-FAK interaction signal did not (Fig. 3, B and C). These data suggests that MID-1 disrupts the MG53-IRS-1 interaction but not MG53-FAK interaction.
FIGURE 3.
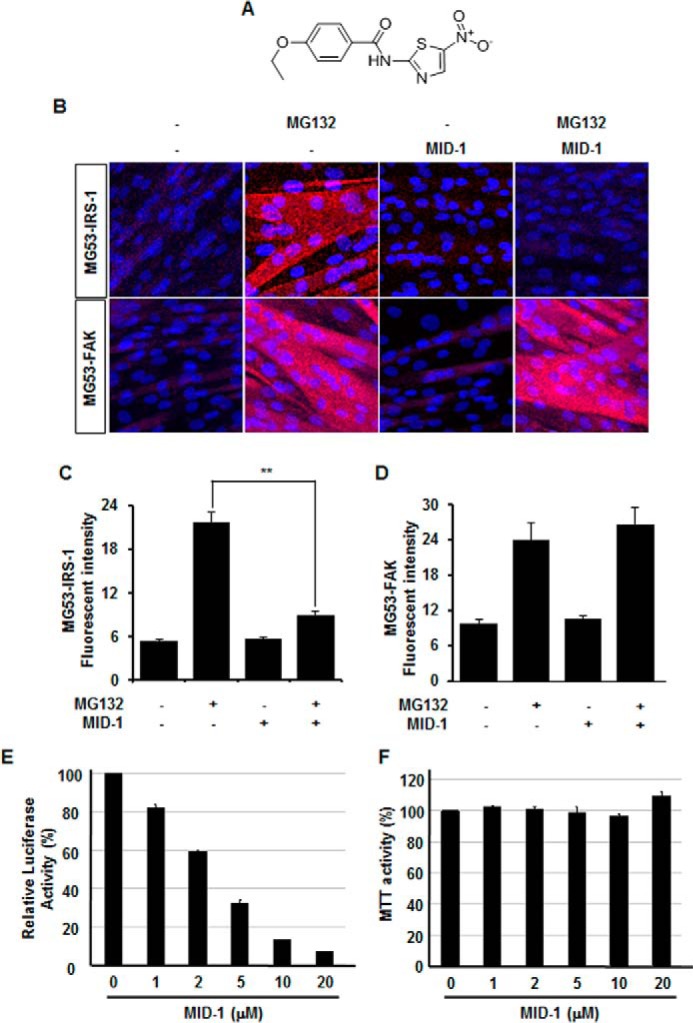
The in situ proximity ligation assay showed that MID-1 disrupts the molecular association of MG53 with IRS-1. The chemical structural formula of MID-1: 4-ethoxy-N-(5-nitrothiazol-2-yl)benzamide (A). 4-Day-differentiated C2C12 myotubes were incubated with different combinations of 2.5 μm MG132 and 5 μm MID-1 for 4 h. The molecular association of MG53 with IRS-1 and FAK was determined by in situ proximity ligation assay (B). The fluorescent signals for MG53-IRS-1 (C) and MG53-FAK (D) interactions were statistically determined from 12 different image fields. t test; **, p < 0.01. The relative luciferase activity and cell survival rate of CLUC-C14A- and NLUC-IRS-1-overexpressing HEK 293 clone 1 were determined after treating the cells with different concentrations of MID-1 for 12 h. The relative luciferase activity (E) and MTT activity (F) were presented as percentages of the control without MID-1 treatment, respectively.
Using co-immunoprecipitation, we then re-confirmed the effect of MID-1 on the MG53-IRS-1 interaction after transient co-expression of C14A and IRS-1 in HEK 293 cells. The exogenous reciprocal immunoprecipitation showed that MID-1 inhibited C14A-IRS-1 interaction (Fig. 4A). Moreover, the MG53-IRS-1 interaction was also inhibited by MID-1 in the presence of MG132 (Fig. 4B). Endogenous co-immunoprecipitation in MG132-treated C2C12 myotubes also showed that MID-1 inhibited the MG53-IRS-1 interaction without affecting the MG53-Cav-3 interaction (Fig. 4C).
FIGURE 4.

MID-1 disrupted the molecular association of MG53 with IRS-1 but not with FAK and Cav-3. HA-C14A (2 μg) and FLAG-IRS-1 (0.5 μg) were transiently overexpressed in different combinations in HEK 293 cells for 48 h. The molecular association of HA-C14A with FLAG-IRS-1 was determined by immunoprecipitation after the cells were treated with or without 10 μm MID-1 for 12 h (A). HA-MG53 (2 μg) and FLAG-IRS-1 (0.5 μg) were transiently overexpressed in different combinations in HEK 293 cells for 48 h (B). The molecular association of HA-MG53 with FLAG-IRS-1 was determined by immunoprecipitation after treating the cells with MG132 (2.5 μm) in the absence or presence of MID-1 (10 μm) for 12 h (A and B). 4-Day-differentiated C2C12 myotubes were treated with different concentrations of MID-1 for 12 h. The molecular associations of MG53 with IRS-1 and Cav-3 were determined by immunoprecipitation (C). HA-C14A (2 μg) was co-transfected along with His-FAK (0.5 μg) or FLAG-UBE2H (0.5 μg) in HEK 293 cells for 48 h. The molecular associations of HA-C14A with His-FAK or FLAG-UBE2H were determined by immunoprecipitation after the cells were treated with or without MID-1 (10 μm) for 12 h (D and E). Myc-MG53 was transiently expressed in HEK 293 cells for 48 h. HEK 293 cells and 4-day-differentiated C2C12 myotubes were incubated with 10 μm MID-1 for 12 h. Whole-cell lysates were obtained from Myc-MG53-overexpressing HEK 293 cells (left panel) and 4-day-differentiated C2C12 myotubes (right panel), treated with 2 μm glutaraldehyde (GA) for 20 min, and analyzed by MG53 immunoblotting (F). HA-MG53 and Myc-MG53 were transiently co-expressed in HEK 293 cells for 48 h. MG53 oligomerization was determined by reciprocal immunoprecipitation (G).
MID-1 Does Not Affect the MG53-FAK Interaction and MG53 Oligomerization
Next, we investigated whether MID-1 affects the MG53-FAK interaction by exogenous reciprocal co-immunoprecipitation after transient co-expression of C14A or MG53 with FAK in HEK 293 cells. As shown in Fig. 4D, MID-1 did not disrupt C14A-FAK or MG53-FAK interactions. We also monitored MG53 oligomerization in Myc-MG53-overexpressing HEK 293 cells and C2C12 myotubes after MID-1 treatment. MG53 oligomerization was determined by immunoblotting after the cells were treated with glutaraldehyde. As shown in Fig. 4F, glutaraldehyde-induced MG53 oligomerization was not affected by MID-1. In addition, we tested MG53 oligomer formation by reciprocal co-immunoprecipitation after HA-MG53 and Myc-MG53 were co-expressed in HEK 293 cells. Fig. 4G shows that MG53 oligomerization was not affected by MID-1.
MID-1 Abolishes MG53-induced IRS-1 Ubiquitination and Degradation
Because MID-1 disrupted the MG53-IRS-1 interaction, MID-1 might abolish MG53-induced IRS-1 ubiquitination and degradation. To address this issue, we monitored MG53-mediated IRS-1 degradation in the presence of MID-1. After HA-MG53 and FLAG-IRS-1 were transiently expressed in HEK 293 cells, the IRS-1 expression level was determined by immunoblotting in the presence of MID-1. As shown in Fig. 5A, IRS-1 protein levels gradually increased with increasing concentrations of MID-1. In addition, MID-1 increased IRS-1 expression level in C2C12 myotubes in a concentration-dependent manner (Fig. 5B). Next, we addressed whether MID-1 could increase half-life time of IRS-1 in 2-day-differentiated C2C12 myotubes. To determine the half-life time of IRS-1, the myotubes were pretreated with MID-1 and further incubated with protein synthesis inhibitor cycloheximide for the indicated time. As shown in Fig. 5C, MID-1 largely increased the half-life time of IRS-1.
FIGURE 5.

MID-1 abrogated MG53-induced IRS-1 ubiquitination and degradation. HA-MG53 (2 μg) and FLAG-IRS-1 (0.5 μg) were transiently co-expressed in HEK 293 cells for 48 h. After the cells were treated with 2.5 μm MG132 or different concentrations of MID-1 for 24 h, MG53 and IRS-1 protein levels were determined by immunoblotting using actin as a loading control (A). 4-Day-differentiated C2C12 myotubes (MT) were treated with 2.5 μm MG132 or different concentrations of MID-1 for 24 h, and the protein levels of IRS-1, MG53 and actin were determined by immunoblotting. Whole-cell lysates of undifferentiated C2C12 myoblasts (MB) were used as a positive control for IRS-1 and a negative control for MG53 (B). Two-day-differentiated C2C12 myotubes were preincubated with 0.1% DMSO or 5 μm MID for 8 h and then treated with cycloheximide (CHX) for 0, 4, 8, 16, and 24 h. IRS-1 and MG53 protein levels were determined by immunoblotting (C). 4-Day-differentiated C2C12 myotubes were treated with 2.5 μm MG132 or 5 μm MID-1 for 12 h. IRS-1 and FAK ubiquitination was determined by ubiquitin (Ub) immunoblotting after IRS-1 and FAK immunoprecipitation (D and E).
We further tested whether MID-1 abolished MG53-induced IRS-1 and FAK ubiquitination in C2C12 myotubes. As shown in Fig. 5, E and F, IRS-1 and FAK ubiquitination was found in the presence of MG132. MID-1 abolished MG53-induced IRS-1 ubiquitination but not MG53-induced FAK ubiquitination. These data indicate that MID-1 specifically abolishes MG53-mediated IRS-1 ubiquitination.
MID-1 Increases Insulin Signaling
Because MID-1 increased the IRS-1 expression level by disrupting the MG53-IRS-1 interaction and then abolishing MG53-mediated IRS-1 ubiquitination, it is tempting to speculate that MID-1 increases insulin signaling. As shown in Fig. 6, A and B, MID-1 increased insulin-elicited IRS-1, Akt, Erk1/2 activation, and PI3K binding with an elevated level of IRS-1 expression.
FIGURE 6.

MID-1 increased insulin signaling and insulin-elicited glucose uptake in C2C12 myotubes. 4-Day-differentiated C2C12 myotubes were incubated with carrier (0.1% DMSO) or 10 μm MID-1 for 24 h and treated with 20 nm insulin for 0, 5, and 10 min. The protein levels of MG53, IRS-1, p-IRS-1, Akt, p-Akt, Erk1/2, p-Erk1/2, IRβ, p-IRβ, and GAPDH were determined by immunoblotting. The molecular interaction between PI3K and IRS-1 was determined by IRS-1 immunoprecipitation (A). The expression levels of p-IRS-1, p-Akt, and p-Erk1/2 were statistically determined using ImageJ from three independent experiments (B). After 3-day-differentiated C2C12 myotubes were incubated with MID-1 for 24 h, the cells were further serum-starved in the presence of carrier or MID-1 for 4 h and then exposed to 100 nm insulin for 30 min. The expression levels of GLUT4 and Cav-3 were determined in WCL and plasma membrane (PM) that were isolated from the myotubes (C). After 4-day-differentiated C2C12 myotubes were incubated with carrier (0.1% DMSO) or 10 μm MID-1 for 4 h, the cells were exposed to 100 nm insulin for 20 min and then incubated with 2-[3H]deoxyglucose for 10 min. Radioactivity was measured from cells and normalized by protein amount. Glucose uptake was presented as cell-associated radioactivity (left panel) and as the percentage of the control (no insulin treatment) (right panel) in both DMSO- and MID-1-treated cells (D). *, significant difference between groups (p < 0.05); **, significant difference between groups (p < 0.01).
Because MID-1 increased insulin signaling, MID-1 might increase insulin-elicited GLUT4 translocation to the sarcolemma and then glucose uptake in C2C12 myotubes. Indeed, MID-1 increased insulin-elicited GLUT4 translocation to the sarcolemma and glucose uptake, as shown in Fig. 6, C and D. Interestingly, MID-1 itself also induced GLUT4 translocation to the sarcolemma and glucose uptake.
MID-1 Enhances Skeletal Myogenesis
Next, we monitored C2C12 skeletal myogenesis after MID-1 treatment. It is tempting to speculate that MID-1 could enhance skeletal myogenesis with elevated IRS-1 protein levels. As determined by myosin heavy chain (MyHC) immunofluorescence, myogenic index, and immunoblotting for myogenic marker proteins such as MyHC and caveolin-3 (Cav-3; Fig. 7), MID-1 enhanced C2C12 myogenesis.
FIGURE 7.
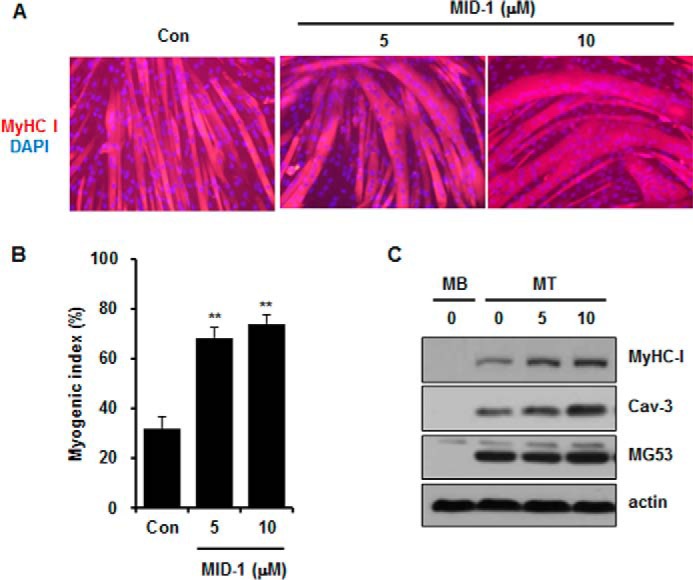
MID-1 enhanced C2C12 myogenesis. One-day-differentiated C2C12 cells were treated with MID-1 (0, 5, or 10 μm) for 24 h and allowed to further differentiate for 2 days. Myogenesis was monitored by MyHC immunofluorescence and DAPI staining (A), myogenic index (B), and immunoblotting for MyHC, caveolin-3, MG53, and actin (C). The myogenic index was determined by counting nuclei number in MyHC-stained cells from 12 different image fields. MB, myoblast; MT, myotube. **, p < 0.01.
Discussion
Striated muscle-specific MG53 acts as a double-edged sword, interacting with many partner proteins such as IRS-1, FAK, caveolin-3, cavin-1, nonmuscle myosin type IIA (NM-IIA), and dysferlin and working as an E3 ligase and membrane repair machinery (21–23). Systemic MG53 ablation abrogates IRS-1 ubiquitination and ameliorates diet-induced insulin resistance. Cai et al. (5) showed that the MG53 protein level is increased 4-fold in the skeletal muscle obtained from animals with metabolic syndromes. These data indicate that MG53 is an excellent therapeutic target for treating insulin resistance. However, skeletal and cardiac muscles lose their capacity for membrane repair in MG53−/− mice, leading to muscular dystrophy. For example, central nuclei, which are a symptom of muscular dystrophy, appear in the skeletal muscle isolated from 16-week-old MG53−/− mice. Thus, MG53 inhibition for ameliorating insulin resistance might lead to muscular dystrophy by preventing membrane repair. To develop a MG53-targeting drug for treating insulin resistance and to overcome MG53 inhibition-induced side effect, the drug should be carefully designed to inhibit only the molecular association of MG53 with IRS-1 without affecting MG53 interaction with other partners such as caveolin-1, cavin-1, and FAK as well as the membrane repair and oligomerization abilities of MG53.
To screen for chemicals disrupting only the molecular association of MG53 with IRS-1, we used the BiLC system because of its lack of background signals and its potential for use in high throughput screening. We used the RING domain-disrupted MG53 mutant (C14A) for the BiLC system because C14A does not induce IRS-1 ubiquitination and degradation but still interacts with IRS-1. The molecular association of C14A with IRS-1 and luciferase activity appeared in HEK 293 cells expressing CLUC-C14A and NLUC-IRS-1 (Fig. 1), indicating that the BiLC system using CLUC-C14A and NLUC-IRS-1 is suitable for screening for MIDs.
Using different experimental approaches such as the BiLC system (Fig. 1 and Table 1), immunofluorescence (Fig. 2), the in situ proximity ligation assay (Fig. 3), and immunoprecipitation (Fig. 4), we demonstrated that MID-1 disrupted the molecular association of MG53 with IRS-1, thereby improving insulin signaling with elevated IRS-1 protein levels in skeletal muscle (Fig. 6). Fig. 8 summarizes a working hypothesis for the mechanism by which MID-1 improves insulin signaling in the skeletal muscle. Because MID-1 disrupts the molecular association of MG53 with IRS-1, MID-1 abrogates IRS-1 ubiquitination and degradation. An increased level of IRS-1 improves insulin signaling, which subsequently increased insulin-elicited IRS-1, Akt, and Erk1/2 activation and glucose uptake. Because MID-1 did not affect the oligomerization of MG53 or the molecular association of MG53 with FAK and caveolin-1, MID-1 might not have any side effects such as muscular dystrophy.
FIGURE 8.
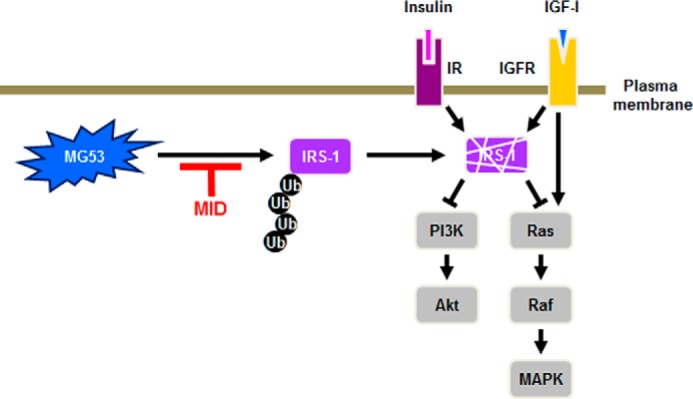
A model explaining how MID-1 increases insulin signaling. MID-1 disrupts the molecular interaction of MG53 with IRS-1 and abolishes MG53-induced IRS-1 ubiquitination and degradation, thus increasing the IRS-1 expression level in skeletal muscle. IGFR, IGF-1 receptor.
Based on MG53-induced IRS-1 ubiquitination and degradation in skeletal muscle (8, 9), we focused on the development of MID as an insulin resistance-treating drug. MID work is important because of the following two reasons. First, the cell biological data of MID might be a cornerstone for developing MID derivatives as an insulin resistance-treating drug. Second, MID is very unique because it is developed based on protein-protein interaction by which few chemicals have been screened.
Because MID-1 did not have good pharmacokinetics (data not shown), we failed to demonstrate that MID-1 ameliorated insulin resistance in vivo. Thus, to develop drug candidates for the treatment of insulin resistance, it is critical to prepare various MID-1 derivatives and test whether these derivatives ameliorate insulin resistance in obesity and diabetes animal models.
Experimental Procedures
Cell Culture and Antibodies
HEK 293T cells and C2C12 myoblasts were purchased from ATCC (Manassas, VA) and grown in growth media (Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA) and 10% fetal bovine serum (Gibco®, Thermo Fisher Scientific)) in a 5% CO2 incubator at 37 °C. C2C12 myoblasts were grown until they reached confluence and were then differentiated to myotubes by incubating them in differentiation media (DMEM with 2% horse serum (Gibco®)) for 3 or 4 days. During C2C12 differentiation, the differentiation medium was refreshed every 2 days. C2C12 myoblasts were used at passage numbers 2–7, and HEK 293 cells were used at passage numbers 20–30.
Anti-IRS-1, anti-caveolin-3, anti-phosphotyrosine, and anti-IRβ antibodies were purchased from BD Transduction Laboratories. Anti-Akt, anti-ERK, anti-phospho-Akt, and anti-phospho-ERK antibodies were obtained from Cell Signaling Technology (Danvers, MA), and anti-MyHC, anti-GAPDH, anti-β-actin, and anti-FLAG antibodies were obtained from Sigma. The anti-HA, ubiquitin, FAK, and phospho-IRβ antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-phospho-IRS-1 and anti-PI3K(p85) antibody were obtained from Merck Millipore (Burlington, MA).
Synthesis of MID-1
1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (4.15 g, 21.7 mmol, 1.2 eq) and 4-dimethylaminopyridine (DMAP) (0.66 g, 5.42 mmol, 0.3 eq) were added to a solution of 4-ethoxybenzoic acid (3.00 g, 18.01 mmol) in N,N-dimethylformamide (180 ml) at 0 °C. After 10 min, 2-amino-5-nitrothiazole (3.15 g, 21.66 mmol, 1.2 eq) was added to the reaction mixture at 0 °C. The resulting mixture was allowed to warm up to room temperature and then stirred overnight. After the solvent was removed by evaporation, the crude residue was diluted with brine and extracted with ethyl acetate (100 ml × 3). The combined organic layers were dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (SiO2, dichloromethane:CH3OH = 79:1) to obtain the desired product (1.51 g, 5.15 mmol) with a 29% yield.
1H NMR (400 MHz, DMSO-d6): δ 13.39 (s, 1H), 8.69 (s,1H), 8.12 (d, J = 8.9 Hz, 2H), 7.08 (d, J = 8.9 Hz, 2H, 4.13 (q, J = 6.7 Hz, 2H), 1.35 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 166.22, 163.52, 163.39, 143.38, 131.44, 123.20, 115.13, 64.34, 15.15; HR-MS (ESI-TOF) m/z for [C12H11N3NaO4S]+ ([M+Na]+): calculated 316.0362, found 316.0361.
Development of the BiLC System and High Throughput Screening of MID Chemicals
To establish the BiLC system that allows a quantitative measurement of protein-protein interaction in cells, the firefly luciferase gene was split into N-terminal (NLUC) and C-terminal (CLUC) fragments, as previously reported (24). To construct the plasmid expressing NLUC-IRS-1, the N-terminal two-thirds of the firefly luciferase gene (corresponding to amino acids 1–398) were cloned into the pcDNA3.1-hyg vector at HindIII/KpnI sites and then the DNA fragment encoding human IRS-1 was inserted into the KpnI/XhoI sites. To construct the plasmid expressing CLUC-MG53 C14A (C14A), the C-terminal one-third of the firefly luciferase gene (corresponding to amino acids 394–551) was cloned into the pcDNA3.1-hyg vector at HindIII/KpnI sites, and then the DNA fragment encoding human C14A was inserted into KpnI/XhoI sites.
Stable 293T cells expressing NLUC-IRS-1 and CLUC-C14A were established by transfection of pcDNA3.1-hyg-NLUC-IRS-1 and pcDNA3.1-hyg-CLUC-C14A followed by selection with hygromycin for 2 weeks. Clonal cell lines were tested for the expression of each of the fused proteins using Western blot analysis with anti-IRS-1 and anti-MG53 antibodies. For high throughput screening of MID chemicals, stable 293T cells expressing NLUC-IRS-1 and CLUC-C14A were lysed in immunoprecipitation lysis buffer (Thermo Fisher Scientific), and the cell lysates (30 μg) were then incubated with chemical compounds for 1 h at 4 °C. The resulting solution was confirmed using luciferase assay (Promega Co., Madison, WI). To validate MIDs in cells, stable cell lines were seeded at a density of 2 × 104 cells/well in 96-well plates. Cells were incubated with MIDs for 24 h, and then luciferase assay was performed. The optical density of the resulting solution was measured in a SpectraMAX I3x multiplate reader (Molecular Devices, Sunnyvale, CA). Each value represents the mean ± S.D. of at least three determinations.
Immunofluorescence and Co-immunoprecipitation
Immunofluorescence staining was performed as previously described (25). For immunoprecipitation, cells were lysed in IP lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm MgCl2, 1 mm CaCl2, 1% Nonidet P-40, 1 mm PMSF, and a protease and phosphatase inhibitor mixture). The whole-cell lysates (500 μg protein) were incubated for 24 h at 4 °C with either anti-IRS-1 or anti-FLAG antibodies and then with a protein A-Sepharose or protein G-agarose beads (Roche Applied Science) slurry for 2 h at 4 °C. The immunoprecipitates were analyzed by immunoblotting. The ubiquitination of IRS-1 and FAK was performed as previously described (10).
Proximity Ligation in Situ Assay
Cells were cultured on collagen-coated confocal dishes and fixed in 4% paraformaldehyde. Then, cells were permeabilized with 0.1% Triton X-100 in PBS and incubated with 5% BSA in PBS for 30 min to avoid nonspecific binding. An in situ proximity ligation assay kit was purchased from Sigma, and the assay was performed as described in the manufacturer's manual. Briefly, the dishes were incubated with primary antibody overnight and then with oligonucleotide-conjugated secondary antibody for 1 h. The dishes were washed and incubated with ligation-ligase solution for 30 min. After ligation, the dishes were further incubated with the amplification solution (nucleotides, fluorescently labeled oligonucleotides, and polymerase) for 90 min. The fluorescently labeled oligonucleotides were hybridized to the amplification product. The fluorescence signal was observed by LSM700 confocal microcopy (Carl Zeiss, Medac, Wedel, Germany).
Cycloheximide Chase Assay
2-Day-differentiated C2C12 myotubes were incubated with 5 μm MID or 0.1% DMSO for 8 h. The cells were then treated with cycloheximide (10 μg/ml, Sigma) and harvested at different time points in radioimmune precipitation assay buffer with proteinase inhibitors. The proteins from each sample were determined by immunoblotting with the indicated antibodies.
Glutaraldehyde Cross-linking Assay
HEK 293 cells and C2C12 cells were harvested with 60 mm octyl-β-d-glucopyranoside in PBS. Whole-cell lysates (100 μg) were mixed with the indicated concentrations of glutaraldehyde and MID-1 and then incubated at 37 °C for 20 min. The cross-linking reaction was stopped by adding 1.5 m Tris-HCl, pH 8.8, and the proteins were separated by SDS-PAGE.
Isolation of Sarcolemma
Plasma membrane was extracted using the detergent-based plasma membrane isolation kit (Thermo Fisher Scientific) according to the manufacturer's protocol. In briefly, differentiated C2C12 myotubes were starved for 4 h and then treated with 100 nm insulin for 30 min after treating with MID-1 for 24 h. After plasma membrane extraction, each sample was determined by Western blot with anti-GLUT4 antibodies (Cell Signaling Technology, #2213).
Glucose Uptake Assay
Briefly, C2C12 myotubes were incubated with 5 μm MID-1 or vehicle (0.1% DMSO) in DMEM after serum starvation with DMEM for 6 h. C2C12 myotubes were incubated in the presence or in the absence of 100 nm insulin for 20 min and then washed 3 times with Krebs-HEPES buffer (1 mm MgSO4, 0.33 mm CaCl2, 137 mm NaCl, 4.7 mm KCl, and 12 mm HEPES, pH 7.4). Glucose uptake was determined by adding 50 μCi/ml 2-deoxy-[3H] glucose (PerkinElmer Life Sciences). After 10 min of incubation, the reaction was stopped with ice-cold PBS, and the cells were washed 3 times with ice-cold PBS. The cells were then lysed with radioimmune precipitation assay buffer, and glucose uptake was assessed using scintillation counting. Nonspecific, 2-deoxy-[3H]glucose uptake was measured by including 100 μm cytochalasin B (Sigma), and glucose uptake was determined by subtracting the nonspecific counts from the total counts and normalizing the protein amount. The assay was performed three times.
Statistical Analysis
Statistical values are presented as the mean ± S.E. A two-tailed Student's t test was used to calculate the p values.
Author Contributions
B.-W. K. and Y.-G. K. supervised and designed the project. H. L. performed the immunofluorescence, proximity ligation assay, and cycloheximide chase assay. J.-J. P. performed co-immunoprecipitation and Western blotting analysis after treating the MID. N. N. and J. S. P. performed the ubiquitination of IRS-1. J. H. and S.-H. K. performed the insulin signaling and glucose uptake. W. Y. S. and H. J. K. performed the synthesis of MID. K. C. and S. C. performed the construction of the BiLC system. Y.-G. K. wrote the paper with help from J.-S. L.
This work was supported by a grant from the Korea of Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C2739; to Y.-G. K.). This work was also supported in part by a Korea University grant (to J.-J. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- IRS-1
- insulin receptor (IR) substrate-1
- MG53
- mitsugumin 53
- MID
- MG53-IRS-1 interaction disruptor
- BiLC
- bimolecular luminescence complementation
- FAK
- focal adhesion kinase
- WCL
- whole cell lysates
- Cav-3
- caveolin-3
- MyHC
- myosin heavy chain
- GLUT4
- glucose transporter type 4
- NLUC
- N-terminal luciferase
- CLUC
- C-terminal
- IP
- immunoprecipitation.
References
- 1. Hotamisligil G. S., Peraldi P., Budavari A., Ellis R., White M. F., and Spiegelman B. M. (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271, 665–668 [DOI] [PubMed] [Google Scholar]
- 2. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., and Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 3. Yu C., Chen Y., Cline G. W., Zhang D., Zong H., Wang Y., Bergeron R., Kim J. K., Cushman S. W., Cooney G. J., Atcheson B., White M. F., Kraegen E. W., and Shulman G. I. (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50230–50236 [DOI] [PubMed] [Google Scholar]
- 4. Tan T., Ko Y. G., and Ma J. (2016) Dual function of MG53 in membrane repair and insulin signaling. BMB Rep. 49, 414–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J. K., Lin P., Thornton A., Zhao X., Pan Z., Komazaki S., Brotto M., Takeshima H., and Ma J. (2009) MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee C. S., Yi J. S., Jung S. Y., Kim B. W., Lee N. R., Choo H. J., Jang S. Y., Han J., Chi S. G., Park M., Lee J. H., and Ko Y. G. (2010) TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 17, 1254–1265 [DOI] [PubMed] [Google Scholar]
- 7. Jung S. Y., and Ko Y. G. (2010) TRIM72, a novel negative feedback regulator of myogenesis, is transcriptionally activated by the synergism of MyoD (or myogenin) and MEF2. Biochem. Biophys. Res. Commun. 396, 238–245 [DOI] [PubMed] [Google Scholar]
- 8. Yi J. S., Park J. S., Ham Y. M., Nguyen N., Lee N. R., Hong J., Kim B. W., Lee H., Lee C. S., Jeong B. C., Song H. K., Cho H., Kim Y. K., Lee J. S., Park K. S., et al. (2013) MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 4, 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song R., Peng W., Zhang Y., Lv F., Wu H. K., Guo J., Cao Y., Pi Y., Zhang X., Jin L., Zhang M., Jiang P., Liu F., Meng S., Zhang X., et al. (2013) Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 494, 375–379 [DOI] [PubMed] [Google Scholar]
- 10. Nguyen N., Yi J. S., Park H., Lee J. S., and Ko Y. G. (2014) Mitsugumin 53 (MG53) ligase ubiquitinates focal adhesion kinase during skeletal myogenesis. J. Biol. Chem. 289, 3209–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai C., Weisleder N., Ko J. K., Komazaki S., Sunada Y., Nishi M., Takeshima H., and Ma J. (2009) Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 284, 15894–15902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim S., Seo J., Ko Y. G., Huh Y. D., and Park H. (2012) Lipid-binding properties of TRIM72. BMB Rep. 45, 26–31 [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Xie W., Zhang Y., Lin P., Han L., Han P., Wang Y., Chen Z., Ji G., Zheng M., Weisleder N., Xiao R. P., Takeshima H., Ma J., and Cheng H. (2010) Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 107, 76–83 [DOI] [PubMed] [Google Scholar]
- 14. Lemckert F. A., Bournazos A., Eckert D. M., Kenzler M., Hawkes J. M., Butler T. L., Ceely B., North K. N., Winlaw D. S., Egan J. R., and Cooper S. T. (2016) Lack of MG53 in human heart precludes utility as a biomarker of myocardial injury or endogenous cardioprotective factor. Cardiovasc. Res. 110, 178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H., Duann P., Lin P. H., Zhao L., Fan Z., Tan T., Zhou X., Sun M., Fu M., Orange M., Sermersheim M., Ma H., He D., Steinberg S. M., Higgins R., Zhu H., John E., Zeng C., Guan J., and Ma J. (2015) Modulation of wound healing and scar formation by MG53 protein-mediated cell membrane repair. J. Biol. Chem. 290, 24592–24603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jia Y., Chen K., Lin P., Lieber G., Nishi M., Yan R., Wang Z., Yao Y., Li Y., Whitson B. A., Duann P., Li H., Zhou X., Zhu H., Takeshima H., Hunter J. C., McLeod R. L., Weisleder N., Zeng C., and Ma J. (2014) Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 5, 4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duann P., Li H., Lin P., Tan T., Wang Z., Chen K., Zhou X., Gumpper K., Zhu H., Ludwig T., Mohler P. J., Rovin B., Abraham W. T., Zeng C., and Ma J. (2015) MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 7, 279ra36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He B., Tang R. H., Weisleder N., Xiao B., Yuan Z., Cai C., Zhu H., Lin P., Qiao C., Li J., Mayer C., Li J., Ma J., and Xiao X. (2012) Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta-Sarcoglycan-deficient hamsters. Mol. Ther. 20, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weisleder N., Takizawa N., Lin P., Wang X., Cao C., Zhang Y., Tan T., Ferrante C., Zhu H., Chen P. J., Yan R., Sterling M., Zhao X., Hwang M., Takeshima M., et al. (2012) Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 4, 139ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 21. Zhu H., Lin P., De G., Choi K. H., Takeshima H., Weisleder N., and Ma J. (2011) Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 286, 12820–12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin P., Zhu H., Cai C., Wang X., Cao C., Xiao R., Pan Z., Weisleder N., Takeshima H., and Ma J. (2012) Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 26, 1875–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim S. C., Kellett T., Wang S., Nishi M., Nagre N., Zhou B., Flodby P., Shilo K., Ghadiali S. N., Takeshima H., Hubmayr R. D., and Zhao X. (2014) TRIM72 is required for effective repair of alveolar epithelial cell wounding. Am. J. Physiol. Lung Cell. Mol. Physiol. 307, L449–L459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paulmurugan R., and Gambhir S. S. (2007) Combinatorial library screening for developing an improved split-firefly luciferase fragment-assisted complementation system for studying protein-protein interactions. Anal. Chem. 79, 2346–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee H., Kim S. H., Lee J. S., Yang Y. H., Nam J. M., Kim B. W., and Ko Y. G. (2016) Mitochondrial oxidative phosphorylation complexes exist in the sarcolemma of skeletal muscle. BMB Rep. 49, 116–121 [DOI] [PMC free article] [PubMed] [Google Scholar]