

Abstract
Calmodulin (CaM) is a Ca2+ binding protein modulating multiple targets, several of which are associated with cardiac pathophysiology. Recently, CaM mutations were linked to heart arrhythmia. CaM is crucial for cell growth and viability, yet the effect of the arrhythmogenic CaM mutations on cell viability, as well as heart rhythm, remains unknown, and only a few targets with relevance for heart physiology have been analyzed for their response to mutant CaM. We show that the arrhythmia-associated CaM mutants support growth and viability of DT40 cells in the absence of WT CaM except for the long QT syndrome mutant CaM D129G. Of the six CaM mutants tested (N53I, F89L, D95V, N97S, D129G, and F141L), three showed a decreased activation of Ca2+/CaM-dependent kinase II, most prominently the D129G CaM mutation, which was incapable of stimulating Thr286 autophosphorylation. Furthermore, the CaM D129G mutation led to bradycardia in zebrafish and an arrhythmic phenotype in a subset of the analyzed zebrafish.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII), calcium, calmodulin (CaM), cell signaling, heart failure, DT40, catecholaminergic polymorphic ventricular tachycardia (CPVT)
Introduction
Ca2+ is the main trigger for muscle contraction (reviewed in Refs. 1 and 2). Ca2+ signaling is highly regulated in cardiomyocytes and controls a number of important proteins governing release and uptake of Ca2+ into the sarcoplasmic reticulum (SR),3 as well as cardiomyocyte contraction. Impaired contractibility and lethal cardiac arrhythmias may result from dysregulated Ca2+ signaling and cycling. One of the main players in Ca2+-mediated effects is calmodulin (CaM), a ubiquitous intracellular Ca2+ receptor involved in a vast number of physiological processes including cell proliferation and muscle activity (reviewed in Ref. 3). Identical CaM proteins are expressed in humans from three independent genes located on different chromosomes (4). It is well known that malfunctioning of several CaM-regulated proteins may cause heart failure including, among many others, the cardiac Ca2+release channel (RyR2) and the L-type Ca2+ channel (reviewed in Ref. 2). In addition, CaM-regulated protein kinases, in particular the Ca2+/CaM-dependent kinase II (CaMKII), have been found to be dysregulated in diverse cardiac disorders (reviewed in Refs. 5–8).
CaM plays a critical role in catecholaminergic polymorphic ventricular tachycardia (CPVT), as well as in long QT syndrome (LQTS) (9, 10). Nyegaard et al. (9) identified a mutation in the CaM gene 1 locus on chromosome 14 of a Swedish family, segregating with a dominantly inherited form of CPVT. This mutation changes residue 53 in the CaM protein from an asparagine to an isoleucine. In addition, a de novo mutation in CaM gene 1 changing residue 97 from asparagine to serine was found by screening CPVT patients. Thus, it was concluded that the CaM genes may be candidates for genetic screening of patients with tachycardia. Using whole exome sequencing of patients with LQTS, Crotti et al. (10) found three other de novo mutations (D130G, which was represented in two patients, and D96V and F142L; (these mutations are numbered D129G, D95V, and F141L, respectively, in this work in line with the nomenclature used in the first article describing arrhythmogenic CaM mutations) (9)) in the CaM genes 1 and 2. An additional inherited CaM 1 mutation F90L (referred to as F89L in this work) was discovered in a family with a history of idiopathic ventricular fibrillation (11). Five novel CaM mutations in the CaM gene 2 have been found in three patients with LQTS (N97S, N97I, and D133H) and two with both LQTS and CPVT features (D131E and Q135P) (12). Two arrhythmogenic CaM mutations, D129G associated with LQTS (13) and A102V associated with CPVT (14), were found in the CaM 3 gene. A recent investigation on the whole exome of 38 elusive LQTS patients revealed five CaM positive cases, of which one had a novel mutation (E140G) (15). In addition, two novel mutations (D131V and D131H), both associated with LQTS, were recently identified (16). Fig. 1 summarizes the currently available information on mutated CaM amino acids associated with arrhythmia. The CPVT mutations exhibit either moderately higher (N53I) or slightly reduced (N97S and A102V) Ca2+ affinities (9, 14), whereas the CaM mutations in LQTS patients all have a high impact on the CaM Ca2+ affinity, likely because of disruption of EF hand 3 or 4 Ca2+ binding (10) (Fig. 1).
FIGURE 1.

Representation of pathogenic CaM variants associated with CPVT, LQTS, or iodiopathic ventricular fibrillation (IVF). The backbone is shown in gray except for the mutated residues where the side chains in stick representation have been included and color-coded either blue or red (red indicates the residue is directly involved in Ca2+ coordination). Ca2+ is shown in yellow space fill presentation. Text boxes show the amino acid conversion, as well as the arrhythmia associated with the mutation and in which of the three calmodulin genes (CALM) the mutation has been identified and references to the discoveries. The CaM model is based on the 1.7 Å structure of Ca2+ bound WT CaM (Protein Data Bank code 1CLL) modified from Ref. 48.
A number of cardiac target proteins are regulated by CaM. So far, the effect of CaM mutants on the interaction with and modulation of the RyR2 and the cardiac L-type channel have been investigated in some detail (17, 18). FRET studies with cardiac SR membranes indicated that CPVT CaM mutants increase binding to the RyR2, enhance its channel opening probability, and lead to spontaneous Ca2+ waves and spark activity, features not seen for LQTS-causing CaM mutants (17). A study using guinea pig ventricular myocytes showed that LQTS mutants induce increased action-potential prolongation, increase Ca2+ transient amplitudes, and suppress the CaM-dependent inactivation of the of L-type channel Cav1.2 (18).
Little is known about the effect of CaM mutants on the function of CaMKII, an important player in cardiac Ca2 signaling. CaMKII regulates the RyR2, Cav1.2, and Nav1.5 in a strictly Ca2+/CaM-dependent manner (reviewed in Ref. 19). Ca2+/CaM activates CaMKII by displacing the autoinhibitory domain of the CaMKII allowing transphosphorylation on position Thr286, which renders the enzyme active (20). Thr286 phosphorylation increases the affinity of CaM to the enzyme 1,000-fold, keeping it active even at low Ca2+ concentrations during a certain time period, which allows the enzyme to act as a chemical memory of previous heartbeats by integrating individual Ca2+ signals (21). Increased activity of CaMKII is implicated in several heart diseases including dilated cardiomyopathy, cardiac hypertrophy, and arrhythmia, mainly because of an increased leak of Ca2+ from the SR and Ca2+-mediated arrhythmias (5, 22).
In this study, our aim was to examine the effect of six CaM mutants (N53I, F89L, D95V, N97S, D129G, and F141L) in an in vitro system with conditional CaM expression in DT40 cells (23). In addition, the goal was to investigate whether the CaM mutants are able to activate CaMKII and to analyze how the mutant with the most pronounced effect in vitro (CaM D129G) affects the heart rhythm of zebrafish. Our study shows that the arrhythmogenic CaM mutants affect the examined functions differentially and indicates that the mutation changing the first Ca2+ coordinating residue from Asp to Gly in EF hand 4 of CaM affects the in vitro parameters to the highest degree and causes an arrhythmogenic phenotype in vivo.
Results
The CaM D129G Mutant Cannot Rescue DT40 Cells Depleted of WT CaM
We have previously established a cellular CaM knock-out/knock-in system with conditional CaM expression (tet off system) (Fig. 2A) (23). This is a unique cellular model system because it allows expressing CaM variants in the absence of endogenous CaM in a vertebrate cell line after 4–5 days of tetracyclin treatment (Fig. 2B). This enabled us to study the effect of CaM mutations on basic cellular processes such as cell growth, viability, and cell cycle regulation, as well as regulation of CaM-dependent signaling pathways and interaction of CaM with its numerous target proteins. In the present study we used this system to investigate the known cardiac failure causing CaM mutants for their potential to support cell growth. Stable expression of the different mutants in a WT CaM background did not affect growth, whereas removing WT CaM in the absence of ectopically expressed CaM blocked cell growth and viability completely. HA tagged CaM was fully able to restore these features as reported before (23). Remarkably, among the tested CaM mutations, the D129G mutant decreased cell growth and viability to a large degree, whereas among the other mutations only CaM F89L or N97S showed an effect on these parameters, however to a much lesser degree as compared with CaM D129G (Fig. 2, C and D). Because none of the mutant proteins were expressed to a higher extent as compared with the wild type and most of them still were able to support cell growth to a major degree (Fig. 2B), we assume that the observed effects were not the result of different levels of mutated CaM. In addition, earlier work (24) showed that lowering CaM concentrations down to 40% does not alter growth properties of DT40 cells. To exclude the possibility that the lack of viability was a clonal artifact, multiple independent D129G-expressing clones were tested (data not shown). The viability did not reflect the clonal diversity in HA-CaM expression because different cell lines with various CaM expression levels gave similar results.
FIGURE 2.

The CaM mutant D129G is not able to replace WT CaM in DT40 cells. The CaM1 gene of the chicken DT40 cells was silenced by disrupting exon 3 (red) by insertion of a tetracyclin-regulatable rat CaM cDNA (green) and the other allele by insertion of a selection marker (23). In this cell line, both alleles of the CaM 2 gene had previously been disrupted (24) rendering the tetracyclin-regulatable rCaM (TetReg-CaM) as the only CaM expressed. Stable transfection of HA-tagged versions of CaM (blue) followed by down-regulation of rCaM enables exchange of WT for HA-tagged WT and mutant CaM. B, Western blot analysis of 25 μg of total protein/lane from DT40 clones showing levels of TetReg-CaM and HA-tagged CaM non-treated (control, upper panel incubated with anti-CaM antibody) and 4 days of tet treatment showing almost complete exchange of WT for mutated HA-tagged CaM (lower panel incubated with both anti-CaM and anti-HA antibodies). Because the CaM antibody has different affinities for the mutant versions of CaM, the signals shown for HA-CaM do not represent the precise amounts of these proteins. C and D, growth curves of cells expressing HA-tagged CaM WT and mutants grown in the absence (C) or presence (D) of tetracyclin (tet). Error bars are S.E. from five independent repetitions. E, bar diagram showing % viable cells at 120-h tet treatment for all clones compared with the control without tet. Error bars are S.E. from five independent repetitions.
CaMKII Activity Is Affected by CaM Mutations to a Various Degree, Most Prominently with CaMD129G
CaMKII plays a central role in a large number of Ca2+/CaM-signaling pathways and is essential for cardiomyocyte functions (25). We therefore asked whether the arrhythmia-causing CaM mutants were capable of activating CaMKII. To do this we used the CaMKII sensor Camui (26), which is based on the CaMKIIα isoform and has been shown to mimic the heart tissue predominant δ-isoform of CaMKII (27). Camui exploits the structural and functional features of CaMKII by containing a fused fluorescent protein on the N and C termini of CaMKII within Förster radius in the inactive state. CaMKII activation leads to separation of the fluorophores and thereby unquenching of the donor mCerulean (CFP). Adding WT CaM increased CFP fluorescence, as did the N53I, N97S, and F141L mutants, whereas the maximal fluorescence as compared with WT CaM was strongly decreased for the F89L and D95V and most prominently for the D129G, as well as for the Ca2+-binding deficient mutants (Fig. 3A). The strongly reduced ability of D129G to activate CaMKII was similar to that of the Ca2+-binding deficient mutants (EF12, EF34, and EF1234 (numbers indicate inactivated Ca2+ binding sites)) (Fig. 3A). In cardiomyocytes the mutant CaMs are expected to exert their effects by competing with WT CaM because only one allele of the three genes expressing CaM is changed (reviewed in Ref. 28). To mimic this situation for CaM D129G, which in all our assays yielded the strongest effect, we mixed CaM D129G and WT CaM in different ratios and measured Camui activation following addition of Ca2+ (Fig. 3B). Exchanging WT for D129G CaM reduced the Camui activation as compared with only reducing WT CaM with buffer (dilution effect), indicating a dominant negative effect of the D129G (Fig. 3B). The CaM mutant with both EF hands 3 and 4 mutated had an even more severe dominant negative effect (Fig. 3B). Because the speed of Ca2+ fluxes in the sarcoplasm regulates the dynamics of the Ca2+ receptor CaM and its interactions with targets and thereby the heart rhythm, we investigated whether the CaM D129G mutant would affect CaMKII in an initial activation phase. Kinetic measurements comparing the activation speed of CaMKII activation in the presence of WT CaM or CaM D129G alone or mixed in a 2:1 ratio showed that CaMKII activation by CaM was significantly reduced when CaM D129G was present (Fig. 3C). These combined data show that the D129G CaM mutant can exert a dominant negative effect on the CaMKII function in vitro.
FIGURE 3.
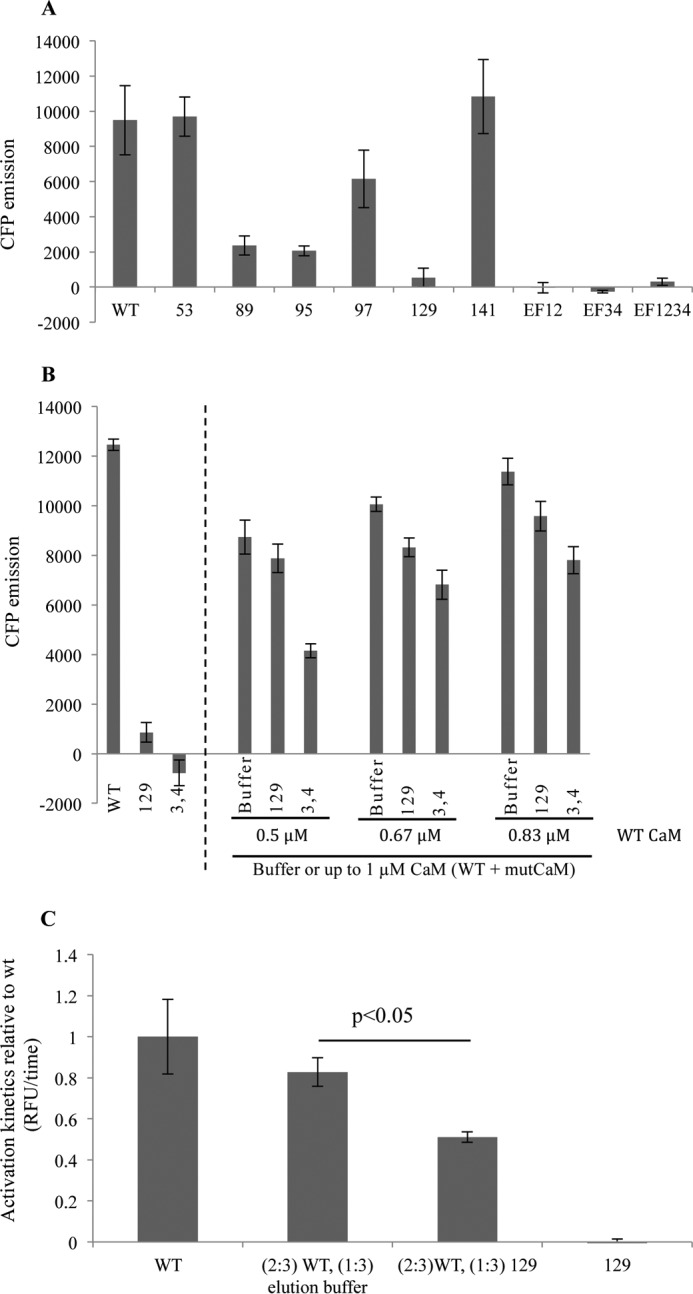
CaM D129G activates CaMKII only to a minor degree and decreases WT CaM-mediated activation. A, recombinant heart disease-related CaMs with the indicated amino acids mutated or WT CaM were incubated with HeLa cell lysate containing the Camui CaMKII sensor, and peak unquenching of mCerulean was measured in the presence of 2.5 μm [Ca2+]. CaM with disrupted Ca2+ binding at EF hands 1 and 2 (EF12), 3 and 4 (EF34), or 1–4 (EF1234) were used as negative controls. B, bar diagram shows the effect of CFP emission on adding 1 μm WT CaM, CaM 129, or EF34 alone (left-hand side), or mixing WT CaM with the mutant CaM proteins (right-hand side). To test different ratios of WT:mutCaM, the mutCaM was added to give a final total CaM concentration (WT + mutCaM) of 1 μm. Addition of buffer served as a control for the effect of reducing WT CaM. C, activation kinetics of CaMKII in the presence of D129G CaM. p < 0.05 as calculated by paired two-tailed analysis. Error bars represent S.E. from three independent experiments.
CaM D129G Cannot Stimulate Phosphorylation of CaMKII Thr286 and Phosphorylation of Downstream Targets Is Markedly Reduced
A small yet consistent Camui activation change by CaM D129G in our in vitro experiment indicated some contribution of the mutant in the activation. To investigate the phosphorylation status of CaMKII in the presence of WT or D129G CaM, we purified ectopically expressed rat CaMKII from HEK cells. CaMKII is well known to become activated by Ca2+/CaM through binding and steric removal of a regulatory domain leading to autophosphorylation of Thr286 (29). Incubation of recombinant CaMKII with D129G CaM led to a diminished autophosphorylation of CaMKII Thr286, as compared with incubation with WT CaM (Fig. 4, A and B). To test whether D129G CaM affected the CaMKII-dependent phosphorylation of downstream targets, we analyzed the in vitro phosphorylation of the CaMKII substrate autocamtide-2 (KKALRRQETVDAL) and found that in the presence of D129G CaM phosphorylation of the peptide was significantly reduced as compared with WT CaM (Fig. 4C). These results along with the Camui data indicate that D129G is able to bind to CaMKII but cannot cause its activation.
FIGURE 4.

Lack of CaMKII Thr286 phosphorylation in the presence of CaM D129G and reduced phosphorylation of a downstream target peptide. Purified CaMKII was stimulated with either WT CaM or D129G CaM under various [Ca2+]. A, representative Western blot analysis of phosphorylation analyzed with an anti-Thr(P)286/287 specific antibody. Ctr indicates control without added Ca2+. B, quantification of Western blot analysis showing Thr(P)286 CamKII to total CaMKII ratios relative to non-Ca2+-stimulated control from three independent experiments. Error bars represents S.D. C, quantitative mass spectrometry analysis of the autocamtide 2 target tryptic peptide (QET(phosp)VDAL, highlighted in blue) following recombinant CaMKIIδ stimulation with either WT or D129G CaM. Measurements were performed in triplicate at 2.5 and 15 min after the reaction was initiated. *, p < 0.05; **, p < 0.005 calculated using either homoscedastic or heteroscedastic two-tailed t test, depending on the statistical value of the F-test (heteroscedastic if p < 0.05). Proteins with t test p values smaller than 0.05 were considered as significantly altered between the two tested conditions. The values are given as fold change of the D129G versus WT CaM. Detected CaMKIIδ tryptic Thr286 phosphopeptide is shown for comparison.
CaM D129G Expression in Zebra Fish Causes Decreased Heart Rate
To examine whether wild type CaM and CaM D129G would elicit different effects on the development and/or function of the embryonic heart, we injected either WT CaM or CaM D129G synthetic RNA into myl7:GFP embryos. In myl7:GFP transgenic zebrafish embryos, GFP expression is driven by the cardiac-specific promoter myl7 (30), allowing in vivo phenotypic analysis and quantitative measurement of cardiac parameters (31). As shown in Fig. 5A, WT or D129G CaM overexpression did not affect the overall development of zebrafish embryos or the morphogenesis of the hearts. After 2 days of development, well patterned hearts capable of propelling circulation through the body were observed in uninjected controls and D129G or WT CaM RNA-injected embryos (Fig. 5A). Although CaM-WT RNA-injected embryos had a heart rate similar to that observed in uninjected control embryos, D129G overexpression reduced the heart rate to ∼83% of the level observed in controls This is a moderate difference, however statistically significant (n = 128, p < 0.001) (Fig. 5B) considering that endogenous CaM is present as well. Interestingly, ∼8% of D129G RNA-injected embryos exhibited an abnormal cardiac rhythm where the atrium beats twice per ventricular contraction (Fig. 5, C and D). Overall, these results demonstrate a significant impact of D129G on the heart rate and conduction of the developing heart in zebrafish.
FIGURE 5.

Impact of CaM D129G on the heart rate and conduction of the developing heart in zebrafish. A, representative images of zebrafish injected with RNA coding for either WT CaM or the mutant D129G, as well as non-injected control (upper panel). The lower panel shows GFP expression in the zebrafish hearts. B, bar diagram showing relative heartbeat normalized to non-injected control embryos. Heartbeat measurements were based on fluorescent intensities from cardiac contractions in non-injected control embryos, WT CaM-injected embryos, and D129G-injected embryos. D129G-injected embryos exhibit a heart rate of 83% compared with non-injected control and CaM-WT-injected embryos. ***, p < 0.001. C, contractions in the atrium and ventricle of a non-injected animal. D, a representative trace from a D129G-injected embryo exhibiting a 2:1 atrium to ventricle beat ratio, observed in ∼8% of D129G-injected embryos (n = 128).
Discussion
In this study we tested the arrhythmogenic CaM mutations N53I, F89L, D95V, N97S, D129G, and F141L in a cell-based system and in a CaMKII activation assay. We found that the CaM mutant D129G stands out from six investigated cardiac arrhythmia-related CaM mutations in interfering with the viability of a genetically modified vertebrate cell line expressing exclusively mutant CaM. Further, CaM D129G showed the strongest effect on CaMKII activity measured in a FRET-based assay and also diminished CaMKII substrate phosphorylation as compared with WT CaM. Based on these results we tested the D129G mutant in vivo. Expression of CaM D129G in zebrafish led to decreased heart rate and in a subpopulation of animals to a 2:1 ratio of atrium to ventricle heart rate denoted as breakdance phenotype.
CaM is the major sensor of Ca2+ signals in all eukaryotic cells regulating a multitude of physiological processes including muscle contraction. In cardiac tissue CaM is involved in the tuning of ion channels including the SR Ca2+ release channel RyR2, potassium, and sodium channels in the sarcolemma directly involved in excitation-contraction coupling (reviewed in Refs. 28 and 32). Several of these ion channels are further regulated indirectly through Ca2+/CaM modulation of cytosolic enzymes such as CaMKII and calcineurin. In addition, CaM regulates transcriptional processes important for proper heart function. One example is the phosphorylation of class II histone deacetylases by the CaM/CaMKII pathway. In the phosphorylated stage these deacetylases are excluded from the nucleus and incapable of repressing genes that are involved in heart hypertrophy (reviewed in Ref. 5). CaM and CaMKII were also shown to affect the heart rhythm by regulating the expression of ion channels involved in excitation contraction coupling (reviewed in Refs. 33 and 34). The recent finding that CaM mutations in one of three expressed CaM genes, which all encode the same protein, may cause several types of heart arrhythmias, mainly CPVT and LQTS, came as a surprise as CaM is perfectly conserved in vertebrates underlining the importance of maintenance of an unchanged primary structure. Even though several laboratories have initiated work to investigate the molecular basis of how CaM mutation cause heart arrhythmia, little is known about the molecular mechanisms affected by CaM mutations in the heart.
We replaced WT CaM with six arrhythmia-causing CaM mutants and found that only CaM D129G affected cell growth and viability to a major degree. This indicated that exchanging aspartic acid at position 129 with glycine renders CaM incapable of supporting basic vertebrate cell functions. One would therefore expect that such a mutation would not be compatible with life. However, it has to be considered that three genes code for an identical CaM protein in mammals and that the D129G mutation was only found in one allele of all the three CaM genes (10, 13, 15). All three CaM genes are transcribed in the heart as measured by quantitative PCR (10). Highest mRNA levels were found for the CaM 3 gene followed by the CaM 2 and the CaM 1 gene in the human left ventricle. However, it is not known how much each transcript contributes to the expression of CaM on the protein level. In addition, it is possible that the different transcripts are used for translation in different subcellular locations in cardiomyocytes in analogy to findings in neurons (35). Therefore, it seems that this mutation can be tolerated for normal human development but would affect heart physiology by a dominant effect over WT CaM. Asp129 is a very important residue because it coordinates Ca2+ in the fourth EF hand Ca2+ binding loop. All other LQTS-related mutations investigated in this report, even though directly at Ca2+ coordinating positions, showed considerably milder effects on Ca2+ affinity (10). Changing an aspartate to a glycine at position 129 may have severe structural consequences because glycine allows the highest flexibility. This is corroborated by an earlier study where we showed that changing Asp129 to Ala did not abolish cell growth and viability (23). In addition, NMR data of CaM D129G from a study by Crotti et al. (10) support the notion that a local structural change along with reduced Ca2+ binding may explain the effect of this mutant in the functions analyzed in our study.
We tested the CaM mutants for their capability to bind and stimulate CaMKII autophosphorylation using the Camui FRET system (36). CaMKII functions in the heart by regulating a number of Ca2+, K+, and Na+ channels through phosphorylation. Its chronic activation may cause heart failure through a variety of mechanisms (5, 32). One crucial target relevant for heart rhythm control and phosphorylated by CaMKII is the RyR2. Phosphorylation of the RyR2 enhances Ca2+ release from the SR disturbing normal Ca2+ cycling (reviewed in Ref. 5). In the heart, the major CaMKII isoform is CaMKIIδ, which is mainly found in the sarcoplasm and acts in regulation of ion channels and Ca2+ handling, whereas CaMKIIβ is found predominantly in the nucleus and regulates hypertrophic gene activity (5). As in other CaMKII isoforms, the active form of CaMKIIδ is stabilized by phosphorylation on Thr286 as well as other post-translational modifications such as oxidation and O-linked glycosylation rendering the enzyme Ca2+/CaM-independent (27). Recent investigations indicate that Thr286 phosphorylation is necessary but not sufficient for full CaMKII activation. Coultrap et al. (37) found that higher than 15–25% activation of CaMKII by preincubation with Ca2+/CaM is an exception and needs a special type of binding by the substrate. Full activation toward regular substrates such as tyrosine hydroxylase and GluR1 requires additional regulation by Ca2+/CaM. Thus, incomplete and substrate-dependent autonomy prevents uncoupling from subsequent regulation. The same group later showed that further stimulation of partially autonomous CaMKII is physiologically important because autonomous CaMKII must be further stimulated by Ca2+/CaM to enhance synaptic strength (38). Molecular mechanisms of this additional Ca2+/CaM-dependent activation of partially autonomous CaMKII have not been established so far.
Hwang et al. (17) tested the available CaM mutants for their potential to affect the RyR2 calcium release channel. Because CaMKII is known to phosphorylate and activate the RyR2 channel, the arrhythmia-causing CaM mutants were also compared with the WT for their potential to regulate CaMKII and thereby have an effect on RyR2 activity. Neither of the mutants led to a decreased CaMKII activity as compared with the WT (17). This is in contrast to our results carried out with the same CaMKII sensor used by Hwang et al. (17). Not only did we see a strong decrease in CaMKII activity with the D129G, but also the D95V and N97S gave significantly reduced (23 and 63%, respectively) CaMKII peak activity (Fig. 3A). Earlier literature indicates that EF hands 3 and 4 in CaM are of crucial importance for CaMKII activation (39), and as position Asp129 is the first Ca2+ coordinating residue in EF loop 4, our results are not surprising. Based on these data, it is surprising that Hwang et al. (17) did not find a difference between mutants affecting EF hands 3 and 4 and WT CaM in CaMKII activation. Crotti et al. (10) found that the C-domain of CaM D129G had a 54-fold lower Ca2+ affinity (kd = 150 μm) as compared with the intact WT CaM C-domain, which would make it unlikely that CaM D129G would be able to fully activate CaMKII at low Ca2+concentration. In the work by Hwang et al. (17), ATP was not added in the CaMKII activation assay. It is therefore questionable whether the CaMKII sensor was autophosphorylated. Our experiments show that the addition of CaM D129G causes a small decrease in Camui FRET indicative of CaM binding; however, binding only occurs under saturating Ca2+ conditions and does not involve Thr286 phosphorylation. From this, we hypothesize that CaM D129G can bind but not stimulate CaMKII autophosphorylation. This leads to a competitive binding responsible for the dominant negative delay in the activation profile of CaMKII (Figs. 3C and 6). The pleiotropic nature of CaMKII, targeting both function and expression of proarrhythmic as well as anti-arrhythmic molecules (reviewed in Ref. 40), makes it difficult to derive a conclusion on the consequences of the D129G CaM defect at the organismal level. It is, however, safe to conclude that the autonomous activation of CaMKII is affected. Studies into molecular mechanism of how CaM mutations affect CaMKII and in particular how they can exhibit a dominant negative effect on CaMKII should be done on a structural level in the future.
FIGURE 6.

Hypothetical model for CaMKII activation delay. Half of the dodecameric holoenzyme has been sketched with the green units representing the regulatory domain and the blue units the hub domain. CaM is in yellow (WT) or orange (D129G), and the red dots represent phosphorylation sites Thr286. The presence of CaM D129G will lead to binding without Thr286 phosphorylation. Because the D129G will occupy a CaM binding site, phosphorylation (drawing 1) and full holoenzyme activation (drawing 2) will be delayed (Fig. 3C).
Based on results obtained in vitro, we then asked whether the D129G CaM, which exhibited the most prominent effect in vitro would also have an effect in vivo. D129G CaM RNA-injected in zebrafish embryos exhibited a major effect on zebrafish heart physiology. Expressing CaM D129G in zebrafish eggs not only altered the heart rhythm but also led to the “breakdance” phenotype characterized by an atrium to ventricle beating ratio of 2:1 of 2-day-old zebrafish. This indicates for the first time that this mutant affects the heart function in an in vivo vertebrate model system. The D129G-induced 2:1 beat phenotype resembles LQTS. This phenotype is comparable with the human long QT syndrome, an arrhythmia caused by a modification of ion channels involved in cardiac repolarization (41). The low penetrance (only 8% of the investigated animals show this phenotype) is likely to be the result of the labile nature of injected RNA. Clearly, generating stable transgenic fish or knock-in D129G fish would help clarify the possible causative relationship between D129G and LQTS. Because it is known that CaM regulates K+ cannels involved in repolarizing current by moving K+ ions outward (42) and mutations in zebrafish K+ channels induce the bradycardia/LQT phenotype (43), it is possible that the observed phenotypes are correlated with failure in Ca2+/CaM regulation of K+ channels either directly or through CaMKII.
Experimental Procedures
DNA Constructs
In this study, six different heart arrhythmia-linked CaM mutations, CaM N53I, CaM F89L, CaM D95V, CaM N97S, CaM F129L, and CaM F141L, were analyzed. The numbers indicate the position of amino acid exchange without the initial methionine. Site-directed mutagenesis was performed in the pCMV_HA-CaM_WT_Eco vector, described in Ref. 23.
For protein expression CaM N53I, CaM F89L, CaM D95V, CaM N97S, CaM F129L, and CaM F141L mutations were made by the quick change method in a pGemex vector. For DT40 cell expression, the pCMV-HA Eco vector providing a short protein tag, earlier shown not to interfere with the function of CaM (23) was used. Mutant Camui constructs were made by PCR techniques in the Camui vector containing full-length CaMKIIα kindly provided by Yasunori Hayashi (36).
Cell Lines
DT40-derived clone ET 1–50 (23) grown in RPMI with 10% FBS and 1% chicken serum served as the parental cell line for generating clones expressing HA-tagged mutant CaMs. Transfection of 5,000,000 cells was performed with 15 μg of linearized pCMV-HA CaM Eco-gpt in 100 μl of electroporation buffer (20 mm HEPES, 135 mmol/liter KCl, 2 mmol/liter MgCl2, 0.5% Ficoll 400, 200 mmol/liter ATP, and GSH 500 mmol/liter, adjusted to pH 7.4 with KOH). Electroporation was done with the Bio-Rad gene pulser system in 1-mm cuvettes at 124 V and 750 microfarad. Immediately afterward, 500 μl of spent medium was added. After 2 min of incubation, the cells were transferred to 2 ml of spent medium. Transfected cells were transferred to a 96-well plate in selection medium prepared by adding the following to the growth medium: 250 mg/liter xanthine, 20 mg/liter hypoxanthine, and 15 mg/liter mycophenolic acid prepared according to the manufacturer's recommendations (Sigma-Aldrich). After 6 days, colonies were transferred to fresh selection medium, and clones were expanded for 3–7 days before freeze stocks were prepared. The clones were tested by Western blot analysis with mouse αHA and mouse αCaM antibody to confirm HA-CaM expression. The specificity of the used antibodies was verified by probing cells that do not contain native CaM or the HA epitope (23).
Growth Curve and Viability Assay
DT40 cell growth was monitored for 7 days with and without tetracycline treatment. The cells were adjusted to 50,000–75,000 cells/ml. Counting and FACS analysis was performed every day. Every second day, the cells were reseeded at 50,000–75,000 cells/ml. Cells pellets were tested by Western blot analysis for the expression of CaM and HA-CaM.
Western Blot Analyses
DT40 cell pellets were lysed in 50 mmol/liter Tris-HCl, pH 7.5, 150 mmol/liter NaCl, 0.5% Nonidet P-40, 2 mmol/liter EDTA, 1 mmol/liter DTT, and protease inhibitor mixture for 20 min on ice followed by centrifugation at 15,000 × g for 10 min at 4 °C. Proteins in lysates were separated by SDS-PAGE (12% polyacrylamide) After blotting, the membrane was treated for 10 min in 0.02% glutaraldehyde. The blot was incubated with the primary antibody mouse αCaM (Millipore, catalog no. 05-173) 1:8,000 or mouse αHA11 (Covance, catalog no.16B12) 1:1,000. For development the Amersham Biosciences ECL Prime Western blotting detection reagent was used, and the blots were analyzed with Bio-Rad Chemie Doc MP. A prestained broad range marker (Thermo Fisher Scientific, catalog no. 26619) was used to verify the size of the observed bands.
Zebrafish
Zebrafish Husbandry and Transgenic Lines
Zebrafish of transgenic line, myl7:GFP were maintained and bred as described previously (44). Full-length D129G and WT CaM cDNA was cloned into pCS2 + 3XFLAG. For mRNA synthesis, plasmids were linearized by NotI and mRNA synthesized using the SP6 mMESSAGE mMachine kit according to the manufacturer's manual (Ambion).
Zebrafish Injections
100 pg of CaM mutant and WT CaM mRNA were injected into one-cell stage embryos collected from crosses of myl7:GFP zebrafish. Embryos were then maintained for further phenotypic analysis at 2 days postfertilization.
Zebrafish Cardiac Imaging and Analysis
Movies of GFP-labeled Tg(myl7:GFP) hearts were taken at 30 frames/s under a Zeiss Stemi SV11 microscope. A custom-designed area scan algorithm calculated pixel density in a user-specified area on the heart, and the value was quantified as a unit of fluorescence intensity over time in frames (30 frames/s). This generated a trace of cardiac contractions, and the data were plotted as cardiac trace over time (s).
CaM Expression and Purification
pGemex-2-CaM constructs were transformed into Escherichia coli competent cells BL-21 (DE3). The cells were incubated at 37 °C until the A600 was 0.8–1, and CaM expression was induced with 100 μmol/liter isopropyl β-d-thiogalactopyranoside. CaM was detected in culture medium after overnight incubation. Culture medium with additional 5 mmol/liter CaCl2 was applied to pre-equilibrated HiTrap Phenyl Sepharose column (GE Healthcare), and CaM elution proceeded according to Ref. 45. All purified CaM proteins used in the Camui/CaMKII experiments were dialyzed overnight against a buffer containing 50 mmol/liter Tris-HCl, pH 8, and 0.1 mmol/liter EGTA using 10,000 molecular weight cutoff SnakeSkin dialysis tubing (Thermo Fisher Scientific).
Camui in Vitro Assay
The Camui mYpet-CaMKIIα-mCerulean construct, kindly provided by Yasunori Hayashi (36), or derived mutants generated in our laboratory were transfected into HeLa cells using Lipofectamine (Invitrogen). HeLa cells grown under standard conditions (DMEM, 10% FBS, 1% penicillin/streptomycin) were seeded at 8,400,000 cells in a T175 flask the day before transfection and harvested 2 days after transfection. Cell lysis was done in 2 ml of buffer (40 mmol/liter HEPES NaOH, pH 8, 5 mmol/liter magnesium acetate, 0.1 mmol/liter EGTA, 0.01% Tween, 1 mmol/liter DTT, and protease inhibitor mixture) and followed by Dounce homogenization. Lysates were cleared by 10 min of centrifugation at 10,000 × g at 4 °C, and aliquots of the supernatant were shock frozen in liquid nitrogen and stored at −80 °C. CFP fluorescence was measured at 37 °C using a Tecan infinite F200 Pro (Thermo Fisher Scientific) and the ex448/em485 filter setting. For activation of Camui, 1 μmol/liter CaM (unless otherwise indicated) and 40 μmol/liter ATP were used. CaCl2 was added to reach the indicated free Ca2+ concentrations as calculated by the Max chelator program (46). Background readings were deducted from all measurements to exclude contribution from lysate components.
Camui Kinetics Measurements
The velocity of the CaM/Camui-CaMKII reaction was calculated using a modified setup of the Camui in vitro assay. CFP fluorescence emitted from Camui containing HeLa cell lysate in a 96-well plate with 1 μm CaM and 40 μm ATP (final concentrations after stimulation) was measured every ∼1 s. After ∼10 s, an automated injector added CaCl2 to reach an estimated final concentration of 200 μm free Ca2+. The background measurements (prior to addition of calcium) were deducted, and velocity of the CFP emission increase was calculated as V = [(Max − B)/2]/t, where V is velocity, Max is the CFP emission (average of 10 measurements at the reaction saturation level), B is the background (average of 9 measurements in the absence of Ca2+), and t is the time (s). A logarithmic regression model equation (y = α *ln(x) ± β) calculated in Microsoft Office Excel was used to calculate the time (t) at the half-maximum ((Max − Β)/2) of the CFP emission.
CaMKII Expression and Purification
His-tagged CaMKII was produced in HEK 293F cells by transfection of constructs using polyethylenimine (Sigma). The cells were grown in DMEM supplemented with 10% FBS (Biochrom AG), 1% penicillin/streptomycin for 3 days after transfection, when cells were harvested by centrifugation at 1,000 × g for 4 min at 4 °C, washed once in PBS, and then lysed in a buffer containing 0.3% Nonidet P-40, 0.5 m NaCl, 20 mm Na3PO4, 30 mm imidazole, and complete protease inhibitor mixture (Roche). Following Dounce homogenization, the lysate was cleared by centrifugation for 30 min at 20,000 × g. The supernatant was incubated overnight with nickel-Sepharose (GE Healthcare) and washed with two volumes of lysis buffer before elution using buffer containing 500 mm imidazole to give a semipurified His-CaMKII solution of unknown CaMKII concentration. The HeLa and HEK 293F cell lines were exclusively used for protein production and tested periodically for mycoplasma infection and phenotypic characteristics.
CaMKII in Vitro Activation
11 μl of semipure His-CaMKII was mixed with 1 μm wild type or mutated CaM, 40 μm ATP and desired concentrations of Ca2+ to give a final volume of 12.5 μl and incubated at 37 °C for 30 min. Proteins in the reaction mix were separated by SDS-PAGE (10% polyacrylamide) and analyzed by Western blot using antibodies against Thr(P)286 CaMKII (Abcam, catalog no. ab32678) and CaMKII (Sigma-Aldrich, catalog no. C6974). Western blots were visualized and analyzed using the ChemiDoc MP bioimaging system (Bio-Rad).
Mass Spectrometry
All chemicals were purchased from Sigma-Aldrich, unless otherwise stated. TiO2 beads were from GL Sciences Inc. (Tokyo, Japan). Endopeptidase Lys-C was from Wako. Sequence grade trypsin was from Promega (Madison, WI). Recombinant human histidine-tagged CaMKIIδ was obtained from Life Technologies. Autocamtide-2 was from Santa Cruz Biotechnology Inc. Okadaic acid was purchased from MP Biomedicals (Thermo Fisher Scientific). Empore C18 extraction disk was from 3M Bioanalytical Technologies (St. Paul, MN). Poros Oligo R3 reversed phase chromatographic material was from PerSeptive Biosystems (Framingham, MA). Reprosil-C18 3-μm beads were purchased from MikroLab Aarhus S/A (Højbjerg, Denmark). All reagents used in the experiments were of sequencing grade. All solutions were made with ultrapure water (ELGA Purelab Ultra water system).
Stimulation of CaMKIIδ Autophosphorylation
Protein concentration was determined by amino acid composition analysis using a Biochrom 30 amino acid analyzer (Biochrom, Cambridge, UK). Autophosphorylation of CaMKIIδ was induced by adding 1.5 μg of CaMKIIδ to a reaction mix containing 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.1% BSA, 1 mm CaCl2, 10 mm MgCl2, 100 μm ATP, 1 μm okadaic acid, and 10 μg of wild type CaM or D129G CaM mutant, respectively, with the addition of 30 μg of substrate peptides (Autocamtide-2; KKALRRQETVDAL). Stimulation of CaMKIIδ was performed for 2.5 and 15 min at 37 °C in a final volume of 54.8 μl. The reaction was stopped by adding reducing agent (10 mm DTT) and endopeptidase Lys-C to the mix, followed by 1 h of incubation at room temperature. Each stimulation condition was performed in triplicate. Peptide digestion was performed by using either Lys-C alone or Lys-C in combination with trypsin. Lys-C only was used for peptides, which would have been too short if digested with trypsin. Lys-C generated peptides were alkylated in 20 mm iodoacetamide for 30 min at room temperature in the dark. Subsequently, the samples were digested with trypsin at an enzyme to substrate ratio of ∼1:50 for 12 h at 37 °C. The samples were acidified to 5% formic acid and desalted using Poros R3 RP column packed in a P200 stage tip with C18 3M plug. The purified peptide samples were dried by lyophilization and labeled using isobaric tagging labeling (iTRAQTM 4-plex; Applied Biosystems, Foster City, CA) as described by the manufacturer. After labeling, all samples were pooled into one tube and dried by vacuum centrifugation to ∼50 μl for phosphopeptide enrichment by TiO2. The TiO2 enrichment of phosphopeptides was performed essentially as previously described (47). Prior to nanoLC-MS/MS analysis, the phosphorylated peptide samples were resuspended in 0.1% TFA and desalted using Poros R3 RP column packed in a P200 stage tip with C18 3M plug. Purified phosphopeptide samples were dried by lyophilization and stored at −80 °C until further analysis.
Author Contributions
M. W. B., J. M. L. C., and T. J. did the planning and directing of the experimental work, as well as writing and commenting; K. K. and M. R. L. carried out phosphorylation assays and mass spectroscopy; K. W. and J.-N. C. performed the zebrafish work; R. T. made the DNA constructs, produced recombinant CaM, and did the DT40 work; and T. Z. performed the Camui in vitro assay and CaMKII in vitro activation.
Acknowledgments
We thank the members of the Berchtold lab, particularly technician Casper Hølmkjær for work on CaM purification, as well as daily lab maintenance.
This work was supported by funds from the Danish Heart Foundation (to J. M. L. C. and M. W. B.), AP Møller Lægefonden (to J. M. L. C.), and the Nilssons, Marshall, Willumsen, and Danielsen Foundations (to M. W. B.) and by Danish Research Council Grant FSS4004-00560 (to M. W. B. and J. M. L. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- SR
- sarcoplasmic reticulum
- CaM
- calmodulin
- CaMKII
- Ca2+/CaM kinase II
- CPVT
- catecholaminergic polymorphic ventricular tachycardia
- LQTS
- long QT syndrome
- RyR2
- ryanodine receptor 2
- CFP
- cyan fluorescent protein.
References
- 1. Berchtold M. W., Brinkmeier H., and Müntener M. (2000) Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 80, 1215–1265 [DOI] [PubMed] [Google Scholar]
- 2. Marks A. R. (2013) Calcium cycling proteins and heart failure: mechanisms and therapeutics. J. Clin. Invest. 123, 46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berchtold M. W., and Villalobo A. (2014) The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta 1843, 398–435 [DOI] [PubMed] [Google Scholar]
- 4. Berchtold M. W., Egli R., Rhyner J. A., Hameister H., and Strehler E. E. (1993) Localization of the human bona fide calmodulin genes CALM1, CALM2, and CALM3 to chromosomes 14q24-q31, 2p21.1-p21.3, and 19q13.2-q13.3. Genomics 16, 461–465 [DOI] [PubMed] [Google Scholar]
- 5. Anderson M. E., Brown J. H., and Bers D. M. (2011) CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 51, 468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Erickson J. R., He B. J., Grumbach I. M., and Anderson M. E. (2011) CaMKII in the cardiovascular system: sensing redox states. Physiol. Rev. 91, 889–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Racioppi L., and Means A. R. (2012) Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J. Biol. Chem. 287, 31658–31665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo M., and Anderson M. E. (2013) Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 113, 690–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nyegaard M., Overgaard M. T., Søndergaard M. T., Vranas M., Behr E. R., Hildebrandt L. L., Lund J., Hedley P. L., Camm A. J., Wettrell G., Fosdal I., Christiansen M., and Børglum A. D. (2012) Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 91, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crotti L., Johnson C. N., Graf E., De Ferrari G. M., Cuneo B. F., Ovadia M., Papagiannis J., Feldkamp M. D., Rathi S. G., Kunic J. D., Pedrazzini M., Wieland T., Lichtner P., Beckmann B. M., Clark T., et al. (2013) Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marsman R. F., Barc J., Beekman L., Alders M., Dooijes D., van den Wijngaard A., Ratbi I., Sefiani A., Bhuiyan Z. A., Wilde A. A., and Bezzina C. R. (2014) A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J. Am. Coll. Cardiol 63, 259–266 [DOI] [PubMed] [Google Scholar]
- 12. Makita N., Yagihara N., Crotti L., Johnson C. N., Beckmann B. M., Roh M. S., Shigemizu D., Lichtner P., Ishikawa T., Aiba T., Homfray T., Behr E. R., Klug D., Denjoy I., Mastantuono E., et al. (2014) Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 7, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reed G. J., Boczek N. J., Etheridge S. P., and Ackerman M. J. (2015) CALM3 mutation associated with long QT syndrome. Heart Rhythm 12, 419–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomez-Hurtado N., Boczek N. J., Kryshtal D. O., Johnson C. N., Sun J., Nitu F. R., Cornea R. L., Chazin W. J., Calvert M. L., Tester D. J., Ackerman M. J., and Knollmann B. C. (2016) Novel CPVT-associated calmodulin mutation in CALM3 (CALM3-A103V) activates arrhythmogenic Ca waves and sparks. Circ. Arrhythm. Electrophysiol. 9, e004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boczek N. J., Gomez-Hurtado N., Ye D., Calvert M. L., Tester D. J., Kryshtal D. O., Hwang H. S., Johnson C. N., Chazin W. J., Loporcaro C. G., Shah M., Papez A. L., Lau Y. R., Kanter R., Knollmann B. C., et al. (2016) Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ. Cardiovasc. Genet. 9, 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pipilas D. C., Johnson C. N., Webster G., Schlaepfer J., Fellmann F., Sekarski N., Wren L. M., Ogorodnik K. V., Chazin D. M., Chazin W. J., Crotti L., Bhuiyan Z. A., and George A. L. Jr. (2016) Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Heart Rhythm 13, 2012–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hwang H. S., Nitu F. R., Yang Y., Walweel K., Pereira L., Johnson C. N., Faggioni M., Chazin W. J., Laver D., George A. L. Jr, Cornea R. L., Bers D. M., and Knollmann B. C. (2014) Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ. Res. 114, 1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Limpitikul W. B., Dick I. E., Joshi-Mukherjee R., Overgaard M. T., George A. L. Jr, and Yue D. T. (2014) Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell Cardiol. 74, 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maier L. S., and Bers D. M. (2007) Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 73, 631–640 [DOI] [PubMed] [Google Scholar]
- 20. DeGrande S., Nixon D., Koval O., Curran J. W., Wright P., Wang Q., Kashef F., Chiang D., Li N., Wehrens X. H., Anderson M. E., Hund T. J., and Mohler P. J. (2012) CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm 9, 2034–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meyer T., Hanson P. I., Stryer L., and Schulman H. (1992) Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 256, 1199–1202 [DOI] [PubMed] [Google Scholar]
- 22. Zhang T., Maier L. S., Dalton N. D., Miyamoto S., Ross J. Jr, Bers D. M., and Brown J. H. (2003) The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919 [DOI] [PubMed] [Google Scholar]
- 23. Panina S., Stephan A., la Cour J. M., Jacobsen K., Kallerup L. K., Bumbuleviciute R., Knudsen K. V., Sánchez-González P., Villalobo A., Olesen U. H., and Berchtold M. W. (2012) Significance of calcium binding, tyrosine phosphorylation, and lysine trimethylation for the essential function of calmodulin in vertebrate cells analyzed in a novel gene replacement system. J. Biol. Chem. 287, 18173–18181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmalzigaug R., Ye Q., and Berchtold M. W. (2001) Calmodulin protects cells from death under normal growth conditions and mitogenic starvation but plays a mediating role in cell death upon B-cell receptor stimulation. Immunology 103, 332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hund T. J., and Mohler P. J. (2015) Role of CaMKII in cardiac arrhythmias. Trends Cardiovasc. Med. 25, 392–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mower A. F., Kwok S., Yu H., Majewska A. K., Okamoto K., Hayashi Y., and Sur M. (2011) Experience-dependent regulation of CaMKII activity within single visual cortex synapses in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 21241–21246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Erickson J. R., Pereira L., Wang L., Han G., Ferguson A., Dao K., Copeland R. J., Despa F., Hart G. W., Ripplinger C. M., and Bers D. M. (2013) Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sorensen A. B., Søndergaard M. T., and Overgaard M. T. (2013) Calmodulin in a heartbeat. FEBS J. 280, 5511–5532 [DOI] [PubMed] [Google Scholar]
- 29. Hudmon A., and Schulman H. (2002) Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 364, 593–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang C. J., Tu C. T., Hsiao C. D., Hsieh F. J., and Tsai H. J. (2003) Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 228, 30–40 [DOI] [PubMed] [Google Scholar]
- 31. Shimizu J., Oka H., Yamano Y., Yudoh K., and Suzuki N. (2016) Cardiac involvement in relapsing polychondritis in Japan. Rheumatology 55, 583–584 [DOI] [PubMed] [Google Scholar]
- 32. Bers D. M. (2008) Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49 [DOI] [PubMed] [Google Scholar]
- 33. Bers D. M., and Grandi E. (2009) Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bers D. M. (2011) Ca2+-calmodulin-dependent protein kinase II regulation of cardiac excitation-transcription coupling. Heart Rhythm 8, 1101–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toutenhoofd S. L., and Strehler E. E. (2002) Regulation of calmodulin mRNAs in differentiating human IMR-32 neuroblastoma cells. Biochim. Biophys. Acta 1600, 95–104 [DOI] [PubMed] [Google Scholar]
- 36. Kwok S., Lee C., Sánchez S. A., Hazlett T. L., Gratton E., and Hayashi Y. (2008) Genetically encoded probe for fluorescence lifetime imaging of CaMKII activity. Biochem. Biophys. Res. Commun. 369, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coultrap S. J., Buard I., Kulbe J. R., Dell'Acqua M. L., and Bayer K. U. (2010) CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J. Biol. Chem. 285, 17930–17937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barcomb K., Buard I., Coultrap S. J., Kulbe J. R., O'Leary H., Benke T. A., and Bayer K. U. (2014) Autonomous CaMKII requires further stimulation by Ca2+/calmodulin for enhancing synaptic strength. FASEB J. 28, 3810–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shifman J. M., Choi M. H., Mihalas S., Mayo S. L., and Kennedy M. B. (2006) Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc. Natl. Acad. Sci. U.S.A. 103, 13968–13973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vincent O., Rainbow L., Tilburn J., Arst H. N. Jr, and Peñalva M. A. (2003) YPXL/I is a protein interaction motif recognized by Aspergillus PalA and its human homologue, AIP1/Alix. Mol. Cell. Biol. 23, 1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kopp R., Schwerte T., and Pelster B. (2005) Cardiac performance in the zebrafish breakdance mutant. J. Exp. Biol. 208, 2123–2134 [DOI] [PubMed] [Google Scholar]
- 42. Roden D. M. (2006) A new role for calmodulin in ion channel biology. Circ. Res. 98, 979–981 [DOI] [PubMed] [Google Scholar]
- 43. Leong I. U., Skinner J. R., Shelling A. N., and Love D. R. (2010) Identification and expression analysis of kcnh2 genes in the zebrafish. Biochem. Biophys. Res. Commun. 396, 817–824 [DOI] [PubMed] [Google Scholar]
- 44. Langenbacher A. D., Huang J., Chen Y., and Chen J. N. (2012) Sodium pump activity in the yolk syncytial layer regulates zebrafish heart tube morphogenesis. Dev. Biol. 362, 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gopalakrishna R., and Anderson W. B. (1982) Ca2+-induced hydrophobic site on calmodulin: application for purification of calmodulin by phenyl-Sepharose affinity chromatography. Biochem. Biophys. Res. Commun. 104, 830–836 [DOI] [PubMed] [Google Scholar]
- 46. Schoenmakers T. J., Visser G. J., Flik G., and Theuvenet A. P. (1992) CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. BioTechniques 12, 870–874, 876–879 [PubMed] [Google Scholar]
- 47. Engholm-Keller K., Hansen T. A., Palmisano G., and Larsen M. R. (2011) Multidimensional strategy for sensitive phosphoproteomics incorporating protein prefractionation combined with SIMAC, HILIC, and TiO2 chromatography applied to proximal EGF signaling. J. Proteome Res. 10, 5383–5397 [DOI] [PubMed] [Google Scholar]
- 48. Chattopadhyaya R., Meador W. E., Means A. R., and Quiocho F. A. (1992) Calmodulin structure refined at 1.7 A resolution. J. Mol. Biol. 228, 1177–1192 [DOI] [PubMed] [Google Scholar]