

Abstract
Fibrosis, driven by inflammation, marks the transition from benign to progressive stages of chronic liver diseases. Although inflammation promotes fibrogenesis, it is not known whether other events, such as hepatocyte death, are required for the development of fibrosis. Interferon regulatory factor 3 (IRF3) regulates hepatocyte apoptosis and production of type I IFNs. In the liver, IRF3 is activated via Toll-like receptor 4 (TLR4) signaling or the endoplasmic reticulum (ER) adapter, stimulator of interferon genes (STING). We hypothesized that IRF3-mediated hepatocyte death is an independent determinant of chemically induced liver fibrogenesis. To test this, we performed acute or chronic CCl4 administration to WT and IRF3-, Toll/Interleukin-1R (TIR) domain-containing adapter-inducing interferon-β (TRIF)-, TRIF-related adaptor molecule (TRAM)-, and STING-deficient mice. We report that acute CCl4 administration to WT mice resulted in early ER stress, activation of IRF3, and type I IFNs, followed by hepatocyte apoptosis and liver injury, accompanied by liver fibrosis upon repeated administration of CCl4. Deficiency of IRF3 or STING prevented hepatocyte death and fibrosis both in acute or chronic CCl4. In contrast, mice deficient in type I IFN receptors or in TLR4 signaling adaptors, TRAM or TRIF, upstream of IRF3, were not protected from hepatocyte death and/or fibrosis, suggesting that the pro-apoptotic role of IRF3 is independent of TLR signaling in fibrosis. Hepatocyte death is required for liver fibrosis with causal involvement of STING and IRF3. Thus, our results identify that IRF3, by its association with STING in the presence of ER stress, couples hepatocyte apoptosis with liver fibrosis and indicate that innate immune signaling regulates outcomes of liver fibrosis via modulation of hepatocyte death in the liver.
Keywords: apoptosis, endoplasmic reticulum stress (ER stress), fibrosis, interferon regulatory factor (IRF), liver injury
Introduction
Fibrosis represents a late stage of liver disease that is common to all chronic liver diseases including viral hepatitis, alcoholic, and non-alcoholic fatty liver disease, biliary liver diseases, and some genetic liver disease. Despite their specific etiologies, common denominators of fibrosis are shared among all these liver diseases, including liver inflammation and hepatocyte death.
Fibrosis results from chronic unresolved liver inflammation and may progress from fibrotic scarring to cirrhosis that ultimately leads to liver failure. Inflammation triggers liver fibrosis via a signaling event in which liver resident macrophages (Kupffer cells, KC) activate hepatic stellate cells (HSCs)2 to deposit collagen (1, 2). Liver dysfunction results from the fibrotic tissue distorting the liver parenchyma. This process involves a dynamic and complex series of multicellular events, involving inflammation and HSC activation. The hepatocyte is primarily responsible for metabolism and detoxification, and as such, it is often exposed to damage because of toxic metabolites and reactive oxygen species. However, the role of hepatocyte death in liver fibrosis has been only partially elucidated. The currently available data demonstrate that hepatocyte apoptosis activates HSCs via paracrine mechanisms. Alternatively, HSCs can be directly activated by apoptotic bodies (3). Apoptosis in hepatocytes results from displacement or down-regulation of the Bcl-2 family of anti-apoptotic proteins and subsequent activation of pro-apoptotic BH3-only proteins such as Bax and Bak (4). These events lead to oligomerization of Bax and Bak, resulting in permeabilization of the outer mitochondrial membrane and execution of the intrinsic apoptotic process (5). Recently, IRF3, a transcription factor that induces IFN-β, has been shown to play a major role in cell death by way of its newly characterized BH3-only domain. IRF3 associated with the pro-apoptotic adaptor Bax and triggered apoptosis in murine embryonic fibroblasts and murine hepatocytes (6–8). However, it is not known whether other key signal transduction events in hepatocytes are required for the pathogenesis of fibrosis. For this reason, we sought to understand the nature of pro-fibrogenic hepatocyte damage and specifically assess the potential role of hepatocyte apoptosis in determining the onset of fibrosis. In this study, we investigated the role of IRF3 in hepatocyte death and liver fibrosis. We used CCl4, a chemical that, in the short term, induces hepatocyte apoptosis (9), which is followed by secondary necrosis (10, 11). When administered repetitively, CCl4 induces liver fibrosis (12). Using two models of CCl4-induced liver injury, we investigated the complex multicellular events associated with fibrosis in a chronic model, as well as the tightly controlled signal transduction events associated with early disruption of homeostasis and cell death in the acute model. We report that hepatocyte-specific endoplasmic reticulum (ER) stress and activation of IRF3 occurs and is mediated by the ER adaptor, STING. Activated IRF3 associates with the pro-apoptotic adaptor Bax and induces hepatocyte death. We initially hypothesized that IRF3 will utilize established pathways of antiviral response, including type I IFNs, to induce hepatocyte apoptosis. However, our results demonstrate that the pro-apoptotic role of IRF3 is independent of its function to induce type I IFNs such as IFN-β. Taken together, our data demonstrate that hepatocyte death is indispensable in liver fibrosis and that the pathogenic role of IRF3 in liver fibrosis is mediated via its pro-apoptotic effect in hepatocytes.
Results
IRF3 Deficiency Attenuates Chronic CCl4-mediated Liver Injury and Fibrosis
Repetitive episodes of liver injury, such as those induced by administration of CCl4 (a chemical inducer of hepatocyte death), result in the development of liver fibrosis, which is a hallmark of advanced and often irreversible liver disease in humans. We previously showed that IRF3-deficient mice were protected from alcohol-induced liver disease and that IRF3 deficiency in hepatocytes prevented fibrogenesis in the liver in alcoholic liver disease (8). We evaluated this hypothesis using a mouse model of liver injury and fibrosis using CCl4 or vehicle, in a 6-week regimen of biweekly intraperitoneal injections. Deficiency of IRF3 significantly attenuated liver damage, as demonstrated by significantly lower levels of serum ALT and substantially improved histological findings compared with CCl4-treated wild-type controls (Fig. 1, A and B). IRF3-deficient mice had decreased TUNEL-positive staining in the liver compared with WT mice, suggestive of decreased apoptosis (Fig. 1C). Interestingly, TUNEL-positive staining was predominantly present in hepatocytes. IRF3-deficient mice showed significant protection from liver fibrosis, indicated by reduced α-smooth muscle actin (α-SMA) immunohistochemistry and Sirius Red staining compared with WT mice (Fig. 1, D and E). The protection from liver fibrosis in IRF3-deficient mice was accompanied by significantly lower expression of Acta2 and Col1a2 (Fig. 1F), encoding α-SMA and type I collagen, and by decreased expression of Ifnb and Isg15, two commonly used markers of IRF3-mediated transcriptional response (Fig. 1G).
FIGURE 1.

IRF3 deficiency attenuates chronic CCl4-mediated liver injury and fibrosis. WT or IRF3-KO mice injected with oil or CCl4 for 6 weeks and sacrificed 48 h after final injection. A and B, liver injury was assessed by serum ALT (A) and H&E staining (B). C, liver apoptosis was assessed via TUNEL staining (black arrows) on paraffin-embedded liver sections. D and E, liver fibrosis was evaluated by Sirius Red staining (D) and α-SMA immunohistochemistry (E), and quantified using ImageJ. F and G, mRNA expression in liver for Acta2 and Col1a2 (F) and Ifnb1 and Isg15 (G) were measured by RT-PCR to assess liver fibrosis and IRF3 activation, respectively. n = 6–7 mice (CCl4-treated, per genotype); 3–4 mice (oil-treated, per genotype).
Acute CCl4 Induces ER Stress and Phosphorylation of IRF3 in Hepatocytes
Disruption of intracellular homeostasis can lead to an accumulation of misfolded proteins associated with the unfolded protein response in the ER, also known as ER stress. Excessive ER stress is associated with pro-apoptotic signaling and can be pathogenic (13, 14). To test whether CCl4-induced acute hepatocyte injury induces an early ER stress response, we analyzed livers of mice at various time points after a single administration of CCl4. We found an early up-regulation of Xbp1 mRNA splicing (sXbp1), a marker of early ER stress (15, 16), at 1 and 2 h after administration of CCl4 (Fig. 2A). Analysis of cell populations from murine livers showed that CCl4 selectively up-regulated Xbp1 mRNA splicing in hepatocytes but not in liver mononuclear cells (LMNCs) (Fig. 2B).
FIGURE 2.
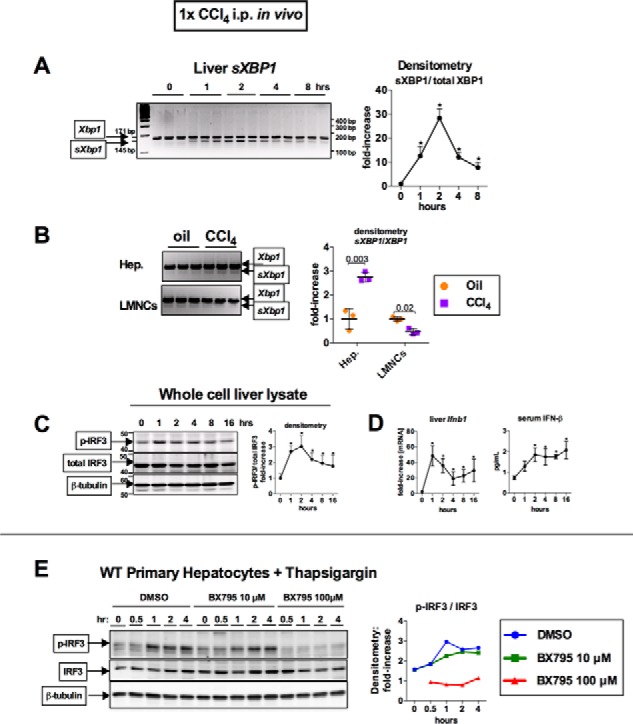
Acute CCl4 induces ER stress and TBK1-mediated phosphorylation of IRF3 in hepatocytes. WT mice received a single injection of corn oil or CCl4. A, at the indicated time points, mice were sacrificed; splicing of Xbp1 mRNA (sXBP1) in the liver, indicating ER stress, was evaluated by PCR; and products were separated on 3% agarose gel. B, 2 h after CCl4 administration, hepatocytes (Hep.), or LMNCs from WT mice were isolated and analyzed for sXBP1 by PCR. C and D, at the indicated time points, phosphorylation of IRF3 was assessed by immunoblotting in liver (C), whereas Ifnb1 mRNA expression in liver and as serum IFN-β were measured by RT-PCR and ELISA, respectively (D). n = 4 mice per time point. E, primary hepatocytes isolated from WT mice were treated with 1 μm thapsigargin and were pretreated with DMSO or BX795 (10 or 100 μm), and analyzed for phospho-IRF3.
Previous reports have shown that ER stress results in IRF3 activation in mouse embryonic fibroblast and that Xbp1 splicing enhances the IFN-β response in immune cells (17, 18). In our study, acute administration of CCl4 led to an early activation of IRF3, a pro-apoptotic BH3-only protein, as seen by the phosphorylation in whole cell liver lysates (Fig. 2C), at the same time as splicing of Xbp1 mRNA (Fig. 2A). The phosphorylation of IRF3 had a functional effect, because liver Ifnb1 mRNA and serum IFN-β, transcriptional targets of IRF3, showed an early up-regulation (Fig. 2D). To test whether direct induction of ER stress can lead to IRF3 phosphorylation, we treated primary hepatocytes with thapsigargin in vitro and found an early increase in phosphorylated IRF3 (Fig. 2E). Furthermore, inhibition of Tank-binding kinase 1 (TBK1), a kinase responsible for IRF3 phosphorylation, with BX795 showed a dose-dependent reduction in IRF3 phosphorylation in thapsigargin-treated hepatocytes (Fig. 2E). Building upon previous findings, our data indicate that acute administration of CCl4 results in hepatocyte-specific ER stress and phosphorylation of IRF3 in vivo and that in vitro inhibition of a kinase upstream of IRF3 inhibited phosphorylation of IRF3 induced by ER stress. Our data support the hypothesis that CCl4 activates IRF3 in hepatocytes via ER stress.
Acute CCl4-induced Liver Injury Is Ameliorated by the Deficiency of IRF3 Independent of TIR Domain-containing Adapter-inducing Interferon-β (TRIF) or TRIF-related Adaptor Molecule (TRAM), or Type I IFN Signaling
Next, we asked whether IRF3 plays a role in liver injury after acute CCl4 administration. We found that IRF3-KO mice were partially protected from liver injury after acute CCl4 administration, as indicated by the significant attenuation in serum ALT compared with WT controls (Fig. 3A) and caspase-3 cleavage in the liver (Fig. 3B). To test whether the pathogenic phenotype of IRF3 involvement was mediated by its function as a transcription factor, we tested mice deficient in type I IFN receptor (IFNAR1) that lack type I IFN signaling and found that IFNAR1-KO mice had no protection from CCl4-mediated liver injury, phosphorylation of IRF3, or apoptotic signaling in the liver compared with WT mice (Fig. 3, C and D). These data suggested that the pathogenic role of IRF3 may be independent of its transcriptional role.
FIGURE 3.

Acute CCl4-induced liver injury is ameliorated by the deficiency of IRF3 or inhibition of TBK1, independent of TRIM or TRAM, type I IFN signaling. A and B, WT or IRF3-KO mice received a single injection of CCl4. Serum ALT levels were assessed (A), and apoptosis was assessed by measuring caspase-3 cleavage in the liver by immunoblot (B). C and D, WT or IFNAR1-KO mice received a single injection of CCl4. Serum ALT levels were assessed (C), and IRF3 activation and apoptosis were assessed by probing for phosphor-IRF3 and caspase-3 cleavage in the liver by immunoblot (D). E and F, WT were pretreated with DMSO or BX795, a TBK1 inhibitor, 2 h before receiving a single injection of corn oil (vehicle) or CCl4. Serum ALT levels were assessed (E), and IRF3 activation and apoptosis were assessed by probing for phosphor-IRF3 and caspase-3 cleavage in the liver by immunoblot (F). G and H, WT or TRIF- or TRAM-KO mice received a single injection of CCl4. G and H, serum ALT levels were assessed (G), and IRF3 activation and apoptosis were assessed by probing for phosphor-IRF3 and caspase-3 cleavage in the liver by immunoblot (H). The mice were bled serially at the indicated time points. n = 8–9 mice (CCl4-treated, per genotype); 3–4 mice (oil-treated, per genotype) (A, C, and G). n = 3–5 mice (CCl4-treated, pretreated with BX795); 3 mice (oil-treated, pretreated with DMSO) (E).
CCl4-induced liver fibrosis activates hepatic stellate cells via TLR4 (19, 20), enhanced by signal transduction emanating from the TLR4 complex induced by gut bacterial products and LPS (1). To investigate whether activation of IRF3 in acute CCl4 liver injury was mediated by the intracellular MyD88-independent TLR4 adaptors TRIF and TRAM), or the kinase TBK1, we first inhibited activity of their downstream kinase, TBK1, by the chemical inhibitor BX795 in vivo. Pretreatment of mice with BX795 significantly attenuated the extent of liver injury and apoptotic signaling in the liver after administration of CCl4 (Fig. 3, E and F). The decrease in phosphorylated IRF3 (Fig. 3F) with BX795 pretreatment further suggests that IRF3 requires activation and phosphorylation of TBK1 to exert a pathogenic effect in CCl4-induced liver injury. In contrast, TRAM- and TRIF-KO mice were each equally as susceptible to acute CCl4 damage and apoptosis in the liver as WT mice (Fig. 3, G and H). Importantly, TRAM- and TRIF-KO mice had similar levels of phosphorylated IRF3 compared with WT mice (Fig. 3H), suggesting that IRF3 activation by acute CCl4 occurs independent of the TLR4 receptor complex and its adaptors. Together, these data suggested that TBK1-mediated activation of IRF3 is pathogenic, leading to increased liver injury. Furthermore, type I IFNs do not have a significant role in acute liver injury after CCl4 administration despite being activated by IRF3. Lastly, activation of IRF3 by acute CCl4 is independent of the canonical MyD88-independent TLR4 adaptor-mediated receptor signaling. Therefore, we hypothesized that activation of IRF3 in the liver by acute CCl4 likely involves different signaling pathways.
CCl4 Induces Early Association of IRF3 with STING in the ER and Association of IRF3 with Bax in the Mitochondria in the Liver
Given the robust protection from acute or chronic CCl4-induced liver injury in mice deficient in IRF3, we next investigated subcellular localization and mechanisms of early IRF3 activation. First, we evaluated the kinetics of IRF3 activation in WT mice upon acute CCl4-mediated injury and found an early phosphorylation of IRF3 at 2 and 4 h in whole cell liver lysates (Fig. 4A). Next, we analyzed the intracellular distribution of IRF3. Analysis of subcellular liver fractions revealed that administration of CCl4 resulted in localization of phosphorylation of IRF3 not only in the cytoplasmic (Fig. 4B) and nuclear extracts (Fig. 4C), but also in the ER (Fig. 4D) and mitochondrial fractions (Fig. 4E). The finding of phosphorylated IRF3 in cytoplasmic and nuclear extracts (Fig. 4, A and B) was consistent with the transcriptional role of IRF3 in induction of Ifnb. However, the finding of phosphorylated IRF3 in the ER (Fig. 4D) and mitochondria (Fig. 4E) was unexpected and prompted us to search for ER- and mitochondria-specific binding partners of IRF3.
FIGURE 4.

Acute CCl4 induces early association of IRF3 with STING in the ER and association of IRF3 with Bax in the mitochondria in the liver. WT mice received a single injection of CCl4. A–E, at the indicated time points, the mice were sacrificed, and phosphorylation of IRF3 was assessed by immunoblotting in liver whole cell lysate (A) or cytoplasmic (B), nuclear (C), ER (D), and mitochondrial (E) extracts. F–H, using immunoprecipitation with anti-total IRF3 antibody for protein pulldown, we evaluated association between total IRF3 and phosphorylated TBK1 or STING in the whole cell (F), mitochondrial (G), and ER (H) extracts. Similarly, we evaluated association between total IRF3 and cleaved caspase-8 p43 fragment or BAX in liver whole cell lysate (I), with representative IgG control shown (2 h after injection). J, at the indicated time points, cleavage of caspase-8 and caspase-3 was assessed by immunoblotting in liver. K, liver injury and apoptosis was assessed by H&E liver histology and TUNEL staining on paraffin-embedded liver sections. n = 4 mice per time point. IP, immunoprecipitation.
It has been reported that during viral infections, IRF3 associates with ER via binding to Stimulator of IFN Gene (STING, alias Tmem173 or MPYS), an adaptor protein residing in the ER membrane. We found that a single administration of CCl4 in vivo resulted in a significant increase in STING in whole cell liver lysates (Fig. 4F) and in ER extracts (Fig. 4H) after a pulldown with IRF3 antibody, suggesting an association between IRF3 and STING in the liver of mice exposed to CCl4.
Based on the recent report that IRF3 plays a key role in virally mediated apoptosis mediated by Bax (6) and on our previous report data that IRF3 is required for hepatocyte apoptosis triggered by FasL (8), we hypothesized that IRF3 would associate with pro-apoptotic proteins involved in the intrinsic, mitochondrial pathway of hepatocytes apoptosis. We found an increased association between IRF3 and Bax at 2 h after administration of CCl4 (Fig. 4I), followed by Casp-8 activation and apoptosis (Fig. 4, J and K). The timing of this association correlates with the increased presence of phosphorylated IRF3 in the mitochondrial fraction, suggesting that only active and phosphorylated IRF3 has a pro-apoptotic role when found in the mitochondria. Moreover, the presence of phosphorylated IRF3 in the mitochondria and its association with BAX coincided with activation and cleavage of the initiator caspase-8 and the executioner caspase-3 (Fig. 4J). Our data suggest that ER stress and phosphorylation of IRF3 (Fig. 2, A and C), which occurred within 1 h, was followed by pro-apoptotic signaling and hepatocyte damage (Fig. 4, I, J, and K), which were established within 2 h. Taken together, our data indicated that following acute administration of CCl4, IRF3 interacts with the ER-associated protein, STING, and with the pro-apoptotic molecule, Bax. Importantly, the association of IRF3 with STING or phospho-TBK1 in the ER occurred within the first 2 h after CCl4 administration (Fig. 4, F–H), concurrent with phosphorylation of IRF3 and up-regulation of Ifnb in the liver (Fig. 2, C and D); all of these events occurred prior to the development of significant liver injury (Fig. 3).
Deficiency in STING, but Not in Type I IFN Signaling, Attenuates Liver Fibrosis
Acute CCl4-induced liver injury resulted in ER stress and IRF3 activation, as well as early localization of IRF3 in the ER in association with STING, an ER-adaptor protein. Furthermore, IRF3 demonstrated a pathogenic role, independent of the TLR4-adaptor complex, in models of acute or chronic CCl4-induced liver injuries, we then hypothesized that activation of IRF3 was through STING after chronic CCl4 administration in mice. In support of this hypothesis, we observed that Tmem173Gt mice, which lack the STING protein (21), had significantly attenuated liver injury induced by repetitive injections of CCl4 (Fig. 5, A and B) to an extent similar to that observed in IRF3-deficient mice (Fig. 1A). Further, STING-deficient mice (Tmem173Gt) showed protection from liver fibrosis (Fig. 5C), as well as a decrease in deposition α-SMA and type 1 collagen protein in the liver (Fig. 5, D and E) and expression of pro-fibrogenic markers (Fig. 5F).
FIGURE 5.

Deficiency in STING, but not in type I IFN signaling, attenuates liver fibrosis. WT or STING-deficient mice (Tmem173Gt) were injected with oil or CCl4 for 6 weeks and sacrificed 48 h after final injection. A and B, liver injury was assessed by serum ALT (A) and H&E staining (B). C, liver fibrosis was evaluated by measuring Sirius Red staining, and the percent-positive area stained was quantified using ImageJ. D and E, liver fibrosis was also evaluated by immunoblot for α-SMA (D) or type 1 collagen (E). F and G, mRNA expression in liver for Acta2 and Col1a2 (F) and Ifnb1 and Isg15 (G) was measured by PCR to assess liver fibrosis and IRF3 activation, respectively. WT or IRF7- or IFNAR1-KO mice injected with oil or CCl4 for 6 weeks and sacrificed 48 h after final injection. H and I, liver injury was assessed by serum ALT (H) and H&E staining (I). J, liver fibrosis was evaluated by Sirius Red staining and quantified using ImageJ. K and L, mRNA expression in liver for Acta2 and Col1a2 (K) and Ifnb1 and Isg15 (L) were measured by RT-PCR. n = 8–9 mice (CCl4-treated, per genotype); 3–4 mice (oil-treated, per genotype).
IRF3 is the transcription factor responsible for type I IFN production, a pro-inflammatory cytokine important in creating an antiviral state. In addition to IRF3, IRF7 is another major regulator of type I IFN production (22). Administration of CCl4 resulted in significantly up-regulated Ifnb1 and the IFN-inducible gene Isg15 in the liver in WT mice, whereas IRF3-deficient mice lacked induction of type I IFN genes (Fig. 1F). However, IRF7 or type I IFNs did not mediate the pathogenic effect of IRF3 in the liver as demonstrated by a lack of protection from liver injury (Fig. 5, H and I) and fibrosis (Fig. 5, J and K) in IRF7- or IFNAR1-KO mice, respectively. The lack of type I IFN induction or signaling in INFAR1-KO mice resulted in aggravated liver fibrosis, as demonstrated by increased Sirius Red area (Fig. 5J) and augmented expression of Acta2 and Col1a2 (Fig. 5K) compared with WT mice, which is consistent with the previously reported anti-fibrotic role of type I IFNs (23, 24). Taken together, these data implied that IRF3, but not IRF7 or type I IFN signaling, plays a pathogenic role in chronic liver injury and fibrosis induced by chronic CCl4 administration. In addition, the data demonstrate the pathogenic role of STING, a binding partner of IRF3 (Fig. 4 and Ref. 25), as a determinant in the development of liver injury and fibrosis in this model.
STING Mediates Pro-apoptotic Activation of IRF3 in a Chronic CCl4-induced Model of Fibrosis
To further characterize the role of STING in mediating the CCl4-induced liver injury in the chronic CCl4 model, we evaluated phosphorylation of TBK1 and IRF3. After 6 weeks of exposure to CCl4, STING-deficient mice had decreased phosphorylation of TBK1 (Fig. 6A), as well as decreased phosphorylation of IRF3 (Fig. 6B) compared with WT mice. Previous reports have studied the pro-apoptotic association of IRF3 with Bax. To test whether STING-mediated activation of IRF3 results in pro-apoptotic signaling, we tested caspase activation in STING-deficient mice using the chronic CCl4-mediated liver injury model. We found that STING-deficient mice (Tmem173Gt) given a 6-week regimen of CCl4 exhibited a significant reduction in the cleavage and activation of the apoptosis initiator caspase-8 (Fig. 6C) and the apoptosis executioner caspase-3 (Fig. 6D) compared with WT mice.
FIGURE 6.

STING mediates pro-apoptotic activation of IRF3 in a chronic CCl4-induced model of fibrosis. WT or STING-deficient mice (Tmem173Gt) were injected with oil or CCl4 for 6 weeks and sacrificed. Whole cell liver lysates were probed for phosphorylated TBK1 (A), phosphorylated IRF3 (B), cleaved caspase-8 (C), and cleaved caspase-3 (D) by immunoblot. n = 8–9 mice (CCl4-treated, per genotype); 3–4 mice (oil-treated, per genotype).
These data were consistent with the association between IRF3, STING, and Bax that we found in the liver following a single exposure to CCl4 (Fig. 4, H and I) and indicated the possibility that ER stress may be a source of IRF3 activation in this model. In addition, these data also supported the possibility that in the chronic model of CCl4-induced liver injury and fibrosis, IRF3 represents an activator of pro-apoptotic signaling in the liver.
Early Pro-apoptotic Activation of IRF3 by CCl4 Is Hepatocyte-specific and Mediated by STING
To further explore the mechanism of cell death and liver injury and to test whether induction of cell death in the acute CCl4 liver injury model was mediated by STING, we evaluated activation of caspase-8 and caspase-3 in mouse livers. We found that caspase-8 and -3 were both activated at 8 and 12 h after CCl4 injection in WT mice (Fig. 7, A and B), whereas the STING-deficient mice (Tmem173Gt) had a complete absence of cleaved caspases (Fig. 7, C and D).
FIGURE 7.

Early pro-apoptotic activation of IRF3 by CCl4 is hepatocyte-specific and mediated by STING. WT or STING-deficient mice (Tmem173Gt) received a single injection of CCl4 or oil (baseline) and sacrificed at indicated time points. A–D, whole cell liver lysates were probed for cleaved caspase-8 (A and B) and cleaved caspase-3 (C and D) by immunoblot. E and F, liver injury was assessed by serum ALT (E) and H&E staining at the indicated time points (F). G and H, deposition of α-SMA 24 h after injection was evaluated by immunoblots from whole cell liver lysate (G), whereas mRNA expression in liver for Acta2 and Col1a2 (H) was assessed by PCR. n = 5–8 mice per time point, per genotype (A–H). I, WT mice received a single injection of CCl4 or oil (baseline) and sacrificed 9 h later. LMNCs, HSCs, or hepatocytes were isolated from livers and probed for phosphorylated IRF3, total IRF3, cleaved caspase-8, and cleaved caspase-3 by immunoblot. n = 3 per condition (LMNCs and HSCs were pooled from 3 mice); n = 1 (hepatocytes isolated from single mouse per condition); the experiment was repeated three times. J, schematic of pro-apoptotic STING and IRF3 activation from CCl4 administration in mice. In CCl4-treated hepatocytes, ER stress results in phosphorylation of TBK1 via STING, followed by phosphorylation of IRF3. IRF3 associates with BAX in the mitochondria through its BH3-only domain, leading to pro-apoptotic caspase-3 activation and hepatocyte apoptosis. After chronic CCl4 administration, hepatocyte apoptosis is associated with secondary necrosis, which results in liver fibrosis.
To better understand the underlying mechanism behind the protection observed in mice deficient in either IRF3 or STING in a chronic CCl4 injury, we tested the dynamics of hepatocyte damage by comparing WT mice with Tmem173Gt mice during an acute CCl4 liver injury. We found protection from liver injury in STING-deficient mice (Tmem173Gt) after CCl4 administration as seen by serum ALT and H&E histology (Fig. 7, E and F). WT mice exhibited extensive necrosis at 12 h, peaking at 24 h, as seen by irregular histological morphology, whereas the STING-deficient mice did not (Fig. 7F). Surprisingly, we found that one dose of CCl4 was sufficient to elicit a pro-fibrogenic response in WT mice as early as 24 h after injection (Fig. 7, G and H). In comparison with WT mice, the STING-deficient mice (Tmem173Gt) were protected from this early pro-fibrogenic response in the liver.
Our data established that CCl4-mediated ER stress occurred primarily in hepatocytes and that phospho-IRF3 after ER stress occurs in primary hepatocytes (Fig. 2, B and C). To assess which liver cell type is responsible for IRF3 phosphorylation and caspase activation after CCl4 administration in vivo, we isolated LMNCs, HSCs, and primary hepatocytes from mouse livers 9 h post CCl4 injection. We found that phosphorylation of IRF3 occurred predominantly in hepatocytes and not in LMNCs or HSCs (Fig. 7I). In assessing cell type-specific contribution to apoptotic signaling, we found increased Casp-8 cleavage in hepatocytes but not in HSCs or LMNCs. Similarly, there was an increase in caspase-3 cleavage predominantly in hepatocytes, although some cleaved caspase-3 was detected in HSCs (Fig. 7I). The phosphorylation of IRF3 and caspase-8 cleavage found exclusively in the hepatocyte population was consistent with our previous findings of TUNEL-positive staining in hepatocytes (Fig. 1C) and in IRF3 association with caspase-8 and BAX in total liver lysates (Fig. 4I).
Taken together, these data demonstrated that activation of IRF3 in acute or chronic liver damage is triggered in hepatocytes via the adaptor STING. Deficiency in either STING or IRF3 resulted in significant decreased hepatocyte death and fibrosis.
Discussion
Previous studies described the role of STING activation in viral infections in immune cells (26–30). In hepatocytes, we demonstrated earlier that STING and IRF3 mediate the pathogenesis in alcoholic liver disease, a role independent of microbial components (8). In the present study, our data demonstrate a hepatocyte-specific role of STING and IRF3 in mediating hepatic apoptosis as an early event in the development of liver fibrosis.
Hepatocytes are responsible for metabolism and for detoxifying portal blood, which can be rich in bacterial products. For this reason, innate immunity plays an important role in the liver; even though this organ is rich in immune cells, parenchymal cells have an abundance of receptors and are capable of initiating immune signaling (31–34). In our study, TBK1-mediated ER stress was detected early after CCl4 administration only in hepatocytes and not in LMNCs. Similarly, phosphorylated IRF3 was detected in hepatocytes exposed to CCl4 in vivo but not in LMNCs or HSCs. Furthermore, in support of our hypothesis of the pro-apoptotic role of IRF3, CCl4-mediated caspase-8 cleavage was detected only in hepatocytes and was absent from LMNCs and HSCs. Lastly, compared with hepatocytes exposed to CCl4, caspase-3 cleavage was reduced in HSCs and absent in LMNCs. The decreased level of caspase-3 cleavage in HSCs compared with hepatocytes is likely due to the absence of concurrent phosphorylated IRF3 and caspase-8 cleavage, suggesting an alternate mechanism of toxicity and cell death signaling independent of IRF3 in HSCs. It is possible that the mechanisms that tightly regulate apoptotic signaling may also be responsible for hepatic stellate cell activation, leading to fibrogenesis.
Regulated IRE1-dependent decay of mRNA, a key component of the unfolded protein response, results in degradation of viral mRNA transcripts and spliced Xbp1 (13, 35). However, there are multiple lines of evidence suggesting that IRF3 activation can result from other forms of noninfectious cell damage that involve Xbp1 splicing (17, 18, 36).
Here we show that STING activation may provide the link between induction of ER stress and IRF3. Moreover, the rapid nature of the phosphorylation of IFR3 and Xbp1 splicing in our experiments caused by an acute CCl4-induced liver injury underscores the importance of STING and its location in the ER.
Viral infections, as well as noninfectious injuries, are associated with induction of cell death signals, emphasizing the importance of the BH3 domain within IRF3 as responsible for its pro-apoptotic properties in the mitochondria. It is also possible that CCl4-induced cell death signaling from STING activation of IRF3 and BAX may trigger ER membrane permeability (37) in addition to the effects on the mitochondrial membrane integrity.
A previous study by Knockaert et al. (38) of CCl4 in mouse livers interestingly found no apoptosis or pro-apoptotic caspase activity at 3 or 24 h, the only two time points they tested. It is important to highlight that our data showed caspase-8 and -3 cleavage peaking between 8 and 12 h in WT mice, whereas apoptotic signaling had resolved by 24 h (Fig. 7, A and B). Importantly, the STING-deficient mice had no detectable caspase-8 or -3 cleavage (Fig. 7, C and D), indicating protection from apoptosis and secondary cell death.
Moreover, the Knockaert et al. (38) study similarly found CCl4-mediated necrosis at 24 h (Fig. 4K and 7F), further supporting our hypothesis that early and extensive pro-apoptotic signaling leads to necrosis, or secondary necrosis, which have overlapping components (39). In support of the study by Roychowdhury et al. (40), our data indicate that an absence of early apoptosis is also associated with an absence in early fibrogenesis and detectable necrosis at the level of liver histology in STING-deficient mice.
Pro-apoptotic signaling may play a beneficial role in the resolution of inflammation after liver injury, specifically when it is targeted to infiltrating immune cells such as neutrophils (41). Uncontrolled parenchymal apoptosis, however, although typically controlled and tightly regulated, may be the tipping point responsible for subsequent necrosis.
As summarized in Fig. 7J, our data suggest that overwhelming levels of IRF3-mediated apoptosis may eventually lead to a form of secondary necrosis. Taken together, our data provide strong evidence that inhibition of parenchymal apoptotic signaling is sufficient to reduce necrosis and amplified liver damage.
In conclusion, our study builds on the previous paradigm of fibrogenesis. The CCl4-induced liver damage, ER stress, and death of hepatocytes are mediated by the engagement of the ER adaptor protein, STING, with phosphorylated IRF3, resulting in activation of the mitochondrial pathway of hepatocyte death. As such, fibrosis is more than a pathology strictly driven by inflammation. Rather, hepatocyte death appears to be a separate key component in the process of liver fibrosis.
Experimental Procedures
Animal Studies
6–8-week-old female C57Bl/6 WT (The Jackson Laboratory, Bar Harbor, ME), IRF3-deficient (IRF3-KO) or type I IFN α/β receptor 1-(IFNAR1)-KO mice (provided by J. Sprent, Scripps Research Institute, La Jolla, CA) and IRF7-KO mice (provided by T. Taniguchi, Tokyo, Japan), all on C57Bl/6 background, were used. The STING-deficient (Tmem173Gt) mice (provided by R. Vance, University of California, Berkeley, CA) and the TRAM- and TRIF-KO mice (provided by S. Akira, Osaka University, Japan) were on the B6.129sf2 background; we used B6.129sf2 WT mice (The Jackson Laboratory) as controls for these strains. Some mice received a single i.p. injection of CCl4, 0.6 μl/gram body weight, diluted 1:3 in corn oil (Sigma). Other mice received a 6-week regiment of CCl4 injections, given twice per week. All animals received proper care in agreement with animal protocols approved by the Institutional Animal Use and Care Committee of the University of Massachusetts Medical School.
In Vitro Experiments
Primary hepatocytes and liver mononuclear cells were isolated as described previously (42). Primary hepatocytes were cultured in Waymouth's medium supplemented with 10% fetal bovine serum and 1% insulin, transferrin, selenium solution. Primary hepatocytes were seeded in 6-well collagen-coated plates (Biocoat; Becton Dickinson, Bedford, MA). Before starting stimulation experiments, hepatocytes were rested for 4 h. Subsequently culture medium was replaced, and stimulation was performed as indicated in the figure legends. The BX795 (10 μm or 100 μm) and thapsigargin (1 μm) were purchased from Sigma.
RNA Analysis
RNA analysis and PCR primers are described in Table 1. sXbp1 PCR was performed, and products were separated on 3% agarose gel as described previously (8).
TABLE 1.
Quantitative PCR primers
| Target gene | Forward primer (5′ → 3′) | Reverse primer (5′ → 3′) |
|---|---|---|
| 18S | gtaacccgttgaaccccatt | ccatccaatcggtagtagcg |
| Tnfa | caccaccatcaaggactcaa | aggcaacctgaccactctcc |
| Ifnb | agctccaagaaaggacgaacat | gccctgtaggtgagggttgatct |
| Isg15 | caggacggtcttaccctttcc | aggctcgctgcagttctgtac |
| Xbp1 | acacgcttgggaatggacac | ccatgggaagatgttctggg |
| Acta2 | acacgcttgggaatggacac | ttcctgaccactagaggggg |
| Col1a2 | ggagggaacggtccacgat | gagtccgcgtatccacaa |
Protein Quantification
Liver whole cell lysates and nuclear preparations were extracted as described previously (8). Mitochondrial and total ER fractions were prepared using specific extraction kits from Imgenex (San Diego, CA) as per the manufacturer's protocol. Immunoblotting was performed as described in Ref. 43. Antibodies specific for phospho-IRF3, phospho-TBK1, STING, Casp-8, Casp-3, cleaved Casp-8, cleaved Casp-3, and Bax were from Cell Signaling (Danvers, MA). Antibodies against the total IRF3, GRP78, and KDEL and control IgG were from Santa Cruz (Santa Cruz Biotechnology, Santa Cruz, CA). β-Actin, β-tubulin, porin, and TATA-binding protein antibodies were from Abcam (Cambridge, MA). The TrueBlot system from eBioscience (San Diego, CA) was used for immunoprecipitation assays and performed as per manufacturer's instructions.
Statistical Analysis
Statistical significance was determined using two-sided t test. Two-way analysis of variance was used in Figs. 3 (A–D) and 7E to determine the global effect of BX795 on serum ALT. The numbers in the graphs indicate global p values. The data are shown as scatter plots of individual data plots ± S.D. to represent error and were considered statistically significant at p < 0.05. *, p < 0.05 versus baseline; **, p < 0.001 versus baseline; #, p < 0.05 versus WT CCl4 condition; ##, p < 0.001 versus WT CCl4 condition. We used SPSS 19.0 (IBM SPSS, Chicago, IL) and GraphPad Prism 6.0 (San Diego, CA) for calculations.
Author Contributions
A. I.-V., J. P., and G. S. designed the research; A. I.-V., J. P., B. G., A. S., P. L., K. K., and D. C. performed the research; E. A. K.-J. and K. A. F. contributed new reagents/analytic tools; A. I.-V., J. P., and G. S. analyzed the data; A. I.-V., J. P., and G. S. wrote the manuscript; and all authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank members of the Szabo Lab and the Programs in Translational Science and Biochemistry and Molecular Pharmacology at the University of Massachusetts Medical School for support.
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grants AA017729 (to G. S.) and PA-12-149 (to A. I.-V.), and National Institute on Alcohol Abuse and Alcoholism Award F31AA025545 (to A. I.-V.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HSC
- hepatic stellate cell
- IRF3
- interferon regulatory factor 3
- STING
- stimulator of interferon genes
- ER
- endoplasmic reticulum
- α-SMA
- α-smooth muscle actin
- IFNAR1
- type I IFN receptor
- TIR
- Toll/interleukin-1R
- TRIF
- TIR domain-containing adapter-inducing interferon-β
- TRAM
- TRIF-related adaptor molecule
- TBK1
- Tank-binding kinase 1
- TLR
- Toll-like receptor
- LMNC
- liver mononuclear cell
- H&E
- hematoxylin and eosin.
References
- 1. Seki E., De Minicis S., Osterreicher C. H., Kluwe J., Osawa Y., Brenner D. A., and Schwabe R. F. (2007) TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 13, 1324–1332 [DOI] [PubMed] [Google Scholar]
- 2. Rivera C. A., Bradford B. U., Hunt K. J., Adachi Y., Schrum L. W., Koop D. R., Burchardt E. R., Rippe R. A., and Thurman R. G. (2001) Attenuation of CCl4-induced hepatic fibrosis by GdCl3 treatment or dietary glycine. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G200–G207 [DOI] [PubMed] [Google Scholar]
- 3. Malhi H., and Gores G. J. (2008) Cellular and molecular mechanisms of liver injury. Gastroenterology 134, 1641–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Danial N. N., and Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 5. Jiang X., and Wang X. (2004) Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 [DOI] [PubMed] [Google Scholar]
- 6. Chattopadhyay S., Marques J. T., Yamashita M., Peters K. L., Smith K., Desai A., Williams B. R., and Sen G. C. (2010) Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 29, 1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chattopadhyay S., Fensterl V., Zhang Y., Veleeparambil M., Yamashita M., and Sen G. C. (2013) Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J. Virol. 87, 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petrasek J., Iracheta-Vellve A., Csak T., Satishchandran A., Kodys K., Kurt-Jones E. A., Fitzgerald K. A., and Szabo G. (2013) STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. U.S.A. 110, 16544–16549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi J., Aisaki K., Ikawa Y., and Wake K. (1998) Evidence of hepatocyte apoptosis in rat liver after the administration of carbon tetrachloride. Am. J. Pathol. 153, 515–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zakim D., and Boyer T. D. (2002) Hepatology: A Texbook of Liver Diseases, 4th Ed., p. 746, Saunders, Philadelphia, PA [Google Scholar]
- 11. Sun F., Hamagawa E., Tsutsui C., Ono Y., Ogiri Y., and Kojo S. (2001) Evaluation of oxidative stress during apoptosis and necrosis caused by carbon tetrachloride in rat liver. Biochim. Biophys. Acta 1535, 186–191 [DOI] [PubMed] [Google Scholar]
- 12. Nakatsukasa H., Nagy P., Evarts R. P., Hsia C. C., Marsden E., and Thorgeirsson S. S. (1990) Cellular distribution of transforming growth factor-β1 and procollagen types I, III, and IV transcripts in carbon tetrachloride-induced rat liver fibrosis. J. Clin. Invest. 85, 1833–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maurel M., Chevet E., Tavernier J., and Gerlo S. (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254 [DOI] [PubMed] [Google Scholar]
- 14. Upton J.-P., Wang L., Han D., Wang E. S., Huskey N. E., Lim L., Truitt M., McManus M. T., Ruggero D., Goga A., Papa F. R., and Oakes S. A. (2012) IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science 338, 818–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 16. Schröder M., and Kaufman R. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 [DOI] [PubMed] [Google Scholar]
- 17. Liu Y.-P., Zeng L., Tian A., Bomkamp A., Rivera D., Gutman D., Barber G. N., Olson J. K., and Smith J. A. (2012) Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J. Immunol. 189, 4630–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu F., Yu X., Wang H., Zuo D., Guo C., Yi H., Tirosh B., Subjeck J. R., Qiu X., and Wang X.-Y. (2011) ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 41, 1086–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paik Y.-H., Schwabe R. F., Bataller R., Russo M. P., Jobin C., and Brenner D. A. (2003) Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 37, 1043–1055 [DOI] [PubMed] [Google Scholar]
- 20. Soares J.-B., Pimentel-Nunes P., Roncon-Albuquerque R., and Leite-Moreira A. (2010) The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 4, 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sauer J.-D., Sotelo-Troha K., von Moltke J., Monroe K. M., Rae C. S., Brubaker S. W., Hyodo M., Hayakawa Y., Woodward J. J., Portnoy D. A., and Vance R. E. (2011) The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79, 688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Servant M. J., Tenoever B., and Lin R. (2002) Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J. Interferon Cytokine Res. 22, 49–58 [DOI] [PubMed] [Google Scholar]
- 23. Roh Y. S., Park S., Kim J. W., Lim C. W., Seki E., and Kim B. (2014) Toll-like receptor 7-mediated type I interferon signaling prevents cholestasis- and hepatotoxin-induced liver fibrosis. Hepatology 60, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petrasek J., Dolganiuc A., Csak T., Nath B., Hritz I., Kodys K., Catalano D., Kurt-Jones E., Mandrekar P., and Szabo G. (2011) Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 53, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y.-T. T., Grishin N. V., and Chen Z. J. (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- 26. Tanaka Y., and Chen Z. J. (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abe T., Harashima A., Xia T., Konno H., Konno K., Morales A., Ahn J., Gutman D., and Barber G. N. (2013) STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 50, 5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishikawa H., Ma Z., and Barber G. N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burdette D. L., Monroe K. M., Sotelo-Troha K., Iwig J. S., Eckert B., Hyodo M., Hayakawa Y., and Vance R. E. (2011) STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia T., Konno H., Ahn J., and Barber G. (2016) Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Reports 14, 282–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao B., Jeong W.-I., and Tian Z. (2008) Liver: An organ with predominant innate immunity. Hepatology 47, 729–736 [DOI] [PubMed] [Google Scholar]
- 32. Akira S., Uematsu S., and Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 33. Deminice R., de Castro G. S., Brosnan M. E., and Brosnan J. T. (2016) Creatine supplementation as a possible new therapeutic approach for fatty liver disease: early findings. Amino Acids 48, 1983–1991 [DOI] [PubMed] [Google Scholar]
- 34. Zhou Z., Xu M.-J., and Gao B. (2016) Hepatocytes: a key cell type for innate immunity. Cell Mol. Immunol. 13, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plumb R., Zhang Z.-R., Appathurai S., and Mariappan M. (2015) A functional link between the co-translational protein translocation pathway and the UPR. eLife 4, 07426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smith J. A., Turner M. J., DeLay M. L., Klenk E. I., Sowders D. P., and Colbert R. A. (2008) Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-β induction via X-box binding protein 1. Eur. J. Immunol. 38, 1194–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X., Olberding K. E., White C., and Li C. (2011) Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 18, 38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knockaert L., Berson A., Ribault C., Prost P.-E., Fautrel A., Pajaud J., Lepage S., Lucas-Clerc C., Bégué J.-M., Fromenty B., and Robin M.-A. (2012) Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab. Invest. 92, 396–410 [DOI] [PubMed] [Google Scholar]
- 39. Vandenabeele P., Galluzzi L., Vanden Berghe T., and Kroemer G. (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 [DOI] [PubMed] [Google Scholar]
- 40. Roychowdhury S., Chiang D. J., Mandal P., McMullen M. R., Liu X., Cohen J. I., Pollard J., Feldstein A. E., and Nagy L. E. (2012) Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl4-induced fibrosis but not steatosis or inflammation. Alcohol. Clin. Exp. Res. 36, 1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fox S., Leitch A. E., Duffin R., Haslett C., and Rossi A. G. (2010) Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petrasek J., Iracheta-Vellve A., Saha B., Satishchandran A., Kodys K., Fitzgerald K. A., Kurt-Jones E. A., and Szabo G. (2015) Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Biol. 98, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iracheta-Vellve A., Petrasek J., Satishchandran A., Gyongyosi B., Saha B., Kodys K., Fitzgerald K. A., Kurt-Jones E. A., and Szabo G. (2015) Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 63, 1147–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]