

Abstract
Viperin is an endoplasmic reticulum-associated antiviral responsive protein that is highly up-regulated in eukaryotic cells upon viral infection through both interferon-dependent and independent pathways. Viperin is predicted to be a radical S-adenosyl-l-methionine (SAM) enzyme, but it is unknown whether viperin actually exploits radical SAM chemistry to exert its antiviral activity. We have investigated the interaction of viperin with its most firmly established cellular target, farnesyl pyrophosphate synthase (FPPS). Numerous enveloped viruses utilize cholesterol-rich lipid rafts to bud from the host cell membrane, and it is thought that by inhibiting FPPS activity (and therefore cholesterol synthesis), viperin retards viral budding from infected cells. We demonstrate that, consistent with this hypothesis, overexpression of viperin in human embryonic kidney cells reduces the intracellular rate of accumulation of FPPS but does not inhibit or inactivate FPPS. The endoplasmic reticulum-localizing, N-terminal amphipathic helix of viperin is specifically required for viperin to reduce cellular FPPS levels. However, although viperin reductively cleaves SAM to form 5′-deoxyadenosine in a slow, uncoupled reaction characteristic of radical SAM enzymes, this cleavage reaction is independent of FPPS. Furthermore, mutation of key cysteinyl residues ligating the catalytic [Fe4S4] cluster in the radical SAM domain, surprisingly, does not abolish the inhibitory activity of viperin against FPPS; indeed, some mutations potentiate viperin activity. These observations imply that viperin does not act as a radical SAM enzyme in regulating FPPS.
Keywords: cholesterol, endoplasmic reticulum (ER), innate immunity, iron-sulfur protein, membrane enzyme, protein degradation, virus
Introduction
Radical S-adenosyl-l-methionine-dependent (SAM)3 enzymes constitute a superfamily of enzymes that use SAM to generate free radicals (1–4). The superfamily is typified by a common CXXXCXXC motif, the cysteinyl residues of which coordinate a [Fe4S4] cluster that is essential for radical generation. These enzymes catalyze a remarkably wide range of reactions involving an equally diverse set of substrates (2). For example, radical SAM-dependent enzymes participate in the biosynthesis of herbicides, antibiotics, vitamins, co-factors such as biotin and thiamin, and various other natural products (2, 5–8). They also function in the modification of ribosomal and transfer RNAs (9, 10) and DNA repair (11) and in the post-translational modification of peptides and proteins (12–14). Sequence analyses indicate that there are potentially thousands of members of the radical SAM superfamily, but to date, relatively few of these enzymes have been isolated and characterized (15, 16). Radical SAM enzymes were until recently thought to be confined to the microbial realm but intriguingly have now been identified in higher aerobic organisms, including plants and animals (3).
Central to the mechanism of all radical SAM enzymes is the generation of a highly reactive 5′-deoxyadenosyl radical (Ado•) (1, 4, 17). This is accomplished through one-electron reduction of SAM by the [Fe4S4] cluster (Fig. 1), which leads to homolytic cleavage of the C-5′–S bond to form Ado• and methionine. Ado• subsequently abstracts a hydrogen atom from the substrate, thereby forming 5′-deoxyadenosine (5′-dA) and a substrate radical that undergoes further chemical reaction. Most radical SAM enzymes consume SAM as a co-substrate during catalysis; however, in a few enzymes, including lysine 2,3-aminomutase (18), spore photoproduct lyase (11), and DesII (7), Ado• formation is reversible, and SAM is regenerated as a co-factor.
FIGURE 1.

General reaction mechanism of radical SAM enzymes. One-electron reduction of SAM by the [Fe4S4] cluster results in generation of methionine and 5′-deoxyadenosyl radical. This reactive radical then abstracts a hydrogen atom from the substrate (R-H) to initiate a diverse range of reactions.
Viperin (Virus inhibitory protein, endoplasmic reticulum-associated, interferon inducible) is an interferon-stimulated gene that forms a component of the innate immune response that is activated upon viral infection (19–21). It is highly conserved in vertebrates and is one of the few interferon-stimulated genes shown to have direct antiviral activity against a number of viruses, including human cytomegalovirus, hepatitis C, influenza A, and HIV. The mechanisms by which viperin mitigates viral infection appear to be dependent on the identity of the virus and are quite variable in nature. In fact, viperin has been shown to interact with a number of cellular and viral proteins, although the specific inhibitory mechanism(s) has yet to be elucidated at the molecular level (20).
Intriguingly, viperin appears to be a radical SAM-dependent enzyme (22). It is one of eight putative radical SAM enzymes identified in humans; four of these appear to be involved in tRNA modifications, two seem to be involved in co-factor biosynthesis, and the remaining two, including viperin, have unknown enzymatic activities (23). The human enzyme is a 361-residue protein that comprises three distinct domains: an N-terminal amphipathic helix thought to play a role in localizing viperin to the endoplasmic reticulum and lipid droplets (19); a large, central domain that contains four motifs characteristic of radical SAM enzymes; and a conserved C-terminal domain that may be involved in [Fe4S4] cluster assembly, the proper folding of viperin, and/or substrate recognition (20, 24). Although no enzymatic function has yet been assigned to viperin, the radical SAM domain has been shown to be important in limiting HIV and Bunyamwera viruses (20). Furthermore, homology modeling suggests that viperin adopts a partial (β/α)6 TIM barrel fold, similar to that of the well-characterized radical SAM enzyme, pyruvate formate-lyase activating enzyme (25). Given the predicted structural similarity of viperin to pyruvate formate-lyase activating enzyme, the molecular targets of viperin may also be proteins or other macromolecules.
The most firmly established target of viperin is farnesyl pyrophosphate synthase (FPPS), which catalyzes the formation of farnesyl pyrophosphate from geranyl pyrophosphate and isopentenyl pyrophosphate (26). Farnesyl pyrophosphate is a key intermediate in the biosynthesis of isoprenoids and cholesterol. Given that many enveloped viruses exploit cholesterol-rich lipid rafts in the cell membrane during the process of viral budding, it was proposed that by inhibiting FPPS (and thereby inhibiting cholesterol production) that viperin prevents viral release from the infected cell (27). Alternatively, through inhibition of FPPS activity and thus the production of isoprenoids, viperin may affect the prenylation state of various cellular proteins that are necessary for viral release. In either case, the mechanism by which viperin reduces cellular FPPS activity is still unknown.
In this study we aimed to investigate how viperin regulates FPPS activity in the cell. We show that viperin reduces FPPS activity by lowering levels of the enzyme within the cell rather than directly inhibiting its catalytic activity. Full-length viperin is able to cleave SAM in an uncoupled fashion to produce 5′-deoxyadenosine. However, surprisingly, mutation of the conserved CXXXCXXC motif reveals that regulation of FPPS levels does not depend on the radical SAM activity of viperin. Further characterization of viperin activity through deletion and substitution experiments shows that the decrease in the cellular level of FPPS is dependent on localization of viperin to the endoplasmic reticulum by its native N-terminal amphipathic α-helix.
Results
Our initial experiments took a reductive approach and focused on studying the interaction of viperin with FPPS using recombinant proteins overexpressed and purified from Escherichia coli. FPPS could be overexpressed and purified from E. coli without difficulty and possessed a specific activity similar to that reported in the literature (28). Viperin lacking the first 50 residues that comprise the N-terminal amphipathic helical domain (designated t-viperin here) was overexpressed as an N-terminal His6-tagged construct and purified using standard approaches (Fig. 2A), similar to those described previously (22), that minimized exposure to air. The [Fe4S4] cluster could be reconstituted by incubating t-viperin with Na2S, Na2S2O4, and Fe(NH3)6SO4 under anaerobic conditions, followed by desalting to remove excess reagents. The UV-visible spectrum of the reconstituted enzyme was similar to that reported previously (22) and characteristic of an iron-sulfur protein (Fig. 2B). In the presence of dithionite and SAM, the reconstituted enzyme catalyzed the slow, uncoupled reductive cleavage of SAM to form 5′-dA (Fig. 2C) as previously reported by Duschene and Broderick (22). Attempts to express the full-length enzyme in E. coli proved unsuccessful; for reasons that are unclear, no protein expression could be detected from cells transformed with the appropriate pET28 expression construct and induced with isopropyl β-d-thiogalactopyranoside.
FIGURE 2.

Characterization of recombinant truncated viperin expressed and purified from E. coli. A, SDS-PAGE of t-viperin (lane 2) after purification using nickel-NTA affinity chromatography; the molecular masses (in kDa) of standard proteins (lane 1) are indicated on the left. B, UV-visible spectrum of purified t-viperin after reconstitution of the [Fe4S4] cluster and desalting. C, HPLC chromatograms demonstrating the reductive cleavage of SAM to form 5′-dA by t-viperin (solid trace) compared with a control reaction with no enzyme. For details see the text.
[Fe4S4]-reconstituted t-viperin and FPPS were incubated together in at equimolar concentrations, ∼10 μm each (10 mm Tris-Cl buffer, pH 8.0, containing 300 mm NaCl and 10% glycerol), in the presence of 5 mm dithionite, 5 mm DTT, and 200 μm SAM under anaerobic conditions at room temperature for 60 min. After the incubation period, the activity of FPPS was determined under anaerobic conditions using [14C]isopentenyl pyrophosphate (IPP) and geranyl-pyrophosphate (GPP) as substrates and following the incorporation of radioactivity into farnesyl-pyrophosphate (29). However, despite multiple trials in which the incubation times and temperature were varied around these initial conditions, no significant change in the activity of FPPS was detected in these experiments. Nor was there any evidence that t-viperin covalently modified FPPS, as judged by ESI-MS analysis of the recovered enzyme. Similarly, the presence of FPPS did not stimulate the reductive cleavage of SAM by t-viperin, as might be expected if FPPS was a substrate for viperin.
Faced with these negative results we concluded that either viperin requires the N-terminal domain for activity against FPPS and/or that additional cellular components are necessary for viperin to interact with FPPS. We therefore pursued our investigation of the effect of viperin on FPPS in a human cell line, HEK293T. We reasoned that this environment should provide: (a) the cellular machinery to correctly install the viperin [Fe4S4] cluster, (b) any additional proteins that may be necessary for its activity, and (c) the membrane structures to which viperin is localized and which may also be important for activity.
Viperin Reduces Cellular Levels of FPPS
Genes encoding full-length viperin and FPPS were introduced into pcDNA3.1 vectors to facilitate constitutive expression from the CMV promoter and equipped with N-terminal FLAG and His tags, respectively, to allow expression levels to be determined by immunostaining. Equal amounts of DNA were used to co-transfect HEK293T cells, and the expression of both proteins followed for 48 h. Expression levels were compared with cells that were singly transfected. Co-expression of viperin and FPPS resulted in a significant reduction (>2-fold) of the intracellular level of FPPS relative to the FPPS only control, FPPS levels were significantly reduced after 48 h compared with when FPPS was expressed on its own (Fig. 3A). An examination of the time course of FPPS expression revealed that under single and co-expression conditions, FPPS levels increased linearly from 24 to 42 h post-transfection (Fig. 3B). However, the rate of accumulation of FPPS in the presence of viperin was ∼3-fold slower. In contrast, expression of viperin was independent of FPPS and reached a maximum at ∼36 h (Fig. 3C).
FIGURE 3.

Reduction of cellular FPPS levels by viperin in co-transfected HEK293T cells. A, Western blotting analysis of viperin and FPPS expression levels as a function of time after transfection. GAPDH was used as the loading control. B, quantification of intracellular levels of FPPS relative to GAPDH as a function of time in the absence and presence of co-expressed viperin. C, quantification of intracellular levels of viperin relative to GAPDH as a function of time in the absence and presence of co-expressed FPPS.
To examine whether the reduction in cellular FPPS levels by viperin was due to reduced transcription of FPPS, we undertook real time quantitative RT-PCR (qRT-PCR) analysis of mRNA levels in HEK293T cells transfected with the genes for FPPS and/or viperin. These experiments showed no significant change in the levels of either FPPS mRNA nor viperin mRNA in co-transfected cells compared with singly transfected cells. We therefore conclude that the regulation of FPPS levels by viperin is very unlikely to occur at the level of gene transcription.
Full-length Viperin Cleaves SAM
We examined the ability of full-length human viperin, expressed in HEK293T cells, to catalyze the reductive cleavage of SAM to produce 5′-dA. Extracts from cells transfected with either viperin and/or FPPS were prepared in Tris-buffered saline containing 1% Triton X-100 under anaerobic conditions in a glove box (Coy Chamber). The extracts were incubated anaerobically at room temperature with 5 mm DTT and 5 mm dithionite for 30 min prior to the addition of 200 μm SAM. After 1 h the nucleotide pool was extracted and analyzed by LC-MS using established protocols (30). No 5′-dA was detected in cell extracts lacking viperin, whereas 5′-dA was readily detected extracts prepared from viperin-expressing HEK293T cells (Fig. 4). However, the amounts of 5′-dA were low: 0.50 ± 0.05 nm, which is less than one turnover based on the concentration of viperin estimated by immunostaining. Interestingly, co-expression of FPPS with viperin had no effect on SAM cleavage (5′-dA = 0.49 ± 0.05 nm). These results mirrored those obtained in our in vitro studies described above and suggested that either viperin may utilize SAM as a true co-factor (rather than a co-substrate) or that radical SAM chemistry is not involved in regulation of FPPS by viperin.
FIGURE 4.

Viperin reductively cleaves S-adenosyl-l-methionine in an uncoupled reaction. HEK293T, amounts of 5′-dA produced when assayed with untransfected HEK293T cells; FPPS, HEK293T cells transfected with FPPS only; Viperin, HEK293T cells transfected with viperin; FPPS+Viperin, HEK293T cells transfected with FPPS and viperin. The data represent the means and standard deviation of three independent biological replicates. For details see the text.
Mutation of Active Site Residues in Viperin
In a complementary approach to depleting the substrate for viperin, we sought to generate inactive viperin variants by mutating each of the conserved cysteine residues within the CXXXCXXC motif to alanine. The C-terminal tryptophan residue (Trp361), which is thought to be an important recognition element for the cellular iron-sulfur cluster assembly proteins, was also mutated to alanine. Each of these mutations abolished the radical SAM activity of viperin and resulted in slightly lower expression levels of viperin in the cells, suggesting that the mutations destabilize the enzyme slightly. (The C87A, C90A, and C83/87/90A mutants were expressed at ∼1.3-, ∼2.6-, and ∼4.5-fold lower levels than WT viperin, respectively; data not shown). Most surprisingly, however, none of the mutations abolished the activity of viperin against FPPS (Fig. 5). Indeed, the C87A, C90A, and triple mutant variants proved to be significantly more effective at reducing the intracellular level of FPPS (∼6-fold) compared with wild-type viperin (∼2-fold). These results demonstrate that the iron-sulfur cluster is not required for viperin to reduce FPPS levels and imply that radical SAM chemistry is not involved in the regulation of FPPS by viperin.
FIGURE 5.
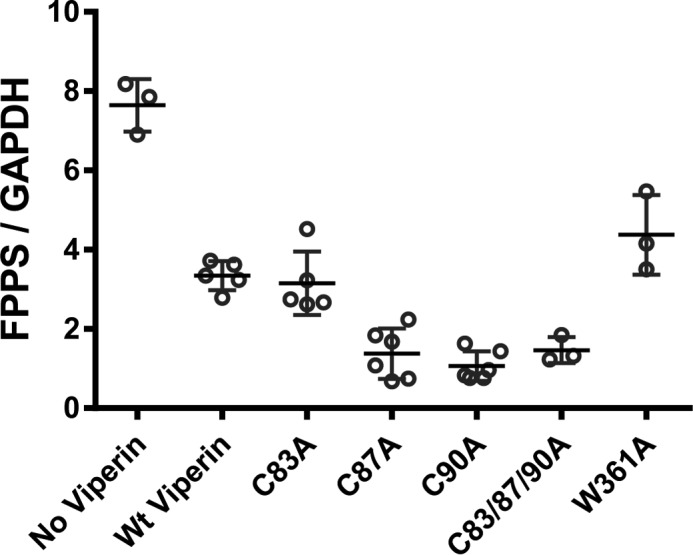
Deletion of cysteinyl ligands to the catalytic [Fe4S4] cluster in viperin does not abolish viperin activity against FPPS. Western blot analysis of cellular levels of FPPS was performed, and the results were quantified and normalized to GAPDH. In all cases, FPPS levels were significantly reduced when co-expressed with wild-type or mutant viperin enzymes; the data are plotted showing the means and standard deviation of at least three independent biological replicates. Reduction of FPPS levels relative to the no viperin control was significant with p values ranging from p = 0.001 to p = 0.0002.
The Native N-terminal Amphipathic α-Helix of Viperin Is Necessary for Reducing Cellular FPPS Levels
To examine whether association of viperin with the ER or lipid droplets is required for activity against FPPS, we employed a truncated version of viperin lacking the first 50 amino acids comprising of the membrane-targeting N-terminal amphipathic α-helix. (This construct retains the N-terminal FLAG tag and is designated TN50 viperin to distinguish it from the bacterially produced truncated enzyme discussed above.) We also constructed a chimeric viperin in which the N-terminal amphipathic helix was replaced with the 30-residue N-terminal membrane-localizing sequence of hepatitis C non-structural protein 5A (NS5A). In previous studies, Weber and co-workers (24) had demonstrated by immunofluorescence imaging that the NS5A sequence relocalizes TN50-viperin to the ER (31), a result that we independently confirmed for the constructs used in this study (data not shown).
We examined the ability of the TN50-viperin and the chimeric NS5A-TN50-viperin to reduce the levels of co-transfected FPPS. Both viperin constructs expressed at similar levels to wild-type enzyme. However, as shown in Fig. 6, neither construct was able to significantly reduce the cellular levels of FPPS. These results suggest that the N-terminal amphipathic helix of viperin is important in facilitating the ability of the enzyme to decrease cellular FPPS levels and that localization to the ER is not of itself sufficient for this activity.
FIGURE 6.

Effect of replacing the ER-localization of viperin on its activity against FPPS. The levels of FPPS were quantified in HEK293T cells transfected with FPPS; co-transfected with wild-type viperin; co-transfected with viperin lacking the N-terminal ER-localization sequence (TN50); and co-transfected with viperin in which the ER-targeting sequence the viral protein NS5A replaces endogenous ER-targeting sequence (NS5A-TN50). Neither the truncated enzyme nor the chimeric enzyme is able to reduce the cellular levels of co-expressed FPPS. The data are plotted showing the means and standard deviation of three independent biological replicates. *, p < 0.05.
Purified FPPS Is Unmodified by Viperin
Lastly, we examined whether viperin catalyzed any covalent modification of FPPS that might alter its activity or lead to its degradation in the cell. His-tagged FPPS was co-transfected with viperin into HEK293T, and the cells were cultured for 48 h. The cells were lysed and FPPS-purified from the lysate using nickel-nitrilotriacetic acid affinity resin. Control experiments were set up where FPPS was purified from HEK293T cells that lacked viperin. Native PAGE analysis (Fig. 7A) revealed no significant difference in the electrophoretic mobility of FPPS isolated from the two preparations that would have been indicative of a post-translational modification. The specific activities of the two enzyme preparations were also compared and found not to differ significantly (Fig. 7B). kcat for FPPS expressed on its own was 20 ± 2.3 min−1, whereas kcat for FPPS co-expressed with viperin was 15 ± 4.0 min−1; these values are similar to those reported previously (28).
FIGURE 7.

FPPS recovered from viperin-expressing cells is un-modified. A, native-PAGE analysis of FPPS purified from HEK293T cells. Lane 1 standard proteins (native molecular masses in kDa indicated on the left); lane 2, FPPS expressed in the absence of viperin; lane 3, FPPS co-expressed with viperin. B, activity of FPPS purified from HEK293T cells in absence and in presence of co-expressed viperin; the small difference in rates is not considered significant. C and D, LC-MS of FPPS purified from HEK293T cells in the absence (C) and presence of co-expressed viperin (D). No significant differences in the spectra are apparent.
LC-ESI-MS analysis of FPPS from each preparation showed no difference in the mass indicative of a covalent modification (Fig. 7, C and D). The observed weight of 42,838 Da differs from the predicted mass of 42,926 Da by 88 Da, which corresponds to the loss of the N-terminal methionine followed by N-terminal acetylation (32). Lastly, no proteolytic fragments of FPPS were evident when FPPS was co-expressed with viperin as judged by immunoblots probed with a polyclonal antibody to FPPS. These observations all indicate that the FPPS remaining in the cell after 48 h is unmodified by viperin.
Discussion
To date, most studies have focused on investigating the antiviral activity of viperin in cell culture-based experiments in which the antiviral activity of the enzyme was assayed either by measuring decreases in viral titer or viral RNA levels. The consensus from these studies is that viperin likely exerts its activity through different pathways that depend on the particular virus and that it interacts with multiple cellular and viral proteins in the process (19, 27, 31, 33, 34). Previous studies, using a truncated version of viperin, recombinantly produced and purified from E. coli, have demonstrated that the enzyme cleaves SAM to form 5′-dA (22), which is the hallmark reaction of radical SAM enzymes. Here we have demonstrated for the first time the ability of the full-length enzyme expressed in human cells to reductively cleave SAM to form 5′-dA. However, less than one turnover was observed, and this presumably represented uncoupled activity. Thus, the relevance of radical SAM chemistry to the physiological function of viperin has remained unclear, in large part because of the lack of detailed information regarding the interaction of viperin with its cellular targets.
Our studies have focused on the interaction of viperin with its best documented target, FPPS (27). Our initial, unsuccessful attempts to demonstrate any effect of viperin on FPPS using purified proteins in vitro necessitated a change of approach to studying their interaction in cellulo. The overexpression of both proteins in HEK293T cells, which may not be considered truly physiological conditions, nevertheless allows the interaction of the proteins to be examined under conditions in which any necessary accessory proteins and co-factors are likely to be present. Furthermore, it was possible to recover FPPS from the cells and biochemically characterize the effect of viperin on the enzyme.
Earlier observations indicated that viperin reduced the activity of endogenously expressed FPPS (27), although whether this occurred by inhibition or altering FPPS levels was unclear. Our experiments demonstrate that viperin exerts its effects by reducing the cellular levels of co-expressed FPPS. However, our observation that mutating the conserved cysteinyl ligands to the catalytic iron-sulfur cluster actually potentiates rather than abolishes the ability of viperin to decrease FPPS levels appears to rule out a radical mechanism in this case. These observations are in accord with previous studies in which mutating the conserved cysteinyl residues failed to abolish the ability of viperin to inhibit the replication of hepatitis C (31) and Dengue (34) viruses.
Given these results, the question arises as to whether viperin really is a radical SAM enzyme. Whereas our results discount a radical mechanism for its interaction with FPPS, we consider that it is still quite possible that radical SAM chemistry plays a role in viperin regulation of other cellular or viral proteins. One candidate activity where radical SAM chemistry may be operating is the restriction of tick-borne encephalitis virus (TBEV) by viperin. Unlike viruses such as influenza A that bud from cholesterol-rich lipid rafts and are thus sensitive to FPPS inhibition, flaviviruses such as TBEV do not require lipid rafts for budding. Indeed it has been shown that inhibition of FPPS by ibandronate has no effect TBEV replication (24).
In contrast to our results with FPPS, it was shown that, whereas localization of viperin to the ER was essential to reduce the titer of TBEV, this was not dependent on identity of the ER-localizing sequence (24). Significantly, mutation of the conserved cysteine residues or the C-terminal tryptophan of viperin, which is important for installation of the [Fe4S4] cluster by CIAO1, abolished viperin inhibition of TBEV replication, indicating that an intact [Fe4S4] cluster was essential for activity. These observations all point to a different mode of action pertaining to viperin activity against TBEV (and other flaviviruses such as Dengue virus and west Nile virus). These observations are consistent with the involvement of radical SAM chemistry, although they do not demonstrate that such chemistry occurs in vivo.
Another pertinent, and better understood, example of a human enzyme in which both radical SAM and non-radical activities have been posited is the ELP3 (elongator protein 3) component of the RNA polymerase II complex (23). The elongator complex comprises six proteins, ELP1–6, and is responsible both for histone acetylation to facilitate transcription and for the modification of the wobble position U-34 of eukaryotic tRNAs by carbamoylmethylation or methoxy-carbonylmethylation at C-5 of uridine (35, 36). ELP3 contains both radical SAM and histone acetyltransferase domains. ELP3 has been shown in vitro to catalyze histone acetylation, but this reaction does not require the iron-sulfur cluster (37). Removal of any of the six ELP components abolishes U-34 modification in eukaryotes, making it hard to assess the role of radical SAM chemistry. However, experiments on an archaeal homolog of ELP3, which functions independently, have provided evidence for a radical SAM-dependent mechanism for uridine carboxymethylation in this organism (38).
Our results provide evidence that the reduction in cellular FPPS levels is due to an increase in protein degradation, rather than transcriptional regulation or covalent modification of FPPS by viperin. Thus, qRT-PCR analysis found no change in FPPS mRNA levels in cells co-transfected with viperin. This makes it unlikely that viperin either regulates transcription of FPPS or catalyzes the degradation of FPPS mRNA. We cannot rule out a mechanism in which viperin regulates FPPS by modifying the mRNA, thereby slowing down translation; indeed many radical SAM enzymes do catalyze nucleic acid modifications. However, we consider this unlikely because viperin is localized to the ER, whereas FPPS is a cytoplasmic enzyme. We think it also unlikely that viperin directly inactivates FPPS because FPPS purified from HEK293T cells co-expressing viperin exhibits the same specific activity as FPPS expressed in the absence of viperin and, furthermore, contains no covalent modifications as assessed by ESI-MS. (Although, of course, one cannot rule out the possibility that inactivated FPPS is very rapidly cleared from the cell.) These observations leave up-regulation of FPPS degradation pathways as the most plausible route by which viperin decreases FPPS levels.
Having ruled out a radical mechanism for FPPS regulation, the biochemical details by which viperin reduces cellular levels of FPPS remain subject to speculation. The N terminus of viperin appears to be essential for viperin activity against FPPS, because replacing this region by a different ER-localizing sequence failed to restore activity. This suggests that the N terminus plays an important role in recognizing FPPS. We hypothesize that viperin may recruit additional, as yet unidentified proteins necessary for FPPS degradation, for example by targeting it for degradation by the proteasome. Interestingly, viperin has been shown to facilitate Lys63-linked ubiquitination of the kinase, IRAK1, by the E3 ubiquitin ligase, TRAF6, which are proteins that play a central role in innate immune signaling (39). Viperin appears to do this by co-localizing these proteins to lipid bodies, and thus it is possible that a similar mechanism may be operating in the case of FPPS. Further investigations are in progress in our laboratory to better define the mechanism(s) of action of this enigmatic enzyme.
Experimental Procedures
Materials
S-Adenosyl-l-methionine, 5′-deoxyadenosine, cycloleucine, and mouse monoclonal FLAG antibody (M2) were obtained from Sigma. Mouse monoclonal GAPDH antibody (6C5) was from EMD Millipore. Mouse monoclonal His6 tag antibody (HIS.H8) was purchased from Thermo Scientific. IPP and GPP were purchased from Isoprenoids. [4-14C]IPP was purchased from PerkinElmer Life Sciences. HEK293T cell line was obtained from ATCC. The pcDNA3.1(+) expression vector was purchased from Invitrogen. FuGENE HD transfection reagent was purchased from Promega. All other reagents and materials were purchased from commercial suppliers and were of the highest grade available.
Cloning of Human Viperin and FPPS
The genes encoding human viperin (GenBankTM accession number AAL50053.1) and FPPS (GenBankTM accession number AAA52423.1) were purchased from GenScript. Both genes were subsequently amplified via PCR and subcloned into the pcDNA3.1(+) vector (Invitrogen). The primers for the pcDNA3.1(+)-viperin construct introduced an N-terminal 3X-FLAG tag (DYKDHDGDYKDHDIDYKDDDDK) and the Kozak consensus sequence (5′-GCCAAC-3′) for downstream protein expression in eukaryotic cells (40). Similarly, the primers for the pcDNA3.1(+)-FPPS construct introduced an N-terminal hexahistidine tag and the necessary Kozak sequence. Two additional N-terminal 3X-FLAG-tagged variants of human viperin were also constructed using standard Gibson assembly cloning methods. The first variant, TN50, featured a deletion of the first 50 amino acids of the N terminus. The second variant, NS5A-TN50, included a replacement of the native N terminus by the first 30 amino acid residues from the hepatitis C virus non-structural 5A (NS5A) protein. The gene encoding the NS5A protein was obtained from AddGene as the pCMV-Tag1-NS5A plasmid. The point mutations described in this paper were introduced using the QuikChange site-directed mutagenesis method (Agilent Technologies, Mississauga, Canada). DNA sequencing confirmed the presence of each gene and the absence of any PCR-induced errors for each construct.
Cloning, Expression, and Purification of N-terminal Truncated Viperin (t-Viperin) in E. coli
A gene, codon-optimized for expression in E. coli, encoding viperin lacking the first 50 amino acids of the N-terminal amphipathic α-helix was purchased from GenScript. The gene construct was housed in a pET28 expression vector and included a N-terminal His6 tag to facilitate purification. Expression of truncated viperin was achieved by standard methods. Briefly, the cells were grown in 2× YT medium containing 50 μg/ml kanamycin at 37 °C with shaking at 150 rpm to an A600 of ∼1.0. The culture was chilled at 4 °C for 30 min, and protein expression was induced by adding isopropyl β-d-thiogalactopyranoside to a final concentration of 0.1 mm. The culture was further incubated at 18 °C with shaking at 150 rpm overnight (∼18 h). The cells were harvested by centrifugation at 5,000 rpm for 30 min at 4 °C. The cell pellets were stored at −80 °C.
The purification of truncated-viperin was performed in a Coy anaerobic chamber (O2 < 25 ppm) or under a nitrogen atmosphere using anoxic buffers unless otherwise noted. The cell pellet from 6 liters of culture was resuspended in 100 ml of lysis buffer (10 mm Tris-Cl, pH 8.0, containing 10% glycerol (v/v), 300 mm NaCl, 5 mm imidazole, 1 mm β-mercaptoethanol, 0.5 mm dithionite, 1 tablet/10 ml complete mini protease inhibitor tablet (Roche), 1 mg/ml lysozyme, and 1 mg/ml benzonase), and the suspension was shaken for 30 min. Cell lysis was completed by minimal sonication outside the Coy chamber. The lysate was taken back into the Coy chamber and allowed to briefly equilibrate with the anoxic environment prior to removal of the cell debris by centrifugation (17,000 rpm, 30 min, 4 °C). The supernatant was loaded onto a 5-ml HIS-Trap column (GE Healthcare) equilibrated in buffer A (10 mm Tris-Cl, pH 8.0, 300 mm NaCl, 10 mm imidazole, 10% glycerol), and the purification was carried out by FPLC outside the Coy chamber with a flow rate of 1 ml/min (the column was kept at 10–15 °C by surrounding it with cold packs). The column was washed with buffer A until the absorbance at 280 nm stabilized, and the protein was eluted with a linear gradient of 0–100% buffer B (10 mm Tris-Cl, pH 8.0, 300 mm NaCl, 500 mm imidazole, 10% glycerol) over 60 min at 1 ml/min. Elution fractions (15 total) were collected in 2.5-ml aliquots into vials followed by the addition of 500 μl of a 30 mm dithionite solution to scavenge oxygen. Fractions containing truncated viperin were pooled, centrifuged to remove precipitated protein, divided into 2-ml aliquots, and stored at −80 °C until ready for reconstitution.
Reconstitution of Truncated Viperin Purified from E. coli
Reconstitution was performed with slight modification of the protocol described by Duschene and Broderick (22). All steps were performed in a Coy chamber (O2 < 25 ppm) using anoxic buffers unless otherwise noted. The day prior to reconstitution, an 8.3-ml PD-10 desalting column was equilibrated with ∼50 column volumes of reconstitution buffer (10 mm Tris-Cl, pH 8.0, 300 mm NaCl, 10% glycerol) to make the column as oxygen free as possible, and the column was left to equilibrate overnight. Approximately 2 mg of purified truncated-viperin stock solution was slowly thawed on a cold pack, and the UV-visible spectrum was recorded using an anaerobic cuvette as a prereconstitution control. The protein was incubated with DTT (final concentration, 5 mm) for 20 min on the cold pack with gentle stirring. FeCl3 (final concentration 150 μm; 6 equivalents) was added dropwise to the stirred solution and incubated for 20 min. Na2S (final concentration 150 μm; 6 equivalents) was added in one aliquot and stirred for 2 h on the cold pack. The cold pack was removed, and the solution was stirred for an additional hour. EDTA (final concentration, 2 mm) was added in one aliquot and stirred for 30 min to chelate any free iron remaining in solution. The solution was then diluted to 2.5 ml with reconstitution buffer and desalted on the PD column. Fractions containing reconstituted t-viperin were dark reddish brown; these were pooled, divided into 500-μl aliquots, and stored at −80 °C. The concentration of protein was estimated using and extinction coefficient ϵ280 = 42,700 m−1 cm−1. The yield of reconstituted protein was typically ∼26 μm (1 mg/ml).
Expression of Human Viperin and FPPS in HEK293T Cells
Overexpression of the full-length recombinant human viperin and FPPS proteins was accomplished through transient transfection of HEK293T cells (cultivated in DMEM supplemented with 10% FBS and 1% antibiotics) with the pcDNA3.1(+)-viperin construct or the pcDNA3.1(+)-FPPS construct, respectively. In addition, co-transfection of both DNA constructs allowed for co-expression of viperin and FPPS within the HEK293T cells. The same transfection protocols were used for expression of each of the viperin variants. Briefly, 2.5 μg of the respective plasmid DNA construct was mixed with FuGENE HD reagent in a 3:1 ratio, incubated at room temperature for 15 min, and then added to HEK293T cells at 40% confluence on a 35-mm dish. The transfected cells were then grown for up to 48 h, gently pelleted, and stored at −80 °C until use. Expression of viperin and FPPS proteins was also examined following growth of HEK293T cells in the presence of cycloleucine, an inhibitor of S-adenosyl-l-methionine synthesis. Transfected HEK293T cells were grown for 48 h in the presence or absence of 20 or 50 mm cycloleucine. In all cases, protein expression was confirmed through Western blotting with the appropriate anti-FLAG (Sigma-Aldrich) and/or anti-His (Thermo Scientific) monoclonal antibodies using GAPDH as a loading control. Chemiluminescent images were acquired on a Bio-Rad ChemiDoc Touch Imager. Where noted, rabbit anti-FPPS polyclonal antibodies (Proteintech) were also used to check for FPPS expression.
Immunofluorescence and Immunoblotting
HEK293T cells were grown to 30% confluence on poly-l-lysine-coated coverslips and then transiently transfected with wild-type viperin, TN50, or NS5A-TN50 plasmid DNA. After 48 h the cells were fixed with 3% paraformaldehyde, permeabilized with 0.5% Triton X-100 diluted in PBS buffer, and stained with mouse monoclonal anti-viperin (Abcam) and rabbit polyclonal anti-calnexin (Abcam) antibodies. Protein localization was visualized with Alexa Fluor 647-conjugated goat anti-mouse (Life Technologies) and Alexa Fluor 488-conjugated goat anti-rabbit (Abcam) antisera. Images were acquired on an Olympus IX-81 confocal microscope. Immunoblotting and imaging of proteins was performed by standard methods using chemiluminescence to visualize proteins. Blots were visualized and band intensities quantified using a Bio-Rad ChemiDoc Touch imaging system. Quantitative measurements of protein expression levels reported here represent the average of at least three independent biological replicates.
qRT-PCR Analyses
HEK293T cells transfected with FPPS, viperin, or both genes were harvested 48 h post-transfection and stored at −80 °C. Total RNA was isolated from each sample using the Aurum Total RNA mini kit (Bio-Rad) following the spin protocol for mammalian cells, with slight modification. Specifically, the DNase digestion time was increased to 2 h, followed by an additional treatment of the eluted RNA samples with a Turbo DNA free kit (Invitrogen) for 2 h at 37 °C. Subsequently, 1 μg of RNA from each sample with an A260/280 nm ratio between 1.8 and 2.2 was reverse transcribed to cDNA using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's protocols. cDNA samples were stored at −20 °C prior to qRT-PCR analysis.
Primers specific for the FPPS and viperin genes, as well as GAPDH and β-actin reference genes, were designed using Geneious R7 software (BioMatters). The forward and reverse sequences of each primer set were as follows: FPPS: 5′-GAAATCGGTCAGACGCTGGA-3′ and 5′-CGCAATCGGCAGGTAGAAAG-3′; viperin: 5′-TGATCCGTGAACGCTGGTTT-3′ and 5′-GGTTTTTCTTGCCCTGACCG-3′; GAPDH: 5′-ACCCACTCCTCCACCTTTGAC-3′ and 5′-TGTTGCTGTAGCCAAATTCGTT-3′; and β-actin: 5′-GCCGGGACCTGACTGACTAC-3′ and 5′-TTCTCCTTAATGTCACGCACGAT-3′.
qRT-PCR was performed using SoFast EvaGreen Supermix (Bio-Rad) on a Bio-Rad CFX96 system to assess the relative expression levels of the FPPS and viperin genes under both single protein expression and co-expression conditions. A minimum of four biological replicates was performed for each expression condition, each with two technical replicates; both no-template controls and no reverse-transcriptase controls were included. The data were analyzed using CFX96TM manager software (Bio-Rad).
Cleavage of SAM by Viperin in Vivo
HEK293T cells overexpressing viperin were harvested from a 10-cm2 dish, resuspended in 200 μl of anoxic Tris-buffered saline (50 mm Tris-Cl, pH 7.6, 150 mm NaCl) containing 1% Triton X-100, sonicated within an anaerobic glovebox (Coy Chamber), and centrifuged at 14,000 × g for 10 min. DTT (final concentration, 5 mm) and dithionite (final concentration, 5 mm) were added to the cell lysate. The assay mixture was incubated at room temperature for 30 min prior to starting the reaction by the addition of SAM (final concentration, 200 μm). The assay was incubated for 60 min at room temperature, after which the reaction stopped by heating at 95 °C for 10 min. The solution was chilled to 4 °C, and the precipitated proteins were removed by centrifugation. The supernatant was then extracted with acetonitrile. Aliquots of 20 μl were subsequently derivatized with benzoyl chloride as described previously (30). The samples were analyzed in triplicate for the presence of 5′-deoxyadenosine by HPLC-tandem mass spectrometry, using an Acquity HSS T3 C18 chromatography column (1 mm × 100 mm, 1.8 μm, 100 Å pore size) on a Waters nanoAcquity ultra performance liquid chromatograph interfaced to an Agilent 6410 triple quadrupole mass spectrometer. Mobile phase A was 100 mm ammonium acetate with 0.15% formic acid. Mobile phase B was acetonitrile. The flow rate was 100 μl/min, and the gradient was as follows: initial, 0% B; 0.01 min, 17% B; 0.5 min, 40% B; 2.99 min, 60% B; 3.00 min, 100% B; 3.99 min, 100% B; 4.00 min, 0% B; 5.00 min, 0% B. Internal standards included benzoylated adenosine, r.t. 3.33 min; 3′-deoxyadenosine, r.t. 3.85 min; and 5′-deoxyadenosine, r.t. 4.05 min. Peaks were integrated using Agilent MassHunter workstation quantitative analysis for triple quadrupole, version B.05.00.
Purification of FPPS from HEK 293T Cells
All purification steps were carried out at 4 °C. Cells overexpressing FPPS or FPPS and viperin (0.5 g wet weight) were resuspended in 2 ml of Tris-buffered saline, 0.1% Tween 20, and 20 mm imidazole (pH 7.5) containing a mix of protease inhibitors (Sigma-Aldrich). The mixture was briefly sonicated followed by centrifugation at 14,000 rpm for 10 min. The supernatant was then mixed with 100 μl of nickel-nitrilotriacetic acid resin, pre-equilibrated with Tris-buffered saline and 20 mm imidazole (pH 7.5). Following a 1-h incubation period, the mix was washed twice with 1 ml of Tris-buffered saline and 20 mm imidazole (pH 7.5) and then twice more with 1 ml of Tris-buffered saline, 20 mm imidazole, and 10% glycerol. FPPS was eluted with 200 μl of Tris-buffered saline, 250 mm imidazole, and 10% glycerol. The protein was concentrated and buffer-exchanged into 10 mm Tris-Cl (pH 8.0) containing 10% glycerol and frozen at −80 °C until use. The purity of the isolated FPPS protein was assessed by SDS-PAGE (10% gel). The Lowry assay method was used to determine the concentration of the protein.
Radiolabeled Assays for FPPS Activity
The catalytic activity of FPPS was monitored through the use of a previously described radiolabeled assay (29) with slight modification. Each 800-μl reaction mixture contained 50 mm Tris-Cl (pH 8.0), 10% glycerol, 2 mm MgCl2, 2 mm DTT, 18 μm IPP, 10.8 Ci/mol [4-14C-IPP], 20 μm GPP, and 4.7 nm FPPS. The reactions were initiated by the addition of FPPS and incubated at 37 °C. Every 4 min, a 100-μl aliquot was removed and quenched with 200 μl of a 4:1 MeOH/HCl solution, vortexed briefly, and incubated at 37 °C for 10 min. Each quenched sample was subsequently extracted with 400 μl of Ligroin and vortexed for 30 s. A 200-μl aliquot of the Ligroin layer was diluted in 4 ml of scintillation mixture followed by scintillation counting on a Beckman LS 6500 scintillation counter. The experiment was repeated in triplicate, and the data were analyzed using Origin, version 8.5 (MicroCal Software, Inc.).
LC-MS Analysis of FPPS
Purified protein samples that were buffer-exchanged into 10 mm Tris-Cl (pH 8.0) containing 10% glycerol were analyzed using an Agilent 6250 Accurate-Mass Q-TOF LC/MS system. The samples (3 μl) were injected into a Zorbax Eclipse Plus C18 column equilibrated with 4.75% acetonitrile and 0.1% formic acid. Proteins were eluted over a 6-min gradient from 4.75% to 95% acetonitrile at a constant flow rate of 0.4 ml/min. Protein masses were obtained using ESI in positive ion mode and analyzed using Agilent MassHunter Quantitative Analysis software.
Author Contributions
C. M., S. G., and G. D. R.-M. conducted most of the experiments and analyzed the results. P. A. M. and R. T. K. conducted the analysis of nucleotides. E. N. G. M. conceived the idea for the project and wrote the paper with C. M. and S. G.
Acknowledgment
We thank Damon Hoff for training and technical advice in confocal microscopy.
This work was supported in part by National Institutes of Health Grants GM 093088 (to E. N. G. M.) and DK 046960 (to R. T. K.), a postdoctoral fellowship from the Natural Sciences and Engineering Research Council of Canada (to C. M.), and National Science Foundation Award DBI-0959823 (to the SMART Center of the University of Michigan). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- SAM
- S-adenosyl-l-methionine
- 5′-dA
- 5′-deoxyadenosine
- Ado•
- 5′-deoxyadenosyl radical
- FPPS
- farnesyl pyrophosphate synthase
- IPP
- isopentenyl pyrophosphate
- GPP
- geranyl pyrophosphate
- NS5A
- hepatitis C virus non-structural protein 5A
- TBEV
- tick-borne encephalitis virus
- qRT-PCR
- quantitative RT-PCR
- ESI
- electrospray ionization
- r.t.
- retention time.
References
- 1. Frey P. A., Hegeman A. D., and Ruzicka F. J. (2008) The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43, 63–88 [DOI] [PubMed] [Google Scholar]
- 2. Roach P. L. (2011) Radicals from s-adenosylmethionine and their application to biosynthesis. Curr. Opin. Chem. Biol. 15, 267–275 [DOI] [PubMed] [Google Scholar]
- 3. Wang J., Woldring R. P., Román-Meléndez G. D., McClain A. M., Alzua B. R., and Marsh E. N. G. (2014) Recent advances in radical SAM enzymology: new structures and mechanisms. ACS Chem. Biol. 9, 1929–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Broderick J. B., Duffus B. R., Duschene K. S., and Shepard E. M. (2014) Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chatterjee A., Li Y., Zhang Y., Grove T. L., Lee M., Krebs C., Booker S. J., Begley T. P., and Ealick S. E. (2008) Reconstitution of thic in thiamine pyrimidine biosynthesis expands the radical sam superfamily. Nat. Chem. Biol. 4, 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fugate C. J., and Jarrett J. T. (2012) Biotin synthase: Insights into radical-mediated carbon-sulfur bond formation. Biochim. Biophys. Acta 1824, 1213–1222 [DOI] [PubMed] [Google Scholar]
- 7. Ruszczycky M. W., Ogasawara Y., and Liu H. W. (2012) Radical SAM enzymes in the biosynthesis of sugar-containing natural products. Biochim. Biophys. Acta 1824, 1231–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Q., van der Donk W. A., and Liu W. (2012) Radical-mediated enzymatic methylation: a tale of two SAMs. Acc. Chem. Res. 45, 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujimori D. G. (2013) Radical SAM-mediated methylation reactions. Curr. Opin. Chem. Biol. 17, 597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Young A. P., and Bandarian V. (2013) Radical mediated ring formation in the biosynthesis of the hypermodified tRNA base wybutosine. Curr. Opin. Chem. Biol. 17, 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang L., Lin G., Nelson R. S., Jian Y., Telser J., and Li L. (2012) Mechanistic studies of the spore photoproduct lyase via a single cysteine mutation. Biochemistry 51, 7173–7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vey J. L., and Drennan C. L. (2011) Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 111, 2487–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davidson V. L., and Liu A. (2012) Tryptophan tryptophylquinone biosynthesis: a radical approach to posttranslational modification. Biochim. Biophys. Acta 1824, 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Flühe L., and Marahiel M. A. (2013) Radical S-adenosylmethionine enzyme catalyzed thioether bond formation in sactipeptide biosynthesis. Curr. Opin. Chem. Biol. 17, 605–612 [DOI] [PubMed] [Google Scholar]
- 15. Coquille S., Roux C., Mehta A., Begley T. P., Fitzpatrick T. B., and Thore S. (2013) High-resolution crystal structure of the eukaryotic hmp-p synthase (ThiC) from Arabidopsis thaliana. J. Struct. Biol. 184, 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hänzelmann P., Hernández H. L., Menzel C., García-Serres R., Huynh B. H., Johnson M. K., Mendel R. R., and Schindelin H. (2004) Characterization of mocs1a, an oxygen-sensitive iron-sulfur protein involved in human molybdenum cofactor biosynthesis. J. Biol. Chem. 279, 34721–34732 [DOI] [PubMed] [Google Scholar]
- 17. Marsh E. N. G., Patterson D. P., and Li L. (2010) Adenosyl radical: reagent and catalyst in enzyme reactions. ChemBioChem 11, 604–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ballinger M. D., Reed G. H., and Frey P. A. (1992) An organic radical in the lysine 2, 3-aminomutase reaction. Biochemistry 31, 949–953 [DOI] [PubMed] [Google Scholar]
- 19. Chin K. C., and Cresswell P. (2001) Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. U.S.A. 98, 15125–15130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Helbig K. J., and Beard M. R. (2014) The role of viperin in the innate antiviral response. J. Mol. Biol. 426, 1210–1219 [DOI] [PubMed] [Google Scholar]
- 21. Seo J.-Y., Yaneva R., and Cresswell P. (2011) Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 10, 534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duschene K. S., and Broderick J. B. (2010) The antiviral protein viperin is a radical SAM enzyme. FEBS Lett. 584, 1263–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landgraf B. J., McCarthy E. L., and Booker S. J. (2016) Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem. 85, 485–514 [DOI] [PubMed] [Google Scholar]
- 24. Upadhyay A. S., Vonderstein K., Pichlmair A., Stehling O., Bennett K. L., Dobler G., Guo J. T., Superti-Furga G., Lill R., Överby A. K., and Weber F. (2014) Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cell. Microbiol. 16, 834–848 [DOI] [PubMed] [Google Scholar]
- 25. Haldar S., Paul S., Joshi N., Dasgupta A., and Chattopadhyay K. (2012) The presence of the iron-sulfur motif is important for the conformational stability of the antiviral protein, viperin. PLoS One 7, e31797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seo J.-Y., and Cresswell P. (2013) Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog. 9, e1003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X., Hinson E. R., and Cresswell P. (2007) The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell. Host. Microbe 2, 96–105 [DOI] [PubMed] [Google Scholar]
- 28. Tsoumpra M. K., Muniz J. R., Barnett B. L., Kwaasi A. A., Pilka E. S., Kavanagh K. L., Evdokimov A., Walter R. L., Von Delft F., Ebetino F. H., Oppermann U., Russell R. G., and Dunford J. E. (2015) The inhibition of human farnesyl pyrophosphate synthase by nitrogen-containing bisphosphonates. Elucidating the role of active site threonine 201 and tyrosine 204 residues using enzyme mutants. Bone 81, 478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kavanagh K. L., Guo K., Dunford J. E., Wu X., Knapp S., Ebetino F. H., Rogers M. J., Russell R. G., and Oppermann U. (2006) The molecular mechanism of nitrogen-containing bisphosphonates as anti osteoporosis drugs. Proc. Natl. Acad. Sci. U.S.A. 103, 7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong J. M., Malec P. A., Mabrouk O. S., Ro J., Dus M., and Kennedy R. T. (2016) Benzoyl chloride derivatization with liquid chromatography-mass spectrometry for targeted metabolomics of neurochemicals in biological samples. J. Chromatogr. A. 1446, 78–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Helbig K. J., Eyre N. S., Yip E., Narayana S., Li K., Fiches G., McCartney E. M., Jangra R. K., Lemon S. M., and Beard M. R. (2011) The antiviral protein viperin inhibits hepatitis c virus replication via interaction with nonstructural protein 5a. Hepatology 54, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonissone S., Gupta N., Romine M., Bradshaw R. A., and Pevzner P. A. (2013) N-terminal protein processing: a comparative proteogenomic analysis. Mol. Cell. Proteomics 12, 14–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nasr N., Maddocks S., Turville S. G., Harman A. N., Woolger N., Helbig K. J., Wilkinson J., Bye C. R., Wright T. K., Rambukwelle D., Donaghy H., Beard M. R., and Cunningham A. L. (2012) HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 120, 778–788 [DOI] [PubMed] [Google Scholar]
- 34. Helbig K. J., Carr J. M., Calvert J. K., Wati S., Clarke J. N., Eyre N. S., Narayana S. K., Fiches G. N., McCartney E. M., and Beard M. R. (2013) Viperin is induced following dengue virus type-2 (denv-2) infection and has anti-viral actions requiring the c-terminal end of viperin. PLoS Negl. Trop. Dis. 7, e2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hawkes N. A., Otero G., Winkler G. S., Marshall N., Dahmus M. E., Krappmann D., Scheidereit C., Thomas C. L., Schiavo G., Erdjument-Bromage H., Tempst P., and Svejstrup J. Q. (2002) Purification and characterization of the human elongator complex. J. Biol. Chem. 277, 3047–3052 [DOI] [PubMed] [Google Scholar]
- 36. Mikiewicz K., Jose L. E., Bento-Abreu A., Fislage M., Taes I., Kasprowicz J., Swerts J., Sigrist S., Versées W., Robberecht W., and Verstreken P. (2011) ELP3 controls active zone morphology by acetylating the elks family member bruchpilot. Neuron 72, 776–788 [DOI] [PubMed] [Google Scholar]
- 37. Greenwood C., Selth L. A., Dirac-Svejstrup A. B., and Svejstrup J. Q. (2009) An iron-sulfur cluster domain in ELP3 important for the structural integrity of elongator. J. Biol. Chem. 284, 141–149 [DOI] [PubMed] [Google Scholar]
- 38. Selvadurai K., Wang P., Seimetz J., and Huang R. H. (2014) Archaeal elp3 catalyzes tRNA wobble uridine modification at c5 via a radical mechanism. Nat. Chem. Biol. 10, 810–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saitoh T., Satoh T., Yamamoto N., Uematsu S., Takeuchi O., Kawai T., and Akira S. (2011) Antiviral protein viperin promotes toll-like receptor 7- and toll-like receptor 9-mediated type i interferon production in plasmacytoid dendritic cells. Immunity 34, 352–363 [DOI] [PubMed] [Google Scholar]
- 40. Kozak M. (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]