

Abstract
Accessory cholera enterotoxin (Ace) of Vibrio cholerae has been shown to contribute to diarrhea. However, the signaling mechanism and specific type of Cl− channel activated by Ace are still unknown. We have shown here that the recombinant Ace protein induced ICl of apical plasma membrane, which was inhibited by classical CaCC blockers. Surprisingly, an Ace-elicited rise of current was neither affected by ANO1 (TMEM16A)-specific inhibitor T16A(inh)-AO1(TAO1) nor by the cystic fibrosis transmembrane conductance regulator (CFTR) blocker, CFTR inh-172. Ace stimulated whole-cell current in Caco-2 cells. However, the apical ICl was attenuated by knockdown of ANO6 (TMEM16F). This impaired phenotype was restored by re-expression of ANO6 in Caco-2 cells. Whole-cell patch clamp recordings of ANO currents in HEK293 cells transiently expressing mouse ANO1-mCherry or ANO6-GFP confirmed that Ace induced Cl− secretion. Application of Ace produced ANO6 but not the ANO1 currents. Ace was not able to induce a [Ca2+]i rise in Caco-2 cells, but cellular abundance of phosphatidylinositol 4,5-bisphosphate (PIP2) increased. Identification of the PIP2-binding motif at the N-terminal sequence among human and mouse ANO6 variants along with binding of PIP2 directly to ANO6 in HEK293 cells indicate likely PIP2 regulation of ANO6. The biophysical and pharmacological properties of Ace stimulated Cl− current along with intestinal fluid accumulation, and binding of PIP2 to the proximal KR motif of channel proteins, whose mutagenesis correlates with altered binding of PIP2, is comparable with ANO6 stimulation. We conclude that ANO6 is predominantly expressed in intestinal epithelia, where it contributes secretory diarrhea by Ace stimulation in a calcium-independent mechanism of RhoA-ROCK-PIP2 signaling.
Keywords: epithelial cell, epithelial cell adhesion molecule (EpCAM), intestinal epithelium, ion channel, membrane, pathogenesis, phosphatidylinositol, signaling
Introduction
Secretory diarrhea caused by enteric infection is a major factor in morbidity and mortality worldwide. In developing countries, the major causes of secretory diarrhea include enterotoxin-producing bacteria such as Vibrio cholerae and enterotoxigenic Escherichia coli (1). The dehydrating diarrhea during cholera is attributed primarily due to the intestinal secretion stimulated by cholera toxin (2). However, two other toxins of V. cholerae that alter short-circuit current (Isc) and/or resistance in Ussing chambers have been identified. These are zonula occludens toxin (3, 4), which acts by disrupting tight junctions, and accessory cholera enterotoxin (Ace)3 (5). Ace is a small amphipathic protein of 96 amino acids without any disulfide bond. Ace bears similarity to the eukaryotic ion-transporting ATPase family especially the transmembrane domain, with the exception that it lacks a nucleotide-binding site (5). Previous in vivo studies showed that after infection by V. cholerae, Ace may contribute to intestinal secretion and diarrhea before the stimulation of the slow-acting cholera toxin (6). It had also been reported earlier that partially purified Ace from ace gene-positive (ace+) V. cholerae strains stimulate Ca2+-dependent Cl−/HCO3− symporters, thereby creating a potential difference across the membrane, which involves both an influx of extracellular Ca2+ across the apical membrane of the cells and intracellular Ca2+ stores (6). Although the mechanism of action of Ace is reported in the literature, a comprehensive study from the pathophysiological point of view is still lacking. We had demonstrated earlier that the biologically active recombinant Ace, purified from a specialized E. coli M15 (pREP4) strain, induced a dose-dependent Isc increase across T84 cell monolayers along with ATP stimulation. This Isc response was significantly inhibited by bumetanide, an inhibitor of the Na,K,2Cl (NKCC) cotransporter, indicating that this current is predominantly carried by chloride ion (Cl−) (7). To further understand the pathophysiological mechanism of action in regulating intestinal ion transport, we sought to define the Ace-mediated signaling pathway in intestinal epithelial cells leading to stimulation of Cl− secretion and the specific channel(s) involved in the process of secretory diarrhea.
CFTR is considered to be the sole luminal Cl− channel responsible for abnormal fluid loss during V. cholerae-mediated diarrhea (8, 9). This absolute role of CFTR as the only apical Cl− channel involved in massive Cl− efflux during diarrhea is hard to imagine, given that other Cl− channels such as the recently discovered anoctamins (also named TMEM16s) are also reported to localize at the luminal surface of intestinal epithelia and contribute to Cl− secretion. Contribution of anoctamins (ANOs) to diarrhea becomes more promising with the finding of mode of action of rotavirus toxin Nsp4. A striking observation was that Nsp4 induces diarrhea in neonatal cystic fibrosis mice lacking expression of CFTR, through the activation of an age- and Ca2+-dependent plasma membrane anion permeability distinct from CFTR (10). The group led by Kunzelmann and co-workers (11) concluded that synthetic Nsp4 peptide uses multiple pro-secretory pathways to induce diarrhea by activating ANO (TMEM16). As a consequence of these observations, ANO has become an object of interest in the field of secretory diarrhea. To date, 10 members of the ANO gene family have been identified in mammals (ANO1–10 or TMEM16a–h and j and k) and predicted to be transmembrane proteins containing eight membrane-spanning helices with the N and C termini located in the cytosol (12–14). Each member is predicted to have multiple isoforms being simultaneously expressed in a given cell (15). Apart from ANO1 and ANO2, ANO6 was also recently shown to be a Ca2+-activated Cl− channel with delayed Ca2+ activation (16, 17). Despite numerous reports on ANO as a Cl− channel, the contribution of specific member(s) of this protein family to Cl− secretion in diarrhea remains obscure. Recently, the group led by Verkman and co-workers (18) reported that ANO1 carries nearly all the CaCC current in salivary gland epithelium but is a minor contributor to total CaCC current in airway and intestinal epithelia. To corroborate this finding, in vivo experiments remain to be conducted. Here, we have analyzed the predominantly expressed ANOs in intestinal epithelial cells that are major contributors to Cl− secretion in secretory diarrhea. The experiments conducted in our present study demonstrated for the first time that essentially ANO6 is able to produce Cl− current by stimulatory effects of phospholipid phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), also commonly known as PIP2, through RhoA activation by recombinant Ace. We have used a combination of electrophysiological, biochemical, molecular biology mutagenesis, and pharmacological approaches along with in vivo mouse ileal loop assay to demonstrate whether alterations in PIP2 levels by the action of Ace affect native ANO6 function in intestinal epithelial cells. Here, we report the dependence of ANO6 function on PIP2 synthesis but no subsequent rise of intracellular calcium [Ca2+]i of Ace action. We further provide evidence that Ace stimulated the RhoA-ROCK-PI(4)P5-kinase (PIP5K) signaling pathway, leading to the synthesis of PIP2, and formed the basis for the activation of ANO6 through an as-yet unknown receptor activation. Moreover, we establish that ANO6 channels possess the PIP2 binding domain in their amino acid sequence that may allow this channel to be activated by changes of PIP2 levels in response to Ace stimulation. Results of point mutations in the N terminus of ANO6, which reduced the binding of PIP2, support the proposed activation mechanism of ANO6. Our data revealed that ANO6 and PIP2 are powerful new additions to the mechanism of secretory diarrhea and have considerable implications for diarrheal disease therapy.
Results
Apical Challenge of Recombinant Ace Protein Induced a Rapid Increase of Isc in Caco-2 Cell Monolayers
Under basal conditions after an equilibrating period of 10 min, the Caco-2 monolayer exhibited an average Isc of 1.35 ± 0.41 μA/cm2. The addition of Ace (1 μm) to the apical bathing solution of Caco-2 cell monolayers caused increases in Isc (Fig. 1A), whereas basolateral addition of Ace had no effect (data not shown) as measured in modified Ussing chambers. Ace increased Isc dose-dependently in Caco-2 cell monolayers to 3.1 ± 1.7, 5.9 ± 1.1, 8.4 ± 0.9, 10.8 ± 1.3, and 12.3 ± 2.1 μA/cm2 at the Ace concentrations of 100, 300, 600, 900, and 1000 nm, respectively (Fig. 1A, top inset). The peak Isc responses to apical addition of recombinant Ace compared with untreated control were 12.3 ± 2.1 μA/cm2 versus 1.35 ± 0.41 μA/cm2. Maximal responses was reached by 12–15 min after the addition of Ace, and the effect persisted for at least 1 h (data not shown here). Subsequent studies of Ace were performed with apical addition only.
FIGURE 1.
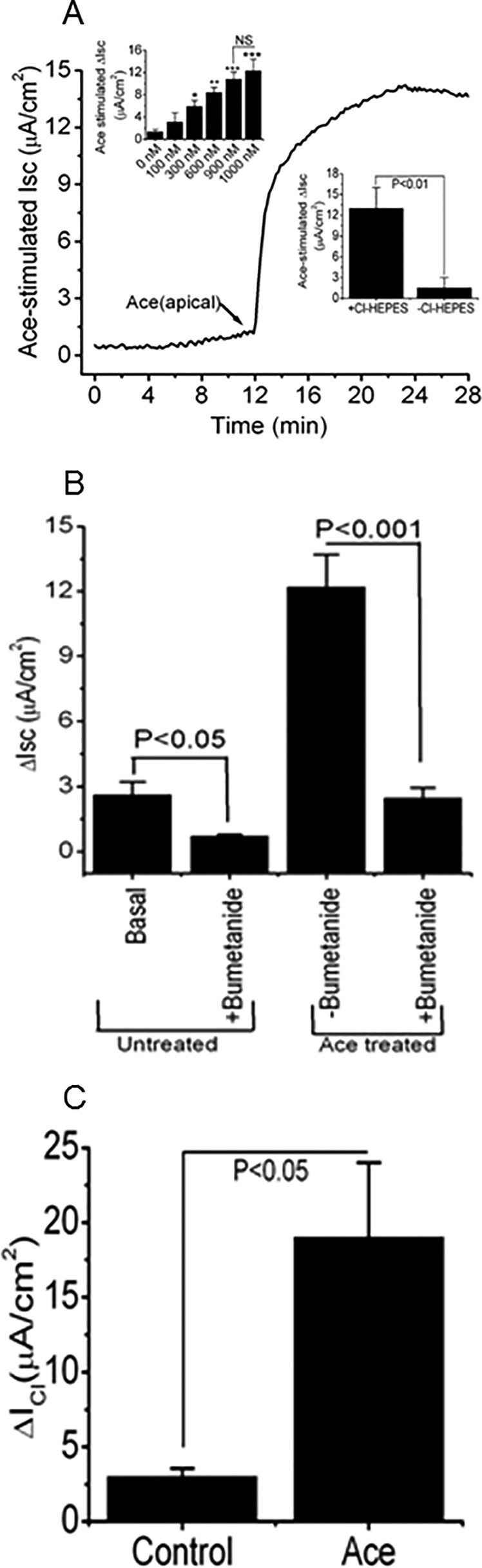
Summarized effects of recombinant Ace stimulation on Cl− current in Caco-2 cell monolayers. A, representative time course of changes in Isc and the effect of different doses of apically applied Ace on the changes in Isc (insets). Each concentration of Ace was added as a single dose to separate monolayers. Values are means ± S.E. n = 3–5. NS, statistically not significant. B, effects of basolateral bumetanide (100 μm) on basal and Ace (1 μm)-stimulated Isc. Summary of data showed average ΔIsc in response to Ace in the absence (−) or presence (+) of bumetanide. Values are means ± S.E. n = 4. C, effects of Ace on apical ICl. After establishment of a basolateral-to-apical Cl− gradient in the high K+ solution by depolarizing the basolateral membrane, addition of Ace (1 μm) evoked an outward current consistent with a secretory Cl− flow from the basolateral-to-apical side. Summary data show the change in ΔICl induced by Ace stimulation. Values are means ± S.E.; n = 5. Error bars represent S.E. *, significant difference compared with control (unpaired t test).
Ionic Basis of the Ace-stimulated Isc
To determine the ionic basis of the Isc response induced by Ace, we examined the effect of Ace in the presence of bumetanide, an NKCC co-transporter inhibitor and ion substitution solution. Pretreatment with bumetanide (100 μm) in the basolateral solution of Caco-2 cell monolayers abolished most of the Ace-induced increase in Isc from 12.02 ± 1.8 μA/cm2. In addition, bumetanide significantly reduced the basal Isc to 1.1 ± 0.02 μA/cm2 from the control value of 2.8 ± 0.04 μA/cm2 (Fig. 1B). To confirm the ionic nature of the Isc, ion substitution experiments were performed. On replacing Cl− on both sides of the preparation by Ace, the Isc value was found to be decreased by ∼86% from 13.1 ± 1.8 to 1.8 ± 0.72 μA/cm2 (Fig. 1A, inset, bottom right). To identify whether the increase of Isc by Ace was involved in the activation of the apical membrane Cl− channels, basolateral membrane was depolarized with high K+ solution in the presence of a basolateral to apical Cl− gradients. Fig. 1C shows that application of Ace elicited a rapid current, which under these conditions represented increased Cl− efflux across the apical membrane down their concentration gradient and showed activation of an apical ICl.
CaCC Inhibitors but Not CFTR Inhibitors Blocked Ace-induced Effects on Isc
Epithelial chloride secretion driven by stimulation of the apical membrane chloride channel has been tested by investigating the effects of chloride channel blockers on Ace-stimulated Isc. CFTRinh-172 (20 μm), an inhibitor of CFTR, was pre-incubated to the apical solution 10 min before the stimulation of Ace, but it did not affect the Ace-evoked increase in Isc, which was 12 ± 2.2 μA/cm2, when compared with the value of experimental 9.3 ± 1.3 μA/cm2 (Fig. 2A, inset). We then re-confirmed whether the mechanism of Cl− secretion following Ace stimulation was not due to CFTR channels; forskolin (FSK, 10 μm), a cAMP elevating agent, was added after Ace stimulation on the apical solution of monolayer. As shown in Fig. 2A, FSK was able to further increase in current even after Ace stimulation that was completely inhibited by CFTRinh-172. FSK had a similar magnitude of Isc response (4 ± 0.81 μA/cm2) when added alone in Caco-2 cell monolayers, which suggests that CFTR was fully activated (supplemental Fig. S1). Interestingly, Ace-evoked Isc was unaltered even in the presence of CFTRinh-172 when added after FSK stimulation, and this suggested the mechanism of Cl− secretion was most likely not to be the CFTR chloride channel (Fig. 2A, inset, top).
FIGURE 2.

Ace-induced Isc and ICl response to CFTR inhibitor and classical CaCC inhibitors NPPB and NFA and ANO inhibitors AO1 and TAO1 in Caco-2 cells. A, representative trace showing the effects of FSK (10 μm) and CFTR inhibitor CFTRinh-172 (20 μm) on Ace-stimulated Isc. CFTRinh-172 inhibited FSK- but not Ace-stimulated Isc in Caco-2 cells (inset). Values are means ± S.E.; n = 3–6. B, effects of cumulative addition of CaCC blockers DIDS (500 μm), TFN (10 μm), AO1 (10 μm), and TAO1 (10 μm) on Ace-induced Isc. AO1 was most effective to inhibit Ace-induced Isc than cumulative addition of different CaCC blockers. C, summary of the inhibitory effects on ICl by various CaCC inhibitors NPPB (100 μm), NFA (100 μm), AO1 (10 μm), TAO1 (10 μm), TFN (10 μm), and TA (100 μm) in Caco-2 cell monolayers. D, mouse ileum. Values are means ± S.E.; n = 5–8. Error bars represent S.E. *, p < 0.01, significant inhibition by blockers (ANOVA). NS, statistically not significant; ND, not determined.
We next examined the effects of chloride channel blockers that are known to block native CaCC on Ace-stimulated Isc in Caco-2 cells. Apical exposure to 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS, 500 μm) reduced the Ace-stimulated Isc by 30% (11 ± 1.5 μA/cm2 versus 7.7 ± 2.1 μA/cm2) (Fig. 2B). The Ace-induced Isc was effectively inhibited after consecutive administration of DIDS + tamoxifen (TFN, 10 μm) + AO1 (10 μm) followed by TFN + DIDS or DIDS alone. Interestingly, exposure to AO1 alone decreased Isc 77% (11 ± 1.5 μA/cm2 versus 2.5 ± 0.5 μA/cm2), whereas the ANO1-specific inhibitor TAO1 (10 μm) had no statistically significant effect on Isc induced by Ace (Fig. 2B).
Ace Increases ICl of Apical Plasma Membrane Sensitive to ANO Inhibitor AO1 and Classical CaCC Inhibitors but Not to ANO1-specific Inhibitor TAO1
To examine whether the sensitivity to widely used classical chloride channel blockers was different, a change in apical ICl was determined in the presence of various chloride channel blockers added on the apical solution after Ace stimulation. Fig. 2C showed the inhibitory effect in ICl responses of Ace to the effective concentrations of classical CaCC blockers. We found that apical application of 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB, 100 μm), niflumic acid (NFA, 100 μm), and AO1 abolished most of the Ace-induced increase in ICl from 17.1 ± 3.1 to 5.3 ± 1.9 μA/cm2, 4.8 ± 2.2, and 5.5 ± 1.3 μA/cm2, respectively in Caco-2 cells. Surprisingly, the Ace-elicited rise in ICl was not affected by ANO1 (TMEM16A)-specific inhibitor TAO1 (Fig. 2C). However, addition of tannic acid (TA, 100 μm) and TFN partially reduced the ICl response to Ace by 35 and 42%, respectively. NPPB, NFA, and AO1 were further studied because of its high inhibition potency and efficacy in mouse ileal tissue for inhibition of Ace-stimulated apical ICl. In fact, in experiments similar to those described in Caco-2 cells, pre-treatment of tissues with mucosal NPPB, NFA, and AO1 reduced the Ace-stimulated Isc significantly by 59.25, 51.44, and 62.55%, respectively (Fig. 2D). However, the ANO1-specific inhibitors TAO1 and TA had no significant inhibitory effect on Ace-stimulated ICl. Taken together, these findings suggest that Ace-evoked ICl on intestinal epithelium depended on ANO channel(s).
Expression of ANO1 and ANO6 in Intestinal Epithelium and Activation by Ace
Because Ace-stimulated currents were only inhibited by classical CaCC inhibitors but not by CFTRinh-172, we speculated that some of the members of the ANO protein family, which had been identified as CaCC (19–22), can be the final effector in the response of intestinal epithelial cells to Ace. The ANO protein family consists of 10 members with tissue-specific patterns of expression (23). To explore whether and which isoforms of the ANO channels were expressed in intestinal epithelia and could contribute to or be accounted for the Ace-induced chloride conductance, qPCR analysis was performed with primers specific for ANO1 and ANO6 genes. qPCR revealed mRNA expression of ANO1 and ANO6 in a different region of mouse intestine and in human Caco-2 cells, with ANO6 having the highest expression level in Caco-2 cells (Fig. 3). In contrast, a more detailed analysis of ANO1 expression indicated little or a lack of its expression in Caco-2 cells, but well detected in mouse small intestine, apart from robust expression in the distal colon. We confirmed the ANO6 expression at the protein level in Caco-2 cells by Western blotting, which indicated that ANO6 is prominently expressed in Caco-2 cells (Fig. 3D). We further studied the distribution of endogenous ANO6 in mouse ileum by immunostaining. Fig. 3D depicts ANO6 expressed at the apical surface of the villus cells as revealed by prominent green fluorescence of ANO6 staining along the plasma membrane of mouse ileum.
FIGURE 3.

ANO6 is expressed abundantly in Caco-2 cells and mouse ileum. Quantitative PCR analysis of ANO1 and ANO6 in human colonic cells T84 and Caco-2 (A) and different parts of mouse intestine as indicated (B and C), which are shown as mRNA levels relative to levels of GAPDH and determined using the difference of PCR cycles to reach a threshold amplification (ΔCT), and the relative amount of the target mRNA are given as 2−ΔCT. CT, cycle threshold. Values are means ± S.E. of triplicate experiments. D, protein from total lysates of T84 and Caco-2 cell monolayers was resolved by 10% SDS-PAGE and immunoblotted with ANO6 antibody. ANO6 protein migrated in all cases with an apparent molecular mass of 106 kDa (top). Paraffin sections of mouse ileum were fixed and stained for ANO6 (green), and xy images were collected by confocal microscopy as described under “Experimental Procedures.” Image is representative of results for three experiments. Significant difference at p < 0.001 was compared between ANO1 and ANO6 (paired t test). NS, statistically not significant; ND, not determined.
Additionally, to confirm the functional significance in response to Ace in the stimulation of Cl− secretion, region-specific changes in Isc were assessed in mouse intestinal tissue to correlate with the expression profile of ANO1 versus ANO6 by Ace stimulation. The increase in Isc activated by Ace was dose-dependent tested in mouse ileum (1–10 μm) and was significantly greater than the change in Isc of control tissue (Fig. 4A). Replacement of Cl− in both apical and the basolateral solutions of mouse intestinal tissue markedly reduced the maximal Isc response to Ace to 26.67 ± 5.33 and 3.78 ± 1,72 μA/cm2, respectively (Fig. 4B).
FIGURE 4.

ANO6 expression correlates with Ace-stimulated Isc change in mouse intestinal epithelium. A, concentration-response showing the increase in Isc following apical treatment with various concentrations of Ace in mouse ileum. The maximum effect was achieved with 5–6 μm Ace. Dose-response curve was created by a non-cumulative single addition to individual tissues. Results are shown as means ± S.E. n = 6–10 tissues from 10 mice were used. B, effect of anion substitution on maximal Isc response to Ace in mouse ileum. Replacement of Cl− in both apical and basolateral solution completely inhibited the maximal Isc response to Ace. Data are means ± S.E., n = 5. C, summary of the effects of Ace on change in Isc of different parts of mouse intestine. Ileum had the highest response to Ace than duodenum, jejunum, and colon. Values are means ± S.E.; n = 4–7. D, representative of Ace-induced Isc after addition of Ace to the apical solution showing the level of recovery after washing out the Ace in mouse ileum. *, significant difference when compared with control to Ace stimulation (paired t test).
To determine whether the increase in Isc was reversible, the apical chamber solution was replaced with Ace free ringer after Ace stimulation. When Ace was washed out, the Isc gradually recovered to nearly the same level as that of basal level. The time course of the recovery was considerably faster to reach the steady level (Fig. 4D). The Ace-stimulated Isc across the duodenum, jejunum, ileum, and colon is presented in Fig. 4C. Ace-stimulated Isc was significantly higher in ileum (24.83 ± 4.2 μA/cm2) than any other regions of mice intestine. These results were correlated with the findings that ileum had higher expression of ANO6 than duodenum, jejunum, and colon examined by qPCR. Thus, the Isc response to Ace is consistent with the expression of ANO6.
Ace Activates Anoctamin-mediated Whole-cell Current, Is It ANO6?
Working from all these data, we predicted that if ANO6 participates in the Ace-activated Isc response in Caco-2 cells and native mouse ileum, then Ace stimulation would produce only ANO6 but not ANO1 currents when expressed heterologously. To test this hypothesis, we performed whole-cell recordings on HEK293 cells transiently transfected with ANO1 and ANO6 constructs. Cells transfected with either mouse ANO1 fused with mCherry (mANO1-mCherry) or mouse ANO6 fused with GFP (mANO6-GFP) were localized to the plasma membrane of HEK293 cells (Fig. 5, A and D). The data presented in Fig. 5 indicate that ANO1 and ANO6 when expressed in HEK293 cells produce Cl− currents upon calcium ionophore A23187 treatment. However, application of Ace did stimulate Cl− current in ANO6-expressing HEK293 cells (25.6 ± 4.0 to 40.44 ± 7.3 pA/pF at +59 mV), but not in ANO1 expressing cells, as measured by whole-cell patch clamp recordings of Cl− currents, and as in the A23187 experiments, currents were inward. We found that in the presence of intracellular calcium (100 nm or 1 μm), Ace failed to display significant currents from ANO1 constructs as presented in Fig. 5B. Importantly, the currents from ANO6 constructs exhibited outward rectification and were identical to whole-cell Cl− current stimulated by Ace from −2.6 ± 0.5 to −6.4 ± 1.4 pA/pF at −83 mV as observed in Caco-2 cells (Fig. 5E). The current-voltage relationship for this channel in Caco-2 is shown in Fig. 9C.
FIGURE 5.
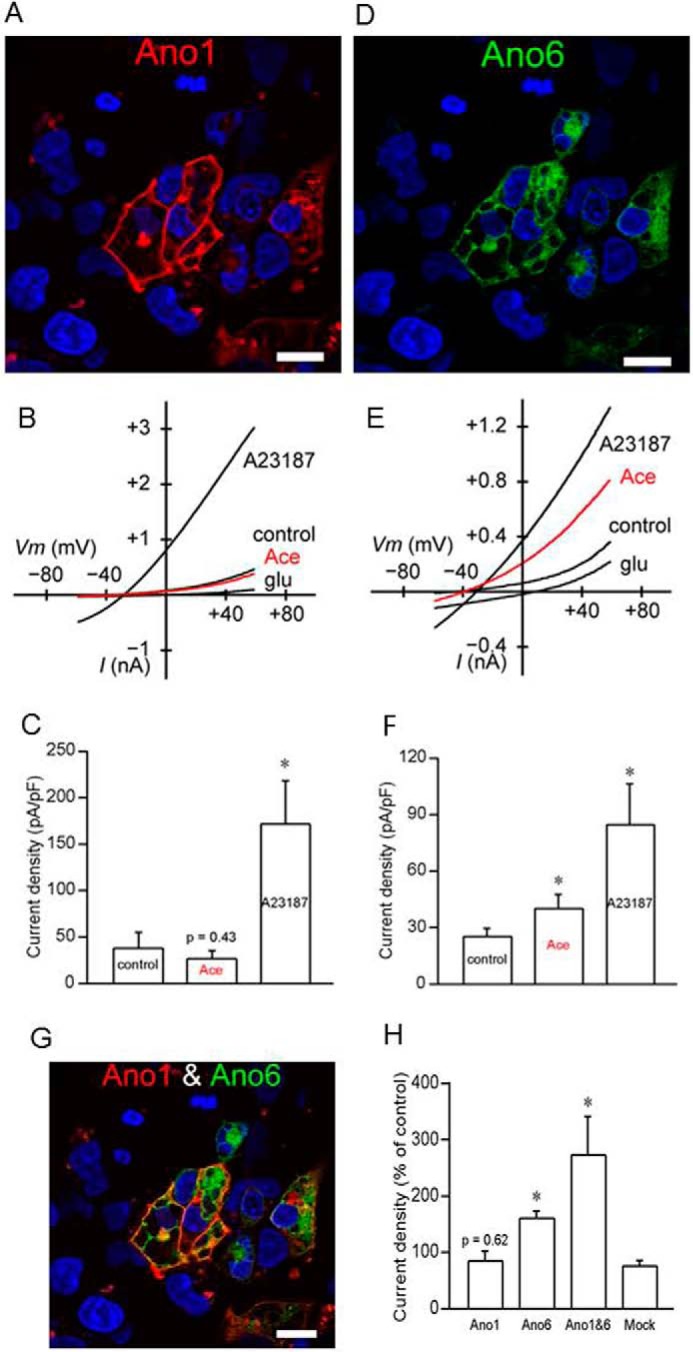
ANO6 is activated by Ace but not the ANO1. HEK293 cells transiently transfected with mcherry-tagged mouse ANO1 (A) and GFP-tagged mouse ANO6 (D) showing membrane expression of ANO1 and ANO6 in these cells. The images were taken using ×63 objective in LSM510 confocal microscope. Bars, 20 μm. Representative whole-cell current traces for ANO1 (B) and ANO6 (E) measured in transfected HEK293 cells treated with either Ace or A23187. Ace stimulated ANO6 but not ANO1 currents and A23187 evoked both of these currents. Average current amplitudes of ANO1 (C) and ANO6 (F) in transiently transfected HEK293 cells in response to Ace and A23187 are shown. ANO6 co-localized with ANO1 at the membrane of transiently transfected HEK293 cells (G). Co-localization augmented the whole-cell current amplitude in these cells (H). Data are presented as mean ± S.E. and tested by one-way ANOVA; *, p < 0.05. For transfection with individual ANO constructs, 2 μg of DNA was used; co-transfection was performed using 1 μg of each ANO constructs.
FIGURE 9.

Quantitative mRNA expression and activation of ANO6 whole-cell current in Caco-2 cells. A, relative expression (normalized by expression of GAPDH) of ANO6 variants in Caco-2 cells; B, mouse intestinal tissues in log scale. Data are means ± S.E. from triplicate experiments. C, whole-cell current response to Ace showing representative current-voltage relationship with gramicidin-perforated patch recording in Caco-2 cells. D, summary graph of current amplitude in response to Ace stimulation. * indicates significant difference at p < 0.05 compared with control (paired t test). E, knocking out of ANO6 mostly abolished Ace-stimulated Isc. Shown are representative time courses on the effects of Ace on Isc in WT, vector control, and ANO6 KO Caco-2 cells (bottom panel); top panel shows the summary of the data from four such monolayers. Values are means ± S.E. F, rescue experiments for ANO6 in Caco-2 cells by re-expression of ANO6 using retroviral expression system described under “Experimental Procedures.” Top panel, Western blotting analysis of proteins in total lysate of wild type, ANO6 KO, ANO6 KI, and in vector control cells. Anti-GAPDH was used as loading control. Bottom panel, summary of the data on Ace-stimulated Isc in ANO6 KO and ANO6 KI cells with WT and vector control. ** indicates significant difference at p < 0.001compared with WT or vector control (paired t test). NS, statistically not significant.
To explore the association between ANO6 and ANO1 that could augment Cl− current upon Ace stimulation, we co-expressed these two constructs together in HEK293 cells. Cells transfected both with mANO1-mcherry and mANO6-GFP were analyzed by confocal microscopy. Coexpression of the two proteins produced intense plasma membrane fluorescence, showing ANO1 and ANO6 expression overlaps each other (Fig. 5G). To test whether the Cl− currents produced by Ace increased when ANO1 and ANO6 coexpressed in HEK293 cells were possibly caused by this interaction, we measured whole-cell currents. Activation of the whole-cell Cl− conductance through Ace stimulation was slightly augmented 2-fold as shown in Fig. 5H. Taken together these results demonstrated that Ace activates ANO6-mediated Cl− currents through the stimulation of ANO6. The results of A23187 experiments in HEK293 cells further indicated that ANO6 generates a Ca2+-dependent Cl− current.
Does Ace Activate Cl− Secretion by Increasing Intracellular Ca2+?
Because the whole-cell patch clamp experiments indicated that Ace stimulated the ANO6 current in HEK293 cells, which is thought to be a CaCC, we speculate that the stimulatory effect of Ace on ANO6 current occurs because the intracellular calcium ([Ca2+]i) increased. To test this hypothesis,[Ca2+]i was measured from single cells using the fluorescent calcium indicator dye fura 2-AM in HEK293 stably expressed with mANO6-mcherry (supplemental Fig. S2) and Caco-2 cells as described earlier (24). Cells grown onto cover glass incubated with Ace failed to show any detectable increase of [Ca2+]i both in HEK293 transfected with mANO6-mcherry and Caco-2 cells as presented in Fig. 6B and supplemental Fig. S2. The membrane-permeable Ca2+ chelator (BAPTA-AM) was studied in the presence or absence of extracellular calcium and tested for inhibitory action, if any or no effect at all, on the Isc induced by Ace in mouse ileum. As shown in Fig. 6A, BAPTA, even in absence of extracellular calcium, had no effect on Isc, suggesting that a calcium-dependent signaling pathway of epithelial chloride secretion does not seem to be involved in the mechanism of Ace action. ATP was used as a secretagogue to induce a rise in intracellular Ca2+ concentration, acting via P2X purinergic receptors. In contrast to the results with Ace, ATP caused a large transient increase in [Ca2+]i in HEK293 and Caco-2 cells. The extracellular solution included 1 mm Ca2+ (see under “Experimental Procedures”). The mean [Ca2+]i values at the peak of the response of 10 μm ATP versus 100 μm was 0.379 ± 0.046 and 0.746 ± 0.087 μm, respectively (supplemental Fig. S2 A). In Caco-2 cells, typically [Ca2+]i began to increase during the application of ATP, peaked rapidly within 30 s, and then returned gradually to baseline levels over the next 1–2 min (Fig. 6A). We found that repeated application of ATP to the cells reproducibly evoked an increase in [Ca2+]i. As illustrated in the example in supplemental Fig. S2, progressively larger amplitude and larger duration responses were evoked by increasing concentrations of ATP. We next considered what the effect could have on the [Ca2+]i rise if Ace and ATP were added together. We found that there were no significant differences in the level of [Ca2+]i rise between stimulation of ATP alone (0.290 ± 0.040 μm) and combination of Ace with ATP (0.305 ± 0.042 μm) tested (supplemental Fig. S2B). Taken together, these results suggested that there may be a Ca2+-independent mechanism of Ace-evoked Cl− secretion through ANO6 stimulation.
FIGURE 6.

Ace stimulated calcium-independent Cl− current and ATP but not Ace increased intracellular calcium in mouse ileum and Caco-2 cells. A, effects of BAPTA-AM on (left panel) and removal of extracellular calcium (right panel) on Ace-stimulated Isc of mouse ileum. BAPTA-AM was applied serosally as indicated. AO1 was applied to ensure that current was ANO-mediated. Traces are representative of four independent experiments. B, time course for Ace + ATP induced increase in [Ca2+]i in Caco-2 cells, as detected by ratiometric Fura2 fluorescence. Stimulation with ATP activates [Ca2+]i rise. Cells were treated with 1 μm Ace followed by the addition of 100 μm ATP. Ace was not able to increase [Ca2+]i, but subsequent addition of ATP evoked a [Ca2+]i rise in these cells. Resting [Ca2+]i of these cells was around 100 nm when measured in the solution contained 10 mm glucose.
Blocking PIP2 Synthesis Inhibits Ace-evoked Chloride Secretion
Summarizing the above results, we demonstrate that the extracellular application of Ace to HEK293 cells transfected with ANO6 elicited a rapid increase in outward whole-cell currents but failed to increase [Ca2+]i. This led us to predict that activation of Cl− secretion by Ace may or may not require a concomitant rise of [Ca2+]i. It is becoming increasingly clear that the lipid components of the membrane are major regulators of the activity of ion channels. Thus, we considered the possibility of phosphoinositols to activate ANO6 upon Ace stimulation. The membrane phospholipid PIP2 has emerged as an important regulator of many ion channels and transporters (25–27). To study the PIP2 regulation of Ace-stimulated ANO6 current, if any, we measured Ace-stimulated Isc in Caco-2 cells and mouse intestinal tissue in the presence of agents that manipulate the change to endogenous PIP2 levels through different routes (PI4K inhibition, PIP2 scavenging). To determine whether the ability of Ace to stimulate ANO6 currents could be attributed to activation of a lipid kinase, we tested whether wortmannin (Wort), at concentrations that block PI4K and PI3K (28), interfered with Ace-induced current of ANO6. As shown in Fig. 7, preincubation of cells for 30 min with 50 μm Wort significantly reduced Ace-evoked Isc (4.8 ± 0.9 μA/cm2 versus 12.2 ± 1.8 μA/cm2) supporting a role for PI4K or PI3K in this process. In contrast, we found a low concentration of Wort (1 μm), where it inhibits only PI3K but not PI4K, which caused little reduction of Ace-stimulated Isc (11.3 ± 1.2 versus 12.3 ± 2.1 μA/cm2; 6.6%), which was similar to DMSO control (5%), suggesting that this process is not mediated by PI3K. To ensure the specific involvement of PI4K, we tested another chemically distinct inhibitor of PI4K, PAO (10 μm). PAO inhibits the synthesis of PIP2 from its precursor phosphatidylinositol, thus lowering the membrane concentration of PIP2 (29, 30). As shown in Fig. 7A, application of PAO caused significant reduction of Ace-evoked Isc ([5.1 ± 0.6 μA/cm2 versus 12.3 ± 2.1 μA/cm2). In addition to PAO, another PIP2 scavenger, PLL (25 μg/ml) depicts similar inhibitory effect on Ace-evoked Isc in Caco-2 cells. To test whether a similar pattern of inhibition exists in mouse intestine, we examined the effect of PAO and PLL on Ace-stimulated Isc in mouse ileum. Pre-incubation of PAO to the mucosal side of mouse intestinal tissue caused large reduction of Ace-evoked Isc (Fig. 7B). When the drug was washed out, the Isc gradually recovered to nearly the same level as that of control (Fig. 7C). However, the time course of the recovery was considerably slower than the control experiment. Together, these data suggest that the reduction of Isc might be attributed to an increase in PIP2 in the membrane. According to this hypothesis, manipulations that deplete PIP2 should accelerate reduction of Isc stimulated by Ace. Indeed, PAO and PLL caused reduction of Ace-evoked Isc in Caco-2 cells and mouse tissue as shown in Fig. 7, A and B, supporting a role for PIP2 in regulating Ace-mediated ANO6 function. Because the cellular level of phosphatidylinositol 4-phosphate is approximately 10 times higher than that of phosphatidylinositol 5-phosphate, the major synthetic pathway for the formation of PIP2 is probably through the activity of PIP5Ks. We also wondered whether cellular depletion of PIP5K would affect Ace-stimulated Isc in Caco-2 cells. Fig. 7E illustrates representative time course (lower panel) and summary of change in Isc (upper panel) in WT and Caco-2 PIP5K KO cells. PIP5K KO cells showed resistance to Ace-stimulated Isc (2.09 ± 0.97 μA/cm2), whereas WT or vector control cells had similar and equivalent change in Isc (12.12 ± 2.69 and 11.02 ± 2.11 μA/cm2). This result confirms the requirement of PIP5K for Isc in response to Ace and suggests that PIP5K is involved in the PIP2-mediated stimulation of Isc. The Western blot in Fig. 7D illustrates complete deficiency of PIP5K protein in Caco-2 PIP5KO cells by CRISPR/Cas9-mediated gene KO.
FIGURE 7.

Inhibition of PIP2 synthesis by lipid kinase inhibitors affects Ace-stimulated ICl. A, pre-incubation with two different concentrations of Wort50 (50 μm), Wort1 (1 μm), PAO (10 μm), and PLL (25 μg/ml) significantly inhibited Isc in Caco-2 cells. NS, statistically not significant. B, following a 30-min incubation in the presence of PAO (20 μm) and PLL (25 μg/ml), Isc was reduced in response to Ace (5 μm) in mouse ileum. C, PIP2 speeds Isc recovery by subsequent washout of PAO. PIP2 modulators were added to both sides of the solutions. Ace was added only on the apical side. D, Western blot (with PIP5K antibody) detection of proteins in total lysate of wild-type Caco-2, vector control, and PIP5K knock out cells. Anti-GAPDH was used to confirm equal loading (n = 3). E, representative time course on the effect of Ace on Isc in PIP5K KO Caco-2, WT, and vector control cells. PIP5K knock-out cells had very little or no effect in response to Ace stimulated Isc. The inset shows the summary of the data from six such monolayers. Values represent means ± S.E. significant difference at *, p < 0.05, or **, p < 0.01when compared with the corresponding control value by ANOVA.
Identification of KR Motifs in ANO6, PIP2 Requires a Unique Conserved Region for the Interaction
Because PIP2 seems to act as an instructive second messenger, inducing ANO6-mediated Cl− secretion in response to Ace stimulation, we then determined whether cellular abundance of PIP2 increased by employing immunofluorescence experiments in Caco-2 cells. After adding Ace, PIP2-specific fluorescence increased at the edge of the cell membrane in comparison with untreated cells (Fig. 8A). As Rho kinase (ROCK) has recently been implicated in the stimulation of PIP2 synthesis, we therefore tested the effect of ROCK inhibitor H1152 on PIP2 synthesis. H1152 dramatically reduced PIP2 fluorescence even in presence of Ace stimulation, suggesting ROCK is required for changing cellular abundance of PIP2 in response to Ace stimulation (Fig. 8A, right). Because ANO6 is involved in Ace-stimulated Cl− secretion by increasing PIP2 synthesis, its PIP2 binding property might be important to elucidate its function. In this context we undertook to identify the consensus sequence for PIP2 binding in ANO6 protein sequence. A firm consensus sequence has not yet emerged, but several important PIP2-binding proteins have clusters of basic/aromatic residues involving stretches of sequence (10–20 residues) loosely denoted as KR motifs containing KK(X)n(K/R)K (31). PIP2 could bind at an interface between the transmembrane domain and the cytoplasmic domain of ANO6. As shown in Fig. 8, B and C, we have identified a conserved KR motif on the N-terminal region is the only region with sequence similarity among ANO6 isoforms recently identified in human and mouse (32). These motifs are present at the proximal positions 280–289, 262–271, 280–289, and 301–310 and at the distal position of 85–95, 67–77, 85–95, and 106–116 in human ANO6 isoforms a, b, c, and d, respectively. We further determined that these KR motifs are located at the proximal positions 302–311 and 281–290 and at the distal positions of 108–118 and 87–97 in mouse ANO6 isoforms a and b. To determine whether physical interaction between PIP2 and ANO6 was promoted by Ace stimulation through the KR motif, in vitro immunoprecipitation experiments were performed in HEK293 cells with plasmids expressing mANO6-EGFP or ANO6-EGFP N-terminally truncated forms that lack distal and/or proximal KR motifs. The cell lysates were then incubated with anti-GFP-protein G-Sepharose beads, and precipitates were analyzed by immunoblotting with anti-PIP2 antibody. As shown in Fig. 8D, ANO6 co-precipitated together with PIP2 from transfected HEK293 cells but was not precipitated from untransfected control cell lysates. Notably, cells stimulated with Ace resulted in a marked increase of the signal intensity produced by PIP2 antibody. These results demonstrated that Ace promotes binding of PIP2 to ANO6 and is consistent with the findings of ex vivo experiments in mouse tissue and Caco-2 cells that the PIP2 manipulator affected Ace-stimulated currents. Interestingly, the results presented in Fig. 8D further demonstrate that deletion of the distal KR motif had very little or no effect on PIP2 binding, although deletion of proximal KR motif in addition to the distal one caused reduction of PIP2 binding, indicating that the N-terminal KR motif plays an important role in PIP2 binding for ANO6. We further constructed site-directed mutants of ANO6 in which the first two conserved lysines (Lys-281 and Lys-282 in proximal and Lys-87 and Lys-88 in distal KR motif) residues and last two conserved arginine and lysine (Arg-289 and Lys-290 in proximal and Arg-96 and Lys-97 in distal KR motif) residues were substituted with asparagines. Mutation of lysine and arginine residues in the proximal KR motif caused reduction of the signal intensity produced by PIP2 antibody. However, mutation of these residues in the distal KR motif had little or no reduction in PIP2 binding, demonstrating that these residues in the proximal KR motif are functionally more important for PIP2 binding to ANO6 than the distal portion of KR motif. Deletion of the distal or proximal KR motif or mutation of lysine and arginine in this motif in the N terminus of ANO6 does not affect surface expression of the channel as presented in supplemental Fig. S3.
FIGURE 8.

Cellular abundance of PIP2 and its potential binding sites in the N terminus among the human and mouse ANO6 variants. A, Caco-2 wild-type cells were stimulated with Ace. Shown are representative images of fixed cells labeled for PIP2 in non-stimulated cells (control, left panel), Ace-stimulated cells (middle panel), and for cells that are pre-treated with H1152 and stimulated with Ace (right panel). Red color represents PIP2 abundance. Scale bars, 10 μm. B, sequence alignments at a comparable position in ANO6 variants showing two similar PIP2-binding motifs are conserved beginning immediately after the putative first membrane spanning domain of human (left) and mouse (right).Typical PIP2-binding motifs consisting of a cluster of basic residues are indicated by the red box. Sequences were obtained from the NCBI protein database. Accession numbers are as follows: variant 1 isoform a, NM_001025356.2 > NP_001020527.2; variant 2 isoform b, NM_001142678.1>NP_001136150.1; variant 3 isoform c, NM_001142679.1 > NP_001136151.1; and variant 4 isoform d, NM_001204803.1>NP_001191732.1. Mouse variant accession numbers are as follows: variant 1 isoform a, NM_001253813.1 > NP_0012407421.1, and variant 2 isoform b NM_175344.4 > NP_780553.2. C, schematic diagram illustrating the topology of human ANO6, highlighting two potential PIP2-binding sites (red box) of the channel N terminus. D, PIP2 co-immunoprecipitates with ANO6 in mANO6-GFP-transfected HEK293 cells whereas truncation mutants led to a significant decrease in the interaction. The cell lysates were incubated with anti-GFP-protein G-Sepharose beads, and inputs and immunoprecipitation eluate fractions were analyzed by Western blotting with either anti-PIP2 with or without Ace stimulation. PIP2 antibodies recognize GFP-tagged ANO6 protein in transfected HEK293 cells. No signal is detected in non-transfected HEK293 cells. This experiment was performed twice. The GFP blot for this experiment was presented to indicate the level of ANO6-GFP precipitation. D and P in truncated protein designate the deletion of distal and both distal and proximal KR motifs. Introduction of four mutations in the proximal and distal KR motifs are as indicated. IB, immunoblot; IP, immunoprecipitation.
ANO6 Alternative Splice Variant Expression in Intestinal Epithelia
Next we examined which isoforms are preferentially expressed to contribute to the increase in Isc. We analyzed differential expression of alternative splice variants of ANO6 in human intestinal cell lines and mouse tissues by RT-qPCR. Fig. 9, A and B, displays different expression patterns of ANO6 transcript variants in Caco-2 cells and mouse intestine. We observed robust expression of variant a in Caco-2 cells, although both variants a and b were broadly expressed in mouse ileum. We also performed whole-cell recording of Caco-2 cells by patch clamp experiments. Ace stimulation produced currents that were outwardly rectifying (Fig. 9C). The mean peak amplitudes of currents at −83 mV was −6.4 ± 1.4 pA/pF (Fig. 9D). This is consistent with the finding that Ace-stimulated ICl was significantly reduced by knockdown of ANO6 in Caco-2 cell monolayers using lenti-shRNA as presented in supplemental Fig. S4. ANO6 knockdown was also verified by qPCR in Caco-2 cells, which indicated a knockdown by 82%. We double confirmed the complete deficiency of ANO6 protein in Caco-2 cells by the CRISPR/Cas9-mediated gene knock-out (KO) technique. We identified one genomic CRISPR/Cas9 target sequence (see Table 1) within exon 1. The appropriate sequence into PX458 vector containing all other elements required for CRISPR/Cas9-mediated KO was described under “Experimental Procedures.” The results shown in Fig. 9F demonstrate ANO6 protein expression by Western blotting and revealed no detectable ANO6 protein in CRISPR/Cas9-treated cells in contrast to vector control or WT Caco-2 cells. To functionally assay for ANO6 deletion, we measured Isc in the Ussing chamber. The CRISPR/Cas9 construct lacking targeting sequence (vector control) displayed the same magnitude of Ace-stimulated Isc to that in WT Caco-2 cells (11.02 ± 2.08 μA/cm2 versus 12.28 ± 1.23 μA/cm2) (Fig. 9E). In contrast, cells transfected with targeting ANO6 sequence showed complete loss of Ace-stimulated ANO6 current (1.34 ± 0.98 μA/cm2). We next tested whether ANO6 function could be rescued to restore the impaired phenotype in the context of Cl− secretion by knock-in (KI) ANO6 gene in Caco-2 cells using platinum retroviral expression system (VPK 301, Cell Biolabs, Inc.). Retroviruses, pseudotyped with the VSVG envelop protein for enhanced stability and efficient infection of Caco-2 cells, were produced and typically yielded considerable transduction rates of ≈70%. After G418 selection, to eliminate nontransduced cells, the retro-mediated expression of ANO6 was measured by immunoblot. The immunoblot results are shown in Fig. 9F and revealed re-expression of ANO6 protein as shown in the 3rd lane. We next examined whether this re-expressed ANO6 protein gained function, and we measured Isc. As shown in the Fig. 9, ANO6 KI cells produced a similar magnitude of Isc when stimulated with Ace. Taken together, these results demonstrate that the impaired phenotype resulted from ANO6 KO in Caco-2 cells not because of any off-target effects but due to ANO6 protein function. Moreover, variant a in human and variant b in mouse ANO6 are likely major contributors of Ace-stimulated Cl− current because of their higher expression than other variants.
TABLE 1.
Genomic sequences of CRISPR/Cas9 targets in Ano6, PIP5K, and RhoA genes
The exon sequences are in red, and target sequences are highlighted in yellow; the underlined NGG sequences are Protospacer Adjacent Motif (PAM), and blue highlighted ATGs are start codons.

RhoA and ROCK Are Required for PIP2 Synthesis for Ace-stimulated Fluid Secretion
Given our hypothesis of Ace stimulation of phosphatidylinositol-4-phosphate 5-kinase, we considered the role of Rho monomeric GTPase, RhoA in Ace-induced Cl− secretion presumably through activation of ANO6. This is because PIP5K is an effector of RhoA as reported in the literature (33, 34). As summarized in Fig. 10A, pretreating cells with C3 toxin (1 μg/ml), which is an inhibitor of RhoA (35), significantly decreased Ace-stimulated ICl from 19.3 ± 3.8 to 6.4 ± 2.6/cm2 in Caco-2 cells. We also confirmed this inhibitory effect of C3 toxin on Ace-stimulated Isc by knocking out RhoA in Caco-2 cells. The Western blots in Fig. 10B illustrate complete depletion of RhoA in Caco-2 cells by the CRISPR/Cas9-mediated gene KO approach. There was no change in the expression of RhoA in vector control (lacking the gene-targeted sequence) cells. The functional consequence of RhoA KO with respect to Ace-stimulated Cl− secretion was tested using these cell lines. As shown in Fig. 10C, the RhoA KO cells exhibited only 20% (2.08 ± 0.65 μA/cm2 versus 10.23 ± 2.46 μA/cm2) of Ace-stimulated Cl− secretion compared with wild-type and vector-transduced cells. We next asked whether RhoA activated ANO6 through signaling via one of its downstream effectors proteins, Rho kinase and MAPK and PI3K cascades. We measured ICl in Caco-2 cells in the presence of H1152 (1 μm), which is an inhibitor of ROCK, MAPK inhibitor PD98059 (10 μm), and PI3K inhibitor Wort (1 μm). Fig. 10A shows typical ICl from voltage clamp experiments performed on Caco-2 cells demonstrating Ace treatment stimulated ICl, which was significantly inhibited by H1152. In contrast, neither the MAPK inhibitor nor the PI3K inhibitors affected Ace-stimulated Isc. Here, we favor the interpretation that ROCK is necessary for RhoA-dependent stimulation of ANO6 in Ace-induced Isc. Because activation of RhoA induces its redistribution to the plasma membrane (36, 37, we re-confirmed the involvement of RhoA in this signaling cascade by measuring RhoA content of cytosolic and membrane fraction of Caco-2 cells treated with Ace. Treatment of cells with Ace induced translocation of RhoA from the cytosolic to the membrane fraction (Fig. 10D), which is a sign of RhoA activation. These data strongly suggest that translocation of RhoA is a consequence of activation by Ace.
FIGURE 10.

RhoA, ROCK, and lipid kinase inhibitors attenuate Ace-induced diarrhea in vivo. A, summary graph of Ace-induced Isc in Caco-2 cell monolayer pre-treatments with C3 toxin (at 1 μg/ml for 4 h in the 6-well plate), H1152 (1 μm), PD98059 (10 μm), and Wort (1 μm) on both sides of the chamber for 20 min before addition of Ace. B, immunoblot analysis of RhoA in WT, vector control, and RhoA KO Caco-2 cells total lysate. Probe for GAPDH was used to ensure equal loading. C, knock-out of RhoA expression abolished Ace-stimulated Isc. Top panel, summary; bottom panel, representative traces of time course experiments showing that RhoA KO inhibited the increase in Isc in response to Ace. Mean ± S.E. were calculated from n = 5 experiments. ** indicates significant difference at p < 0.01 compared with WT or vector control (paired t test). ND, not determined. D, representative immunoblot from three independent experiments demonstrate Ace stimulation increases membrane amount of RhoA. Typical Western blotting probed with anti-RhoA antibody containing the plasma membrane fraction from Caco-2 cells. E, fluid accumulation ratio in closed mouse loops by Ace stimulation in the presence of ROCK inhibitor H1152 (1 μm), lipid kinase inhibitors PAO (100 μm), PLL (25 μg/ml), and CaCC blockers. F, photographs of representative mouse ileal loops at 6 h after luminal installation of Ace with or without inhibitors. Data represent the means ± S.E. n = 10 mice per group.* indicates significant difference at p < 0.05 when compared with control or with Ace stimulation by ANOVA; NS, statistically not significant.
In Vivo Evidence of the Alteration of PIP2 Levels Involving RhoA-ROCK Signaling by Ace Action Affects Native ANO6 Function to Induce Diarrhea
To provide proof of concept that Ace induced secretory diarrhea in vivo stimulating ANO6 channels due to alteration of PIP2 levels through RhoA-ROCK signaling, we tested the effect of the ANO-specific inhibitor AO1, ANO1-specific inhibitor TAO1, and PAO, a PIP2 synthesis inhibitor, PLL, a PIP2 scavenger, and ROCK inhibitor, H1152, on Ace-stimulated diarrhea in the mouse model (Fig. 10E) described by us previously (38). The intestinal loops inoculated with Ace were filled with clear fluid and markedly distended after 6 h. A false-positive reaction rarely occurred after injection of saline. As shown in Fig. 10F, fluid accumulation caused by Ace was significantly inhibited by PAO, PLL, and ROCK, AO1, NFA inhibitor. However, ANO1 inhibitor TAO1 and DIDS partially reduced Ace-induced fluid accumulation, which was not statistically significant. These results clearly demonstrate that modulation of intracellular PIP2 levels may contribute to the Ace-induced secretory diarrhea through an AO1 and/or a TAO1-sensitive chloride channel. Together, all these results are consistent with PIP5K being downstream of RhoA and ROCK in a signaling pathway leading to activation of ANO6 for fluid secretion by Ace stimulation.
Discussion
Diarrhea occurs either because of decreased absorption or increased secretion or both of water and electrolytes such as Na+ and Cl−. Earlier studies had shown that purified Ace from ace+ V. cholerae strains is a Ca2+-dependent agonist that acts at the apical membrane of polarized intestinal epithelial cells (T84 models) to stimulate anion secretion (6). Our results, however, contradict their findings that recombinant Ace stimulated a calcium-independent Cl− secretion in intestinal epithelial cells. Evidence that Ace-stimulated Cl− secretion is not due to a rise of [Ca2+]i is based on the following: 1) application of Ace did not raise [Ca2+]i in mouse ANO6 fused to mCherry-expressing HEK293 cells or in Caco-2 cells; 2) in the presence of intracellular calcium (100 nm or 1 μm), Ace failed to display significant whole-cell currents from ANO1-expressing HEK293 cells in our patch clamp study; 3) in the absence of extracellular calcium and in the presence of BAPTA-AM (a widely used [Ca2+]i chelator), Ace was able to stimulate Isc in mouse tissue (supplemental Fig. S5). The results of Trucksis et al. (6) are potentially important. However, we suspect that their results depend on partially purified Ace from ace+ Vibrio culture. There was no direct evidence for the rise of [Ca2+]i by Ace, as their conclusion was predicted only on the basis of Ussing chamber data from T84 monolayers.
In our study, we found that Ace increases Cl− secretion across the apical membrane predominantly through ANO6 that greatly contributes to secretory diarrhea. This is consistent with prior reports of its secretory activity in animal models (5, 17, 39). Using human epithelial colorectal adenocarcinoma cells, Caco-2 which possessed the machinery for Cl− transport, we showed that Ace acutely stimulated Cl− secretion. This was supported by the findings that Ace stimulated an apical Cl− conductance, and the Ace-induced increase in Isc was blocked by the NKCC co-transporter inhibitor bumetanide in Caco-2 cells, or replacement of Cl− completely inhibited the Ace-stimulated Isc in the mouse ileum. Ace added to the apical solution produced a dose-dependent increase in the Isc response, whereas the basolateral addition did not have any effect, suggesting that Ace receptors were predominantly located at the apical membrane. We did not identify the Ace receptor in this communication, but a role of G protein-coupled receptor-linked receptor mediated RhoA signaling of Ace or a receptor that could stimulate RhoA signaling will be the focus of our future study. In addition, the Ace-induced increase of Isc was blocked by various Cl− channel blockers, including native CaCC blockers, NPPB, NFA, and AO1 but not by the CFTR inhibitor CFTRinh-172 or the ANO1(TMEM16A)-specific inhibitor TAO1. As the present results showed that the Ace-induced Cl− current was nearly completely inhibited by NPPB, AO1, and NFA and partially by DIDS, it was most likely that one of the members of the ANO family was the primary target of the Ace-stimulated Cl− secretion. Indeed, our data identify ANO6 as the candidate channel of Ace-stimulated Cl− secretion. We observed that Ace activated a strong outwardly rectifying current that peaked after 2–4 min in HEK293 cells transiently expressing mANO6-GFP as well as in Caco-2 cells. In our study, mANO6 was characterized by an outwardly rectifying current in asymmetrical solutions. (Curiously, mANO6-GFP activated with a time-independent manner.) Surprisingly, Ace was not able to increase either [Ca2+]i or to activate ANO1 currents measured in Caco-2 cells and HEK293 cells transiently expressing mANO1-mCherry, respectively. We did not find support for our hypothesis that the Ace-stimulated ANO6-mediated Cl− secretion was a calcium-dependent phenomenon. However, treatment with the calcium ionophore A23187 induced strong outwardly rectifying ANO1 and ANO6 currents in these HEK293 cells, which is similar to the findings of other researchers (17). There is controversy about ANO6 localization. A recent report claimed that ANO3–7 are intracellular proteins (40). This is contrary to our present results that demonstrate clear plasma membrane expression for both ANO1 and ANO6 in HEK293 cells. We have further confirmed the membrane expression of endogenous ANO6 in Caco-2 monolayers and mouse intestinal tissue (Fig. 3). These results are consistent with the finding of Schreiber et al. (23), which demonstrates plasma membrane expression of most ANOs, including ANO6 in HEK293 cells. To conclude this section, our data provide evidence that ANO6 constitutes a Ca2+-activated Cl− channel (because A23187 was activated) and express in the plasma membrane that was activated by Ace in a calcium-independent mechanism. It is not surprising to conclude that the Ace response does not involve CFTR. This is because treatment of Caco-2 cells with a maximum concentration of Ace to maximally activate Isc did not prevent a further increase of Isc by forskolin (FSK), a cAMP-elevating agent that stimulates CFTR-dependent Cl− secretion. CFTRinh-172 reduced the FSK-stimulated Isc only and had no effect on Ace-stimulated Isc or ICl as shown in Fig. 2A, suggesting that Ace may target an apical Cl− channel different from CFTR for increasing Cl− secretion. Indeed, we found that Ace stimulated ANO6 in Caco-2 cells for Cl− secretion because Ace stimulated both whole-cell Cl− current and Isc, which was reduced due to KO of ANO6 in Caco-2 cells (Fig. 9, C–E). This additional KO approach of CRISPR/Cas9 increased confidence in data relating to an shRNA effect of the ANO6 gene silencing and thus a similar phenotype in Caco-2 cells. The rescue experiments presented in Fig. 9F confirm the phenotype (dysfunction of Cl− secretion) we see in Caco-2KO cells due to ANO6. This is because the impaired phenotype was restored by re-expression of the ANO6 protein in Caco-2 cells by retroviral expression system.
We next delineated the Ca2+-independent signaling pathway by which Ace could activate ANO6 for Cl− secretion. There is growing appreciation that in addition to being a substrate for phospholipase C, PIP2 is also a regulator of various ion channels, including KCNQ-encoded channels, transient receptor potential 1 (TRPC1), Kir, and epithelial sodium channel (26, 41). We hypothesized that it could be the PIP2-dependent signaling in close proximity to ANO6 that matters for channel activation rather than the calcium-dependent mechanism. The idea of the PIP2-dependent mechanism being relevant for activation of ANO6 was supported by our three key findings. The first is that inspection of the sequence of the N-terminal domain of ANO6 revealed two potential binding motifs called KR motifs contain KK(X)n(K/R)K at comparable positions among mouse and human variants, which are similar to the motifs identified in gelsolin and villin (42). The second and key finding is the in vitro experiments of ANO6 binding to PIP2 isolated from mANO6-GFP-expressing HEK293 cells provide a candidate mechanism for the Ace stimulation of ANO6 Cl− current (Fig. 8). The interaction between ANO6 and PIP2 was increased in ANO6-expressing HEK293 cells when stimulated by Ace as presented in Fig. 8D. Moreover, deletion of the proximal KR motif and mutation of the first and last conserved residues (Lys-281, Lys-282, Arg-289, and Lys-290) largely diminished this signal intensity produced by PIP2 antibody. The third is Ace stimulation increased cellular abundance of PIP2-specific immunofluorescence (Fig. 8A) and modulators (PIP2 synthesis blockers, scavengers) used to manipulate PIP2 levels predictably diminish the Ace evoked ANO6 currents in Caco-2 cells and native tissue as well as in mouse loop experiments. Because mutation of the conserved residues of the proximal KR motif largely diminished the PIP2 signal, it therefore is likely that these residues at the proximal KR motif may participate in ANO6-PIP2 interactions. Our results seem to indicate that ANO6 is exclusively regulated by PIP2 and that disruption of the PIP2 synthesis or PIP2-binding sites results in dysfunctional or non-functional ANO6 channels. The feature we emphasize here is that PIP2 binding is a mechanism to activate ANO6, which subsequently increase fluid accumulation inducing Cl− secretion by Ace stimulation. But what could underlie Ace-mediated stimulation of PIP2 synthesis? We extend the in vitro understanding by looking at involvement of the Rho monomeric GTPase and its downstream target PIP5K, the enzyme that catalyzes the formation of PIP2, and thus, RhoA signaling potentially could be a critical regulator of cellular PIP2 level to the close proximity of ANO6 for Ace stimulation of Cl− secretion. The reduction of Isc stimulated by Ace in presence of RhoA inhibitor, C3 toxin, or KO of RhoA protein expression provided evidence that RhoA may mediate an important regulatory function of Cl− secretion. In unstimulated cells, RhoA is localized predominantly in the cytosol with usually only a small fraction associated with the plasma membrane (43, 44). To provide further evidence for a role of RhoA, Ace stimulation was able to stimulate redistribution of RhoA to the plasma membrane fraction as evident from our Western blotting analysis (Fig. 10D). These data strongly suggest that translocation of RhoA is a consequence of activation by Ace stimulation for Cl− secretion. This is consistent with the findings that blocking RhoA effector ROCK by H1152 reduced both cellular abundance of PIP2, Isc, and fluid accumulation in Caco-2 cells and mouse loop experiments. Because PIP5K is an effector of RhoA and ROCK (33, 34), we tested whether this lipid kinase is involved in RhoA regulation of Cl− secretion in Caco-2 cells. There is an appreciable reduction of Isc by Wort, PAO, and PLL in response to Ace. Most importantly, deficiency of PIP5K protein expression caused complete reduction of Ace-stimulated Isc in Caco-2 cells (Fig. 7, D and E). Together, these data suggest that PIP5K being downstream of RhoA and ROCK in a signaling pathway leads to activation of ANO6 current. Further studies indicating that ANO6 may associate with Rho-GEF-RhoA-PIP5K by forming a complex are required. Excitingly, our very preliminary data gave such hint of speculation (supplemental Fig. S5). We suspect that ANO6 function is determined by co-ordinated and dynamic interactions with at least RhoA, which not only regulated ANO6 but also affected other protein (Rho-GEF, PIP5K) associations with and regulation of ANO6 in a co-ordinated manner, which will be addressed in a future paper.
Based on these results, we propose a model in Fig. 11 that schematically represents the potential mechanism by which PIP2 influences the ANO6 function by Ace stimulation in intestinal epithelium for Cl− secretion to induce diarrhea. This study identifies a novel role for ANO6 and further suggests that one primary mechanism for regulating ANO6 is by alteration of cellular PIP2 levels and association with a physical interaction of the proximal KR motif for intestinal Cl− secretion. In conclusion, our data provide the first evidence to support a specific role of RhoA in the Ace-PIP2-ANO6-Cl− secretion linkage. Our data revealed that PIP2 and ANO6 are powerful and new additions in the mechanisms of secretory diarrhea that have considerable implications for diarrheal disease therapy.
FIGURE 11.
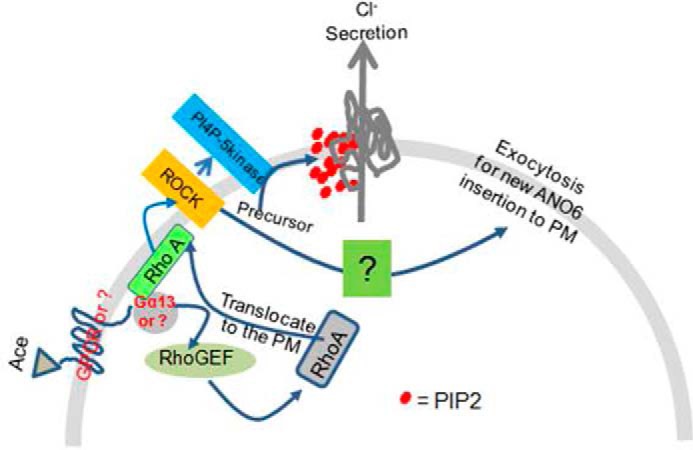
Model for the role and regulation of ANO6 to induce diarrhea by Ace stimulation. Ace-mediated activation of RhoA-ROCK influences function of PIP5K, the enzyme that catalyzes the formation of PIP2, which after subsequent interaction to the N-terminal proximal KR motif became the key mechanism to acutely activate ANO6 for Cl− secretion to induce diarrhea.
Experimental Procedures
Materials
Unless otherwise stated, all chemicals used in this study were obtained from Sigma. Cell culture media and fetal bovine serum (FBS) were purchased from Cell Clone (catalogue number cc3021) and Hi Media (catalogue number RM9970), respectively. cDNA synthesis reagents were purchased from Invitrogen (catalogue number 11904-018), and Real time PCR Master Mix was from Applied Biosystems (catalogue number 4309155). Penicillin/streptomycin was obtained from Invitrogen. TRIzol (catalogue number 15596-026), FITC (catalogue number A11036), and Alexa Fluor 568-conjugated secondary antibody were from Invitrogen. C3 toxin (1 μg/ml, catalogue number CT04) was purchased from Cytoskeleton Inc. GAPDH was obtained from Cell Signaling Technology. PIP2 (catalogue number ab2335), ANO6 (catalogue number ab113777), anti-GFP (catalogue number ab69314), PIP5K (ab137441), and RhoA(ab54835) antibodies were purchased from Abcam (Cambridge, MA). T16A(inh)-AO1 (TAO1, catalogue number 613551) and AO1 (catalogue number 208293) were purchased from Calbiochem. H1152 (catalogue number 2414) was purchased from Tocris.
Cell Culture, cDNA, shRNA, and Transfection
Caco-2 and HEK293 cells were obtained from our institutional core facility, which was originally purchased from American Type Culture Collection (ATCC, Manassas, VA). Caco-2 cells were routinely maintained in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Caco-2 cells between passages 6 and 28 were seeded onto polycarbonate membrane, 12-mm Snapwell-permeable support cell culture inserts (0.4 μm pore size; Costar, catalogue no. 3407) and grown for 10–14 days during which time the media were changed every 48 h. Monolayer resistance was determined using an EVOM Ohmmeter with STX2 electrodes (World Precision Instruments Inc.). Monolayers were considered polarized and mounted in an Ussing chamber when resistance was equal to or greater than 250 ohms/cm2. For whole-cell ANO1 and ANO6 current measurements, HEK293 cells were plated on cover glass and transfected alone or together with cDNA encoding mouse ANO1 or ANO6 tagged with mCherry (monomeric Cherry) and GFP (green fluorescence protein), respectively. The ANO1-mCherry construct was made by replacing EGFP with mCherry as described in Ref. 45. These constructs were obtained from Dr. Shigekazu Nagata of Kyoto University, Japan. Endogenous localization of ANO6 was determined in Caco-2 cells and mouse ileum using anti-ANO6 antibody.
CRISPR/Cas9 and RNA Interference
For ANO6 KO by CRISPR-Cas9 or lenti-shRNA, we used the CRSIPR/Cas9 technique for KO of ANO6, RhoA, and PIP5K genes in the Caco-2 cell line, individually. Briefly, the target sequences were designed using the CRISPR website. The target sequences of various genes are shown in the Table 1. The target sites without the PAM sequences were cloned between two BbsI sites of the vectors pSpCas9 (BB)-2A-GFP (PX458) or pSpCas9 (BB)-2A-puro (PX459). Both the vectors contained two expression cassettes, a human codon-optimized SpCas9 with GFP or puromycin and the single guide RNA, which is a fusion of crRNA and tracrRNA, following the protocol described in Ref. 46. We picked the successful clone by sequencing with the primer named U6 (Table 2). ANO6 targeting sequence was cloned in the vector pX458, although both RhoA and PIP5K targeting sequences were cloned in vector pX459. Caco-2 cells were grown to 70% confluence in a 6-well plate and 10 μg of DNA were transfected with Lipofectamine 2000. ANO6 targeting sequence was cloned in the pX458 vector that, when transfected in Caco-2 cells, exhibited considerable expression of GFP after 48 h of transfection. Cell expressing GFP were sorted by flow cytometry with FAC StAR plus flow cytometer (BD Biosciences). In contrast, cells were selected in the presence of puromycin (10 μg/ml) after RhoA and PIP5K targeting vector transfections. The knockouts of ANO6, RhoA, and PIP5K genes were confirmed by Western blotting as well as by function in Ussing chamber experiments. Control cells transfected the empty vectors and were selected in the same manner mentioned above.
TABLE 2.
Sequences of primers used for cloning and sequencing into vector px458 or px459
| Gene name | Species | Primer sequence |
Length | |
|---|---|---|---|---|
| 5′ → 3′ forward sequence | 5′ → 3′ reverse sequence | |||
| bases | ||||
| U6 | Human | GCCTATTTCCCATGATTCC | 19 | |
| ANO6 | Human | CACCGACGACGACGATGGGGATAT | AAACATATCCCCATCGTCGTCGTC | 24, 24 |
| RHOA | Human | CACCGAACTATGTGGCAGATATCG | AAACCGATATCTGCCACATAGTTC | 24, 24 |
| PIP5K | Human | CACCGCTATTTCGAATGCCATTCCG | AAACCGGAATGGCATTCGAAATAGC | 25, 25 |
In addition to the ANO6 gene KO by the CRISPR/Cas9 technique, the RNA Interference Consortium (TRC) lentivirus-based short hairpin RNAs (shRNAs) were used for their capacity to knock down the ANO6 protein in Caco-2 cells according to our previously described method (38). To trigger the ANO6 gene silencing, we used the clone providing a mean knockdown level of 0.89 within the lentivirus plasmid vector pLKO.1-puromycin obtained from Sigma (TRCN 0000133946, TRCN 0000134710, and TRCN 0000134775). The target sequence for shRNA directed against human ANO6 is 5′-CCGGCCTTGGATCTTATCAGGAAATCTCGAGATTTCCTGATAAGATCCAAGGTTTTTTG-3′. Stable cell lines of Caco-2 with expression of ANO6 knocked down were generated by infecting cells with respective gene-specific lenti-shRNA particles, and positively transduced cells were selected with 10 μg/ml puromycin-containing medium. mRNA expression in transduced cells was evaluated by quantitative PCR. The lenti-shRNA construct specific for GFP was transduced in Caco-2 cells and served as a negative control. This pLKO.1 GFP shRNA construct was a gift from David Sabatini (Addgene plasmid 30323), and the target sequence is 5′-GCAAGCTGACCCTGAAGTTCAT-3′.
Methods
Cloning, Expression and Purification of Ace
V. cholerae classical strain O395 was used for amplification of the ace gene by PCR using the primers 5′-GTAGGCGGATCCATGCTTATG-ATGGACCCC-3′ and 5′-TCTACGGTCGACGCGCCGTGATGAATAAAG-3′ as we described previously (7). Plasmid DNA from selected clones with an N-terminal hexahistidine tag was isolated and transformed into E. coli M15 (pREP4) cells for protein expression. Expression of Ace was induced by the addition of 0.5 mm isopropyl β-d-thiogalactopyranoside to an E. coli M15 (pREP4) cell culture with an A595 of 0.3, for 4 h at 37 °C. The protein obtained in the supernatant was loaded into a pre-equilibrated nickel-agarose resin (Qiagen). The protein was finally eluted in 20 mm imidazole-containing buffer, and the purity of the protein was checked on a 10% Tris-Tricine electrophoresis gel under denaturing conditions and biological activity in an Ussing chamber as described before (7).
Mutagenesis and Construction of ANO6 in pMXs-IRES-Neo Retroviral Expression
Mutagenesis was performed by PCR with Phusion High-Fidelity DNA polymerase (Thermo Scientific). To truncate the N terminus at distal and/or proximal KR motifs of ANO6, we designed upstream primers that had an NheI restriction site and start codon, 5′-AAGCTAGCATGAGACAAGCATACGAATCTAACC-3′, to truncate a distal KR motif and 5′-AAGCTAGCATGTATTACGGCGAGAAGATTGG-3′ to truncate both KR motifs. To create point mutations at Lys and Arg in KR motifs, we designed mutagenic primers and used a QuikChange site-directed mutagenesis kit (Stratagene). The DNA fragment containing the mutation was excised by restriction enzymes and subcloned into pcDNA3.1(+). To express wild-type ANO6 without a fluorescent tag, we designed the downstream primer 5′-TTTTCTCGAGTCATTCGAGTTTTGGCCGCAC-3′, which had stop codon and XhoI restriction site, and subcloned into pIRES2-AcGFP1 (Clontech) or pMXs-IRES-Neoretroviral vector (catalogue no. RTV-105, Cell Biolabs, Inc., San Diego). The mutation was confirmed by sequencing with ABI 3100 Genetic Analyzer. For ANO6 rescued experiments, the mANO6 gene was cloned in pMXs-IRES-Neo Retroviral vector between EcoRI and XhoI restriction sites. This plasmid was then transfected into the HEK293 cell line along with envelope plasmid (pVSVG) using our established protocol for production of viral particles (38). The supernatant from the transfected cells was removed 24–36 h later and applied to ANO6 KO Caco-2 cells. Transduced cells were selected and maintained in the presence of the appropriate antibiotics (G418 sulfate, Invitrogen) (400 μg/ml). Caco-2 cells were grown in DMEM supplemented with 5% FBS.
Animal and Tissue Preparation
Overnight fasting of C57BL/6 male mice, 6–8 weeks of age, was done in all experiments. Animal experiments and protocols used to demonstrate the Ussing chamber method and ileal loop experiments were approved by the Institutional Animal Ethics Committee (NICED/CPCSEA/AW/(225)/IAEC-MH/3). Following euthanasia by CO2 inhalation, the mouse intestine was removed by sharp dissection but not pulling the intestine from its mesenteric attachment to avoid damaging the epithelium, as described elsewhere (47). The intestinal section was opened longitudinally along the mesenteric border so that the anti-mesenteric mucosa will be situated in the aperture of the chamber. After completion of sero-musculature stripping, done on the top of an ice-cold glass plate as we described previously (48), the mucosa was typically mounted on the pins of sliders for snapwell chambers (catalogue number P2304; area 0.30 cm2; Physiologic Instrument Inc.) and bathed on both mucosal (apical) and serosal (basolateral) sides with HEPES-buffered solution for measurement of transepithelial transport of ions.
Electrophysiology
Measurement of Transepithelial Short-circuit Current
Caco-2 cells grown on snapwell inserts and section of mice ileum were mounted in an Ussing chamber, and transepithelial potential differences were clamped to 0 mV using VCC MC6 multichannel voltage-current clamp amplifier as described previously (24, 38). The Isc was continuously recorded using Ag-AgCl electrodes in 3 m KCl agar bridges. Apical and basolateral solutions were maintained at 37 °C by heated water jackets of metallic bar and were separately perfused and oxygenated with 100% O2. The bath Ringer solution contained the following (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES adjusted to pH 7.4. The change in Isc induced by the treatment was expressed as the difference from the baseline to the steady state.
Measurement of Apical Membrane Chloride Conductance
The apical membrane ICl was investigated in Caco-2 cell monolayers and mouse ileum according to the method described by Schultheiss et al. (49). In brief, the basolateral membrane was depolarized with a solution containing high potassium concentration for 40–50 min. Thus, the ICl current was measured in the presence of a serosal-to-mucosal Cl− gradient with the following bath solution (in mm): apical, 107 potassium gluconate, 4.5 KCl, 25 NaHCO3, 1.8 Na2HPO4, 0.2 NaH2PO4, 5.75 calcium gluconate, 1.0 MgSO4, and 1glucose; and basolateral, 111.5 KCl, 25 NaHCO3, 1.8 Na2HPO4, 0.2 NaH2PO4, 1.25 CaCl2, 1.0 MgSO4, and 12 glucose. This procedure allows for the measurement of changes in the apical anion conductance. Under these conditions, there was an agonist-induced ICl as Cl− ions from the cell moved down the concentration gradient through the Cl− channels in the apical membrane.
Whole-cell Current Recording by Patch Clamp
Heterologously transfected ANO1 and ANO6 into HEK293 cells were superfused with Tyrode's solution containing the following (in mm): 140 NaCl, 0.33 NaH2PO4, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 5.5 glucose, 5 HEPES/NaOH, pH 7.4. To measure Ace-induced current, extracellular Na+ was substituted by NMDG+ and the “NMDG-Cl” bath solution contained the following (in mm): 150 NMDG-Cl, 1 CaCl2, 5 HEPES/NMDG, pH 7.4. To determine the Cl− permeability of the outward current, all chloride salts were iso-osmotically replaced by respective glutamate salts. To induce the Ca2+-activated Cl− currents, the Ca2+ ionophore A23187 (10 μm) was applied in “NMDG-Cl” bath. The patch clamp pipettes were filled with a cesium-glutamate pipette solution containing the following (in mm): 130 cesium glutamate, 10 CsCl, 1 MgCl2, 0.5 EGTA, 10 glucose, 10 HEPES/CsOH, pH 7.4. Whole-cell currents in Caco-2 cells were recorded using gramicidin-perforated patch methods. Gramicidin D was dissolved in dimethyl sulfoxide at 20 mg/ml and then diluted in a standard KCl-rich perforated patch solution to a final concentration of 0.1 mg/ml. The standard KCl-rich perforated patch solution contained the following (in mm): 150 KCl, 10 HEPES/KOH, pH 7.4. Caco-2 cells were superfused with a standard bathing solution contained the following (in mm): 145 NaCl, 5 KCl, 1 CaCl2, 5 HEPES/NaOH, pH 7.4. The membrane potential was corrected for the liquid junction potential at the tip of the patch pipette in the bathing solution, and for that at the tip of the indifferent reference electrode filled with bathing solution and placed in the bath. Gramicidin-perforated patch recording was started after the stabilization of the capacitive currents. Experiments were carried out at 23–26 °C. Whole-cell currents were recorded using EPC 800 patch clamp amplifier (HEKA, Lambrecht, Germany). The amplifier was driven by Clampex 9 (Molecular Devices, Sunnyvale, CA). Subsequent current analysis was performed using Clampfit 9 (Molecular Devices).
Real Time PCR
Quantitative RT-PCR (qPCR) was performed using StepOnePlusTM real time PCR system (Applied Biosystems, Foster City, CA) as described previously (38). Briefly, total RNA were extracted from Caco-2 cell monolayer or mouse intestinal tissue using TRIzol reagent according to the manufacturer's instructions and served as the template for cDNA preparation using superscript cDNA preparation kit. The thermal cycler was programmed as follows: step 1, one cycle, 50 °C for 2 min; step 2, one cycle, 95 °C for 10 min; step 3, 45 cycles, 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s; step 4 (dissociation; ramp rate, 2%), 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15 s. Amplification reactions were performed in a final volume of 25 μl containing cDNA, 300 nm each of forward and reverse primers listed in Table 3 for ANO1 and ANO6 and SYBR Green PCR Master Mix. To control for specific PCR products, a dissociation curve was generated after the end of the last cycle. Quantitative expression among ANO6 variants in Caco-2 and mouse tissue were also performed using forward and reverse primes listed in Table 4.
TABLE 3.
List of sequence of forward and reverse primers of Ano1 and Ano6 used for qPCR in this study
| Gene name | Species | Primer sequence |
Length | |
|---|---|---|---|---|
| 5′ → 3′ forward | 5′ → 3′ reverse | |||
| bases | ||||
| ANO1 | Human | GAGGAGTACGAGGGCAACCT | CCGACTCCGTGGATTTTAGT | 20, 20 |
| ANO6 | Human | CCATCATGCAAGGAATAGCA | TGTCCGACGTGAAAGCTATG | 20, 20 |
| Ano1 | Mouse | CGACTGGCATTTGTCATTGT | ATCAGGGATCACCCAGTCC | 20, 19 |
| Ano6 | Mouse | GGGAACAGGATTACCATCTGC | AAGTGACGAACCCAAACTGAA | 21, 21 |
TABLE 4.
List of primer sequences of Ano6 variants used for qPCR in this study
These sequences were adopted from Ref. 32.
| Name of Ano6 variants | Species | Primer sequence |
Length | |
|---|---|---|---|---|
| 5′ → 3′ forward | 5′ → 3′ reverse | |||
| bases | ||||
| Variant 1 | Human | ACGATGGGGATATCGTGTTGG | AAACAATGCGGCTTCTGGTG | 20, 21 |
| Variant 2 | Human | CCCCTTTTGTTCTGCTGTGG | AAACAATGCGGCTTCTGGTG | 20, 20 |
| Variant 3 | Human | GCCTATGGGCAAACTGGGAT | GCCAAGATCATGCCACTGTG | 20, 20 |
| Variant 5 | Human | GGTGATGTTCCTGCCTCCAG | AAACAATGCGGCTTCTGGTGT | 20, 20 |
| Variant 1 | Mouse | AGGATGGAGACATTGGTGATGT | AACCAGTTGAGGTTGCCAAAG | 22, 21 |
| Variant 2 | Mouse | TGCTGAACATGGAGCTGGAG | ATAGGTGCACAGCACTTCCC | 20, 20 |
Mouse Ileal Loop Experiment
The ileal loop experiment was performed in male mice of 6–8 weeks old by a modified rabbit ileal loop assay originally described by De and Chatterje (50). Following gut sterilization, the animals were kept fasting for 24 h prior to surgery and fed only water ad libitum. Anesthesia was induced by a mixture of ketamine (35 mg per kg of body weight) and xylazine (5 mg per kg of body weight). A laparotomy was performed, and an experimental loop of 5 cm length was constricted at the terminal ileum by tying with non-absorbable silk. The following fluids were instilled in each loop by means of a tuberculin syringe fitted with a disposable needle through the ligated end of the loop as the ligature is tightened: Ace, PBS (negative controls), Ace + AO1, Ace + TAO1, Ace + PAO, Ace + PLL, and Ace + H1152. The intestine was returned to the peritoneum, and the mice were sutured. Mice were allowed to recovery before being returned to their cages. After 6 h, these animals were sacrificed by euthanasia by CO2 inhalation, and the loops were excised. The fluid from each loop was collected, and the ratio of the amount of fluid contained in the loop with respect to the length of the loop (fluid accumulation ratio, gram/cm) was calculated as a reflection of efficacy of Ace.
Immunofluorescence Study
Method used for immunofluorescence study was originally described in our previous paper (38). Briefly, transfected HEK293 cells grown onto a fluoro dish (FD 35, World Precision Instruments) were examined on a Zeiss LSM 710 laser-scanning confocal microscope (×20 objective, oil immersion). For staining of mouse intestinal tissue sections, the intestine was dissected, and the ileum was rinsed with ice-cold saline and fixed in 3% paraformaldehyde prior to standard paraffin embedding as described. Briefly, individual sections were heat-fixed and deparaffinized followed by microwave treatment in 0.01 m sodium citrate solution for antigen recovery. Endogenous peroxidase activity was blocked by a 30-min preincubation in H2O2 (Sigma). Sections were then washed in PBS and blocked with 5% normal goat serum in PBS for 30 min at room temperature. Subsequently, sections were incubated with primary ANO6 antibody diluted (1:100) in 5% normal goat serum in PBS for 1 h at room temperature followed by FITC-conjugated goat secondary antibody, and images were obtained using a Zeiss LSM 710 confocal microscope. For PIP2 fluorescence study, Caco-2 cells grown onto transwell inserts were treated with Ace for 10 min before being fixed on ice in 3% paraformaldehyde solution in PBS. Cell monolayers were incubated with PIP2-specific mouse monoclonal antibody following our previously published protocol for confocal study (38).
Measurement of Intracellular Ca2+ Concentration
HEK293 or Caco-2 cells were grown on glass bottom dishes. Cells were loaded with 3μm Fura2-AM (Dojindo, Kumamoto, Japan) in culture medium without serum for 30–45 min in the dark at 37 °C. The cells were perfused with a standard bath solution containing the following (in mm): 115 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 25 NaHCO3, 10 glucose, 10 HEPES/NaOH, pH 7.4. The solution was equilibrated with 5% CO2 in O2. The temperature was kept at 37 °C. Fura2 fluorescence images were monitored at a 510-nm emission with alternating excitation at 340 and 380 nm using Aquacosmos imaging system equipped with a cooled CCD camera (Hamamatsu Photonics, Hamamatsu, Japan). The ratio of the intensities excited at 340 and 380 nm was used to indicate the relative changes in [Ca2+]i. All measurements shown are representative of a minimum of three independent experiments with no fewer than 40 cells calculated in each study.
Immunoprecipitation and Western Blotting
Protein was isolated from untransfected or transfected with a GFP-tagged mouse ANO6-expressing HEK293 cells in RIPA (Sigma) lysis buffer containing 1% protease inhibitor mixture. Prior to the addition of mouse monoclonal GFP antibody (Sigma), protein lysates were pre-cleared with protein G-agarose beads (Pierce). Incubation of the pre-cleared protein lysates with antibody was performed overnight at 4 °C, and the protein-antibody complex was immobilized with the addition of 20 μl of a 50% slurry of protein G-agarose beads for 3 h at 4 °C. The beads were washed five times in wash buffer, and after the last washing step, beads were mixed in ×2 Laemmli sample buffer. For Western blotting analysis, the 10% SDS-polyacrylamide gel was transferred to a polyvinylidene difluoride membrane (Bio Rad). Membranes were incubated with anti-PIP2 (1:100) or anti-ANO6 (1:500) antibodies overnight at 4 °C. Proteins were visualized using goat anti-mouse IgG conjugated to horseradish peroxidase (HRP), and 1:1000 dilution and blots were visualized using chemiluminescence (Pierce).
Statistical Analyses
Statistical analyses were performed using Origin 7.5 software (Origin Lab). Data were subjected to t test (for paired or unpaired samples as appropriate) and analysis of variance (ANOVA) for statistical analysis. p ≤ 0.05 was considered as significant difference with * indicating p ≤ 0.05, ** indicating p ≤ 0.01, and *** indicating ≤0.001.
Author Contributions
I. A. S., M. H., J. A., and K. M. H. conducted most of the experiments, analyzed the results, and wrote the paper. K. M. H. conceived the idea of this project. M. H. conducted experiments on ANO1 and ANO6 whole-cell current measurements in HEK293 and Caco-2 cells by patch clamp recordings and intracellular calcium measurements by ratiometric imaging system, deletion, and mutagenesis of the KR motif. P. G., T. S., and P. S. conducted experiments on real time PCR and Western blottings. J. A., R. B., and T. C. conducted experiments on gene cloning for Ace purification. J. A. conducted experiments on mouse tissue for electrophysiological study in an Ussing chamber and some Western blottings. T. C., P. C., and M. K. C. provided intellectual input for writing the paper. D. G. did experiments for CRISPR/Cas9-mediated genes KO in Caco-2 cells.
Supplementary Material
Acknowledgments
We gratefully acknowledge the help and research facilities provided by the Director, National Institute of Cholera and Enteric Diseases (ICMR), Kolkata, India. We acknowledge Sohini Basu (summer student, National Institute of Cholera and Enteric Diseases), for help in carrying out mouse ileal loop experiments.
This work was supported in part by Government of India, Ministry of Science and Technology, Department of Biotechnology Grant BT/HRD/35/02/07/2009 (to K. M. H.), Japan Society for the Promotion of Science KAKENHI Grant 24790226 (to M. H.), Indian Council of Medical Research, Government of India (to I. A. S., J. A.), and a Department of Science and Technology fellowship (to P. S.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S5.
- Ace
- accessory cholera enterotoxin
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- ANOVA
- analysis of variance
- PAO
- phenyl arsine oxide
- PLL
- poly-l-lysine
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- ANO
- anoctamin
- Wort
- wortmannin
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CaCC
- calcium-activated chloride channel
- NKCC
- Na,K,2Cl
- FSK
- forskolin
- DIDS
- 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid
- TFN
- tamoxifen
- NPBB
- 5-nitro-2-(3-phenylpropylamino)benzoic acid
- NFA
- niflumic acid
- TA
- tannic acid
- qPCR
- quantitative PCR
- pF
- picofarad
- [Ca2+]i
- intracellular calcium
- BAPTA-AM
- 1, 2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- PIP5K
- phosphatidylinositol 4-phosphate 5-kinase
- ROCK
- Rho kinase
- EGFP
- enhanced GFP
- KI
- knock-in
- lenti-shRNA
- lentivirus-based short hairpin RNA
- NMDG
- N-methyl-d-glucamine.
References
- 1. Walker C. L., Rudan I., Liu L., Nair H., Theodoratou E., Bhutta Z. A., O'Brien K. L., Campbell H., and Black R. E. (2013) Global burden of childhood pneumonia and diarrhoea. Lancet 381, 1405–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sears C. L., and Kaper J. B. (1996) Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 60, 167–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fasano A., Baudry B., Pumplin D. W., Wasserman S. S., Tall B. D., Ketley J. M., and Kaper J. B. (1991) Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. U.S.A. 88, 5242–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fasano A., and Uzzau S. (1997) Modulation of intestinal tight junctions by zonula occludens toxin permits enteral administration of insulin and other macromolecules in an animal model. J. Clin. Invest. 99, 1158–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trucksis M., Galen J. E., Michalski J., Fasano A., and Kaper J. B. (1993) Accessory cholera enterotoxin (Ace), the third toxin of a Vibrio cholerae virulence cassette. Proc. Natl. Acad. Sci. U.S.A. 90, 5267–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trucksis M., Conn T. L., Wasserman S. S., and Sears C. L. (2000) Vibrio cholerae ACE stimulates Ca2+-dependent Cl−/HCO3− secretion in T84 cells in vitro. Am. J. Physiol. Cell Physiol. 279, C567–C577 [DOI] [PubMed] [Google Scholar]
- 7. Chatterjee T., Mukherjee D., Dey S., Pal A., Hoque K. M., and Chakrabarti P. (2011) Accessory cholera enterotoxin, Ace, from Vibrio cholerae: structure, unfolding, and virstatin binding. Biochemistry 50, 2962–2972 [DOI] [PubMed] [Google Scholar]
- 8. Kunzelmann K. (2001) CFTR: interacting with everything? News Physiol. Sci. 16, 167–170 [DOI] [PubMed] [Google Scholar]
- 9. Xiao Q., Yu K., Perez-Cornejo P., Cui Y., Arreola J., and Hartzell H. C. (2011) Voltage- and calcium-dependent gating of TMEM16A/ANO1 chloride channels are physically coupled by the first intracellular loop. Proc. Natl. Acad. Sci. U.S.A. 108, 8891–8896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morris A. P., Scott J. K., Ball J. M., Zeng C. Q., O'Neal W. K., and Estes M. K. (1999) NSP4 elicits age-dependent diarrhea and Ca2+-mediated I− influx into intestinal crypts of CF mice. Am. J. Physiol. 277, G431–G444 [DOI] [PubMed] [Google Scholar]
- 11. Ousingsawat J., Mirza M., Tian Y., Roussa E., Schreiber R., Cook D. I., and Kunzelmann K. (2011) Rotavirus toxin NSP4 induces diarrhea by activation of TMEM16A and inhibition of Na+ absorption. Pflugers Arch. 461, 579–589 [DOI] [PubMed] [Google Scholar]
- 12. Galietta L. J. (2009) The TMEM16 protein family: a new class of chloride channels? Biophys. J. 97, 3047–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartzell C., Putzier I., and Arreola J. (2005) Calcium-activated chloride channels. Annu. Rev. Physiol. 67, 719–758 [DOI] [PubMed] [Google Scholar]
- 14. Hartzell H. C., Yu K., Xiao Q., Chien L. T., and Qu Z. (2009) Anoctamin/TMEM16 family members are Ca2+-activated Cl− channels. J. Physiol. 587, 2127–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duran C., Thompson C. H., Xiao Q., and Hartzell H. C. (2010) Chloride channels: often enigmatic, rarely predictable. Annu. Rev. Physiol. 72, 95–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grubb S., Poulsen K. A., Juul C. A., Kyed T., Klausen T. K., Larsen E. H., and Hoffmann E. K. (2013) TMEM16F (anoctamin 6), an anion channel of delayed Ca2+ activation. J. Gen. Physiol. 141, 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimizu T., Iehara T., Sato K., Fujii T., Sakai H., and Okada Y. (2013) TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am. J. Physiol. Cell Physiol. 304, C748–C759 [DOI] [PubMed] [Google Scholar]
- 18. Namkung W., Phuan P. W., and Verkman A. S. (2011) TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J. Biol. Chem. 286, 2365–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., and Galietta L. J. (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594 [DOI] [PubMed] [Google Scholar]
- 20. Yang Y. D., Cho H., Koo J. Y., Tak M. H., Cho Y., Shim W. S., Park S. P., Lee J., Lee B., Kim B. M., Raouf R., Shin Y. K., and Oh U. (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215 [DOI] [PubMed] [Google Scholar]
- 21. Schroeder B. C., Cheng T., Jan Y. N., and Jan L. Y. (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stephan A. B., Shum E. Y., Hirsh S., Cygnar K. D., Reisert J., and Zhao H. (2009) ANO2 is the cilial calcium-activated chloride channel that may mediate olfactory amplification. Proc. Natl. Acad. Sci. U.S.A. 106, 11776–11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schreiber R., Uliyakina I., Kongsuphol P., Warth R., Mirza M., Martins J. R., and Kunzelmann K. (2010) Expression and function of epithelial anoctamins. J. Biol. Chem. 285, 7838–7845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoque K. M., Woodward O. M., van Rossum D. B., Zachos N. C., Chen L., Leung G. P., Guggino W. B., Guggino S. E., and Tse C. M. (2010) Epac1 mediates protein kinase A-independent mechanism of forskolin-activated intestinal chloride secretion. J. Gen. Physiol. 135, 43–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hilgemann D. W., Feng S., and Nasuhoglu C. (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, re19. [DOI] [PubMed] [Google Scholar]
- 26. Suh B. C., and Hille B. (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu. Rev. Biophys. 37, 175–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suh B. C., and Hille B. (2005) Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 15, 370–378 [DOI] [PubMed] [Google Scholar]
- 28. Nakanishi S., Catt K. J., and Balla T. (1995) A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. U.S.A. 92, 5317–5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wiedemann C., Schäfer T., and Burger M. M. (1996) Chromaffin granule-associated phosphatidylinositol 4-kinase activity is required for stimulated secretion. EMBO J. 15, 2094–2101 [PMC free article] [PubMed] [Google Scholar]
- 30. Várnai P., and Balla T. (1998) Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell Biol. 143, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barret C., Roy C., Montcourrier P., Mangeat P., and Niggli V. (2000) Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP2) binding site in the NH2-terminal domain of ezrin correlates with its altered cellular distribution. J. Cell Biol. 151, 1067–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scudieri P., Caci E., Venturini A., Sondo E., Pianigiani G., Marchetti C., Ravazzolo R., Pagani F., and Galietta L. J. (2015) Ion channel and lipid scramblase activity associated with expression of TMEM16F/ANO6 isoforms. J. Physiol. 593, 3829–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weernink P. A., Meletiadis K., Hommeltenberg S., Hinz M., Ishihara H., Schmidt M., and Jakobs K. H. (2004) Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J. Biol. Chem. 279, 7840–7849 [DOI] [PubMed] [Google Scholar]
- 34. Yang S. A., Carpenter C. L., and Abrams C. S. (2004) Rho and Rho-kinase mediate thrombin-induced phosphatidylinositol 4-phosphate 5-kinase trafficking in platelets. J. Biol. Chem. 279, 42331–42336 [DOI] [PubMed] [Google Scholar]
- 35. Sakai N., Chun J., Duffield J. S., Wada T., Luster A. D., and Tager A. M. (2013) LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 27, 1830–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhattacharya M., Babwah A. V., and Ferguson S. S. (2004) Small GTP-binding protein-coupled receptors. Biochem. Soc. Trans. 32, 1040–1044 [DOI] [PubMed] [Google Scholar]
- 37. Jaffe A. B., and Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 38. Sheikh I. A., Koley H., Chakrabarti M. K., and Hoque K. M. (2013) The Epac1 signaling pathway regulates Cl− secretion via modulation of apical KCNN4c channels in diarrhea. J. Biol. Chem. 288, 20404–20415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anvari S., Najar Peerayeh S., Behmanesh M., and Boustanshenas M. (2012) Biological activity of recombinant accessory cholerae enterotoxin (ace) on rabbit ileal loops and antibacterial assay. Cell J. 14, 209–214 [PMC free article] [PubMed] [Google Scholar]
- 40. Duran C., Qu Z., Osunkoya A. O., Cui Y., and Hartzell H. C. (2012) ANOs 3–7 in the anoctamin/Tmem16 Cl− channel family are intracellular proteins. Am. J. Physiol. Cell Physiol. 302, C482–C493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alexander S. P., Benson H. E., Faccenda E., Pawson A. J., Sharman J. L., Spedding M., Peters J. A., Harmar A. J., and CGTP Collaborators (2013) The concise guide to pharmacology 2013/14: ligand-gated ion channels. Br. J. Pharmacol. 170, 1582–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu F. X., Sun H. Q., Janmey P. A., and Yin H. L. (1992) Identification of a polyphosphoinositide-binding sequence in an actin monomer-binding domain of gelsolin. J. Biol. Chem. 267, 14616–14621 [PubMed] [Google Scholar]
- 43. Adamson P., Paterson H. F., and Hall A. (1992) Intracellular localization of the P21rho proteins. J. Cell Biol. 119, 617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boivin D., and Béliveau R. (1995) Subcellular distribution and membrane association of Rho-related small GTP-binding proteins in kidney cortex. Am. J. Physiol. 269, F180–F189 [DOI] [PubMed] [Google Scholar]
- 45. Suzuki J., Fujii T., Imao T., Ishihara K., Kuba H., and Nagata S. (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 288, 13305–13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clarke L. L. (2009) A guide to Ussing chamber studies of mouse intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1151–G1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hoque K. M., Rajendran V. M., and Binder H. J. (2005) Zinc inhibits cAMP-stimulated Cl secretion via basolateral K-channel blockade in rat ileum. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G956–G963 [DOI] [PubMed] [Google Scholar]
- 49. Schultheiss G., Lán Kocks S., and Diener M. (2005) Stimulation of colonic anion secretion by monochloramine: action sites. Pflugers Arch. 449, 553–563 [DOI] [PubMed] [Google Scholar]
- 50. De S. N., and Chatterje D. N. (1953) An experimental study of the mechanism of action of Vibrio cholerae on the intestinal mucous membrane. J. Pathol. Bacteriol. 66, 559–562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.