

Abstract
Salmonella pathogenesis studies to date have focused on Salmonella typhimurium, and the pathogenesis of a second major serotype, Salmonella enteritidis, is poorly understood. Salmonella spp. possess effector proteins that display biochemical activities and modulate host functions. Here, we generated a deletion mutant of the effector AvrA, S.E-AvrA−, and a plasmid-mediated complementary strain, S.E-AvrA−/pAvrA+ (S.E-AvrA+), in S. Enteritidis. Using in vitro and in vivo infection models, we showed that AvrA stabilizes epithelial tight junction (TJ) proteins, such as ZO-1, in human intestinal epithelial cells. Transepithelial electrical resistance was significantly higher in cells infected with S.E-AvrA+ than in cells infected with S.E-AvrA−. Inhibition of the JNK pathway suppresses the disassembly of TJ proteins; we found that enteritidis AvrA inhibited JNK activity in cells infected with wild type or S.E-AvrA+ strains. Therefore, Enteritidis AvrA-induced ZO-1 stability is achieved via suppression of the JNK pathway. Furthermore, the S.E-AvrA− strain led to enhanced bacterial invasion, both in vitro and in vivo. Taken together, our data reveal a novel role for AvrA in S. Enteritidis: Enteritidis AvrA stabilizes intestinal TJs and attenuates bacterial invasion. The manipulation of JNK activity and TJs in microbial-epithelial interactions may be a novel therapeutic approach for the treatment of infectious diseases.
Keywords: c-Jun N-terminal kinase (JNK), infection, inflammation, Salmonella enterica, tight junction
Introduction
Salmonella spp. are Gram-negative, facultative anaerobes, and intracellular pathogens in both humans and animals that pose a major public health problem worldwide. It is estimated that more than 1 million people acquire a Salmonella infection as a foodborne illness and that such infections contribute to >350 deaths annually in the United States (1) and 93.8 million illnesses and 155,000 deaths worldwide each year (2). In the past 20 years, Salmonella enterica serotype enteritidis (S.Enteritidis)3 has become one of the most common serotypes reported in human cases of salmonellosis worldwide, despite ongoing implementation of targeted control and prevention measures (3–5). In humans, S. Enteritidis salmonellosis causes symptoms such as abdominal pain, diarrhea, nausea, vomiting, fever, and headache (6). S. Enteritidis is also increasingly reported in cases of invasive and extraintestinal infections, such as septicemia, arthritis, endocarditis, meningitis, and urinary tract infections (7–13). Most Salmonella pathogenesis studies to date have focused on Salmonella typhimurium; thus, the pathogenesis of another major serotype, Salmonella Enteritidis, is poorly understood.
Salmonella species possess a range of effector proteins that are translocated into host cells via a type III secretion system (14, 15). These bacterial effector proteins display a large repertoire of biochemical activities and modulate the function of host regulatory molecules, including tight junction (TJ) proteins and their upstream regulators (16–20). AvrA is a Salmonella effector protein secreted by the Salmonella pathogenicity island 1 (SPI-1) type III secretion system. The AvrA protein in S. typhimurium is an anti-inflammatory effector that processes acetyltransferase activity toward specific host MAPKKs and inhibits the host JNK/AP-1 and NF-κB signaling pathways (21–25).
The intestinal epithelium is formed by a single layer of epithelial cells that separates the intestinal lumen from the underlying lamina propria. The space between these cells is sealed by TJs, which regulate the permeability of the intestinal barrier (26). TJs are the most apical component of intercellular junctional complexes formed by integral membrane proteins such as occludin, claudin family members, and junctional adhesion molecules. The cytoplasmic domain of occludin and claudins binds to peripheral membrane proteins known as the zonula occludens (ZO), such as ZO-1, ZO-2, and ZO-3 (27, 28), which separate the apical and basolateral cell surfaces to provide a barrier function, inhibit solute and water flow through the paracellular space, and inhibit bacterial invasion (29). Signaling pathways involved in the assembly, disassembly, and maintenance of TJs are controlled by numerous signaling pathways, such as the protein kinase C, MAPK, myosin light chain kinase, and Rho GTPase pathways (30–33).
The JNKs were originally identified based on their activation in response to environmental stress stimuli, such as bacterial infection. JNK binds and phosphorylates c-Jun within its transcriptional activation domain, and this signaling pathway generally contributes to inflammatory responses (34). The JNK pathway plays a role in regulating TJ integrity in different epithelial cells (25, 35, 36). Previous studies on AvrA were based on S. typhimurium and were performed using in vitro and mouse models (37). However, the role of the bacterial effector AvrA in S. Enteritidis remains unknown. We hypothesized that S. Enteritidis AvrA stabilizes intestinal epithelial TJs and decreases bacterial invasion. Using S. Enteritidis-infected intestinal epithelial cells and a mouse colitis model, we found that enteritidis AvrA may reprogram the regulation of TJs in eukaryotic cells to benefit the pathogen.
Results
Establishment of an AvrA Deletion Mutant and a Plasmid-mediated Complemented Strain in S. Enteritidis
We first established an AvrA deletion mutant, S.E.-AvrA−, in S. Enteritidis strain C50336 using λ-Red-mediated recombination and complemented by plasmid pBR322-AvrA (Fig. 1). Our PCR data showed that the AvrA gene was deleted in the S.E-AvrA− mutant and restored in the complemented strain S.E-AvrA+ (Fig. 1A). RT-PCR verification of the AvrA gene transcription further confirmed the successful generation of the AvrA mutant and the complemented strain, with the GMK bacterial reference gene for normalization (Fig. 1B). Our Western blot data showed that the AvrA protein was absent in the mutant strain (S.E-AvrA−) and overexpressed in the complemented strain (S.E-AvrA+) (Fig. 1C). Ponceau S staining indicated even protein loading in each lane. The growth characteristics of the strains were determined in LB liquid medium; there was no difference between the AvrA mutant or complemented strains and the wild type C50336 strain (Fig. 1D). The mutant and complemented strains were characterized biochemically using the API 20E identification kit, which revealed no differences between the mutant and complemented strains relative to the wild type strain (Fig. 1E).
FIGURE 1.

Construction of the AvrA deletion mutant and complemented strains from S. Enteritidis strain C50336. The AvrA deletion mutant was constructed using the λ-Red-mediated recombination system and complemented by plasmid pBR322-AvrA. Bacteria were grown in liquid LB medium at 37 °C for 12 h, and harvested bacteria were used in the following studies. A, PCR verification of the C50336 AvrA mutant and its complementation. The wild type strain harbors the complete AvrA gene (909 bp); the wild type PCR product amplified by the primer AvrA-e is 2136 bp (left panel), and the PCR product from S.E.-AvrA− (AvrA gene deleted) is 1357 bp (right panel). The PCR fragment amplified by primer AvrA-r in the complemented strain (right panel, 1591 bp) is identical to that in the wild type plasmid pBR322-AvrA. The small band in the S. E-AvrA+ lane corresponds to DNA amplified from the deleted bacterial AvrA. B, RT-PCR verification of AvrA gene transcription in the mutant and complemented strains; GMK was used as the bacterial reference transcript. C, Western blot detection of AvrA protein expression in the mutant and complemented strains and relative protein intensity, as shown by Ponceau S staining. D, growth curves of wild type S. Enteritidis C50336, the AvrA mutant S.E-AvrA− and the complemented strain S.E-AvrA+. Bacteria were grown in liquid LB medium at 37 °C for 12 h with agitation, and the A600 values of triplicate cultures in LB medium were determined in 1-h intervals. The data are reported as the means ± S.D. of three independent experiments. E, biochemical tests of S. Enteritidis wild type, AvrA mutant, and AvrA complementary strain using the Api 20E identification kit. Sterile water was used as control.
Enteritidis AvrA Stabilizes Epithelial TJ Proteins and Alters Transepithelial Electrical Resistance in Vitro
ZO-1 is a TJ protein on the surface of intestinal epithelial cells. In human intestinal epithelial Caco-2 BBE cells, ZO-1 protein levels were decreased after S.E-AvrA− infection compared with wild type S.E or S.E-AvrA+ infection (Fig. 2A). Densitometry analysis showed a significant difference between cells infected with S.E-AvrA− and those infected with either wild type S.E or S.E-AvrA+ (Fig. 2, B and C). In contrast, there were no significant changes in the TJ proteins occludin, claudin-1, and claudin-7 or the adhesion junction protein E-cadherin with or without S. Enteritidis colonization (Fig. 2A). We observed similar trends for ZO-1 following S.E-AvrA−, S.E-AvrA+, or wild type S.E infection in HCT116 and SKCO15 cells (Fig. 2, D and E). RT-PCR data showed no change in ZO-1 mRNA expression (Fig. 2, F–H).
FIGURE 2.

S. Enteritidis AvrA stabilizes epithelial TJ proteins and inhibits JNK pathway activation in epithelial cells. AvrA expression stabilizes the TJ protein ZO-1 and inhibits the JNK pathway in a model of bacteria-infected human epithelial cells. A, Western blot shows the tight junction proteins ZO-1, claudin-1, claudin-7, and occludin; the adhesion junction protein E-cadherin; and JNK in human epithelial Caco-2 BBE cells after colonization with S. Enteritidis wild type, AvrA mutant, or AvrA-complemented strains. B and C, the relative density of ZO-1 (B) and p-SAPK/JNK (54 kDa) (C) was determined using Quantity One 4.6.2 software (Bio-Rad). D and E, Western blot shows tight junction and adhesion junction protein expression, as well as JNK expression in human epithelial HCT116 cells (D) and SKCO15 cells (E). Epithelial cells were colonized with S. Enteritidis wild type, AvrA mutant, or AvrA-complemented strains. F–H, RT-PCR shows the relative ZO-1 mRNA level in epithelial Caco-2 (F), HCT116 (G), or SKCO15 cells (H) treated with different Salmonella strains (n = 6). *, p ≤ 0.05; **, p ≤ 0.01.
We further determined the distribution of ZO-1 protein in Caco-2 BBE cells by immunofluorescence. As shown in Fig. 3, ZO-1 localization was disrupted in S.E-AvrA−-infected cells but was unchanged in S.E-AvrA+ or wild type S.E-infected cells. There was no change in the adhesion junction protein E-cadherin with or without S. Enteritidis colonization (see Fig. 6E).
FIGURE 3.

S. Enteritidis AvrA stabilizes ZO-1 structure and changes the TEER. Caco-2 BBE cells were cultured on an insert and treated with different Salmonella strains. At the indicated times, the changes in ZO-1 and the TEER were measured. A, immunofluorescence shows the normal localization of ZO-1 in Caco-2 BBE control cells and the disruption following infection by AvrA-mutant bacteria. There were no significant changes in other TJ proteins, such as occludin, claudin-1, and claudin-7, or in the adhesion junction protein E-cadherin between S. E-AvrA− and the wild type or complemented strains. B, high TEER is evident in Caco-2 BBE cells after colonization with wild type or AvrA complemented strains. TEER was measured at the indicated times, and multiple comparisons of mean TEER values were performed using two-way ANOVA and generalized linear mixed models. The mean differences between the TEER values for the S.E-AvrA− mutant and either the S.E-AvrA+ or wild type strain were significant for follow-up measurements at 1–8 h (n = 6). **, p ≤ 0.01.
FIGURE 6.

S. Enteritidis AvrA inhibits the JNK pathway and stabilizes ZO-1 in the mouse colon. AvrA stabilizes ZO-1 and inhibits the JNK pathway in a streptomycin pretreatment mouse model of enteric salmonellosis. A, Western blot shows that the tight junction protein ZO-1 decreased and p-JNK increased in the S. E-AvrA−-infected mouse colon; claudin-7, occluding, and E-cadherin expression was not changed in the mouse colon after infection with different strains of S. Enteritidis. B and C, the relative density of ZO-1 (B) and p-SAPK/JNK (C) was determined using Quantity One 4.6.2 (n = 5). *, p ≤ 0.05; **, p ≤ 0.01. D, RT-PCR confirmed that the srelative ZO-1 mRNA level did not change in the mouse colon after infection with different S. Enteritidis strains (n = 6). *, p ≤ 0.05; **, p ≤ 0.01. E, immunofluorescence shows the normal localization of ZO-1 (indicated by yellow arrows) in the control mouse colon and the disrupted localization (indicated by red arrows) following infection with AvrA mutant bacteria (top row). There was no significant difference in claudin-7 (middle row), E-Cadherin (bottom row) localization between the S.E-AvrA− strain and the wild type or complemented strains.
The disruption of TJs results in greater intestinal epithelial cell permeability (38). We next measured the transepithelial electrical resistance (TEER) in human epithelial Caco-2 BBE cells (Fig. 3B). There was a dramatic decrease in TEER 1–8 h after S. Enteritidis infection, and this decrease was greater in S.E-AvrA−-infected cells than in S.E-AvrA+ or wild type S.E-infected cells (Fig. 3B). There was a significant difference between S.E-AvrA−-infected cells and wild type S.E or S.E-AvrA+-infected cells 2–8 h after S. Enteritidis infection (Fig. 3B).
Enteritidis AvrA Inhibits Cellular JNK Activation
ZO-1 is regulated by the JNK signaling pathway (25, 39, 40). Using Western blotting, we found that JNK signaling was activated, as indicated by the marked increase in JNK phosphorylation, in Caco-2 BBE, HCT116, and SKCO15 cells following S. Enteritidis colonization (Fig. 2). There was greater JNK phosphorylation in S.E-AvrA−-infected cells than in wild type S.E or S.E-AvrA+-infected cells. JNK pathway activation induces IL-8 production in epithelial cells (41), and JNK pathway-dependent expression of this pro-inflammatory cytokine is shown in Fig. 4. In parallel with p-JNK, IL-8 levels in culture supernatant were higher 6 h after colonization with S.E-AvrA− compared with wild type S.E and S.E-AvrA+. Together, these results show that AvrA inhibits JNK activation in epithelial cells upon infection by S. Enteritidis. ZO-1 protein levels were inversely correlated with phosphorylated JNK levels. In S.E-AvrA−-infected cells, increased JNK phosphorylation correlated with reduced ZO-1 expression, whereas in wild type S.E or S.E-AvrA+-infected cells, ZO-1 protein levels did not change.
FIGURE 4.

S. Enteritidis AvrA deletion leads to more bacterial invasion in human epithelial cells, and AvrA inhibits the production of the JNK pathway-dependent pro-inflammatory cytokine IL-8. A, human epithelial Caco-2 BBE cells were grown on an insert, colonized with an equal number of the indicated bacteria for 30 min, and washed with HBSS; then the total number of associated Salmonella bacteria (Salmonella adhesion) was determined. B, after washing with HBSS, cells were incubated in DMEM containing gentamicin (100 μg/ml) for 30 min, and the number of internalized Salmonella (Salmonella invasion) was then determined. C–E, human epithelial Caco-2 BBE (C), HCT116 (D), and SKCO15 cells (E) were colonized with an equal number of the indicated bacteria for 30 min, washed with HBSS, and incubated in DMEM containing gentamicin (100 μg/ml) for 4 h; the culture supernatant was harvested, and IL-8 levels in the supernatant were measured by ELISA (n = 6). *, p ≤ 0.05; **, p ≤ 0.01.
Enteritidis AvrA Deletion Leads to Enhanced Bacterial Invasion
The disruption of TJs results in greater intestinal epithelial cell permeability, as shown by the TEER changes in the epithelial cells following colonization by S. Enteritidis. Next, we determined the efficiency of bacterial invasion in human epithelial Caco-2 BBE cells. As shown in Fig. 4B, the S.E-AvrA−-colonized cells showed an increased intracellular bacterial load compared with those colonized with wild type S.E or S.E-AvrA+. Therefore, we conclude that disrupting TJs also results in enhanced invasion of epithelial cells by S. Enteritidis.
Enteritidis AvrA Deletion Leads to Enhanced Pathological Lesions in the Mouse Cecum
To study the role of the S. Enteritidis protein AvrA in an in vivo model of natural intestinal infection, we used the streptomycin pretreatment mouse model of enteric salmonellosis (42, 43). 6–8-week-old female C57BL/6 mice were pretreated with streptomycin for 24 h before infection with wild type S. Enteritidis C50336, S.E-AvrA−, or S.E-AvrA+ by oral gavage.
Mice cecal histopathology was graded with a semiquantitative scale (43); morphological changes were observed following S. Enteritidis infection, including goblet cell loss, polymorphonuclear cell infiltration into the lamina propria, and epithelial damage (Fig. 5A). Mice infected with S.E-AvrA− showed a marked increase in the pro-inflammatory response compared with mice infected with the wild type S.E or S.E-AvrA+ strain (Fig. 5, B and C).
FIGURE 5.
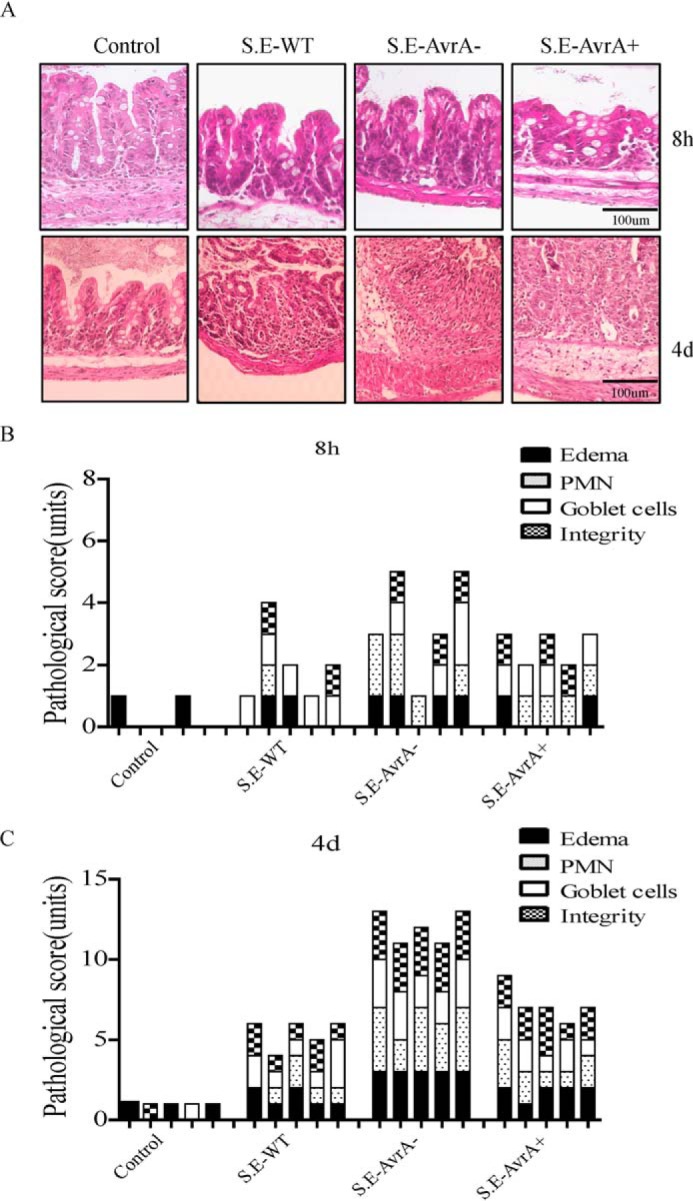
Infection with the S. Enteritidis AvrA deletion mutant leads to a more severe inflammatory response in the mouse cecum. Streptomycin pretreatment mouse model of enteric salmonellosis was performed. A, H&E staining of cecal mucosa sections from mice at 8 h (upper row) and 4 days (lower row) following oral infection with the indicated Salmonella strain. B and C, the semiquantitative inflammatory scores for murine cecal mucosa from H&E staining of sections at 8 h (B) and 4 days (C) post-infection encompassed individual scores for edema, polymorphonuclear cell infiltration, decreased goblet cell number, and epithelial layer integrity, as described under “Experimental Procedures.”
Enteritidis AvrA Stabilizes the TJ Protein ZO-1 and Inhibits the JNK Pathway in the Mouse Colon
In vitro, we found that the S. Enteritidis protein AvrA inhibits the JNK pathway and stabilizes the TJ protein ZO-1. In colonic samples from S.E-AvrA−-infected mice, ZO-1 protein expression was significantly decreased (Fig. 6, A and B) in parallel with increased JNK phosphorylation compared with samples from wild type S.E or S.E-AvrA+-infected mice (Fig. 6, A and C). No changes were observed in the expression levels of claudin-7, occluding, or E-cadherin (Fig. 6A). RT-PCR showed that ZO-1 mRNA levels in the mouse colon were not altered by S.E-AvrA−, S.E-AvrA+ or wild type S.E infection (Fig. 6D). Immunofluorescence analysis of mouse colonic tissues further confirmed that ZO-1 localization was disrupted in S.E-AvrA−-infected mice compared with S.E-AvrA+- or wild type S.E-infected mice (Fig. 6E). Similar to the Western blotting results, there was no change in claudin-7 or E-cadherin (Fig. 6E) by immunofluorescence.
AvrA Deletion Leads to Increased Bacterial Invasion and Inflammation in the Mouse Colon
Next, we investigated bacterial invasion in the mouse colon. In our model, bacterial colonization was measureable in the mouse colon. There was a significantly higher bacterial load in S.E-AvrA−-colonized colon tissue than in wild type S.E or S.E-AvrA+-colonized colon tissue (Fig. 7A), whereas there was no evidence of Salmonella in the uninfected control mice.
FIGURE 7.

S. Enteritidis AvrA deletion leads to enhanced bacterial invasion and pro-inflammatory cytokines in the mouse colon. A, invasion of Salmonella in the mouse colon at 4 days following oral infection with the indicated Salmonella strain. B and C, Salmonella were counted on MacConkey agar media plates (n = 5–7). *, p ≤ 0.05; **, p ≤ 0.01. The serum levels of the pro-inflammation cytokines IL-6 (B) and TNF-α (C) in mice infected with the indicated Salmonella strain were measured by Luminex. D and E, local colon mRNA levels of IL-6 (D) and TNF-α (E) in mice infected with the indicated Salmonella strain were measured by RT-PCR (n = 6). *, p ≤ 0.05; **, p ≤ 0.01.
We also measured serum levels of pro-inflammatory cytokines in S. Enteritidis-infected mice, using Luminex assays (Fig. 7, B and C). The pro-inflammatory cytokines IL-6 (Fig. 7B) and TNF-α (Fig. 7C) were not increased at 8 h after Salmonella infection. However, after 4 days, the concentrations of IL-6 and TNF-α were markedly increased in S.E-AvrA−-infected mice compared with wild type S.E or S.E-AvrA+-infected mice. We confirmed the changes in pro-inflammatory cytokine expression at the mRNA level using RT-PCR: IL-6 (Fig. 7D) and TNF-α (Fig. 7E). mRNA expression was significantly increased in S.E.-AvrA−-infected colons. Our results indicate that S. Enteritidis AvrA decreases bacterial invasion and reduces inflammation.
Inhibition of the JNK Pathway Abolishes the Effect of AvrA Deletion on ZO-1
To confirm whether AvrA stabilizes the TJ protein ZO-1 via the JNK pathway, we treated Caco-2 BBE cells with a JNK inhibitor (SP600125) to determine whether inhibiting the JNK pathway destabilizes ZO-1 in S.E-AvrA−-infected cells. As shown in Fig. 8 (A–C), after treatment with the JNK inhibitor, phosphorylated JNK was nearly undetectable in S.E-AvrA−, S.E-AvrA+, or wild type S.E-infected cells. However, the JNK inhibitor did not disrupt ZO-1 expression in S.E-AvrA−-infected cells. RT-PCR supported the observation that the JNK inhibitor had no effect on ZO-1 mRNA expression (Fig. 8D). Inhibiting the JNK pathway decreased the expression of the JNK-dependent pro-inflammatory cytokine IL-8, even after Salmonella infection, and no difference was observed among the S.E-AvrA−, S.E-AvrA+, or wild type S.E strains post-infection (Fig. 8E). Together, AvrA-induced suppression of the JNK pathway and TJ protein disassembly increases ZO-1 stability following S. Enteritidis infection.
FIGURE 8.
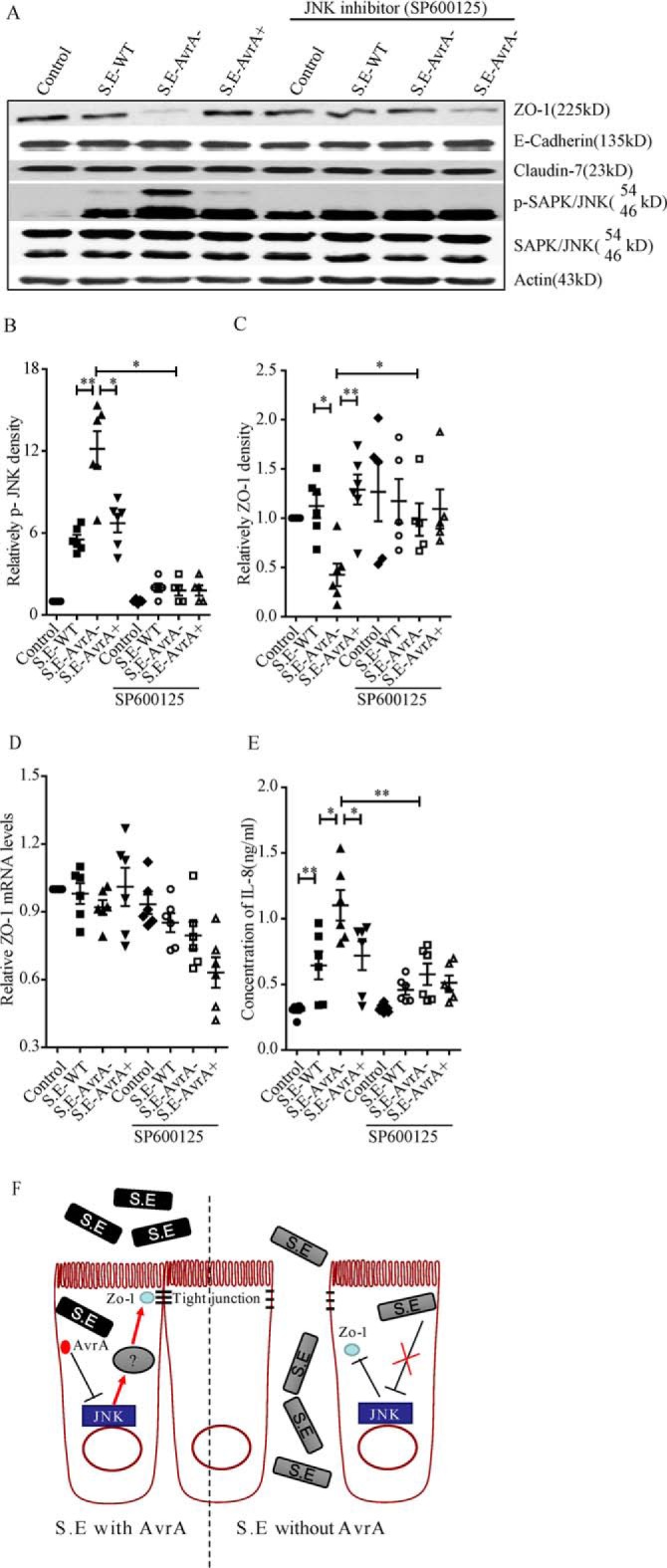
AvrA-mediated regulation of ZO-1 expression is dependent on the SAPK/JNK pathway. The effect of AvrA on ZO-1 expression was abolished by the SAPK/JNK inhibitor (SP600125) in Caco-2 BBE cells. A, cells were treated with SP600125 (50 μm) for 1 h, colonized with S. Enteritidis for 30 min, and incubated for 30 min in DMEM with gentamycin. Western blot shows that in S. E-AvrA−-infected cells treated with SP600125, ZO-1 expression was restored, whereas p-JNK levels were decreased. B and C, relative density analysis indicated a significant difference in ZO-1 (B) and p-JNK (C) in cells treated with the JNK inhibitor relative to untreated cells. D and E, ZO-1 mRNA levels (D) and IL-8 production (E) in Caco-2 BBE cells treated with the JNK inhibitor and infected with different Salmonella strains (n = 6). *, p ≤ 0.05; **, p ≤ 0.01. F, a working model of the role of AvrA in regulating TJs by blocking JNK activity.
Discussion
In the current study, we report that the S. Enteritidis effector protein AvrA stabilizes the TJ protein ZO-1, thus preserving intestinal epithelial permeability and decreasing bacterial invasion. Bacterial AvrA-mediated regulation of host TJs involves blocking the JNK signaling pathway, which was demonstrated in vitro and in vivo (Fig. 8F). The S. Enteritidis protein AvrA inhibits the JNK pathway in epithelial cells, enhances TJ barrier function by stabilizing ZO-1 expression at the protein level, increases cell permeability, reduces bacteria invasion, and reduces the inflammatory response in mice. The JNK inhibitor SP600125 abolished the stabilizing effect of AvrA on ZO-1.
Most Salmonella pathogenesis studies to date have focused on S. Typhimurium. In this study, we are the first to report that the S. Enteritidis effector protein AvrA inhibits the JNK pathway, thereby preventing intestinal epithelial barrier disruption, and normalizes the expression of the TJ protein ZO-1. There are numerous serotypes of Salmonella; different serotypes are found in different animal hosts or environments, and they may cause different symptoms in humans (6). Previous mouse studies have shown significant pathological changes in the intestine at 8 h after infection with wild type S. Typhimurium (20, 21, 37, 44, 45). In the current study, we found that mice did not show marked pathological or inflammatory changes 8 h after infection with S. Enteritidis. Instead, body weight loss occurred 1–2 days later than in S. Typhimurium colitis mice. We could detect significant increased serum IL-6 2 h after infection with wild type S. Typhimurium but not in the mice infection with S. Enteritidis for 8 h. All of the above results suggest that mice react differently when infected with S. Enteritidis or S. Typhimurium, despite nearly identical disease symptoms.
The JNK inhibitor SP600125 stabilizes epithelial barrier function by regulating claudin expression in murine and human epithelial cells (35, 46). In the present study, we found that SP600125 enhanced TJ function by abolishing the effect of Salmonella AvrA on the TJ protein ZO-1. The Salmonella protein AvrA is an anti-inflammatory effector that possesses acetyltransferase activity toward specific host MAPKKs and inhibits the host JNK/AP-1 and NF-κB signaling pathways (21–25). Previous studies on S. typhimurium showed that AvrA targets ZO-1 expression by blocking the JNK/AP-1 pathway and by targeting occludin expression (37, 47). In the current study, we did not observe significant regulation of occludin by S. Enteritidis. Our previous study showed that AvrA of S. Typhimurium increased TJ proteins, including claudin-1 and claudin-2, but these claudins were not regulated by S. Enteritidis AvrA in this study. The adherens junction protein E-cadherin was decreased in the colon of mice treated with wild type S. Typhimurium (19). JNK also regulates adherens junctions and barrier function in keratinocytes (48). In the present study, E-cadherin levels were not changed in the mouse colon after S. Enteritidis infection. AvrA inhibited the JNK pathway and enhanced TJs by stabilizing ZO-1 but not the adherens junction protein E-cadherin. This indicates that S. Enteritidis AvrA enhances TJ barrier function by selectively regulating ZO-1 expression in intestinal epithelial cells, which is different from the role of S. typhimurium AvrA. There was no obvious change in ZO-1 expression at the transcriptional level after S. Enteritidis infection, but ZO-1 protein levels were noticeably changed. JNK activation leads to tyrosine phosphorylation of ZO-1 and the disruption of TJs. We speculate that AvrA-mediated stabilization of ZO-1 occurs at the post-translational level. Salmonella spp. possess a range of effector proteins; different serotypes may secrete different proteins, and the resulting interactions with effector proteins may differentially influence host cellular function. The observed differences between S. Enteritidis AvrA and S. Typhimurium AvrA in terms of the regulation of TJ proteins may be due to interactions with different effectors.
Two p-JNK bands were detected by Western blotting. S. Enteritidis AvrA inhibited the 54-kDa p-JNK band to a greater extent than the 46-kDa p-JNK band, whereas S. Typhimurium AvrA inhibited both p-JNK bands. We also examined downstream molecules in the JNK pathway, including FoxOs and c-Jun. FoxO and total c-Jun levels were not changed by AvrA, whereas p-c-Jun was regulated by AvrA (data not shown). Similar to ZO-1, the activation of p-c-Jun by AvrA was abolished by treatment with the JNK inhibitor. Although Typhimurium AvrA targets multiple pathways, we found that the Enteritidis AvrA-mediated regulation of SPAK/JNK is specific.
Taken together, our data reveal a novel role of AvrA in S. Enteritidis in the stabilization of TJs and the attenuation of bacterial invasion in intestinal epithelial cells. We also identified differences between S. Enteritidis and S. Typhimurium, including delayed pathological changes during S. Enteritidis infection, different changes in tight junctions and adherens junction protein E-cadherin, and delayed responses in inflammatory cytokines. Bacterial effector proteins paralyze or reprogram eukaryotic cells to benefit pathogens. Our findings indicate an important role of S. Enteritidis effector AvrA, which influences host cellular function and bacterial invasion during infection and inflammation. The manipulation of JNK activity and TJs in microbial-host interactions may be a novel therapeutic approach for the treatment of infectious diseases.
Experimental Procedures
Animals and Ethics Statement
C57BL/6 mice (female, 6–8 weeks) were obtained from The Jackson Laboratory (Bar Harbor, ME). All animal work was approved by the Rush University and University of Illinois at Chicago Committee on Animal Resources. Euthanasia was accomplished via sodium pentobarbital administration (100 mg/kg body weight, i.p.) followed by cervical dislocation. All procedures were conducted in accordance with the approved guidelines of the Committees on Animal Resources.
Bacterial Strains and Growth Conditions
The S. Enteritidis WT strain C50336 was obtained from the Chinese National Institute for the Control of Pharmaceutical and Biological Products. The S. Enteritidis AvrA mutant strain S.E-AvrA− derived from C50336 and the AvrA complemented strain S.E-AvrA+ were constructed in this study. The S. typhimurium wild type strain SL1344 (SB300) and the Salmonella AvrA mutant strain SB1117 derived from SL1344 were provided by Dr. Galan (24). The bacteria and plasmids used in this study are listed in Table 1. Bacterial growth conditions were as follows: non-agitated microaerophilic bacterial cultures were prepared by inoculating 10 ml of LB broth with 0.01 ml of a stationary phase culture, followed by overnight incubation (>18 h) at 37 °C (49).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strains/plasmids | Characteristics | References |
|---|---|---|
| Strains | ||
| S.E-WT | Salmonella Enteritidis wild type CMCC(B)50336 | Chinese National Institute for the Control of Pharmaceutical and Biological Products |
| S.E-AvrA− | C50336 AvrA-deficient mutant | This study |
| S.E-AvrA+ | C50336 AvrA-deficient mutant carrying pBR322-AvrA (Ampr) | This study |
| SB300 | S. typhimurium wild type SL1344 | |
| SB1117 | SL1344 AvrA-deficient mutant | |
| Plasmids | ||
| pKD3 | Cmr; Cm cassette template | Ref. 50 |
| pKD46 | Ampr, λ-Red recombinase expression | Ref. 50 |
| pCP20 | Ampr, Cmr; Flp recombinase expression | Ref. 50 |
| pBR322-AvrA | pBR322 carrying the entire AvrA gene (Ampr) | This study |
Construction of the AvrA Deletion Mutant and Complemented Strains
The AvrA deletion mutant was constructed using the λ-Red-mediated recombination system, as described previously (50). The primers used in this study are listed in Table 2. Briefly, the chloramphenicol (Cm) cassette was amplified from the pKD3 plasmid using primers AvrA-cm forward and AvrA-cm reverse to include 39-bp homology extensions from the 5′ and 3′ ends of the AvrA gene. The PCR products were purified and introduced into the pKD46 plasmid containing S. Enteritidis C50336 by electroporation. Recombinant 50336-Cm bacteria were screened and selected on both Cm and ampicillin resistance LB agar plates. Allelic replacement of AvrA by the Cm cassette was verified by PCR screening (primers: AvrA-e forward and AvrA-e reverse) and DNA sequencing. Subsequently, the Cm cassette of 50336-Cm was excised by introducing the Flp recombinase-expressing vector pCP20. The full-length AvrA gene was cloned from S. Enteritidis strain C50336 using primers AvrA-r forward and AvrA-r reverse and ligated into plasmid pBR322. The recombined plasmid pBR322-AvrA was introduced into the AvrA mutant strain, and the transformants were screened and selected on ampicillin resistance LB agar plates. The AvrA mutant and complemented strains were confirmed by PCR, AvrA DNA sequencing, quantitative PCR, and Western blotting. Biochemical tests were performed using the API 20E identification kit (bioMerieux SA, Lyon, France) in accordance with the manufacturer's protocol.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′) |
|---|---|
| AvrA-e forward | CAGGCCACAAAAGAAAAC |
| AvrA-e reverse | ATAACAGCCGCATCAAAC |
| AvrA-r forward | CCCAAGCTTATCTACCTCCCGGTGTCA |
| AvrA-r reverse | CGGGATCCTTGCGGGCGTCTATCTGT |
| AvrA-cm forward | AGGCAATATATTGAATCTGAAAAGTTAAAGATGATATTTGTGTAGGCTGGAGCTGCTTC |
| AvrA-cm reverse | TTTCCTCTGGCAGGCAACCTTATAATTTCATTACGATTTATGGGAATTAGCCATGGTCC |
| AvrA-q forward | AGAGTTATGGACGGAAAGAC |
| AvrA-q reverse | AAATACCGCATTCAGAAGAG |
| GMK forward | CACTCAGGTTTCCGTTTCA |
| GMK reverse | CACTTGCTCAATGGTTTCG |
| mIL-6 forward | CTGCAAGAGACTTCCATCCAG |
| mIL-6 reverse | AGTGGTATAGACAGGTCTGTTGG |
| hZo-1 reverse | TGGTGTCCTACCTAATTCAACTCA |
| hZo-1 forward | CGCCAGCTACAA ATATTCCAACA |
| mZo-1 reverse | ATCTGGCTCCTCTCTTGCCAACTT |
| mZo-1 forward | AGGTCTTCGCAGCTCCAAGAGAAA |
| mTNF-α forward | CCCTCACACTCAGATCATCTTCT |
| mTNF-α reverse | GCTACGACGTGGGCTACAG |
Cell Culture
Human epithelial Caco-2 BBE, HCT116 and SKCO15 cells were maintained in DMEM supplemented with 10% FBS, streptomycin-penicillin, and l-glutamine. Monolayers of Caco-2 BBE and SKCO15 cells were grown on permeable supports (0.33 or 4.67 cm2, 0.4-mm pore; Costar, Cambridge, MA).
Cell Treatment with the JNK Inhibitor SP600125
The JNK inhibitor SP600125 (50 mm; EMD Biosciences, San Diego, CA) was added directly to the culture medium 1 h before Salmonella treatment. Caco-2 BBE cells pretreated with SP600125 were incubated with Salmonella for 30 min, washed three times in Hanks' balanced salt solution (HBSS), incubated in DMEM containing 100 μg/ml gentamicin and 50 μm SP600125 for 30 min, and harvested. Protein levels were determined by Western blotting.
Streptomycin Pretreated Mouse Model
Water and food were withdrawn 4 h before oral gavage with 7.5 mg/mouse streptomycin. Afterward, animals were supplied with water and food. 20 h after streptomycin treatment, water and food were once again withdrawn for 4 h before the mice were infected with 1 × 108 CFU of Salmonella (100-μl bacterial suspension in HBSS) or treated with sterile HBSS (control) by oral gavage, as previously described (42). At the indicated times post-infection, the mice were sacrificed, and intestinal tissue samples were removed for analysis.
Bacterial Colonization
Polarized human epithelial cells (Caco-2 BBE, HCT116, and SKCO15 cells) were grown in DMEM with 10% FBS. At 90–100% confluence, the cells were colonized with an equal number of the indicated bacteria for 30 min, washed with HBSS, and incubated in DMEM containing gentamicin (100 μg/ml) for 30 min. The first 30 min of incubation allowed bacteria to contact the epithelial cell surface and inject the effectors into the host cells. After extensive HBSS washing, extracellular bacteria were washed away. Incubation with gentamicin inhibited bacterial growth. To determine bacterial adhesion to cells, the cells were washed 30 min after colonization with prewarmed HBSS and then incubated with shaking in HBSS containing Triton X-100 (1%) in a cold room. To ascertain bacterial invasion, 30 min after bacterial colonization, the cells were incubated for an additional 30 min in DMEM with gentamicin, washed and incubated with shaking in HBSS containing Triton X-100 in a cold room. Bacterial CFU were determined by plating diluted cell lysates onto MacConkey agar culture plates (Difco Laboratories, Inc.) and incubating the cultures at 37 °C overnight.
Immunoblotting
Mouse epithelial cells were scraped and lysed in lysis buffer (1% Triton X-100, 150 mm NaCl, 10 mm Tris, pH 7.4, 1 mm EDTA, 1 mm EGTA, pH 8.0, 0.2 mm sodium orthovanadate, and protease inhibitor mixture), and the protein concentration was measured using protein assay kits (Bio-Rad). Cultured cells were rinsed twice in ice-cold HBSS and lysed in protein loading buffer (50 mm Tris, pH 6.8, 100 mm dithiothreitol, 2% SDS, 0.1% bromphenol blue, and 10% glycerol), and the remaining cells were scraped off the dish and sonicated to shear DNA and reduce the sample viscosity. Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes; nonspecific sites were blocked with 5% BSA in TBST (50 mm Tris, 150 mm NaCl, and 0.05% Tween 20 adjusted to pH 7.6 with HCl), and the membranes were then incubated with dilutions of primary antibody as recommended by the manufacturer. The primary antibodies included the following: anti-ZO-1(1:1000, 33–9100), anti-claudin-1 (1:1000, 71–7800), anti-claudin-7 (1:1000, 34–9100), and anti-occludin (1:1000, 33–1500) (Invitrogen); anti-phospho-SAPK/JNK (Thr183/Tyr185, 1:1000, 9251), anti-SAPK/JNK (1:1000, 9258) (Cell Signaling, Beverly, MA); anti-E-cadherin (1:1000, 610405; BD Bioscience, Franklin Lakes, NJ), anti-Villin (1:1000, SC-7672; Santa Cruz Biotechnology, Inc., Santa Cruz, CA); and anti-β-actin (1:2000, A1978; Sigma-Aldrich). The membranes were washed and incubated with HRP-conjugated secondary antibody (anti-mouse, 1:5000, 31430; anti-rabbit, 1:1000, 31460; anti-goat, 1:1000, R-21459; Invitrogen). The membranes were then washed again, treated with ECL Western blotting substrate (Thermo Scientific), and visualized on X-ray film. The membranes that were sequentially probed with more than one antibody were stripped in stripping buffer (Thermo Scientific) before reprobing. No other bands were detected on the blots with antibodies E-Cadherin, Claudin-1, Claudin-7, Occludin Villin, and β-actin. ZO-1 had two bands (225 and 48 kDa) detected by Western blotting. At a target molecular mass of ∼225 kDa, there was no nonspecific band found. The other band has low molecular mass (48 kDa). For JNK and p-JNK, it is known two bands will be detected: 46 and 54 kDa. All bands detected on blots were consistent with production information from the companies and other scientific reports which studied particular protein with antibodies ZO-1 (20, 51–53), E-Cadherin (20, 39, 52, 53), Claudin-1 (20, 53, 54), Claudin-7 (47, 54), Occludin (52), Villin (45, 55, 56), JNK, p-JNK (40, 53, 57, 58), and β-actin (59).
Immunoblotting for AvrA
Bacteria were lysed in lysis buffer (50v Tris, pH 8.0, 150v NaCl, 5 mm EDTA with a complete Mini protease inhibitor mixture (1 tablet/10 ml, Roche), and 1% Triton X-100), and sonicated. Equal amounts of total proteins were loaded, separated by SDS-PAGE, and processed for immunoblotting with custom-made AvrA antibody, as reported in our previous studies (19). The 15-amino acid peptide CGEEPFLPSD-KADRY was designed based on the AvrA sequence amino acids 216–230 (GenBankTM accession no. AE008830).
Immunofluorescence
Colonic tissues from mice were freshly isolated and embedded in paraffin wax after fixation with 10% neutral buffered formalin. Immunofluorescence was performed on paraffin-embedded sections (5 μm). After preparation as previously described, the slides were incubated for 1 h in blocking solution (2% BSA and 1% goat serum in HBSS) to reduce nonspecific background. The tissue samples were incubated with the indicated primary antibodies, including ZO-1 (1:100, 33–9100), claudin-7 (1:100, 34-9100; Invitrogen), and E-cadherin (1:100, 610405; BD Biosciences), overnight at 4 °C. Samples were then incubated with secondary antibodies (anti-mouse AF488, A-11029, anti-rabbit AF594, A-11037; Invitrogen), for 1 h at room temperature. Tissues were mounted with the SlowFade antifade kit (s2828; Life Technologies) and coverslipped, and the edges were sealed to prevent drying. Specimens were examined with a Zeiss laser scanning microscope 710 (Carl Zeiss, Inc., Oberkochen, Germany). Fluorescence images were analyzed for ZO-1 distribution using image analysis software (LSM 710 META, version 4.2; Carl Zeiss). Each analysis was performed in triplicate for each tissue section on a total of 10 images/mouse sample (n = 5).
Quantitative Pathological Analysis
Segments of the cecum and colon were fixed and embedded in paraffin according to standard procedures. Cryosections (5 μm) were mounted on glass slides, air dried for 2 h at room temperature, and stained with hematoxylin and eosin (H&E). Pathological evaluations were performed by two pathologists in a blinded manner. Based on an earlier study (42, 60), a scoring scheme for quantitative pathological analysis of cecal inflammation in H&E-stained sections (5 μm) was followed.
Submucosal Edema
Submucosal edema was scored as follows: 0, no pathological changes; 1, mild edema (submucosa: 0.20-mm-wide and accounting for 50% of the diameter of the entire intestinal wall (tunica muscularis to epithelium)); 2, moderate edema (submucosa: 0.21–0.45-mm-wide and accounting for 50–80% of the diameter of the entire intestinal wall); and 3, profound edema (submucosa: 0.46-mm-wide and accounting for 80% of the diameter of the entire intestinal wall). Submucosa widths were determined by quantitative microscopy and represent the average of 30 evenly spaced radial measurements of the distance between the tunica muscularis and the lamina muscularis mucosae.
PMN Infiltration into the Lamina Propria
Polymorphonuclear granulocytes (PMNs) in the lamina propria were enumerated in 10 high power fields (400× magnification; field diameter, 420 μm), and the average number of PMNs/high power field was calculated. The scores were defined as follows: 0, <5 PMNs/high power field; 1, 5–20 PMNs/high power field; 2, 21–60 PMNs/high power field; 3, 61–100 PMNs/high power field; and 4, >100 PMNs/high power field. Transmigration of PMNs into the intestinal lumen was consistently observed for situations with >60 PMNs/high power field.
Goblet Cells
The average number of goblet cells per high power field (magnification, 400×) was determined in 10 different regions of the cecal epithelium. Scoring was as follows: 0, >28 goblet cells/high power field (magnification, 400×; in the cecum of normal SPF mice, we observed an average of 6.4 crypts/high power field, and the average crypt consisted of 35–42 epithelial cells, of which 25–35% were differentiated into goblet cells); 1, 11–28 goblet cells/high power field; 2, 1–10 goblet cells/high power field; and 3, <1 goblet cell/high power field.
Epithelial Integrity
Epithelial integrity was scored as follows: 0, no detectable pathological changes in 10 high power fields (400× magnification); 1, epithelial desquamation; 2, erosion of the epithelial surface (gaps of 1–10 epithelial cells/lesion); and 3, epithelial ulceration (gaps of 10 epithelial cells/lesion; at this stage, there is generally granulation tissue below the epithelium). We averaged the two independent scores for submucosal edema, PMN infiltration, goblet cells, and epithelial integrity for each tissue sample. The combined pathological score for each tissue sample was determined as the sum of these averaged scores. The combined pathological score ranged between 0 and 13 arbitrary units and covered the following levels of inflammation: 0, intestinal intact without any signs of inflammation; 1–2, minimal signs of inflammation (this was frequently found in the ceca of SPF mice; this level of inflammation is generally not considered a sign of disease); 3–4, slight inflammation; 5–8, moderate inflammation; and 9–13, profound inflammation.
IL-8 Enzyme-linked Immunosorbent Assay
IL-8 was measured in cell culture medium using a sandwich ELISA method. Briefly, goat anti-human IL-8 antibody (1 μg, AB-208-NA; R&D Systems) was coated on plastic wells at 4 °C overnight. Then culture medium and recombination human IL-8 standards were added to each well. After washing, the plates were incubated with rabbit anti-human IL-8 antibody (1 μg, ab7747; Abcam, Cambridge, UK), washed again, and then incubated with peroxidase-conjugated anti-rabbit IgG antibody (214-1516; KPL Inc., Gaithersburg, MD). Following complete washing, KPL SureBlue peroxidase substrate (52-00-01) was added. The A450 values were determined after stop solution was added to the wells.
Real Time Quantitative PCR Analysis
Total RNA was extracted from mouse epithelial cells or cultured cells using TRIzol reagent (Invitrogen). RNA integrity was verified by electrophoresis. Reverse transcription of RNA was performed using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's protocol. The cDNA was subjected to quantitative real time PCR using a CFX Connect real time system (Bio-Rad) and SYBR Green Supermix (Bio-Rad). All expression levels were normalized to β-actin levels in the same sample. The percentage of expression was calculated as the ratio of the normalized value of each sample to that of the corresponding untreated control sample. All real time PCR analyses were performed in triplicate.
Statistical Analysis
All data are presented as the means ± S.D. All statistical tests were two-sided. p values of less than or equal 0.05 were considered statistically significant. Differences between two samples were analyzed using Student's t test. Multiple comparisons of mean TEER (Fig. 3B) were performed using two-way ANOVA, and p values were adjusted using Tukey's method. Because we were interested in comparing the mean differences among the wild type, S.E-AvrA+ and S.E-AvrA− groups, the group effects at each time point of 0, 0.5, 1, 2, 3, 4, 5, 6, 7, or 8 h were tested using generalized linear mixed models. The pairwise treatment group differences among the wild type, S.E-AvrA+ and S.E-AvrA− groups at each time point were compared using Tukey's method to adjust p values to ensure accurate results. For fluorescence intensity data, one-way ANOVA was conducted to analyze the mean differences among the control, S.E-AvrA+, wild type, and S.E-AvrA− groups, and the p values were adjusted using Tukey's method. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC).
Author Contributions
Z. L. and Y.-G. Z. were responsible for data acquisition, analysis, and interpretation and drafting of the manuscript. Z. L. and J. S. wrote the main manuscript text and prepared figures. Y. X. was responsible for statistical analysis and drafting of the manuscript. X. J. and X. X. were responsible for technical or material support and drafting of the manuscript. J. S. was responsible for the study concept and design, critical revision of the manuscript for important intellectual content, and study supervision. All authors reviewed the manuscript.
This work was supported by NIDDK, National Institutes of Health Grant 1R01DK105118-01, a Swim Across America Cancer Research Award, the University of Illinois at Chicago Cancer Center (to J. S.), and Projects of International Cooperation and Exchanges National Natural Science Foundation of China Grant 31320103907. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- S. Enteritidis
- Salmonella enterica serotype enteritidis
- TJ
- tight junction
- ZO
- zonula occludens
- TEER
- transepithelial electrical resistance
- S. Typhimurium
- S. typhimurium serotype typhimurium
- Cm
- chloramphenicol
- HBSS
- Hanks' balanced salt solution
- H&E
- hematoxylin and eosin
- PMN
- polymorphonuclear granulocyte
- ANOVA
- analysis of variance.
References
- 1. Scallan E., Hoekstra R. M., Angulo F. J., Tauxe R. V., Widdowson M.-A., Roy S. L., Jones J. L., and Griffin P. M. (2011) Foodborne illness acquired in the United States: major pathogens. Emerg. Infect. Dis. 17, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Majowicz S. E., Musto J., Scallan E., Angulo F. J., Kirk M., O'Brien S. J., Jones T. F., Fazil A., Hoekstra R. M., and International Collaboration on Enteric Disease “Burden of Illness” Studies (2010) The global burden of nontyphoidal Salmonella gastroenteritis. Clin. Infect. Dis. 50, 882–889 [DOI] [PubMed] [Google Scholar]
- 3. Braden C. R. (2006) Salmonella enterica serotype enteritidis and eggs: a national epidemic in the United States. Clin. Infect. Dis. 43, 512–517 [DOI] [PubMed] [Google Scholar]
- 4. Hendriksen R. S., Vieira A. R., Karlsmose S., Lo Fo Wong D. M., Jensen A. B., Wegener H. C., and Aarestrup F. M. (2011) Global monitoring of Salmonella serovar distribution from the World Health Organization Global Foodborne Infections Network Country Data Bank: results of quality assured laboratories from 2001 to 2007. Foodborne Pathog. Dis. 8, 887–900 [DOI] [PubMed] [Google Scholar]
- 5. Lane C. R., LeBaigue S., Esan O. B., Awofisyo A. A., Adams N. L., Fisher I. S., Grant K. A., Peters T. M., Larkin L., Davies R. H., and Adak G. K. (2014) Salmonella enterica serovar enteritidis, England and Wales, 1945–2011. Emerg. Infect. Dis. 20, 1097–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Centers for Disease Control and Prevention (2007) Salmonella serotype enteritidis infections among workers producing poultry vaccine: Maine, November-December 2006. MMWR Morb. Mortal Wkly. Rep. 56, 877–879 [PubMed] [Google Scholar]
- 7. Ghosh T. S., and Vogt R. L. (2006) Cluster of invasive salmonellosis cases in a federal prison in Colorado. Am. J. Infect. Control 34, 348–350 [DOI] [PubMed] [Google Scholar]
- 8. Mutlu H., Babar J., and Maggiore P. R. (2009) Extensive Salmonella enteritidis endocarditis involving mitral, tricuspid valves, aortic root and right ventricular wall. J. Am. Soc. Echocardiogr. 22, 210. [DOI] [PubMed] [Google Scholar]
- 9. Gordon M. A., Graham S. M., Walsh A. L., Wilson L., Phiri A., Molyneux E., Zijlstra E. E., Heyderman R. S., Hart C. A., and Molyneux M. E. (2008) Epidemics of invasive Salmonella enterica serovar enteritidis and S. enterica serovar typhimurium infection associated with multidrug resistance among adults and children in Malawi. Clin. Infect. Dis. 46, 963–969 [DOI] [PubMed] [Google Scholar]
- 10. Katsenos C., Anastasopoulos N., Patrani M., and Mandragos C. (2008) Salmonella enteritidis meningitis in a first time diagnosed AIDS patient: case report. Cases J. 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi H., Hall G. S., Tuohy M. J., Knothe U., Procop G. W., and Bauer T. W. (2009) Bilateral periprosthetic joint infection caused by Salmonella enterica serotype enteritidis, and identification of Salmonella sp. using molecular techniques. Int. J. Infect. Dis. 13, e463–466 [DOI] [PubMed] [Google Scholar]
- 12. Morpeth S. C., Ramadhani H. O., and Crump J. A. (2009) Invasive non-Typhi Salmonella disease in Africa. Clin. Infect. Dis. 49, 606–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tena D., González-Praetorius A., and Bisquert J. (2007) Urinary tract infection due to non-typhoidal Salmonella: report of 19 cases. J. Infect. 54, 245–249 [DOI] [PubMed] [Google Scholar]
- 14. Dalbey R. E., and Kuhn A. (2012) Protein traffic in Gram-negative bacteria: how exported and secreted proteins find their way. FEMS Microbiol. Rev. 36, 1023–1045 [DOI] [PubMed] [Google Scholar]
- 15. Ramos-Morales F. (2012) Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol. 2012, 1–36 [Google Scholar]
- 16. Misselwitz B., Barrett N., Kreibich S., Vonaesch P., Andritschke D., Rout S., Weidner K., Sormaz M., Songhet P., Horvath P., Chabria M., Vogel V., Spori D. M., Jenny P., and Hardt W. D. (2012) Near surface swimming of Salmonella typhimurium explains target-site selection and cooperative invasion. PLoS Pathog. 8, e1002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGhie E. J., Hayward R. D., and Koronakis V. (2001) Cooperation between actin-binding proteins of invasive Salmonella: SipA potentiates SipC nucleation and bundling of actin. EMBO J. 20, 2131–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guttman J. A., and Finlay B. B. (2009) Tight junctions as targets of infectious agents. Biochim. Biophys. Acta 1788, 832–841 [DOI] [PubMed] [Google Scholar]
- 19. Boyle E. C., Brown N. F., and Finlay B. B. (2006) Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell. Microbiol. 8, 1946–1957 [DOI] [PubMed] [Google Scholar]
- 20. Liao A. P., Petrof E. O., Kuppireddi S., Zhao Y., Xia Y., Claud E. C., and Sun J. (2008) Salmonella type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PLoS One 3, e2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones R. M., Wu H., Wentworth C., Luo L., Collier-Hyams L., and Neish A. S. (2008) Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3, 233–244 [DOI] [PubMed] [Google Scholar]
- 22. Ye Z., Petrof E. O., Boone D., Claud E. C., and Sun J. (2007) Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 171, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collier-Hyams L. S., Zeng H., Sun J., Tomlinson A. D., Bao Z. Q., Chen H., Madara J. L., Orth K., and Neish A. S. (2002) Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF-κB pathway. J. Immunol. 169, 2846–2850 [DOI] [PubMed] [Google Scholar]
- 24. Du F., and Galán J. E. (2009) Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog. 5, e1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naydenov N. G., Hopkins A. M., and Ivanov A. I. (2009) c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. Cell Cycle 8, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 26. Ulluwishewa D., Anderson R. C., McNabb W. C., Moughan P. J., Wells J. M., and Roy N. C. (2011) Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 141, 769–776 [DOI] [PubMed] [Google Scholar]
- 27. Kojima T., Kokai Y., Chiba H., Osanai M., Kuwahara K., Mori M., Mochizuki Y., and Sawada N. (2001) Occludin and claudin-1 concentrate in the midbody of immortalized mouse hepatocytes during cell division. J. Histochem. Cytochem. 49, 333–340 [DOI] [PubMed] [Google Scholar]
- 28. Chiba H., Osanai M., Murata M., Kojima T., and Sawada N. (2008) Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 1778, 588–600 [DOI] [PubMed] [Google Scholar]
- 29. Gumbiner B. M. (1993) Breaking through the tight junction barrier. J. Cell Biol. 123, 1631–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farhadi A., Keshavarzian A., Ranjbaran Z., Fields J. Z., and Banan A. (2006) The role of protein kinase C isoforms in modulating injury and repair of the intestinal barrier. J. Pharmacol. Exp. Ther. 316, 1–7 [DOI] [PubMed] [Google Scholar]
- 31. Basuroy S., Seth A., Elias B., Naren A. P., and Rao R. (2006) MAR interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem. J. 393, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turner J. R., Rill B. K., Carlson S. L., Carnes D., Kerner R., Mrsny R. J., and Madara J. L. (1997) Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 273, C1378–C1385 [DOI] [PubMed] [Google Scholar]
- 33. Walsh S. V., Hopkins A. M., Chen J., Narumiya S., Parkos C. A., and Nusrat A. (2001) Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology 121, 566–579 [DOI] [PubMed] [Google Scholar]
- 34. Ip Y. T., and Davis R. J. (1998) Signal transduction by the c-Jun N-terminal kinase (JNK): from inflammation to development. Curr. Opin. Cell Biol. 10, 205–219 [DOI] [PubMed] [Google Scholar]
- 35. Carrozzino F., Pugnale P., Féraille E., and Montesano R. (2009) Inhibition of basal p38 or JNK activity enhances epithelial barrier function through differential modulation of claudin expression. Am. J. Physiol. Cell Physiol. 297, C775–C787 [DOI] [PubMed] [Google Scholar]
- 36. De Walle J. V., Sergent T., Piront N., Toussaint O., Schneider Y. J., and Larondelle Y. (2010) Deoxynivalenol affects in vitro intestinal epithelial cell barrier integrity through inhibition of protein synthesis. Toxicol. Appl. Pharmacol. 245, 291–298 [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y., Wu S., Ma J., Xia Y., Ai X., and Sun J. (2015) Bacterial protein AvrA stabilizes intestinal epithelial tight junctions via blockage of the c-Jun N-terminal kinase pathway. Tissue Barriers 3, e972849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen M. L., Ge Z., Fox J. G., and Schauer D. B. (2006) Disruption of tight junctions and induction of proinflammatory cytokine responses in colonic epithelial cells by Campylobacter jejuni. Infect. Immun. 74, 6581–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konno T., Ninomiya T., Kohno T., Kikuchi S., Sawada N., and Kojima T. (2015) c-Jun N-terminal kinase inhibitor SP600125 enhances barrier function and elongation of human pancreatic cancer cell line HPAC in a Ca-switch model. Histochem. Cell Biol. 143, 471–479 [DOI] [PubMed] [Google Scholar]
- 40. Kojima T., Fuchimoto J., Yamaguchi H., Ito T., Takasawa A., Ninomiya T., Kikuchi S., Ogasawara N., Ohkuni T., Masaki T., Hirata K., Himi T., and Sawada N. (2010) c-Jun N-terminal kinase is largely involved in the regulation of tricellular tight junctions via tricellulin in human pancreatic duct epithelial cells. J. Cell. Physiol. 225, 720–733 [DOI] [PubMed] [Google Scholar]
- 41. Hubert A., Cauliez B., Chedeville A., Husson A., and Lavoinne A. (2004) Osmotic stress, a proinflammatory signal in Caco-2 cells. Biochimie 86, 533–541 [DOI] [PubMed] [Google Scholar]
- 42. Barthel M., Hapfelmeier S., Quintanilla-Martínez L., Kremer M., Rohde M., Hogardt M., Pfeffer K., Rüssmann H., and Hardt W. D. (2003) Pretreatment of mice with streptomycin provides a Salmonella enterica serovar typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71, 2839–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vijay-Kumar M., Wu H., Jones R., Grant G., Babbin B., King T. P., Kelly D., Gewirtz A. T., and Neish A. S. (2006) Flagellin suppresses epithelial apoptosis and limits disease during enteric infection. Am. J. Pathol. 169, 1686–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu X., Lu R., Wu S., and Sun J. (2010) Salmonella regulation of intestinal stem cells through the Wnt/β-catenin pathway. FEBS Lett. 584, 911–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu X., Wu S., Xia Y., Li X. E., Xia Y., Zhou Z. D., and Sun J. (2011) Wingless homolog Wnt11 suppresses bacterial invasion and inflammation in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G992–G1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kojima T., Yamaguchi H., Ito T., Kyuno D., Kono T., Konno T., and Sawada N. (2013) Tight junctions in human pancreatic duct epithelial cells. Tissue Barriers 1, e24894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y. G., Wu S., Xia Y., and Sun J. (2013) Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS One 8, e58606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee M. H., Koria P., Qu J., and Andreadis S. T. (2009) JNK phosphorylates β-catenin and regulates adherens junctions. FASEB J. 23, 3874–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCormick B. A., Colgan S. P., Delp-Archer C., Miller S. I., and Madara J. L. (1993) Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J. Cell Biol. 123, 895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muramatsu R., Kuroda M., Matoba K., Lin H., Takahashi C., Koyama Y., and Yamashita T. (2015) Prostacyclin prevents pericyte loss and demyelination induced by lysophosphatidylcholine in the central nervous system. J. Biol. Chem. 290, 11515–11525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu S., Yi J., Zhang Y. G., Zhou J., and Sun J. (2015) Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol. Rep. 3, e12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shiou S. R., Yu Y., Chen S., Ciancio M. J., Petrof E. O., Sun J., and Claud E. C. (2011) Erythropoietin protects intestinal epithelial barrier function and lowers the incidence of experimental neonatal necrotizing enterocolitis. J. Biol. Chem. 286, 12123–12132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghulé V. V., Gray C., Galimberti A., and Anumba D. O. (2012) Prostaglandin-induced cervical remodelling in humans in the first trimester is associated with increased expression of specific tight junction, but not gap junction proteins. J. Transl. Med. 10, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shaked H., Hofseth L. J., Chumanevich A., Chumanevich A. A., Wang J., Wang Y., Taniguchi K., Guma M., Shenouda S., Clevers H., Harris C. C., and Karin M. (2012) Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. U.S.A. 109, 14007–14012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu S., Zhang Y. G., Lu R., Xia Y., Zhou D., Petrof E. O., Claud E. C., Chen D., Chang E. B., Carmeliet G., and Sun J. (2015) Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 64, 1082–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Uto T., Fukaya T., Takagi H., Arimura K., Nakamura T., Kojima N., Malissen B., and Sato K. (2016) Clec4A4 is a regulatory receptor for dendritic cells that impairs inflammation and T-cell immunity. Nat. Commun. 7, 11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu Q., Tao B., Liu G., Chen G., Zhu Q., Yu Y., Yu Y., and Xiong H. (2016) Thromboxane A2 receptor inhibition suppresses multiple myeloma cell proliferation by inducing p38/c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK)-mediated G2/M progression delay and cell apoptosis. J. Biol. Chem. 291, 4779–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patino-Lopez G., Aravind L., Dong X., Kruhlak M. J., Ostap E. M., and Shaw S. (2010) Myosin 1G is an abundant class I myosin in lymphocytes whose localization at the plasma membrane depends on its ancient divergent pleckstrin homology (PH) domain (Myo1PH). J. Biol. Chem. 285, 8675–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Madsen K., Cornish A., Soper P., McKaigney C., Jijon H., Yachimec C., Doyle J., Jewell L., and De Simone C. (2001) Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 121, 580–591 [DOI] [PubMed] [Google Scholar]