

Abstract
The small molecule metal ion chelators bipyridine and terpyridine complexed with Zn2+ (ZnBip and ZnTerp) act as CCR5 agonists and strong positive allosteric modulators of CCL3 binding to CCR5, weak modulators of CCL4 binding, and competitors for CCL5 binding. Here we describe their binding site using computational modeling, binding, and functional studies on WT and mutated CCR5. The metal ion Zn2+ is anchored to the chemokine receptor-conserved Glu-283VII:06/7.39. Both chelators interact with aromatic residues in the transmembrane receptor domain. The additional pyridine ring of ZnTerp binds deeply in the major binding pocket and, in contrast to ZnBip, interacts directly with the Trp-248VI:13/6.48 microswitch, contributing to its 8-fold higher potency. The impact of Trp-248 was further confirmed by ZnClTerp, a chloro-substituted version of ZnTerp that showed no inherent agonism but maintained positive allosteric modulation of CCL3 binding. Despite a similar overall binding mode of all three metal ion chelator complexes, the pyridine ring of ZnClTerp blocks the conformational switch of Trp-248 required for receptor activation, thereby explaining its lack of activity. Importantly, ZnClTerp becomes agonist to the same extent as ZnTerp upon Ala mutation of Ile-116III:16/3.40, a residue that constrains the Trp-248 microswitch in its inactive conformation. Binding studies with 125I-CCL3 revealed an allosteric interface between the chemokine and the small molecule binding site, including residues Tyr-37I:07/1.39, Trp-86II:20/2.60, and Phe-109III:09/3.33. The small molecules and CCL3 approach this interface from opposite directions, with some residues being mutually exploited. This study provides new insight into the molecular mechanism of CCR5 activation and paves the way for future allosteric drugs for chemokine receptors.
Keywords: allosteric regulation, C-C chemokine receptor type 5 (CCR5), computer modeling, G protein-coupled receptor (GPCR), small molecule, 7-transmembrane helix receptor, activation mechanism
Introduction
CCR5 is one of 19 human chemokine receptors and thereby belongs to the protein family of seven-transmembrane helix (7TM)2 G protein-coupled receptors (GPCRs). The human chemokine system additionally comprises around 50 endogenous chemokine ligands, which together with their receptors organize leukocyte trafficking. A chemokine receptor can have several chemokine ligands, and a single chemokine can bind to several receptors, properties that confer redundancy to the system (1). At the same time, the system's components are spatially and temporally organized and characterized by receptor, ligand, and tissue bias (2, 3), implying that a chemokine interacting with a given receptor in a certain tissue in fact relays a very specific and non-redundant signal (4). The chemokine system is investigated as a target for treating acute and chronic inflammations, allergies, and autoimmune diseases but also for cancer growth and metastasis, angiogenesis, and HIV infection (5).
Chemokines are 8–12-kDa large peptides that are divided into four groups according to the position of conserved cysteines: CC-chemokines (25 members), CXC-chemokine (18 members), XC-chemokines (XCL1 and XCL2), and CX3CL1 (1). These cysteines form disulfide bridges with cysteines in the chemokine core domain, which itself consists of an N-loop, a three-stranded β-sheet, and a C-terminal α-helix. The N-terminal residues in front of the first cysteine thereby remain unstructured and flexible (6). Recently, two crystal structures of chemokine receptors in complex with a chemokine ligand were revealed: CXCR4 in complex with the viral chemokine vMIP-II (7) and the viral chemokine receptor US28 in complex with CX3CL1 (8). These structures confirmed the overall binding mode of chemokines to their receptors, whereby the chemokine core interacts with extracellular receptor domains, such as the receptor N terminus and extracellular loop (ECL) 2, whereas the flexible chemokine N terminus protrudes into the transmembrane receptor area. This agrees with the suggested “pseudo”-two-step model that roughly separates the chemokine-receptor interaction into an affinity-providing step 1 (chemokine core-extracellular receptor domains) and an activation-inducing step 2 (chemokine N terminus-transmembrane receptor domain) (9, 10).
Furthermore, crystal structures of chemokine receptors with various ligands have been solved and show different binding sites for each ligand. In CXCR4, the small molecule antagonist IT1t binds to a site in the minor binding pocket (delimited by TM-1 to -3 and -7 (11)), whereas the peptide-based CVX15 binds in the major binding pocket (delimited by TM-3 to -7) (12). Maraviroc in CCR5 spans the major and minor binding pockets (13), as suggested for many other small molecule CC-chemokine receptor antagonists (14).
A comparison of the binding modes of small molecules and chemokines shows that an overlap in binding sites may arise within the transmembrane receptor domain, which is targeted by the chemokine N termini and the small molecule ligands to varying extents. Thus, although small molecules traditionally are considered to bind allosterically to the larger orthosteric chemokine ligands (15), they might in fact overlap with the chemokine N terminus. A chemokine-dependent allosteric behavior of small molecules has, for example, been shown in CCR1. There, the binding of CCL3 was enhanced by the small molecule agonists metal ion chelator complexes, highlighting an allosteric binding mode, whereas another chemokine, CCL5, was displaced with equimolar affinities (16). CCL5 and the metal ion chelator complexes were affected by the same transmembrane receptor mutations, pointing to an overlap in binding sites that results in the observed competitive binding pattern for CCL5 and the small molecules. It has furthermore been discussed whether an allosteric binding mode should be pursued in the development of chemokine receptor antagonists. In general, although allosteric binding allows for the modulation of a chemokine-mediated response, and thereby depends on the presence and level of the chemokine, it might not confer strong enough antagonism to reach clinical efficacy (15, 17). More knowledge about the molecular pharmacology of chemokine receptors and their signaling outcomes under healthy and pathological conditions is therefore needed to design ligand- and receptor-specific or broad inhibitors or allosteric modulators with distinct signaling properties.
CCR5 gained prominence as an HIV co-receptor after it was found that the deletion variant CCR5Δ32 provides resistance to HIV infection by abrogating normal receptor expression in homo- and heterozygotes (18–20). This has initiated strong drug developmental efforts, resulting in the marketing of maraviroc as a CCR5 antagonist and HIV entry inhibitor in 2007 (17). Here we investigate the binding mode of agonistic allosteric modulators to CCR5. We have previously described the metal ion chelator complex ZnBip (Zn2+ in complex with 2,2′-bipyridine) as a small molecule agonist and allosteric enhancer of CCL3 binding to CCR5 (and CCR1) but a competitor for CCL5 binding (16, 21). By screening 20 chelator analogs, we identified ZnTerp (Zn2+ in complex with 2,2′:6′,2″-terpyridine), which acts more strongly than ZnBip in both agonistic activity and allosteric modulation, and ZnClTerp (Zn2+ in complex with 4′-chloro-2,2′:6′,2″-terpyridine), which was a pure allosteric enhancer of CCL3 binding but did not activate CCR5 (22). We also investigate the structural basis for agonistic and allosteric properties in CCR5. We use computational modeling to predict the binding sites of the metal ion chelator complexes and confirm these in vitro by receptor activation and 125I-CCL3 binding assays in 23 receptor mutants. We thereby describe the molecular mechanism for small molecule-mediated activation and allosteric modulation in CCR5.
Results
Activity of Metal Ion Chelator Complexes
As shown previously, ZnTerp is a very efficacious agonist at CCR5 with a higher potency than ZnBip when measuring inositol 1,4,5-trisphosphate (IP3) formation in transiently transfected COS-7 cells expressing CCR5 and the chimeric G protein GαΔ6qi4myr (Gqi4myr) that translates a Gαi coupling to a Gαq readout (Fig. 1, A and B, and Table 1) (22). As expected, the agonistic activity of both metal ion chelator complexes depends on complex formation between the chelator and Zn2+. To test the ligands' ability to induce Gαi activation more directly (in the absence of the chimeric G protein), we measured cAMP production in CHO cells stably transfected with CCR5 (Fig. 1, D–F), which were induced to produce cAMP with forskolin. The chemokines CCL3 and CCL5 inhibited forskolin-induced cAMP production (i.e. they induced Gαi activation and inhibition of adenylyl cyclase) (Fig. 1D). Also, ZnBip acted as an agonist through this pathway with a potency of 4.6 μm (Fig. 1E); the level of this activity depended on complex formation between Bip and Zn2+ (Fig. 1F). Similarly, we observed a specific activity of CCL3, CCL5, and ZnBip in a Ca2+-imaging assay in the same cells (Fig. 1, G–I).
FIGURE 1.

A, structures of ZnBip and ZnTerp; B, activity of ZnBip (black circle) and ZnTerp (black square) at CCR5 as measured in an IP3 assay (see “Experimental Procedures” for details). Non-complexed controls are shown in white (Zn2+, Terp, and Bip shown as circles, squares, and diamonds, respectively). Data are normalized to maximal response induced by chemokine at CCR5 (n ≥ 3). C, ability of ZnBip and ZnTerp to enhance the binding of 125I-CCL3 to CCR5, as determined in a heterologous competition binding assay. Data are normalized to the homologous binding curve (n ≥ 3). For B and C, see Table 1 and 3, respectively. D–F, cAMP turnover experiments in stably transfected CHO cells representing inhibition of adenylyl cyclase in response to Gαi activation. D, activity of 0.1 μm CCL3 and CCL5 in untransfected cells (controls) and stably transfected CCR5-expressing cells. *, p < 0.1; **, p < 0.01 as calculated by the Mann-Whitney test (t test for unpaired non-parametric data). E, specific normalized cAMP turnover induced by ZnBip in CCR5-expressing cells (background subtracted); 100% represents the lowest observed level of cAMP in the absence of forskolin stimulation, and 0% is the initial cAMP level in the assay induced by 10 μm forskolin (see “Experimental Procedures” for details). D and E, n ≥ 3. F, direct readout from one representative cAMP turnover assay of three experiments for ZnBip, Zn2+, and Bip in CCR5-CHO cells (arbitrary units). G and H, fluorescence-based Ca2+ assays in stably transfected CHO cells; one representative experiment of three is shown. The arrow indicates when ligands were added (at 80 s). Shown is the activity of 0.1 μm CCL3 and CCL5 (G) or 1 mm ZnBip (H) in untransfected cells and stably transfected CCR5-expressing cells. I, activity of 1 mm ZnBip in comparison with 1 mm Zn2+ and 1 mm Bip alone in stably transfected CCR5-expressing cells.
TABLE 1.
Activation of CCR5 and mutant receptors by CCL3, CCL5, ZnBip and ZnTerp
The name and position of mutants according to the Ballesteros/Weinstein (left) and Baldwin/Schwartz numbering system are given. The surface expression of each mutant was determined by ELISA using N-terminally Flag-tagged receptors. The activity of ligands was measured in an IP3 assay in COS-7 cells co-transfected with the receptor and the promiscuous G protein Gqi4myr. EC50 (activity) values are given in log and nm/μm. Fmut is the factor presenting the -fold decrease of EC50 for the mutant compared to WT CCR5. The number of experiments (n) is given in parantheses.
Allosteric Properties of Metal Ion Chelator Complexes
Like ZnBip, ZnTerp was previously shown to act as an allosteric enhancer of CCL3 binding to CCR5 with an affinity (Ki) higher than that of ZnBip (Fig. 1C) (22). A third high affinity ligand exists for CCR5, namely CCL4, which is more closely related to CCL3 than to CCL5. Homologous competition binding experiments revealed a KD of 1.4 nm for CCL4 (Fig. 2, A and B), and consistent with the closer structural homology between CCL3 and CCL4, CCL3 displaced 125I-CCL4 with high affinity (Ki of 3.7 nm (i.e. very similar to the Kd of 4.5 nm; see Table 2)), whereas CCL5 was not able to displace CCL4 with high affinity (Ki of 0.13 μm) (Fig. 2C). None of the metal ion chelator complexes enhanced the binding of 125I-CCL4 (Fig. 2, D–F) to the same extent as the binding of 125I-CCL3 (Fig. 1C). In fact, they acted oppositely, with weak displacement for ZnBip (Ki value of 290 μm) and weak enhanced binding for ZnTerp, with a Ki of 1.8 μm and maximal enhancement of 160% (compared with 670% for CCL3) (Fig. 2, D and E, respectively). Thus, consistent with previous observations (16, 21), this shows that the allosteric property of metal ion chelators is chemokine-dependent.
FIGURE 2.

Homo- and heterologous competition binding assays with 125I-CCL4. A, average of original CCL4 binding data in cpm; binding to COS-7 cells transiently transfected with CCR5 (drawn line) or untransfected COS-7 cells (stippled line). B, normalized curve of A. C, heterologous binding curves against CCL3 (squares) and CCL5 (triangles). Note that CCL5 has a lower Ki than CCL3. D–F, heterologous competition binding against metal ion chelator complexes and single complex components (Zn2+, chelator) for ZnBip (D), ZnTerp (E), and ZnClTerp (F). Only ZnTerp (E) seems to be slightly able to enhance the binding of CCL4, yet to maximal levels much lower than what was observed for CCL3 (Fig. 1). For all curves, n ≥ 3.
TABLE 2.
Homologous radioactive competition binding assays for 125I-CCL3
The name and position of mutants according to the Ballesteros/Weinstein (left) and Baldwin/Schwartz numbering system are given. KD values are given in log and nm. Fmut is the factor presenting the -fold decrease of KD for the mutant compared to WT CCR5. Bmax is given in fmol/100.000 cells. The number of experiments (n) is given in parentheses.

Computational Modeling
We decided to explore the binding sites of ZnBip and ZnTerp in CCR5. Due to their agonistic nature, we built an active-like CCR5 model in MODELLER using the crystal structure of the constitutively active chemokine receptor US28 (PDB entry 4XT1) (Fig. 3, A–C). In comparison with the inactive CCR5 crystal structure in complex with maraviroc (PDB entry 4MBS), our model shows conformational changes and active-like characteristics; the extracellular part of TM-1 is moved slightly toward TM-7, the extracellular part of TM-2 is slightly tilted away from TM-1, ECL-1 is tilted further outward, ECL-3 is tilted further inward, and most characteristically for the active-like conformation, the intracellular part of TM-6 is tilted away from the center of the TM-bundle (Fig. 3, A and B). Furthermore, all binding pocket residues are slightly altered in their position, and a tighter aromatic interaction is observed between Tyr-108III:08/3.32, Phe-109III:09/3.33, and Phe-112III:12/3.36 in TM-3. Furthermore, the side chains of Tyr-244VI:09/6.44, Trp-248VI:13/6.48, and Tyr-251VI:16/6.51 in TM-6 are rotated slightly downward toward the intracellular receptor side (Fig. 3C) (the residue position according to the Baldwin-Schwartz and Ballesteros-Weinstein numbering system is given in superscript the first time a residue is mentioned (23, 24)).
FIGURE 3.

The active-like CCR5 model and docking poses of ZnBip (green) and ZnTerp (orange). A–C, comparison of the inactive CCR5 crystal structure (PDB entry 4MBS; light blue) in complex with maraviroc (yellow) and our active-like CCR5 model (gray) in complex with ZnBip (green). A, overall structure as seen from the membrane. B, pocket and extracellular domains as seen from the extracellular side. C, major binding pocket residues as seen from TM-5 with TM-4 and -5 removed for clarity. D, receptor-ZnTerp binding site as seen from TM-4/-5 to highlight the shape of the lower cavity in the major binding pocket to which only ZnTerp can reach. This lower cavity is lined by Tyr-108, Phe-109, Phe-112, Trp-248, and Tyr-251. E, comparable view of the receptor-ZnBip binding site. F, comparison of the Zn2+ chelator binding modes using an overlay view.
The chelators were then docked to a model in which the suggested metal ion anchor Glu-283VII:06/7.39 (16, 21, 22) has been manually set to coordinate a Zn2+ ion. A water molecule is positioned in the minor binding pocket and bridges Tyr-37I:08/1.39, Tyr-108, Glu-283, and the Zn2+ ion. Both Bip and Terp dock to the same area and largely overlap in their binding sites. Importantly, the major binding pocket of CCR5 becomes very narrow between Tyr-108, Phe-109, and Tyr-251 before extending to a small lower cavity bordered by Phe-112 and Trp-248. The additional pyridine ring of Terp protrudes into this lower cavity of the major binding pocket (Fig. 3D).
Both Bip and Terp make van der Waals contacts with Tyr-108, Phe-109, and Tyr-251 in the major binding pocket, π-π stacking with Tyr-37 and Trp-86II:20/2.60 in the minor binding pocket, and cation-π interactions with Arg-168Cys-10 in ECL-2a. Terp additionally interacts with Phe-112 and Trp-248 deep in the major binding pocket (Fig. 3, D–F).
Probing the CCR5-bound Zn2+-Terp Complex
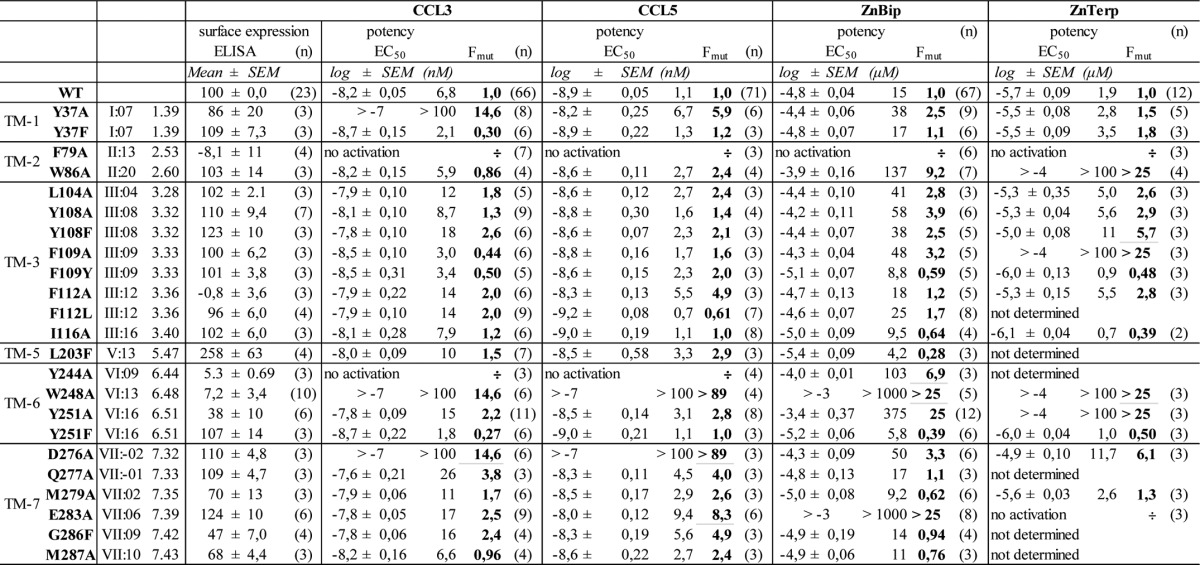
The modeling suggested two alternative poses for Terp in CCR5, one with all three nitrogen atoms coordinated to Zn2+ and another with pyridyl in the lower cavity rotated away from Zn2+. This prompted us to investigate the need for a tridentate interaction. To this end, Terp analogues were synthesized with the terminal pyridyl shifted to the meta- (mTerp) or para-position (pTerp) (25, 26) or replaced by phenyl (PhBip) (27, 28) (Fig. 4). Thus, all analogues were unable to form a tridentate complex with Zn2+ but otherwise as closely as possible preserved the features of Terp.
FIGURE 4.
Role of the additional pyridyl ring for ZnTerp. The tridentate coordination of zinc in ZnTerp allowed by the ortho-position of the nitrogen atom of the third pyridyl ring is not possible for PhBip, mTerp, or pTerp, which can thereby not form tridentate complexes with Zn2+. The relevance of these compounds as CCR5 ligands is compared using the IP3 assay.
The potency of each of the Terp analogues on CCR5 in complex with Zn2+ was determined in the IP3 assay described above (Fig. 4). None of the three Terp analogues gave a significant effect alone. In complex with Zn2+, mTerp exhibited a tendency toward weak agonism at 100 μm, pTerp was inactive, and PhBip showed inverse agonism at 100 μm. Thus, the results strongly support a tridentate complex of Terp with Zn2+.
Receptor Mutagenesis
To validate the predicted binding conformations of ZnBip and ZnTerp, we performed ligand-mapping experiments with a library comprising 23 receptor mutants (Fig. 5). We first assessed the cell surface expression of each mutant by an ELISA technique with antibodies against a FLAG tag inserted in the N terminus. We then assessed the ability of the endogenous chemokines CCL3 and CCL5 and the metal ion chelator complexes ZnBip and ZnTerp to activate each mutant receptor in an assay measuring IP3 in COS-7 cells transiently expressing the mutants and the chimeric G protein, Gqi4myr (Table 1). Finally, homologous competition binding experiments with 125I-CCL3 were performed (Table 2; for controls and homologous curves, see supplemental Fig. 1). Most of the mutants showed WT-like cell surface expression, except F79A, F112A, Y244A, and W248A, which showed absent (F79A and F112A) or highly reduced (Y244A and W248A) surface expression (Table 1). These mutants were also characterized by impaired signaling properties. F79A and W248A could not be activated by any ligand. Y244A could not be activated by CCL3 or CCL5 but could be activated by ZnBip (Table 1), and F112A showed highly reduced efficacy for all ligands (not shown). F112A, Y244A, and W248A, but not F79A, bound CCL3, albeit with reduced Bmax values (Table 2). Thus, F79A is a completely disintegrated receptor, Ala substitution of Trp-248 results in a non-signaling receptor, and Y244A selectively impairs chemokine-induced signaling. The remaining mutants only selectively impaired the signaling properties of single ligands, which verifies the general integrity of our mutant library and allows for validation of the proposed ZnBip and ZnTerp binding site.
FIGURE 5.

Receptor mutagenesis in activity assays. The helical wheel diagram of CCR5 in the center shows all residues of the extracellular halves of helices. The residues that were mutated are shown in white on black; boldface circled residues are conserved among class A 7TM receptors. All mutations included in this study are listed beside their respective helices. This figure only presents data for those in boldface type. A–D, activity of ZnBip (left lanes, circles) and ZnTerp (right lanes, squares) at selected receptor mutants as measured in an IP3 assay. Data are normalized to the maximal chemokine-induced response at each mutant (n ≥ 3). A, relevant mutations in TM-3 of the major binding pocket: Y108A/F and F109A. B, relevant mutants in TM-6 of the major binding pocket: Y251A/F. C, relevant mutants in the minor binding pocket: Y37A/F and W86A. D, Ala mutation of the metal ion anchor Glu-283 in TM-7.
Functional Mapping of the CCL3 and CCL5 Binding Sites
In addition to the above-mentioned residues important for receptor integrity and chemokine-mediated activation (Phe-79, Phe-112, Tyr-244, and Trp-248), we found the potency of CCL3 and CCL5 to be impaired by Y37A in TM-1 and by D276A and Q277A in TM-7. Interestingly, CCL5 depended to a smaller degree on Y37A (5.9-fold reduced potency) than CCL3 (>15-fold reduced potency). A potency decrease for CCL5, but not CCL3, was also observed for E283A and G286F (Table 1). Thus, through this screen, we identified receptor residues commonly used by both CCL3 and CCL5 and residues used by each chemokine distinctly.
Functional Mapping of ZnBip and ZnTerp Binding Sites
Computational modeling suggested ZnBip to interact with Tyr-108, Phe-109, and Tyr-251 in the major binding pocket and Tyr-37 and Trp-86 in the minor binding pocket (Fig. 3). Mutagenesis of aromatic residues in the major and minor binding pocket indeed supports this binding mode (Fig. 5, A–C). Thus, Y251A completely abrogates ZnBip signaling (Fig. 5B), and Y108A and F109A result in a 3.9- and 3.2-fold decreased potency, respectively (Fig. 5A). Interestingly, Y108F and Y251F do not yield WT-like potency or efficacy (Fig. 5, A and B), highlighting a role of the OH groups. This was also suggested from the computational modeling, where the OH group of Tyr-108 coordinates Zn2+, whereas that of Tyr-251 does not directly interact with ZnBip. Furthermore, W86A in the minor binding pocket decreased the potency by 9.2-fold, and a minor effect (2.5-fold) was observed for Y37A, which is rescued by Y37F (Fig. 5C).
ZnTerp overlaps in its binding site with that of ZnBip and additionally interacts with Phe-112 and Trp-248 (Fig. 3F). Our mutagenesis data agree with such a binding pose, because mutants Y108A, F109A, and Y251A in the major binding pocket abrogate the activity of ZnTerp (Fig. 5, A and B). Also here the OH group of Tyr-108, but not that of Tyr-251, is important for ZnTerp activity, because Y108F does not yield WT-like potency. W86A results in >50-fold reduced potency of ZnTerp, and Y37A, although not affecting potency, is crucial for its efficacy. Interestingly, ZnTerp showed WT-like activity at Y37F, emphasizing the importance of the aromatic moiety at this position (Fig. 5C). As discussed above, F112A is characterized by low surface expression and reduced efficacies for all ligands, including ZnBip and ZnTerp. ZnTerp in fact displays WT-like potency at F112A, and its dependence on Phe-112 therefore could not be confirmed. Furthermore, ZnBip showed WT-like activity at F112L, highlighting that an aromatic residue in this position is not absolutely required for this ligand. W248A is a signaling-deficient receptor, and we therefore cannot draw conclusions regarding its role for ZnTerp (Table 1) (29).
Finally, Ala mutation of the proposed metal ion anchor Glu-283 abrogates the activity of ZnTerp in addition to ZnBip, for which this was previously shown (Fig. 5D) (21). The rest of the mutants showed no or only minor effects (i.e. <3-fold; Table 1) on the potency of ZnBip or ZnTerp and were in fact not suggested as interaction partners from our in silico modeling. Only D276A decreased the potency of ZnBip and ZnTerp by 3.3- and 6.1-fold, respectively (Table 1).
Effect of Receptor Mutagenesis on the Allosteric Modulation by ZnBip and ZnTerp
After having identified and validated the binding site of ZnBip and ZnTerp, we went on to describe the structural basis for their allosteric modulation of CCL3 by performing binding studies with 125I-CCL3 on selected mutants. The metal ion anchor Glu-283 was crucial for the activity of ZnBip and ZnTerp, whereas F109A selectively impaired ZnTerp (Fig. 5, A and D). F109A and E283A also abrogated the allosteric modulation by ZnBip and ZnTerp (Fig. 6, A and B). Furthermore, Ala mutation of Trp-248, which was suggested to selectively interact with ZnTerp, nearly abrogated the CCL3 binding-enhancing ability of ZnTerp but had no effect on ZnBip (Table 3). Interestingly, the other mutants in the major binding pocket (Y108A, F112A, and Y251A) had no effect in binding assays (Table 3), whereas they all affected the agonistic function of ZnBip and ZnTerp (Fig. 5, A and B, and Table 1). Finally, we observed a ligand-dependent role of Tyr-37 and Trp-86. Similar to the functional assays (Fig. 5C), the allosteric modulation by ZnBip was mainly affected by W86A with a 6.3-fold decreased Ki compared with WT, but not by Y37A (Fig. 6, C and D). In contrast, Y37A, but not W86A, impaired the allosteric modulation by ZnTerp (Fig. 6, C and D), whereas both mutants impaired the activity of ZnTerp (Fig. 5C). Altogether, this highlights a general importance of Phe-109 in the allosteric modulation by metal ion chelator complexes and a selective role of Tyr-37 for ZnTerp and of Trp-86 for ZnBip.
FIGURE 6.

Receptor mutagenesis in heterologous competition binding assays with 125I-CCL3. The ability of ZnBip (white circle) or ZnTerp (black square) to enhance the binding of 125I-CCL3 to WT (stippled lines) or four different mutants was assessed in heterologous competition binding experiments. A, E283A; B, F109A; C, Y37A; D, W86A. Data are normalized to the homologous binding curve (n ≥ 3).
TABLE 3.
Heterologous radioactive competition binding assays with 125I-CCL3 as tracer and ZnBip, ZnTerp or ZnClTerp as competitor
Note that the metal ion chelator complexes enhance the binding of 125I-CCL3 and thus do not act as classical competitors. The name and position of mutants according to the Ballesteros/Weinstein (left) and Baldwin/Schwartz numbering system are given. Ki values are given in log and μm. Fmut is the factor presenting the -fold decrease of Ki for the mutant compared to WT CCR5. ZnClTerp displaces 125I-CCL3 from Y37A. The number of experiments (n) is given in parentheses.
Binding Site of the Allosteric Enhancer ZnClTerp
We next included ZnClTerp, which has a chloro-substituent at the central pyridine ring as the only difference from ZnTerp (Fig. 7C) and which acts as a positive allosteric modulator of CCL3 binding without agonistic properties (22) (Fig. 7, A and B). Furthermore, it bound allosterically to CCL4, neither enhancing nor displacing this chemokine (Fig. 2F). Docking reveals that the chelator moiety 4′-chloro-2,2′:6′,2″-terpyridine (ClTerp) binds to the same site as Bip and Terp, consisting of Tyr-108, Phe-109, Phe-112, Trp-248, and Tyr-251 in the major binding pocket and Tyr-37 and Trp-86 in the minor binding pocket (Fig. 7D). The chloro-substituent points upward toward the extracellular surface, where it interacts with Phe-109. In binding studies with 125I-CCL3, mutations of residues in the major binding pocket have largely the same effect on ZnClTerp as on the two other metal ion chelator complexes. Thus, F109A and E283A abrogated CCL3 enhancement, whereas Tyr-108, Phe-112, and Tyr-251 play no role or only a minor role (Table 3). The selective role of minor binding pocket residues that was observed for ZnBip and ZnTerp is also seen for ZnClTerp, which, similar to ZnTerp, is only impaired by Y37A (Fig. 8A) and not by W86A (Table 3). In fact, ZnClTerp was turned into a competitor of 125I-CCL3 at Y37A and, in addition, acted as an antagonist of CCL5- and ZnBip-mediated activation with potencies of 0.74 and 0.73 μm, respectively (Fig. 8B). CCL3 did not activate Y37A and therefore was not used as an activating ligand in the antagonism experiments.
FIGURE 7.
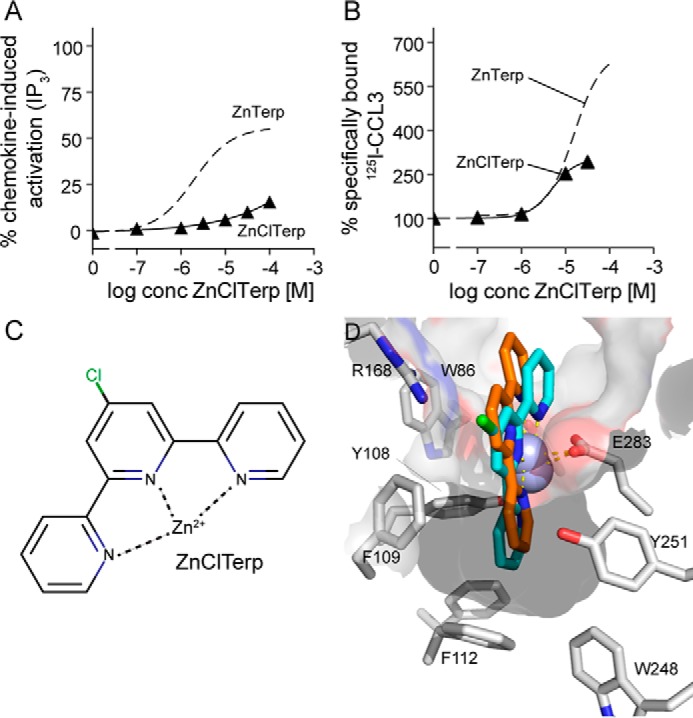
Structure, function, and docking of ZnClTerp. A, activity of ZnClTerp (black triangle) in comparison with ZnTerp (stippled line) at CCR5 as measured in an IP3 assay (see “Experimental Procedures” for details). Data are normalized to maximal response induced by chemokine at WT (n ≥ 3). B, ability of ZnClTerp (black triangle) in comparison with ZnTerp (stippled line) to enhance the binding of 125I-CCL3 to CCR5 as determined in a heterologous competition binding assay. Data are normalized to the homologous binding curve (n ≥ 3). C, structure of ZnClTerp. D, docking of ZnClTerp (cyan) in comparison with ZnTerp (orange) to the active-like CCR5 model, including residues in the vicinity of the chloro-substituent of ZnClTerp.
FIGURE 8.
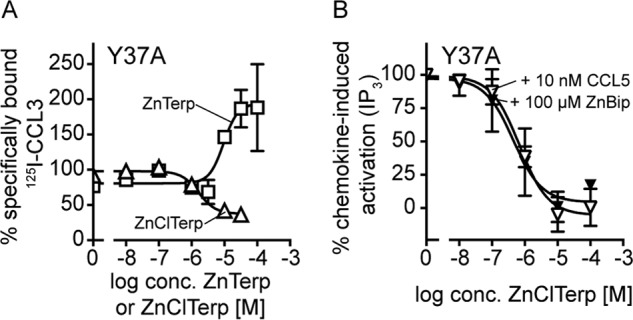
The effect of mutation Y37A on the allosteric action and activity of ZnClTerp. A, heterologous competition binding of 125I-CCL3 to Y37A competed by ZnClTerp (triangle) in comparison with ZnTerp (square). Data are normalized to the homologous binding curve (n ≥ 3). B, ZnClTerp can antagonize ZnBip-induced (black triangle) and CCL5-induced (white triangle) activity of Y37A, as measured in an IP3 assay. Data are normalized to maximal response induced by chemokine or ZnBip at Y37A (n ≥ 3).
Finally, the binding orientation of ZnClTerp reveals a possible mechanism for its loss of function. In comparison with ZnTerp, the overall geometry of the ZnClTerp complex does not allow favorable aromatic interactions between the major binding pocket-occupying pyridyl ring and Trp-248 (Fig. 7D), a residue that is central for the chemokine- (29, 30) and small molecule-mediated activation of CCR5 (Table 1). In fact, we observe that W248A only slightly impaired the maximal level of CCL3 enhancement by ZnClTerp in accordance with the low surface expression of this mutant receptor. This is in contrast to ZnTerp, where W248A almost completely abrogated the enhancement of CCL3 binding (Fig. 9A). The independence of ZnClTerp from Trp-248 and its deficient interaction with this residue could thus explain the lack of activity. Ile-116III:16/3.40 has previously been shown to function as a gate for the rotation of Trp-248 during activation of CCR5 (29). We therefore tested whether ZnClTerp could be turned into an agonist if this gate were removed. Indeed, at I116A, ZnClTerp gained activity and 2.9-fold higher potency than ZnTerp at WT CCR5 (Fig. 9B).
FIGURE 9.
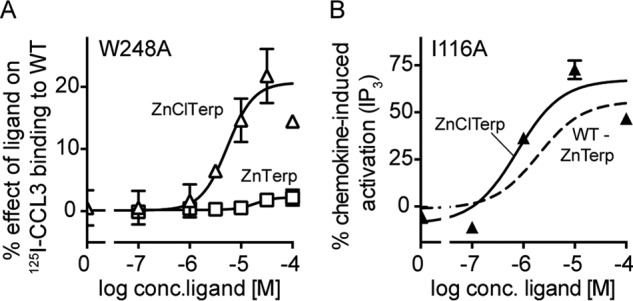
Interactions in the major binding pocket determine activity profiles of ZnTerp and ZnClTerp. A, heterologous competition binding of 125I-CCL3 to W248A competed by ZnTerp (squares) and ZnClTerp (triangles). Data are normalized to the maximal effect observed for the ligand on WT (n > 3). B, activity of ZnClTerp (triangle) in comparison with ZnTerp (stippled line) on I116A as measured in an IP3 assay (see “Experimental Procedures” for details). Data are normalized to maximal response induced by chemokine at I116A (n ≥ 3).
Discussion
We herein describe the structural basis for CCR5 activation by small molecule agonists, the metal ion chelator complexes ZnBip, ZnTerp, and ZnClTerp. ZnTerp has a higher potency than ZnBip due to its deeper anchorage in the major binding pocket, whereas ZnClTerp lacks intrinsic activity, presumably due to an altered positioning in this pocket. The allosteric enhancement of CCL3 by all three ligands arises from a positive modulation of an allosteric interface between the chelator and chemokine binding sites comprising Phe-109 in TM-3 and aromatic residues in the minor binding pocket. Metal ion site engineering has been used for decades to predict helical connectivity, initially described in the tachykinin receptors NK1 and NK3 (31–33) and followed up by various receptors (34–36), hereunder the two CXC-chemokine receptors ORF74 encoded by human herpesvirus 8 (HHV8) (37, 38) and CXCR3 (39). In contrast to the above-mentioned chemokine receptors, the metal ion chelator complex site in CCR5 is not engineered but rather naturally occurring (21).
CCR5 Activation Mechanism and Activity Profile of ZnBip, ZnTerp, and ZnClTerp
The activation of 7TM receptors is controlled by a number of conserved residues that constitute microswitches and change rotameric state or interaction partners upon receptor activation and thereby transduce an extracellular signal into an intracellular response. The most important microswitches are 1) an arginine at position III:26/3.50 that is part of the DRY motif and interacts with the adjacent AspIII:25/3.49 in the inactive conformation or TyrV:24/5.58 in the active conformation; 2) TyrVII:20/7.53 of the NPXXY motif in TM-7, which interacts with aromatic residues in the intracellular small helix 8 or in the receptor core in the inactive and active conformation, respectively; and 3) the so-called toggle switch TrpVI:13/6.48 of the CWXP motif in TM-6 (30, 40, 41). Upon receptor activation, this tryptophan is suggested to rotate toward TM-5 (30, 40, 41), which allows for an outward movement of TM-6 on the intracellular side, a conformational change that indeed was observed in active-like crystal structures of the β2-adrenergic receptor and US28 (8, 42, 43). However, a rotation of TrpVI:13/6.48 was not observed in these structures, and in fact TrpVI:13/6.48 is only conserved in ∼71% of all 7TM receptors. Thus, whereas the movement of TM-6 upon receptor activation is by now generally accepted, the role of TrpVI:13/6.48 seems to be more subtle and less universal.
For CCR5, we have previously shown that CCL3- and CCL5-mediated activation depends on TrpVI:13/6.48, which is Trp-248 (29, 30). Here we find that also small molecule-mediated activation depends on Trp-248, which allows us to propose a central role of Trp-248 for the activation mechanism of CCR5. Furthermore, Steen et al. (29, 30) showed that the rotamer switch of Trp-248 is linked to a slight movement of Tyr-244VI:09/6.44 located one helical turn below, which in turn is controlled by Ile-116 in TM-3 on the opposite site of the major binding pocket. Consistent with this, Tan et al. (13) described Trp-248 together with Tyr-244 as relays of receptor activation in their crystal structure of CCR5. We suggest that the interaction of ZnTerp with Trp-248 accounts for its higher potency compared with ZnBip, which does not directly interact with Trp-248 yet still acts as agonist. ZnClTerp interacts in an impaired manner with Trp-248 (Fig. 7D), and its lack of activation could be due to its inability to induce a correct stabilization of this residue, therefore hindering the conformational change of Trp-248 required for receptor activation. This is confirmed by the observation that ZnClTerp gains activity at I116A (i.e. when the gating function of Tyr-244 and Trp-248 is released).
An Allosteric Interface between CCL3 and the Metal Ion Chelator Complex Binding Site
According to the two-step model of chemokine-mediated receptor activation (9, 10), the chemokine core and extracellular receptor domains mediate binding of chemokines to their receptors. For CCL3, this involves the receptor N terminus and residues in ECL-2 (including Arg-168) and ECL-3 but also residues at the top of TM-5 and -6 (reviewed in Refs. 9 and 10). A TXP motif in TM-2 and the surrounding non-polar residues play a role for the second activity-inducing step (44, 45). Govaerts et al. (44) showed that a serine, cysteine, or threonine two residues before the conserved proline in TM-2 fortifies the proline-induced kink via a hydrogen bond from their side chain to the main chain in the turn below. They also found that an entire hydrophobic network around the TXP motif, among others comprising residues Phe-85II:19/2.58, Leu-104III:04/3.28, Phe-109III:09/3.33, and Phe-112III:12/3.36, is important for CCL3-mediated receptor activation (46). We previously showed the importance of Asp-276 for CCL3-mediated activation (21) and in the present study extend this observation to Tyr-37 and Gln-277.
We identify the metal ion chelator binding sites in CCR5 as consisting of Tyr-37, Trp-86 (minor binding pocket), Arg-168 (ECL-2a), Tyr-108, Phe-109, and Tyr-251 (major binding pocket) and, for ZnTerp and ZnClTerp, additionally Phe-112 and Trp-248 (lower cavity of the major binding pocket). Thus, this site lies directly beneath the CCL3 binding site in CCR5, and both ligand types interact with some of the same residues, such as Tyr-37, Phe-109, and Arg-168. We therefore identify an allosteric interface located on the verge of the CCL3 and metal ion chelator complex binding sites. Whereas metal ion chelator complexes approach this allosteric interface from their transmembrane receptor domain, CCL3 approaches it from the extracellular side (Fig. 10). Changing the receptor at this allosteric interface alters the direction and nature of the allosteric interaction. Thus, F109A abrogates the positive allosteric character and leaves the metal ion chelator complexes as pure agonists with no effect on CCL3 binding. Y37A even turns ZnClTerp into a competitive displacer and an antagonist and induces an inactive receptor conformation, with which the agonist CCL3 has an inherently low affinity. Arg-168, also part of both the chemokine and metal ion chelator site, is in our active-like model rotated downward to the metal ion chelator ligand, whereas it points outward into the extracellular space in the antagonist-bound CCR5 crystal structure (13).
FIGURE 10.

Allosteric interface between small molecule and chemokine binding sites. A, schematic drawing of receptor (TM-1 to -5); B, the extracellular half of our CCR5 model. The parts of CCR5 that were shown to bind CCL3 are presented in light blue, and parts of CCR5 involved in CCL3-mediated activation are shown in dark blue and include the TXP motif in TM-2. The approximate small molecule binding site in CCR5 is shown in yellow. The border between the chemokine and small molecule site forms the allosteric interface (green). The hydrophobic core of CCR5 that confers receptor integrity and is essential for the receptor activation mechanism is shown in red.
Ligand-directed Biased Action of Small Molecules in CCR5, a Model for Future Biased Drugs
It should also be noted that any allosteric interface is ligand-specific and depends on the binding site and induced receptor conformations for both the endogenous ligand and the allosteric modulator. This is highlighted by the finding that ZnBip, ZnTerp, and ZnClTerp are differently influenced by alterations in the minor binding pocket (Y37A and W86A; Figs. 5C and 8 and Tables 1 and 3). The situation is also entirely different for CCL5, which is not enhanced in its binding by metal ion chelator complexes and interacts differently with CCR5 than CCL3 (21). For example, only CCL5 directly interacts with the metal ion anchor residue Glu-283VII:06/7.39. A glutamate in position VII:06/7.39 is found in 74% of chemokine receptors, and because it is involved in the binding of most current chemokine receptor antagonists (14), position VII:06/7.39 might give rise to an overlap with the chemokine site (9). In addition to CCL5, this was, for example, shown for the interaction of CCL2 with CCR2 (47) and other CC-chemokine receptors (reviewed in Ref. 9). Furthermore, we find CCL5 to have a lower dependence on Tyr-37 than CCL3. We propose Tyr-37 to be part of the allosteric interface between metal ion chelator complexes and CCL3, and a different interaction of CCL5 with Tyr-37 might thus explain the lack of positive allosteric enhancement of this chemokine by metal ion chelator complexes.
A third chemokine interacting with CCR5 is CCL4. CCL4 is known to bind to the receptor N terminus and ECL-2, and also residues within the transmembrane receptor domain contribute to CCL4 binding, including Tyr-37, Tyr-108, Asp-276, Glu-283, and Met-287 (reviewed in Ref. 9). Tyr-37 and Tyr-108 are part of the allosteric interface proposed for CCL3 and metal ion chelator complexes and might be utilized similarly by CCL4 and ZnTerp for allosteric enhancement. A dual interaction with Glu-283 does, however, favor a competitive binding behavior toward CCL4, which might explain the absent (for ZnBip and ZnClTerp) and low level of positive allosteric enhancement (for ZnTerp) observed for CCL4. Rational design of allosteric modulators in general requires detailed insights into the interaction of each chemokine with its receptor(s) but at the same time will allow us to manipulate those interactions for singular chemokine-receptor pairings.
A Hydrophobic Core in CCR5 Maintains Receptor Integrity and Relays Signal Transduction
In addition to their central role for CCR5 activation, Tyr-244 and Trp-248 are also important for receptor integrity, as illustrated by the low surface expression of Y244A and W248A (Table 1). We mutated the entire set of aromatic residues in the main binding pocket of CCR5 and identified two additional residues with importance for receptor activation and integrity: Phe-79 and Phe-112 (Table 1). F79A could not be detected at the cell surface and was not activated by any ligand; nor could it bind CCL3. Also, F112A displayed very low surface expression and similarly lowered efficacies and Bmax for CCL3. Interestingly, both residues are located deeply in the main binding crevice, at the same level as Tyr-244 and Trp-248. Thus, Phe-79 is located centrally in TM-2 two helical turns below Trp-86 and directly underneath Tyr-108 in TM-3. Phe-112 is located in TM-3 and points directly toward Trp-248. The model shows that Phe-79, Phe-112, Tyr-244, and Trp-248 pack tightly and form a hydrophobic core at the center of the receptor and the base of the transmembrane binding pocket (Fig. 10). We find this hydrophobic core to be important for maintaining structural receptor integrity. Its deep location at the bottom of the transmembrane binding pocket also suggests a role in relaying signal transduction. This was proven for Tyr-244 and Trp-248 (13, 29, 30), and we now identify Phe-79 in TM-2 as central for transducing activation signals in the minor binding pocket.
Antagonism in CCR5
Like the majority of small molecule ligands for chemokine receptors (14), maraviroc is positively charged and engages in a salt bridge with the chemokine receptor-conserved Glu-283. It furthermore interacts with Tyr-37, Thr-195, and Thr-259, and its phenyl ring reaches deeply into the major binding pocket interacting with Tyr-108, Phe-109, Phe-112, Trp-248, and Tyr-251 (13). Therefore, the binding site of maraviroc and the metal ion chelator complexes identified here overlap. However, maraviroc differently influences the allosteric interface, as indicated by the different rotation of Arg-168 (pointing outward) and its opposite property (being an antagonist). Other small molecule CCR5 antagonists (e.g. TAK-779, aplaviroc, SCH-C, vicriviroc, and YM-370749) also bind across both subpockets (major and minor) and interact with Glu-283 (48–50). Two of them, TAK-779 and aplaviroc, were also suggested to directly interact with Trp-248 (48, 49). Furthermore, the antagonist aplaviroc makes extensive interactions with ECL-2b (i.e. the part downstream of the disulfide bridge) (21), whereas our agonists or allosteric modulators here interact with ECL-2a. Together, this highlights how different ligands can fit the same overall transmembrane binding pocket yet have distinct allosteric interfaces and provoke different outcomes: inhibition, activation, and allosteric modulation. This outcome also depends on the applied chemokine. Aplaviroc, for example, can fully inhibit CCL3- and CCL5-mediated activation as a result of its antagonistic activity and ability to stabilize an inactive receptor conformation. However, whereas aplaviroc also fully displaces CCL3, it cannot fully overcome CCL5 binding (15, 51). This reflects the different binding modes of these chemokines to CCR5, which differ, among others, in terms of utilizing receptor residues Tyr-37 and Glu-283 (Table 1). Furthermore, the receptor N terminus and ECL-2 are the domains contributing most strongly to chemokine affinity, and it is not surprising that a small molecule antagonist binding to the transmembrane receptor domain cannot sterically compete with binding to these regions. These insights illustrate that small molecule drugs can be tailored to modify the interactions of specific chemokine-receptor pairs, a concept that holds much promise because it allows us to achieve a currently unexploited level of control over the chemokine system.
The current lack of success in producing clinically efficacious anti-inflammatory drugs targeting chemokine receptors (4, 17, 52) remains as a reminder that we have not yet fully understood the complexity of the chemokine system. In contrast to the previous view of this system as being redundant, it now seems that it is finely tuned and displays ligand, receptor, and tissue bias (2–4). It is also evident that the interaction between chemokines and receptors is better described by a “pseudo”-two-step mechanism consisting of multiple steps (10). The present study describes the molecular basis for CCR5 activation and the complexity of allosteric interactions in chemokine receptors. This knowledge is central for future rational design of specific ligand types, such as allosteric or overlapping antagonists, biased ligands, or modulators of chemokine function.
Experimental Procedures
Materials
Human CCL3 was purchased from Peprotech. 125I-CCL3 was produced in house. ZnCl2, Bip, Terp, ClTerp, and DMSO were purchased from Sigma-Aldrich and used without further purification. Synthesis of PhBip was performed as described previously (27), as was the intermediate for both 2,2′:6′,3″-terpyridine (mTerp) and 2,2′:6′,4″-terpyridine (pTerp). Both mTerp and pTerp were synthesized following a procedure published previously (25). The structure for all synthesized compounds was confirmed by 1H and 13C NMR and high resolution mass spectrometry. The highest concentrations of metal ion chelator complexes were 20 mm for ZnBip and 2 mm for ZnTerp/ZnClTerp and were made from 0.2 or 0.02 m ZnCl2 in water and 100 mm Bip or 10 mm Terp/ClTerp in DMSO, respectively, and were supplemented with water and 70% ethanol. The ratio of Zn2+/chelator was 1:2 to ensure full complexation of Zn2+. Dilutions were made in water. CCR5 was cloned in-house from a leukocyte DNA library. The promiscuous G protein Gqi4myr was kindly provided by Evi Kostenis (University of Bonn). Myo[3H]inositol (PT6-271), iodine-125, and 125I-CCL4 were purchased from PerkinElmer Life Sciences.
Molecular Biology
Receptor mutations were introduced by the PCR overlap extension technique or the QuikChange technique (Agilent Technologies) using WT CCR5. All reactions were carried out using Pfu polymerase (Stratagene) under conditions recommended by the manufacturer. The mutations were cloned into pcDNA3.1+ for use in IP3 and binding assays (Invitrogen) or into pcDNA3.1+ with M1 tag for ELISA. All constructs were verified by restriction endonuclease digestion and DNA sequencing (GATC Biotech).
Computational Modeling and Ligand Docking
A CCR5 model was generated using the recently crystallized active-like structure of US28 in complex with its partial inverse agonist CX3CL1 (PDB entry 4XT1) (8). 50 models were generated using MODELLER version 9.14 (53), and the five best models were chosen for subsequent docking experiments, based on the MODELLER objective function (DOPE), the GA341 score, and the overall orientation of conserved residues in the binding pocket of the models. The N and C termini of CCR5 were not considered during model generation, whereas the structural waters of US28 were preserved. Model refinement was performed in Vega ZZ version 3.1 (54) through 500 steps of conjugate gradient (CG) minimization in the SP4 force field while keeping the protein backbone fixed. The overall quality of the refined models was valuated using PROCHECK, ERRAT, and Verify 3D. All docking experiments were performed in AutoDock version 4.2.6 (55), using a force field adjusted for metal ions (56). Docking was carried out on the five best-scoring models, using manually constrained Zn2+-containing receptors, generated as follows. Zn2+ was placed in the vicinity of Glu-2837.39/VII:06 and relaxed in the binding site through 500 steps of CG in the SP4 force field, followed by 1000 CG steps of binding site optimization within a 10-Å radius of Zn2+. Default parameters for the flexible ligand GA-LS docking were employed for all docking experiments, using a 46 × 46 × 46-Å3 docking grid centered on Zn2+. 50 runs of GA-LS were performed for each ligand/receptor model pair. Results were clustered, and the lowest energy cluster representatives were chosen for further analyses. The binding sites of the best scoring ligand/receptor pairs were subsequently minimized through 500 steps of CG minimization using the SP4 force field, while keeping the protein backbone fixed, and further globally optimized through 500 steps of Monte Carlo simulation, as implemented in ICM version 3.8 (Molsoft LLC, San Diego, CA).
Transfection and Tissue Culture
COS-7 cells were grown in DMEM with Glutamax (Invitrogen) supplemented with 10% FBS, 180 units/ml penicillin, and 45 μg/ml streptomycin at 37 °C in a 10% CO2, 90% air-humidified atmosphere. Transfection of cells was carried out by the calcium phosphate precipitation method (57, 58). Briefly, plasmid DNA (20 μg of receptor cDNA and 30 μg of Gqi4myr for IP3 assays or 40 μg of receptor cDNA for 125I-CCL3-binding assays) was mixed with TE buffer (10 mm Tris-HCl, 2 mm EDTA-Na2, pH 7.5) and 30 μl of calcium chloride (2 m) to a total volume of 480 μl and was then added to the same amount of HEPES-buffered saline (280 mm NaCl, 50 mm HEPES, 1.5 mm Na2HPO4, pH 7.2). The mixture was allowed to precipitate for 45 min at room temperature, after which the precipitate and 300 μl of chloroquine (2 mg/ml) in 10 ml of culture medium was added to the 6 × 106 COS-7 cells seeded the day before. Transfection was stopped after 5 h by replacing with fresh medium, and cells were incubated overnight.
ELISA
Cells were transfected with M1-tagged WT or mutant receptors as described above. The following day, 35,000 cells/well were seeded in 96-well plates, which 24 h later were washed in Tris-buffered saline (TBS; 0.05 m Tris base, 0.9% NaCl, pH 7.6), fixed in 3.7% formaldehyde for 15 min at room temperature, washed three times in TBS, and incubated in TBS with 2% BSA for 30 min. The cells were then incubated for 2 h with anti-FLAG M1-antibody (Sigma-Aldrich) at 2 μg/ml in TBS with 1 mm CaCl2 and 1% BSA. After three washes with TBS supplemented with 1 mm CaCl2, the cells were incubated with goat anti-mouse HRP-conjugated antibody at 0.8 μg/ml (Thermo Fisher Scientific) for 1 h. After three additional washes, the immunoreactivity was revealed by the addition of TMB Plus substrate (Kem-En-Tec Diagnostics, Taastrup, Denmark). The reaction was stopped with 0.2 m H2SO4 after ∼5 min. Absorbance was measured at 450 nm on a Wallac Envision 2104 Multilabel Reader (PerkinElmer Life Sciences).
Functional Scintillation Proximity IP3 Assay (SPA)
One day after transfection, COS-7 cells (35,000 cells/well) were incubated with myo-[3H]inositol (5 μl/ml, 2 μCi/ml) in 0.1 ml of medium overnight in a 96-well plate. The following day, cells were washed twice in PBS and were incubated in 0.1 ml of Hanks' balanced salt solution (Invitrogen) supplemented with 10 mm LiCl at 37 °C in the presence of various concentrations of ligands for 90 min. Assay medium was then removed, and cells were extracted by the addition of 50 μl of 10 mm formic acid to each well, followed by incubation on ice for 30–60 min. The [3H]inositol phosphates in the formic acid cell lysates were thereafter quantified by adding yttrium silicate-poly-d-Lys-coated SPA beads. Briefly, 35 μl of cell extract was mixed with 80 μl of SPA bead suspension in H2O (12.5 μg/μl) in a white 96-well plate. Plates were sealed, agitated for at least 30 min, and centrifuged for 5 min at 402 relative centrifugal force. SPA beads were allowed to settle and react with the extract for at least 8 h before radioactivity was determined using a Packard Top Count NXTTM scintillation counter (PerkinElmer Life Sciences). All determinations were made in duplicate. Normalized and averaged curves of at least three experiments are shown in the figures. Unless otherwise stated, 100% accounts for the maximal chemokine-induced response observed at the given mutant, or WT when chemokines were inactive at the mutant, and 0% is the signaling of the given receptor (mutant or WT) in the absence of any ligand. These overall readouts have previously been used effectively for CCR5, CXCR4, and other chemokine receptors (21, 22, 59, 60).
cAMP Turnover Assay
CHO cells stably transfected with CCR5 WT or naive CHO cells were grown in HAMF12 supplemented with 10% FBS, 180 units/ml penicillin, 45 μg/ml streptomycin, and 0.5 mg/ml gentamicin at 37 °C in a 5% CO2, 90% air-humidified atmosphere. The HitHunter cAMP assay for small molecules (DiscoveRx) was used. Briefly, 35,000 cells were seeded in 96-well plates in 0.1 ml of cell culture medium. The following day, cells were washed once in 200 μl of Hepes-buffered saline, and 94 μl of Hepes-buffered saline supplemented with 1 mm isobutylmethylxanthine was added. After a 30-min incubation at 37 °C, 1 μl of forksolin was added to a final concentration of 10 μm, and cells were further incubated for 15 min at 37 °C. Then 5 μl of ligands were added. Following a 30-min incubation at 37 °C, the buffer was aspirated, cells were washed once in 100 μl of prewarmed PBS, and 30 μl of PBS were added. The procedure was then followed according to the manufacturer's instructions (i.e. by the addition of 15 μl of antibody solution, 60 μl of cAMP Working Detection Solution, incubation at room temperature for 60 min, and the addition of 60 μl of cAMP Solution A). Plates were agitated lightly at room temperature for 60 min, and luminescence was measured on a Wallac Envision 2104 Multilabel Reader (PerkinElmer Life Science Products).
Ca2+ Release Assay
CHO cells stably transfected with CCR5 WT or naive CHO cells were grown under the same conditions as for the cAMP turnover assay. For an experiment, cells were seeded on an 8-well NuncTM Lab-TekTM Chambered Coverglass (Thermo Scientific) and grown until 80% confluence. Cells were washed once with 200 μl of wash buffer prewarmed to 37 °C (Hanks' balanced salt solution (Gibco, Thermo Fisher Scientific) supplemented with 20 mm HEPES (Invitrogen), 1 mm CaCl2, 1 mm MgCl2, and 0.7 mg/μl probenicid (Life Technologies, Thermo Fisher Scientific)). Subsequently, 200 μl/well of prewarmed loading buffer (wash buffer supplemented with 0.2% Fluo-4 (Life Technologies, Thermo Fisher Scientific)) were added. Cells were incubated for 30 min at 37 °C in 5% CO2 in the dark. After incubation, cells were washed twice with 200 μl of prewarmed wash buffer, and 150 μl of 37 °C prewarmed cell medium was added. The cells were treated with CCL3 (0.1 μm), CCL5 (0.1 μm), ZnBip (1 mm), zinc (1 mm), or Bip (1 mm) and respective buffer controls. Changes in calcium concentrations were recorded with a Zeiss LSM 780 confocal microscope recording with 1 frame/s at 488 nm excitation. Ligands were added after the fluorescence signal reached a baseline and was stable.
Iodination of CCL3
17 μg (∼2 nmol) of carrier-free CCL3 (R&D Systems, Bio-Techne Corp.) were dissolved in 10 μl of iodination buffer (300 mm phosphate buffer, pH 7.4). 4 μl of iodine-125 (PerkinElmer Life Sciences, NEX033A) were added. For the reaction, 6 × 5 μl of a 3 μg/ml chloramine T solution in 300 mm phosphate buffer, pH 7.4, were added while occasionally stirring and at 1-min intervals. After 6 min, the reaction was stopped by the addition of 400 μl of water with 0.1% trifluoroacetic acid. The reaction mixture was then purified on a C18 column with an acetonitrile gradient from 20 to 80% over ∼45 min.
125I-CCL3 and 125I-CCL4 Competition Binding Assays
6 × 106 COS-7 cells were transfected with 40 μg of receptor cDNA and transferred to culture plates 1 day after transfection. The number of cells seeded per well was determined by the apparent expression efficiency of the receptors and was aimed at obtaining 5–10% specific binding of tracer. The number of cells thus ranged from 10,000 to 300,000. Two days after transfection, cells were assayed by competition binding for 3 h at 4 °C using 10–15 pm 125I-CCL3 or 125I-CCL4 plus unlabeled ligand in 0.2 ml (24-well plates) or 0.3 ml (12-well plates) of 50 mm Hepes buffer, pH 7.4, supplemented with 1 mm CaCl2, 5 mm MgCl2, and 0.5% (w/v) bovine serum albumin. After incubation, cells were washed quickly two times with 4 °C binding buffer supplemented with 500 mm NaCl. Nonspecific binding was determined in the presence of 0.1 μm unlabeled CCL3 or CCL4, respectively. Determinations were made in duplicate.
Author Contributions
S. K., O. L., V. D., and A. S. conducted the in vitro experiments and analyzed assays. R. M. A. and A. P. conducted the computational modeling under the supervision of T. M. F. T. U. designed metal ion chelators and edited the manuscript. M. B. designed and synthesized metal ion chelators. S. K. wrote the first draft of the manuscript. M. M. R. conceived the idea for the project.
Supplementary Material
Acknowledgment
We thank Maibritt Baggesen for excellent technical assistance.
This work was supported in whole or part by the AP Møller Maersk Foundation, the Aase and Einar Danielsen Foundation, and the Hørslev Foundation. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. 1.
- 7TM
- seven-transmembrane helix receptor
- ECL
- extracellular loop
- GPCR
- G protein-coupled receptor
- TM
- transmembrane helix
- ZnBip
- Zn2+ in complex with 2,2′-bipyridine
- ZnClTerp
- Zn2+ in complex with 4′-chloro-2,2′:6′,2′′-terpyridine
- ZnTerp
- Zn2+ in complex with 2,2′:6′,2″-terpyridine
- IP3
- inositol 1,4,5-trisphosphate
- PhBip
- 6-phenyl-2,2′-bipyridine
- mTerp
- 2,2′:6′,3″-terpyridine
- pTerp
- 2,2′:6′,4″-terpyridine
- ClTerp
- 4′-chloro-2,2′:6′,2″-terpyridine
- PDB
- Protein Data Bank
- Gqi4myr
- GαΔ6qi4myr
- CG
- conjugate gradient
- SPA
- scintillation proximity IP3 assay.
References
- 1. Bachelerie F., Ben-Baruch A., Burkhardt A. M., Combadiere C., Farber J. M., Graham G. J., Horuk R., Sparre-Ulrich A. H., Locati M., Luster A. D., Mantovani A., Matsushima K., Murphy P. M., Nibbs R., Nomiyama H., et al. (2014) International Union of Pharmacology: LXXXIX: update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 66, 1–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amarandi R.-M., Hjortø G. M., Rosenkilde M. M., and Karlshøj S. (2016) Probing biased signaling in chemokine receptors. Methods Enzymol. 570, 155–186 [DOI] [PubMed] [Google Scholar]
- 3. Steen A., Larsen O., Thiele S., and Rosenkilde M. M. (2014) Biased and G protein-independent signaling of chemokine receptors. Front. Immunol. 5, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Proudfoot A. E. I., Bonvin P., and Power C. A. (2015) Targeting chemokines: Pathogens can, why can't we? Cytokine 74, 259–267 [DOI] [PubMed] [Google Scholar]
- 5. Viola A., and Luster A. D. (2008) Chemokines and their receptors: drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 48, 171–197 [DOI] [PubMed] [Google Scholar]
- 6. Allen S. J., Crown S. E., and Handel T. M. (2007) Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820 [DOI] [PubMed] [Google Scholar]
- 7. Qin L., Kufareva I., Holden L. G., Wang C., Zheng Y., Zhao C., Fenalti G., Wu H., Han G. W., Cherezov V., Abagyan R., Stevens R. C., and Handel T. M. (2015) Structural biology: crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burg J. S., Ingram J. R., Venkatakrishnan A. J., Jude K. M., Dukkipati A., Feinberg E. N., Angelini A., Waghray D., Dror R. O., Ploegh H. L., and Garcia K. C. (2015) Structural biology: structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thiele S., and Rosenkilde M. M. (2014) Interaction of chemokines with their receptors: from initial chemokine binding to receptor activating steps. Curr. Med. Chem. 21, 3594–3614 [DOI] [PubMed] [Google Scholar]
- 10. Kufareva I., Salanga C. L., and Handel T. M. (2015) Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol. 93, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenkilde M. M., Benned-Jensen T., Frimurer T. M., and Schwartz T. W. (2010) The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol. Sci. 31, 567–574 [DOI] [PubMed] [Google Scholar]
- 12. Wu B., Chien E. Y. T., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., and Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan Q., Zhu Y., Li J., Chen Z., Han G. W., Kufareva I., Li T., Ma L., Fenalti G., Li J., Zhang W., Xie X., Yang H., Jiang H., Cherezov V., et al. (2013) Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosenkilde M. M., and Schwartz T. W. (2006) GluVII:06: a highly conserved and selective anchor point for non-peptide ligands in chemokine receptors. Curr. Top. Med. Chem. 6, 1319–1333 [DOI] [PubMed] [Google Scholar]
- 15. Scholten D. J., Canals M., Maussang D., Roumen L., Smit M. J., Wijtmans M., de Graaf C., Vischer H. F., and Leurs R. (2012) Pharmacological modulation of chemokine receptor function. Br. J. Pharmacol. 165, 1617–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jensen P. C., Thiele S., Ulven T., Schwartz T. W., and Rosenkilde M. M. (2008) Positive versus negative modulation of different endogenous chemokines for CC-chemokine receptor 1 by small molecule agonists through allosteric versus orthosteric binding. J. Biol. Chem. 283, 23121–23128 [DOI] [PubMed] [Google Scholar]
- 17. Proudfoot A. E. I., Power C. A., and Schwarz M. K. (2010) Anti-chemokine small molecule drugs: a promising future? Expert Opin. Investig. Drugs 19, 345–355 [DOI] [PubMed] [Google Scholar]
- 18. Dean M., Carrington M., Winkler C., Huttley G. A., Smith M. W., Allikmets R., Goedert J. J., Buchbinder S. P., Vittinghoff E., Gomperts E., Donfield S., Vlahov D., Kaslow R., Saah A., Rinaldo C., et al. (1996) Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene: Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273, 1856–1862 [DOI] [PubMed] [Google Scholar]
- 19. Samson M., Libert F., Doranz B. J., Rucker J., Liesnard C., Farber C. M., Saragosti S., Lapoumeroulie C., Cognaux J., Forceille C., Muyldermans G., Verhofstede C., Burtonboy G., Georges M., Imai T., et al. (1996) Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722–725 [DOI] [PubMed] [Google Scholar]
- 20. Liu R., Paxton W. A., Choe S., Ceradini D., Martin S. R., Horuk R., MacDonald M. E., Stuhlmann H., Koup R. A., and Landau N. R. (1996) Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377 [DOI] [PubMed] [Google Scholar]
- 21. Thiele S., Steen A., Jensen P. C., Mokrosinski J., Frimurer T. M., and Rosenkilde M. M. (2011) Allosteric and orthosteric sites in CC chemokine receptor (CCR5), a chimeric receptor approach. J. Biol. Chem. 286, 37543–37554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thiele S., Malmgaard-Clausen M., Engel-Andreasen J., Steen A., Rummel P. C., Nielsen M. C., Gloriam D. E., Frimurer T. M., Ulven T., and Rosenkilde M. M. (2012) Modulation in selectivity and allosteric properties of small-molecule ligands for CC-chemokine receptors. J. Med. Chem. 55, 8164–8177 [DOI] [PubMed] [Google Scholar]
- 23. Schwartz T. W. (1994) Locating ligand-binding sites in 7TM receptors by protein engineering. Curr. Opin Biotechnol. 5, 434–444 [DOI] [PubMed] [Google Scholar]
- 24. Ballesteros J. A., and Weinstein H. (1995) in Receptor Molecular Biology (Sealfon S. C., ed) pp. 366–428, Academic Press, Inc., New York [Google Scholar]
- 25. Zhao L.-X., Sherchan J., Park J. K., Jahng Y., Jeong B.-S., Jeong T. C., Lee C.-S., and Lee E.-S. (2006) Synthesis, cytotoxicity and structure-activity relationship study of terpyridines. Arch. Pharm. Res. 29, 1091–1095 [DOI] [PubMed] [Google Scholar]
- 26. Ioachim E., Medlycott E. A., Polson M. I., and Hanan G. S. (2005) Synthesis of a novel series of 6,6′-disubstituted 4,4′-bipyrimidines by radical anion coupling: new π-accepting ligands for coordination chemistry. Eur. J. Org. Chem. 2005, 3775–3780 [Google Scholar]
- 27. Klein A., Rausch B., Kaiser A., Vogt N., and Krest A. (2014) The cyclometalated nickel complex [(Phbpy) NiBr](Phbpy− = 2,2′-bipyridine-6-phen-2-yl): synthesis, spectroscopic and electrochemical studies. J. Organometallic Chem. 774, 86–93 [Google Scholar]
- 28. Liu K. M., Liao L. Y., and Duan X. F. (2015) Iron catalyzed oxidative assembly of N-heteroaryl and aryl metal reagents using oxygen as an oxidant. Chem. Commun. 51, 1124–1127 [DOI] [PubMed] [Google Scholar]
- 29. Steen A., Sparre-Ulrich A. H., Thiele S., Guo D., Frimurer T. M., and Rosenkilde M. M. (2014) Gating function of isoleucine-116 in TM3 (position III:16/3.40) for the activity state of the CC-chemokine receptor 5 (CCR5). Br. J. Pharmacol. 171, 1566–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Steen A., Thiele S., Guo D., Hansen L. S., Frimurer T. M., and Rosenkilde M. M. (2013) Biased and constitutive signaling in the CC-chemokine receptor CCR5 by manipulating the interface between transmembrane helix 6 and 7. J. Biol. Chem. 288, 12511–12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elling C. E., Nielsen S. M., and Schwartz T. W. (1995) Conversion of antagonist-binding site to metal-ion site in the tachykinin NK-1 receptor. Nature 374, 74–77 [DOI] [PubMed] [Google Scholar]
- 32. Rosenkilde M. M., Lucibello M., Holst B., and Schwartz T. W. (1998) Natural agonist enhancing bis-His zinc-site in transmembrane segment V of the tachykinin NK3 receptor. FEBS Lett. 439, 35–40 [DOI] [PubMed] [Google Scholar]
- 33. Holst B., Elling C. E., and Schwartz T. W. (2000) Partial agonism through a zinc-ion switch constructed between transmembrane domains III and VII in the tachykinin NK(1) receptor. Mol. Pharmacol. 58, 263–270 [DOI] [PubMed] [Google Scholar]
- 34. Thirstrup K., Elling C. E., Hjorth S. A., and Schwartz T. W. (1996) Construction of a high affinity zinc switch in the κ-opioid receptor. J. Biol. Chem. 271, 7875–7878 [DOI] [PubMed] [Google Scholar]
- 35. Elling C. E., Thirstrup K., Holst B., and Schwartz T. W. (1999) Conversion of agonist site to metal-ion chelator site in the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 96, 12322–12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elling C. E., Frimurer T. M., Gerlach L.-O., Jorgensen R., Holst B., and Schwartz T. W. (2006) Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J. Biol. Chem. 281, 17337–17346 [DOI] [PubMed] [Google Scholar]
- 37. Rosenkilde M. M., Kledal T. N., Bräuner-Osborne H., and Schwartz T. W. (1999) Agonists and inverse agonists for the herpesvirus 8-encoded constitutively active seven-transmembrane oncogene product, ORF-74. J. Biol. Chem. 274, 956–961 [DOI] [PubMed] [Google Scholar]
- 38. Rosenkilde M. M., David R., Oerlecke I., Benned-Jensen T., Geumann U., Beck-Sickinger A. G., and Schwartz T. W. (2006) Conformational constraining of inactive and active states of a seven transmembrane receptor by metal ion site engineering in the extracellular end of transmembrane segment V. Mol. Pharmacol. 70, 1892–1901 [DOI] [PubMed] [Google Scholar]
- 39. Rosenkilde M. M., Andersen M. B., Nygaard R., Frimurer T. M., and Schwartz T. W. (2007) Activation of the CXCR3 chemokine receptor through anchoring of a small molecule chelator ligand between TM-III, -IV, and -VI. Mol. Pharmacol. 71, 930–941 [DOI] [PubMed] [Google Scholar]
- 40. Schwartz T. W., Frimurer T. M., Holst B., Rosenkilde M. M., and Elling C. E. (2006) Molecular mechanism of 7TM receptor activation: a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519 [DOI] [PubMed] [Google Scholar]
- 41. Nygaard R., Frimurer T. M., Holst B., Rosenkilde M. M., and Schwartz T. W. (2009) Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 30, 249–259 [DOI] [PubMed] [Google Scholar]
- 42. Rasmussen S. G. F., Choi H.-J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., et al. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rasmussen S. G. F., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T. A., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Govaerts C., Blanpain C., Deupi X., Ballet S., Ballesteros J. A., Wodak S. J., Vassart G., Pardo L., and Parmentier M. (2001) The TXP motif in the second transmembrane helix of CCR5: a structural determinant of chemokine-induced activation. J. Biol. Chem. 276, 13217–13225 [DOI] [PubMed] [Google Scholar]
- 45. Govaerts C., Bondue A., Springael J.-Y., Olivella M., Deupi X., Le Poul E., Wodak S. J., Parmentier M., Pardo L., and Blanpain C. (2003) Activation of CCR5 by chemokines involves an aromatic cluster between transmembrane helices 2 and 3. J. Biol. Chem. 278, 1892–1903 [DOI] [PubMed] [Google Scholar]
- 46. Blanpain C., Doranz B. J., Bondue A., Govaerts C., De Leener A., Vassart G., Doms R. W., Proudfoot A., and Parmentier M. (2003) The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J. Biol. Chem. 278, 5179–5187 [DOI] [PubMed] [Google Scholar]
- 47. Mirzadegan T., Diehl F., Ebi B., Bhakta S., Polsky I., McCarley D., Mulkins M., Weatherhead G. S., Lapierre J. M., Dankwardt J., Morgans D. Jr., Wilhelm R., and Jarnagin K. (2000) Identification of the binding site for a novel class of CCR2b chemokine receptor antagonists: binding to a common chemokine receptor motif within the helical bundle. J. Biol. Chem. 275, 25562–25571 [DOI] [PubMed] [Google Scholar]
- 48. Maeda K., Das D., Ogata-Aoki H., Nakata H., Miyakawa T., Tojo Y., Norman R., Takaoka Y., Ding J., Arnold G. F., Arnold E., and Mitsuya H. (2006) Structural and molecular interactions of CCR5 inhibitors with CCR5. J. Biol. Chem. 281, 12688–12698 [DOI] [PubMed] [Google Scholar]
- 49. Seibert C., Ying W., Gavrilov S., Tsamis F., Kuhmann S. E., Palani A., Tagat J. R., Clader J. W., McCombie S. W., Baroudy B. M., Smith S. O., Dragic T., Moore J. P., and Sakmar T. P. (2006) Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology 349, 41–54 [DOI] [PubMed] [Google Scholar]
- 50. Saita Y., Kodama E., Orita M., Kondo M., Miyazaki T., Sudo K., Kajiwara K., Matsuoka M., and Shimizu Y. (2006) Structural basis for the interaction of CCR5 with a small molecule, functionally selective CCR5 agonist. J. Immunol. 177, 3116–3122 [DOI] [PubMed] [Google Scholar]
- 51. Maeda K., Nakata H., Koh Y., Miyakawa T., Ogata H., Takaoka Y., Shibayama S., Sagawa K., Fukushima D., Moravek J., Koyanagi Y., and Mitsuya H. (2004) Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J. Virol. 78, 8654–8662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schall T. J., and Proudfoot A. E. I. (2011) Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 11, 355–363 [DOI] [PubMed] [Google Scholar]
- 53. Webb B., and Sali A. (2014) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–5.6.32 [DOI] [PubMed] [Google Scholar]
- 54. Pedretti A., Villa L., and Vistoli G. (2004) VEGA: an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 18, 167–173 [DOI] [PubMed] [Google Scholar]
- 55. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Santos-Martins D., Forli S., Ramos M. J., and Olson A. J. (2014) AutoDock4(Zn): an improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 54, 2371–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rosenkilde M. M., Cahir M., Gether U., Hjorth S. A., and Schwartz T. W. (1994) Mutations along transmembrane segment II of the NK-1 receptor affect substance P competition with non-peptide antagonists but not substance P binding. J. Biol. Chem. 269, 28160–28164 [PubMed] [Google Scholar]
- 58. Kissow H., Hartmann B., Holst J. J., Viby N.-E., Hansen L. S., Rosenkilde M. M., Hare K. J., and Poulsen S. S. (2012) Glucagon-like peptide-1 (GLP-1) receptor agonism or DPP-4 inhibition does not accelerate neoplasia in carcinogen treated mice. Regul. Pept. 179, 91–100 [DOI] [PubMed] [Google Scholar]
- 59. Brandish P. E., Hill L. A., Zheng W., and Scolnick E. M. (2003) Scintillation proximity assay of inositol phosphates in cell extracts: high-throughput measurement of G-protein-coupled receptor activation. Anal. Biochem. 313, 311–318 [DOI] [PubMed] [Google Scholar]
- 60. Thiele S., Mungalpara J., Steen A., Rosenkilde M. M., and Våbenø J. (2014) Determination of the binding mode for the cyclopentapeptide CXCR4 antagonist FC131 using a dual approach of ligand modifications and receptor mutagenesis. Br. J. Pharmacol. 171, 5313–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.