

Abstract
Drug delivery systems are required for drug targeting to avoid adverse effects associated with chemotherapy treatment regimes. Our approach is focused on the study and development of plant virus-based materials as drug delivery systems; specifically, this work focuses on the tobacco mosaic virus (TMV). Native TMV forms a hollow, high aspect-ratio nanotube measuring 300 × 18 nm with a 4 nm-wide central channel. Heat-transformation can be applied to TMV yielding spherical nanoparticles (SNPs) measuring ~50 nm in size. While bioconjugate chemistries have been established to modify the TMV rod, such methods have not yet been described for the SNP platform. In this work, we probed the reactivity of SNPs toward bioconjugate reactions targeting lysine, glutamine/aspartic acid, and cysteine residues. We demonstrate functionalization of SNPs using these chemistries yielding efficient payload conjugation. In addition to covalent labeling techniques, we developed encapsulation techniques, where the cargo is loaded into the SNP during heat-transition from rod-to-sphere. Finally, we developed TMV and SNP formulations loaded with the chemotherapeutic doxorubicin, and we demonstrate the application of TMV rods and spheres for chemotherapy delivery targeting breast cancer.
Keywords: Drug delivery, Tobacco mosaic virus, Nanoparticles, Nanorods, Breast cancer
1. Introduction
In the United States, approximately 300,000 women will be diagnosed with breast cancer this year and more than 40,000 of those will die from the disease. Chemotherapy has limited success due to adverse effects such as congestive heart failure [1–5]. Nanotechnologies have improved treatment because nanoparticle delivery systems offer better safety and drug efficacy. Many different classes of nanomaterials are undergoing development and some approaches have advanced to clinical use. Examples include liposomal formulations Doxil® or albumin-based formulation Abraxane®. Currently available nanoparticle-delivery systems increase safety of the treatment; however they often do not increase efficacy due to inefficient carrier tissue penetration and drug release [6]. Therefore, more research is required to develop and assess novel drug delivery system and strategies. Our research is focused on the development and application of nanomaterials derived from plant viruses manufactured through farming in plants.
Mammalian virus-based nanoparticles for gene therapy and oncolytic virotherapy are undergoing clinical trials [7–9], so the potential of virus-based materials for medical applications has been recognized. There are many novel virus-based materials in the pipeline, with plant viruses typically considered safer in humans than mammalian viruses [10]. Plant viruses do not infect or replicate in mammals. They can be administered at doses of up to 100 mg (1016 particles) per kg body weight without clinical toxicity [11,12]. We have shown that biomaterials derived from the plant virus, tobacco mosaic virus (TMV), can be delivered intravenously and do not induce hemolysis or coagulation [13]. Further, the protein-based carriers exhibit rapid tissue clearance (hours) from non-target organs [13]. The pharmacokinetics of plant viruses is tunable through PEGylation achieving half-lives of minutes-to-hours [14]. Like other nanomaterials, unmodified TMV is moderately immunogenic, but the immunogenicity of protein-based carriers [15,16], such as plant viruses [17], can be attenuated by polymer coating. These properties make this biodegradable platform a promising strategy for drug delivery.
Viruses have naturally evolved to deliver cargos to specific cells and tissues; we seek to repurpose this natural ability to deliver cargos for drug delivery targeting cancer. Advantages of the bio-inspired drug delivery system include the production through molecular farming in plants, which is highly scalable and therefore provides a realistic nanotechnology for translation. For example, Medicago Inc. already produces several virus-like particles in plants at pharmaceutical scale [18–21]. The protein-based materials are genetically encoded, therefore these materials offer a high degree of reproducibility and monodispersity; each particle is a clone of another; this degree of quality control is still challenging to achieve with synthetic systems.
TMV was the first virus to be discovered more than 100 years ago and it has served as a research tool for structural biology and virology since [22]. TMV-based materials are undergoing development for diverse applications as battery electrodes and light harvesting systems [23,24], as well as tissue engineering [25], molecular imaging [26], vaccines [27], and as demonstrated in this work, drug delivery. Native TMV forms high aspect ratio rods measuring 300 × 18 nm with a 4 nm-wide central channel. Each particle consists of a ssRNA genome encapsidated into 2130 identical copies of a coat protein unit. The length can be tailored through a bottom-up RNA-templated self-assembly approach and TMV shape-switching into spherical nanoparticles can be achieved through thermally-induced process [13,28,29].
While the development pipeline for nanoparticle platform technologies continues to progress rapidly, the fundamental nanomaterial–cell interactions have been studied only in a limited way and its relationship to drug efficacy is largely unknown. Size, shape, composition and surface chemistry of the nanocarrier impact its biodistribution, cell interaction, and intracellular trafficking. Some fundamentals are understood: mammalian cell membranes are negatively charged, therefore positively charged nanomaterials interact more strongly compared to negatively charged counterparts [30–32]. PEGylation is an accepted strategy to camouflage nanoparticles and inhibit (or reduce) cell binding or uptake; and receptor targeting is an effective strategy to enhance cell binding and/or induce tissue-specificity [33]. Based on the abundance of spherical nanomaterials, the effect of size on endocytosis is well understood: through competition between hydrodynamic driving force and receptor diffusion kinetics, the optimum radius for cellular uptake lies at r = 30 nm (viruses were used as model systems in several of these studies) [30,31,34–40]. Nevertheless, data remain elusive with regard to understanding the effects of nanoparticle shape: while some data indicate that higher aspect ratio (AR5) materials show faster uptake kinetics [31,32]. Others reported the opposite: spheres and low aspect ratio objects have enhanced cell interaction compared to high aspect ratio nanorods [41–44]. The TMV and SNP platforms provide a tool set of different-shaped protein-based nanoparticles and their side-by-side studies are expected to provide further insights into the design space ‘shape’ and how to utilize it in the design of next-generation drug delivery systems.
In the present studies, we considered TMV rods and spheres as a platform for drug delivery. While the chemistries on TMV rods are well-established, enabling functionalization of the interior or exterior surface with a variety of payloads [28,45,46]; bioconjugate chemistries have not yet been established using the SNP platform technology. The availability of chemical modification procedures would enable functionalization of SNPs with targeting ligands and medical cargo for molecular imaging [29,45] or drug delivery (this paper), as well as for vaccine development [47]. A recent study showed that SNPs could be non-covalently modified through adsorption of foreign proteins and epitopes on the SNP surface, however, these modifications are nonspecific and driven through electrostatic and hydrophobic interactions [47]. The availability of bioconjugate chemistry would streamline the functionalization of SNPs through targeted, covalent approaches and also enable functionalization with small cargos such as contrast agents and therapeutics. Chemical modification strategies would therefore complement the ‘modification by adsorption’ techniques. In addition to covalent labeling techniques, we present encapsulation techniques, where the cargo is loaded into the SNP during heat-transition from rod-to-sphere. Finally, we demonstrate the application of TMV rods and spheres for chemotherapy delivery targeting breast cancer.
2. Experimental section
2.1. Materials
Unless otherwise noted, chemicals and materials were obtained from Fisher Scientific.
2.2. TMV nanomanufacturing
2.2.1. Propagation
TMV was propagated in Nicotiana benthamiana plants and recovered, with a yield of 5 mg TMV per gram infected leaf material, using established extraction methods [28]. The concentration of TMV from plant extracts was determined by UV/vis spectroscopy (ε260 nm = 3.0 mg−1 mL cm−1) and virus integrity was verified by TEM imaging.
2.2.2. Thermal transition of TMV rods into spherical nanoparticles (SNPs)
Thermal transition from TMV rods to SNPs was carried out by heating TMV rods (0.3 mg mL−1 in H2O) for 60 s at 96 °C using a Peltier thermal cycler and then recovering concentrated particles by centrifugation at 160,000 ×g for 1 h. The methods were as we previously described [29,48].
2.3. TMV and SNP bioconjugation chemistry
2.3.1. Lysine modification
Lysine reactivity of SNPs was tested with Sulfo-Cyanine5 NHS ester (Lumiprobe). Sulfo-Cy5 NHS (1, 2, 5, 10, 20, and 40 M equivalents per coat protein (eq)), in 0.1 M potassium phosphate buffer pH 7.0 was added to 1 mg/mL SNPs at room temperature overnight. The reaction was purified by ultracentrifugation at 160,000 ×g for 1 h and analyzed (see below).
2.3.2. Cysteine modification
SNP cysteine reactivity was tested using Sulfo-Cyanine5 maleimide (Lumiprobe). Sulfo-Cy5 maleimide (1, 2, 5, 10, 20, and 40 M equivalents per coat protein (eq)), in 0.1 M potassium phosphate buffer pH 7.0 was added to 1 mg/mL SNPs at room temperature overnight. The reaction mix was purified by ultracentrifugation at 160,000 ×g for 1 h and analyzed (see below).
2.3.3. Glutamic/aspartic acid modification
Carboxylic acids were activated using hydroxybenzotriazole (HOBt) and ethyldimethylpropyl carbodiimide (EDC) to form amide bonds with the primary amine from Cyanine5 amine (Lumiprobe). Here, HOBt:EDC:amine ratio was set at 1:2.5:5, the molar equivalents tested were: 0.625:1.25:2.5, 1.25:2.5:5, 2.5:5:10, 5:10:20, and 10:20:40. SNPs (1 mg/mL) were reacted with HOBt:EDC:amine overnight at room temperature in 0.1 M HEPES buffer pH 7.0. The reaction was purified by ultracentrifugation at 160,000 ×g for 1 h and analyzed (see below).
2.3.4. Conjugation of Cy5 or doxorubicin to TMV and SNP
For drug delivery studies, doxorubicin hydrochloride (DOX, Indofine Chemical Company) and for imaging studies amine-Cyanine5 (Cy5) was conjugated to TMV and SNP targeting carboxylic acid functional groups on the TMV/SNP; TMV or SNP (1 mg/mL) was reacted with HOBt:EDC:DOX (10:20:40 eq per CP) overnight at room temperature in 0.1 M HEPES buffer pH 7.0. The reaction was purified by ultracentrifugation at 160,000 ×g for 1 h and analyzed (see below).
2.3.5. Biotin conjugation to SNPs
For lysine modification, SNPs (1 mg/mL in 0.1 M potassium phosphate buffer pH 7.0) were added to NHS-biotin (LifeTechnologies, 20217) at 20 eq and reacted overnight at room temperature. For cysteine modification, SNPs (1 mg/mL in 0.1 M potassium phosphate buffer pH 7.0) were added to maleimide-PEG2-biotin (LifeTechnologies) at 20 eq and reacted overnight at room temperature. For modification targeting carboxylic acid (Glu and Asp), SNPs (1 mg/mL in 0.1 M HEPES buffer pH 7.0) were added to amine-PEG2-biotin (LifeTechnologies) at 20 eq with 10 eq EDC and 5 eq HOBt and reacted overnight at room temperature. Click chemistry protocols were used: for exterior modification of TMV, TMV was reacted with a diazonium salt generated from 3-ethynylaniline (25 M equivalents (eq), pH = 8.5, 30 min) to incorporate a terminal alkyne. Then copper-catalyzed azide-alkyne cycloaddition (CuAAC) was performed to attach azide-biotin (Lumiprobe). Alkyne-labeled TMV (2 mg/mL) in 0.1 M potassium phosphate buffer (pH 7.0) was mixed with azide-biotin (2 eq), aminoguanidine (2 mM), ascorbic acid (2 mM) and copper sulfate (1 mM) for 30 min on ice. To establish click chemistry protocols for SNPs, the following steps were performed: TMV was modified with an alkyne on the interior channel using propargylamine (50 eq) with ethyldimethylpropyl carbodiimide (EDC, 100 eq) and n-hydroxybenzotriazole (HOBt, 50 eq) for 24 h. The alkyne-modified TMV rod was then transitioned into SNPs. Then the alkyne-SNP underwent click addition of azide-biotin. All biotin bioconjugation reactions were purified by ultracentrifugation at 160,000 ×g for 1 h and analyzed (see below).
2.3.6. DOX encapsulation
Encapsulation of DOX was accomplished by adding DOX (5 eq per CP) to TMV rods (0.3 mg mL−1 in H2O) and incubating at room temperature for 5 min. Thermal transition from TMV rods to SNPs was carried out by heating the mixture for 30 s at 96 °C using a Peltier thermal cycler and then recovering concentrated particles by centrifugation at 160,000 ×g for 1 h. DOX loading was confirmed by measuring UV–vis absorbance at 480 nm (extinction coefficient ~ 11,500 cm−1 M−1).
2.4. Particle characterization
2.4.1. UV/vis spectroscopy
A Thermo Scientific NanoDrop 2000 Spectrophotometer was used to determine the concentration of TMV/SNP and conjugated fluorophores and DOX using the TMV-specific or fluorophore- and DOX-specific extinction coefficients and Beer–Lambert law.
2.4.2. Transmission electron microscopy (TEM)
Drops of TMV rods or SNPs (0.2 mg mL−1 in 5 μL deionized water) were placed on formvar coated copper TEM grids, adsorbed for 5 min, washed with deionized water, and negatively stained with 2% (w/v) uranyl acetate for 2 min. Samples were examined by using a Zeiss Libra 200FE transmission electron microscope operated at 200 kV.
2.4.3. Immunogold labeling
TMV samples were dried on TEM grids, washed with 10 mM sodium phosphate buffer pH 7.0 and floated on a drop of 1% (w/v) bovine serum albumin (BSA) in Tris-buffered saline pH 7.4 plus 0.1% (v/v) Tween-20 (TBST) for 30 min. Samples were equilibrated with 0.1% (w/v) BSA for 5 min before binding for 1 h with an anti-biotin labeled 10 nm gold nanoparticles. The grids were then washed in TBST, then three to five water washes, and finally staining with 2% (v/v) uranyl acetate for 1 min. The grids were imaged by TEM using a Zeiss Libra 200FE transmission electron microscope operated at 200 kV.
2.4.4. Scanning electron microscopy (SEM)
Samples (0.2 mg mL−1 in deionized water) were dried onto silicon wafers (Ted Pella) and then mounted on the surface of an aluminum pin stub using double-sided adhesive carbon disks (Agar Scientific). The stubs were then sputter-coated with gold (or palladium) in a high-resolution sputter coater (Agar Scientific, Ltd.) and transferred to a Hitachi 4500 scanning electron microscope. The diameter of the SNPs was determined using ImageJ (imagej.nih.gov/ij/); at least 50 particles on three separate images were measured and average diameter and standard deviation were calculated.
2.4.5. SDS-PAGE and Western blotting
Protein subunits were analyzed on denaturing 4–12% NuPAGE gels (Invitrogen) using 1 × 3- (N-morpholino)propanesulfonic acid (MOPS) running buffer (Invitrogen) and 10 μg of sample. After separation, the gel was photographed using an AlphaImager (Biosciences) imaging system before and after staining with Coomassie Blue, or further processed for Western blotting. To detect biotinylated particles, 10 μg samples were separated on a 4–12% NuPAGE Bis-Tris gel using MOPS buffer (see above). After separation, the proteins were transferred onto a nitrocellulose membrane (Thermo Scientific) using NuPAGE transfer buffer (Invitrogen). The membrane was blocked at room temperature for 1 h using 0.1 M TBS (pH 7.6) containing 5% (w/v) skimmed milk powder and 0.05% (w/v) Tween 20. Detection was carried out using alkaline phosphatase-conjugated streptavidin (Sigma-Aldrich) (1:1000) in blocking buffer solution. Alkaline phosphatase activity was detected using the BCIP/NBT liquid substrate system (Sigma-Aldrich).
2.4.6. Dynamic light scattering (DLS)
DLS measurements were performed with the use of the 90plus instrument (Brookhaven Instruments Corporation) at the scattering angle h = 90°. Reported values are average of multiple measurements consisting for total measurement time of 2.5–5.0 min. Hydrodynamic diameters (Dh) and polydispersity indices (PDI) were calculated using manufacturers’ software.
2.5. In vitro analysis
2.5.1. Cell culture
MCF-7 cells (ATCC) were maintained in minimal essential medium (MEM) at 37 °C in a 5% CO2 humidified atmosphere. MDA-MB-231 cells (ATCC) were maintained in RPMI-1640 medium at 37 °C in a 5% CO2 humidified atmosphere. The medium was supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), and 1% (v/v) penicillin–streptomycin. All reagents were obtained from Gibco.
2.5.2. Flow cytometry
MDA-MB-231 or MCF-7 cells (500,000 cells in 200 μL medium per well) were added to an untreated 96-well v-bottom plate. The TMV or SNP (Cy5-labeled) was added at a concentration of 100,000 particles/cell in triplicate and incubated 6 h at 37 °C in a 5% CO2 humidified atmosphere. Following incubation, the cells were pelleted at 500 ×g for 4 min and washed three times using FACS buffer (1 mM EDTA, 1% (v/v) FBS, and 25 mM HEPES, pH 7.0 in Ca2+ and Mg2+ free PBS). The cells were then fixed in 2% (v/v) paraformaldehyde in FACS buffer for 10 min at room temperature and washed another three times. Analysis was carried out using the BD LSR II or BD FACS Aria flow cytometer, and 10,000 gated events per sample were collected.
2.5.3. Cell viability assay
Cells (10,000 cells in 100 μL medium per well) were seeded in a sterile, tissue culture-treated, 96-well clear bottom plate overnight at 37 °C in a 5% CO2 humidified atmosphere. Cells were washed and TMV/SNP as well as PBS and free DOX controls were added in medium (at concentrations ranging from 0.1–10 μM concentration DOX) and incubated overnight. Unbound particles were removed and cells were washed. The cells were then incubated in fresh medium for three days at 37 °C in a 5% CO2 humidified atmosphere prior to cell viability analysis using an MTT cell proliferation assay. The assay was carried out following the manufacturer’s instructions (ATCC).
3. Results
3.1. Engineering SNPs through bioconjugate chemistry and encapsulation protocols
3.1.1. Probing SNPs with fluorescent labels to quantify label incorporation
TMV was propagated in N. benthamiana plants and obtained at yields of 5 mg TMV per gram infected leaf tissue [28]. Thermal transition from TMV rods to SNPs was carried out by heating TMV rods for 60 s at 96 °C using a thermal cycler [29,48]. The size of SNPs was determined using dynamic light scattering (DLS) and scanning electron microscopy (SEM) studies. The size of SNPs correlates to the protein concentration of TMV in solution; here we chose a protein concentration of 0.3 mg/mL yielding SNPs with a diameter of approximately 50 nm, which equates to a single TMV rod transitioned into a SNP (larger SNPs are likely the result of fusions of multiple TMV rods during the thermal shape transitioning process) [49]. It should be noted that while TMV rods utilized in these experiments contain the single stranded RNA genome, SNPs obtained after heat transition are generally RNA-free [49].
While the structure of TMV rods is available at 3.6 Å resolution [50] allowing the use of software such as Chimera (http://www.cgl.ucsf.edu/chimera/) and PyMol (https://www.pymol.org/) to probe the structure for the availability of solvent-exposed amino acid, this information is not available at the present time for SNPs. The most commonly utilized amino acids in bioconjugation protocols are lysine (amine-functional group), aspartic and glutamic (carboxylate-functional group), and cysteine (thiol-functional group) [51]. Analysis of the TMV CP amino acid sequence (NCBI entry VCTMVU) indicates the presence of two (2) lysine side chains (K54 and K69), one (1) cysteine residue (C38), and fifteen (15) glutamic and aspartic acids (E23, E50, E96, E89, E107, E132, E146 and D20, D65, D67, D77, D89, D100, D116, D117). Therefore, we sought to probe the reactivity of these amino acids using standard bioconjugation protocols targeting these amino acids (Fig. 1).
Fig. 1.
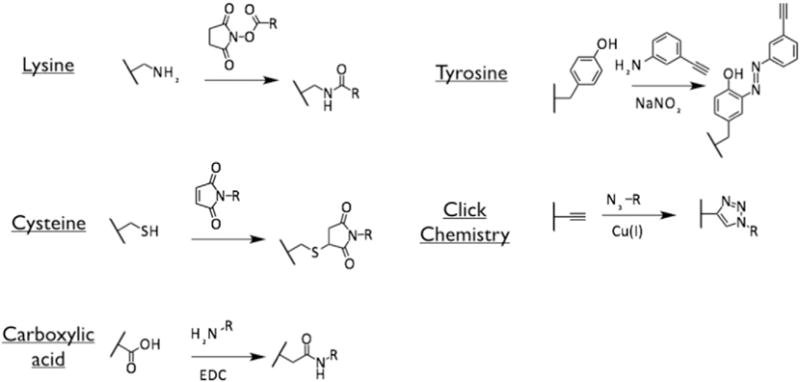
Bioconjugate chemistries targeting lysine, cysteine, aspartic/glutamic acids (carboxylic acids) and tyrosine on TMV and SNP-based nanoparticle formulations (see Sections 2.1.1 and 2.1.2).
Labeling reactions were first carried out using Cy5 dye (this section), then labeling studies were carried out using the affinity tag biotin (see Section 3.1.2) and the chemotherapeutic doxorubicin (DOX, see Section 3.1.3). We found that chemistries targeting amines, thiols, or carboxylic acids were successful resulting in efficient incorporation of fluorophores into the SNP-based platform (Fig. 2).
Fig. 2.
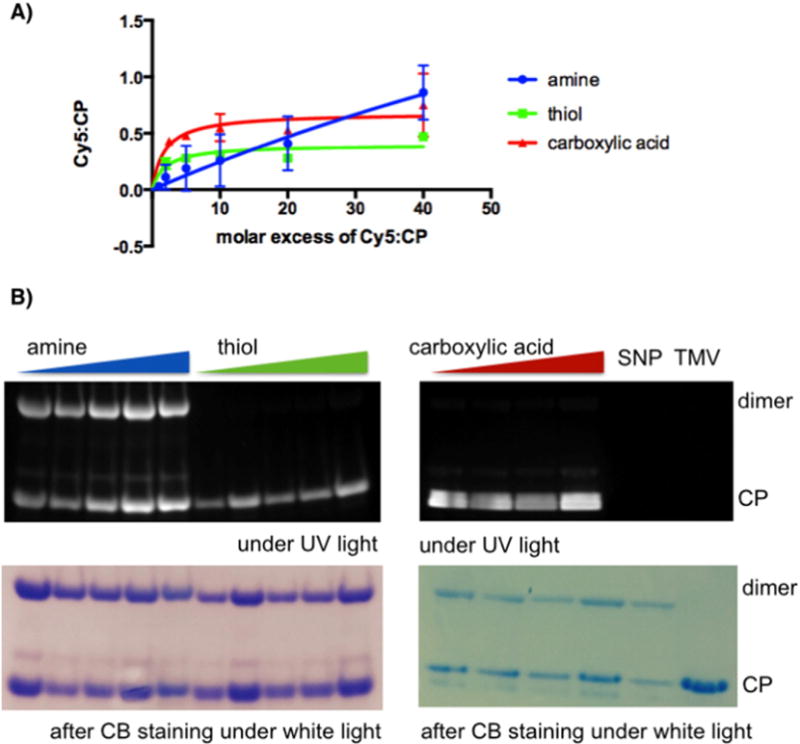
A) Quantification of Cy5 attachment to SNP coat proteins (CPs) as a function of molar excess used. Averaged values from three independent experiments are shown error bars represent the standard deviation. Prism® 6.0b software was used to analyze and plot the data. B) SDS-PAGE of Cy5-labeled and native SNP and TMV visualized under UV light (to detect the Cy5 label) and under white light after Coomassie Blue (CB) staining (to detect the protein). Cy5-labeled SNPs after modification of amines (blue), thiols (green), and carboxylic acids (red) were analyzed where SNPs were probed with molar excess of 2, 5, 10, 20, 40 Cy5 per CP for amine- and thiol-selective reactions and 5, 10, 20, 40 Cy5 per CP targeting carboxylates.
SNPs were incubated with Cy5 fluorophores using NHS-, maleimide, or amine-functional probes. To evaluate the degree of labeling that could be achieved, SNPs were probed with various molar excess of Cy5 per coat protein (CP, ranging from 1:1 to 40:1 Cy5:CP, see Materials and Methods). After completion of the reaction, SNPs were purified from excess reagents and the degree of labeling was determined based on the Beer–Lambert law using the Cy5-specific extinction coefficient. The SNPs used in our studies exhibited an averaged diameter of 50 nm, therefore we assume that each SNP represents one thermally transitioned TMV and therefore consists of approximately 2000 CPs [49]. The amount of protein per SNP was determined through a combination of Lowry protein assays and SDS-PAGE using TMV standards of known concentrations; it should be noted that the concentrations and ratio of Cy5:CP are approximations and should not be interpreted as absolute values.
The averaged values of Cy5-to-CP ratios obtained per molar excess used from three independent experiments are shown in Fig. 2. Each bioconjugation chemistry yielded fluorescent nanoparticles incorporating the Cy5 dye through covalent bonding, as confirmed by UV/vis spectroscopy and SDS-PAGE (Fig. 2). Thiol-selective chemistries reached a plateau at ~0.4 Cy5:CP (i.e., every other protein is labeled with a Cy5); using carboxylic acid-specific reactions, up to 0.7 Cy5:CP was incorporated; and amine-selective chemistries achieved almost complete labeling (0.9 Cy5:CP), however only at large molecular excess. While thiol- and carboxylate-specific chemistries reached maximum labeling efficiency using an excess of ~10 Cy5 per CP, a plateau could not be reached using amine-selective chemistries, indicating that more than one lysine side chain may be reactive. Labeling reactions using an excess of higher than 40 Cy5:1 CP is experimentally challenging because the hydrophobic dyes tend to aggregate at high concentrations also inducing nanoparticle aggregation; conjugation at higher molar excess was therefore not pursued any further.
Overall, data indicate that using a molar excess of 10× of the label per CP, thiol and amine-targeted chemistries yield SNPs in which every second-to-third CP carries the label; at the 10× excess carboxylate-selective chemistries yield SNPs in which every second SNP is modified. And, under forcing conditions, using a high molecular excess, amine-based chemistries drive incorporation of labels on each CP per SNP. However, it should be noted that these experiments are averaged values and multiple labels could be attached to a single CP while other CPs within the assembly remain label-free.
The covalent attachment of the fluorescent label was confirmed using denaturing gel electrophoresis (SDS-PAGE, Fig. 2B) and structural integrity and particle stability were confirmed using a combination of TEM and SEM imaging pre- and post-labeling (not shown). SNPs proved to remain intact throughout the labeling and purification procedures. Differences in stability of SNPs labeled at different functional groups were not noted.
Separation of denatured CP in SDS-PAGE confirmed that the labels were indeed covalently attached to the CPs and matched the migration pattern of the CPs (Fig. 2B). It was interesting to note that for SNPs free CP and CP dimers were detectable on Coomassie Blue-stained gels; this pattern was reproducible for Cy5-SNPs, when the dye was conjugated to lysine side chains; however, when labeled at cysteine- and aspartic/glutamic acid residues only the monomer but not the dimer is fluorescent. It is not clear whether the dimer formation is part of the nature of the SNP or an artifact induced during preparation of the samples for the SDS-PAGE; dimers cannot be reduced through addition of β-mercaptoethanol, indicating that disulfide bridges may not be the only dimer-forming mechanism; data may suggest that carboxylic acid also plays a role in the formation or stabilization of the dimer complexes.
3.1.2. Probing SNPs with biotin to gain spatial information on label presentation
Having demonstrated that SNPs can be functionalized through chemical bioconjugation, we set out to gain insights into the spatial display of the labels. Specifically, we determined whether SNP-conjugated labels were solvent-accessible, presented on the surface of the nanoparticles. To do this, we chose biotin as a label, because biotin can be detected and imaged using immunogold staining techniques.
The following steps were carried out: first, wild type (wt)-TMV was transitioned into SNPs. SNPs were then probed with NHS-biotin, maleimide-biotin, or amine-biotin using the above-described chemistries targeting lysine, cysteine, and aspartic/glutamic acids yielding K-biotin-SNP, C-biotin-SNP, and D/E-biotin-SNP, respectively (Fig. 3). Successful labeling of the coat proteins with biotin was confirmed by Western blotting (not shown). Immunogold-staining using gold-labeled anti-biotin antibodies and TEM imaging confirmed decoration of SNPs with solvent (i.e., antibody)-accessible biotins. Imaging indicated a high density of biotins displayed on the surface of D/E-biotin-SNP versus K-biotin-SNP and C-biotin-SNP (with K-biotin-SNP > C-biotin-SNP, Fig. 3).
Fig. 3.
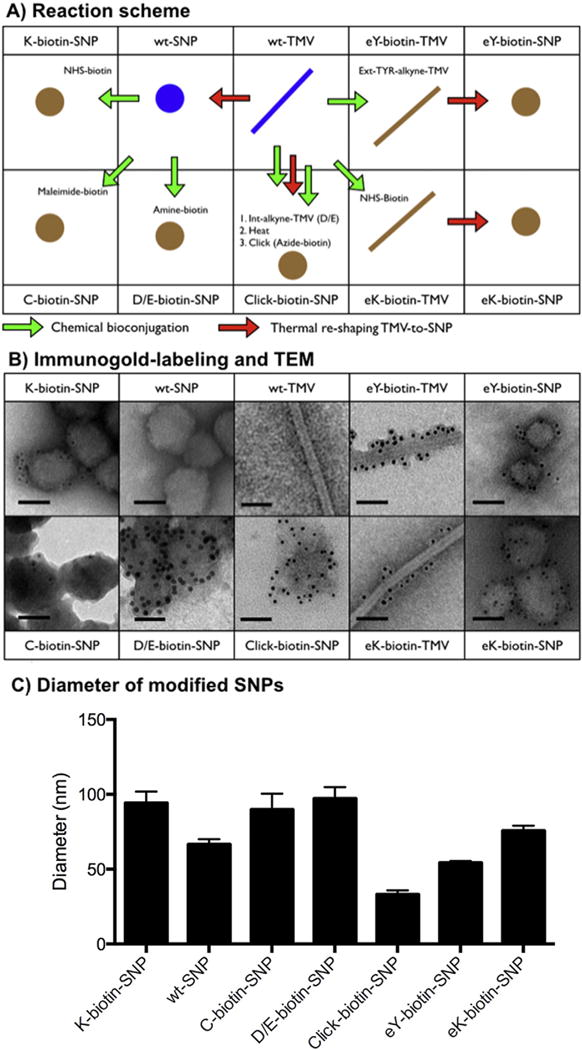
A) Schematic detailing the reactions leading to biotinylated TMV and SNP formulations, and B) corresponding TEM images of negative (UAc)-stained samples after immunogold staining using a gold-labeled anti-biotin antibody; the scale bar is 50 nm. C) Averaged diameter and standard deviation of the modified SNPs as determined by quantitative SEM image analysis.
In parallel with these studies, we also investigated the spatial distribution of labels when biotin was conjugated to the exterior surface of TMV rods prior to transition to SNPs; wt-TMV and a lysine-added mutant of TMV (S152K, TMVlys) that displays a reactive amine-functional lysine group at the solvent-exposed C-terminus of the coat protein [52] were considered. Biotin was conjugated to the exterior surface of wt-TMV and TMVlys yielding eY-biotin-TMV and eK-biotin-TMV, respectively (for reaction schemes see also Fig. 1). Then SNPs were formed through thermal transition yielding eY-biotin-SNP and eK-biotin-SNP. Immunogold staining confirmed the presence of labels of the surface of the SNPs accessible to the antibodies. It should be noted that not necessarily all labels are displayed on the exterior surface, but using the immunogold-staining we can only detect labels accessible to the antibody.
Lastly, we sought to establish whether click chemistry protocols could be applied to SNPs. We carried out the following steps: TMV was modified with an alkyne on the interior channel using propargylamine and EDC:HOBt (see Materials and Methods). The alkyne-modified TMV rods were then transitioned into SNPs through heat treatment followed by click addition of azide-biotin. Again, immunogold-staining and TEM imaging confirmed presence of biotin-displaying SNPs (click-biotin-SNP, Fig. 3).
We would like to note that the immunogold-labeling studies are not quantitative. However, these studies provide insights that the chemical modifiers introduced, are, at least in part, solvent exposed. Therefore, the described chemistries could be applied to introduce surface ligands to induce stealth or targeting properties.
In parallel with the imaging studies, we validated the size distribution of wt-SNP versus chemically modified SNPs. A quantitative SEM imaging study was performed and the size distribution was determined by measuring at least 50 particles on three separate images using ImageJ software (Fig. 3C). D/E-biotin-SNP, K-biotin-SNP, and C-biotin-SNP were prepared through chemical bioconjugation of wt-SNPs (the same batch of particles was used in this experiment). Overall, we find that the size distribution of the individual nanoparticle formulations is rather narrow. Nevertheless, we noted a batch-to-batch variation of about ~35%. The SNPs were prepared batch-by-batch using the thermocycler method. We also developed a mesofluidic method for large-scale nanomanufacture [29], and in future studies, we will investigate methods to negate the batch-to-batch variability.
In summary, these studies confirm that SNPs serve as a robust and versatile platform for chemical modification strategies, including click chemistry; modification of SNPs and surface ligand display can be achieved pre- or post-shape transitioning.
3.1.3. Drug conjugation to and encapsulation into TMV and SNPs
For drug delivery studies, doxorubicin was either encapsulated into SNPs during heat transition or covalently attached to TMV rods and SNPs (see Materials and Methods). Covalent attachment was achieved through activation of the primary amine group of DOX with NHS and EDC followed by coupling to TMV’s carboxylic acids. Reactive glutamic side chains (4260 per TMV) are presented on the solvent-accessible interior channel. TEM and SEM imaging confirmed the structural integrity of the samples (data not shown). UV/visible spectroscopy was used to determine the payload attached or encapsulated; we found approximately 5400 ± 10% DOX per TMV. The slightly increased loading of 5400 DOX per TMV vs. the ~4200 available carboxylates on the interior channel, maybe explained by non-specific DOX adsorption onto the protein carrier. Nonetheless, the absorption of DOX to TMV/SNP was stable: when samples were dialyzed against water or buffer, DOX was not released over extended time periods (days).
DOX-loaded SNP particles were obtained either by heat-transition of TMV rods into spheres, yielding DOXSNP, or through non-covalent encapsulation of DOX into SNPs during the thermal re-shaping process, yielding eDOXSNP. Both methods gave rise to comparable yields but larger batch-to-batch variations, DOXSNP and eDOXSNP were loaded with 800–1800 DOX per SNP (see also Discussion below).
3.2. Drug delivery targeting breast cancer cells: HER2 and TNBC
3.2.1. TMV– and SNP–breast cancer cell interactions
To evaluate the effectiveness of the drug delivery approach, we selected two breast cancer cell lines as proof-of-concept; MDA-MB-231 and MCF-7 cells. Flow cytometry protocols were used to evaluate the TMV– and SNP–cell interactions. For these studies, Cy5-labeled formulations were used; the particles were prepared as previously reported [45]: in brief, TMV rods were used and labeled with Cy5 using a combination of carbodiimide coupling targeting interior glutamic acids to introduce alkyne ligation handles, followed by introduction of an azide-functional Cy5 using CuAAC chemistry. The obtained Cy5TMV was then heat-converted into Cy5SNPs. Cy5TMV/SNP were added to cells at a ratio of 100,000 particles/cell and incubated for 6 h, then unbound nanoparticles were removed and cells were analyzed by flow cytometry (Fig. 4). The uptake rates between the different cell lines differed, for MDA-MB-231 the entire cell population shifted indicating uniform uptake throughout — in contrast, for MCF-7 cells only a subset (~20% for TMV, and 30% for SNP) of the cells took up the nanoparticles. Nevertheless, we found that longer incubation times and higher nanoparticle-to-cell ratios enhance cell uptake, therefore achieving effective cargo delivery and cell killing (see below).
Fig. 4.
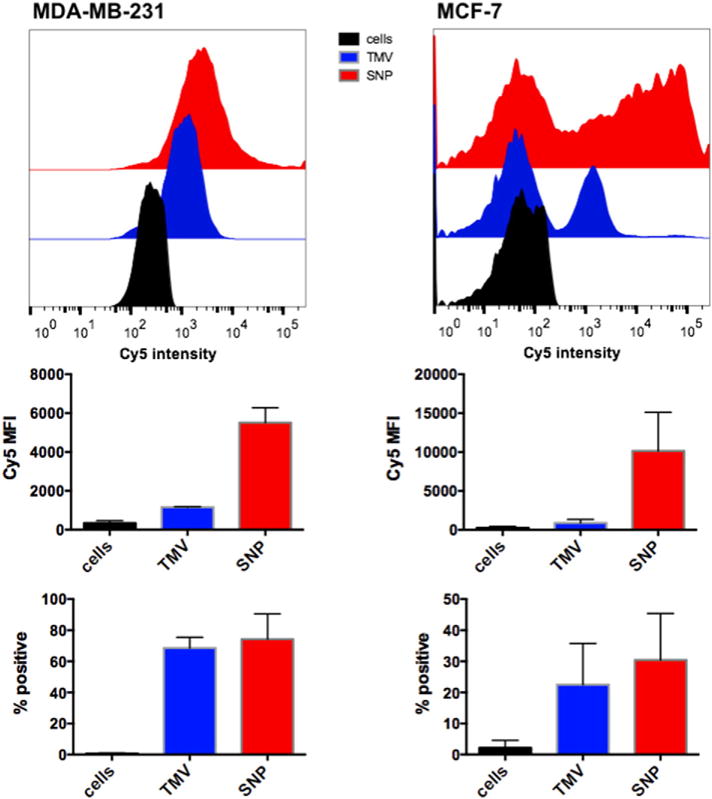
TMV– and SNP–cell interactions using MDA-MB-231 and MCF-7 cells; Cy5-labeled nanoparticle formulations (TMV shown in blue, SNP shown in red) were incubated with cells in medium for 6 h, then washed, fixed, and analyzed using BD LSR II or BD FACS Aria flow cytometer, and 10,000 gated events per sample were collected (all samples were analyzed in triplicates). Data were plotted and analyzed using FlowJo® 10.6 software; the mean fluorescence intensity (Cy5 MFI) or the percentage TMV/SNP-positive cells are plotted (error bars show the standard deviation).
3.2.2. Efficacy of the drug delivery approach
To confirm the efficacy of the drug delivery approach and determine the IC50 values for the different TMV/SNP formulations, cell killing was determined in vitro using MTT cell viability assay. MCF-7 and MDA-MB-231 cells were considered, and cells were incubated with free DOX, DOX conjugated to TMV and SNP (DOXTMV and DOXSNP) as well DOX encapsulated into SNPs (eDOXSNP). The IC50 values for free DOX after 72 h exposure for MCF-7 and MDA-MB-231 were determined at IC50MCF-7 = 0.64 ± 0.06 μM and IC50MDA-MB-231 = 0.97 ± 0.02 μM, respectively; this is in good agreement with published reports [53–55]. Studying the TMV and SNP formulations, we found that overall efficacy is maintained after drug conjugation or encapsulation (in)to the proteinaceous nanoparticle carriers (Fig. 5); a trend was observed in which SNPs with conjugated or encapsulated DOX showed enhanced efficacy over the DOXTMV formulations. For MDA-MB-231 cells, IC50 values were 1.41 ± 0.05 μM for DOXTMV versus 1.13 ± 0.04 μM for DOXSNP and 0.61 ± 0.07 μM for eDOXSNP; for MCF-7 cells IC50 values were 0.55 ± 0.09 μM for DOXTMV versus 0.29 ± 0.13 μM for DOXSNP and 0.38 ± 0.06 μM for eDOXSNP. This is consistent with enhanced cell uptake of the spherical versus elongated nanoparticle formulations (see Fig. 4). It should be noted that while cell uptake rates were significantly enhanced for SNPs over TMV, the differences in drug efficacy comparing the two nanoparticle-shapes were less profound. This may be reflected by the fact that a considerably higher nanoparticle:cell ratio was used in the drug delivery experiment compared to the cell uptake studies: for efficacy studies a range of 5 × 105 to 3 × 107 TMV/SNP:1 cell was used, while cell uptake studies were carried out using 1 × 105 TMV/SNP:1 cell (see Materials and Methods). While cell uptake studies indicated that only up to ~30% of the MCF-7 cell population tested positive for TMV/SNP, the higher nanoparticle:cell ratio and longer incubation time (24 h vs. 6 h) used in the cell killing assay is also expected to enhance cell uptake resulting in potent efficacy in the MCF-7 cells (as well as MCDA-MB-231).
Fig. 5.
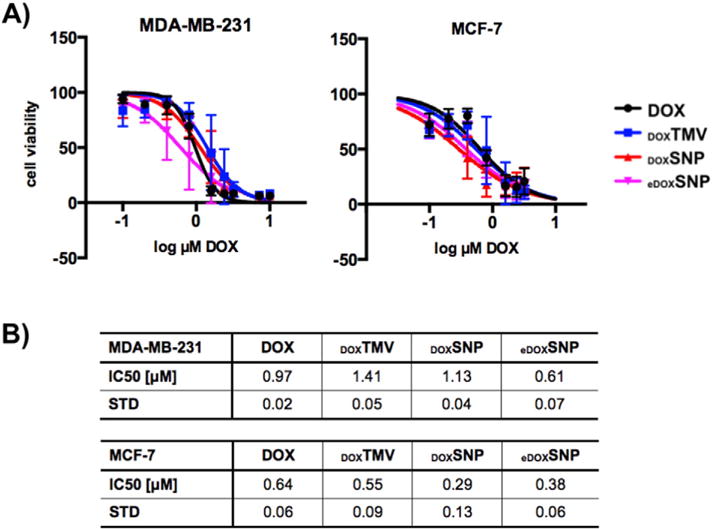
A) Cell viability of MDA-MB-231 and MCF-7 cells after treatment with DOX (black), DOXTMV (blue), DOXSNP (red), and eDOXSNP (pink). Cell viability was determined after 72 h using MTT assay; dose-dependency was evaluated testing a concentration range of 0.1–10 μM normalized to DOX. Data shown are averaged data from three biological replicas (each performed in triplicates); error bars indicate the standard deviation. B) IC50 values and standard deviation comparing DOX, DOXTMV, DOXSNP, and eDOXSNP. Data were analyzed and graphed using Prism® v6.0b software.
Effective cell killing of the covalently-delivered DOX for the DOXTMV and DOXSNP formulations can be explained by the intracellular fates of the protein-based nanocarrier: in previous cellular trafficking experiments we have shown that TMV is internalized by cancer cells through endocytosis and targeted to the endolysosome, where the free drug is released following degradation of the proteinaceous carrier [56,57].
4. Discussion
We developed bioconjugate chemistries as well as encapsulation techniques enabling the functionalization of TMV-derived SNPs for efficient payload delivery. SNPs and rod-shaped TMVs were loaded with the chemotherapeutic drug doxorubicin; cell targeting and cell killing were demonstrated using two breast cancer cell lines (MDA-MB-231 and MCF-7).
While a recent study demonstrated that SNPs could be functionalized with proteins and peptides through non-specific adsorption driven by electrostatic and hydrophobic interactions [47], the availability of bioconjugate chemistry streamlines the functionalization of SNPs through targeted, covalent approaches; most importantly the developed bioconjugation and encapsulation techniques facilitate the loading of small chemical cargos such as contrast agents and therapeutics. Fluorescence and biotin labels were conjugated for structure–function studies; while the fluorophores were employed for quantitative studies, the biotin labeling in combination with immunogold TEM studies gave insights into the spatial distribution, i.e. it was confirmed that at least some of the conjugated labels (but unlikely all of the labels) were presented on the surface of the SNPs. It was found that 40–90% of the CPs per SNP incorporated the labels using chemistries specific to amine-, carboxylate-, or thiol-functional groups. Using a 10-fold excess of the label of interest, the most efficient reaction was targeted to carboxylic acids using carbodiimide coupling. Quantitative studies were in agreement with qualitative immunogold TEM studies. This is not surprising considered that the TMV CP amino acid sequence (NCBI entry VCTMVU) indicates the presence of two (2) lysine side chains, one (1) cysteine residue, and fifteen (15) glutamic and aspartic acids. The SNPs were found to remain structurally sound, even when undergoing multiple reactions including Cu(I)-catalyzed azide-alkyne cycloaddition chemistry (Figs. 2, 3).
While the atomic coordinates are available for the TMV platform, the structure of the SNPs remains to be elucidated. Initial work has shown that while the coat proteins do not denature during the thermal transition, considerable conformational changes occur within the coat proteins during the TMV-to-SNP transition [58]. The transition into SNPs is accompanied by an almost complete loss of the α-helical protein structure and emergency of a β-barrel sheet structure, supporting a protein conformation allowing for more dense packing into SNPs. So it is clear that there are structural differences comparing TMV and SNPs. The availability of bioconjugate chemistries, as we describe, might be an asset toward future structural studies of the SNP complex; understanding the presentation and orientation of the available reactive groups would enable structure-based design of therapeutics. For example, surface display of ligands would be of interest when considering the development of stealth coatings for improved pharmacokinetics or incorporation of targeting ligands to confer tissue specificity; on the other hand, drug encapsulation inside the carrier may be preferred over surface display to avoid systemic side effects.
Doxorubicin was conjugated and encapsulated using TMV rods and SNPs. While bioconjugation of DOX to TMV rods showed good batch-to-batch consistency, resulting in 5400 ± 10% DOX per TMV, SNP functionalization resulted in a larger degree of batch-to-batch variability with 800–1800 DOX per SNP loaded through either heat conversation of covalently-labeled TMV or encapsulation during rod-to-sphere transition. These studies highlight the larger variability of the SNP chemistry compared to the TMV rod chemistry. Future studies will investigate whether manufacturing could be streamlined using the mesofluidic method that we previously developed [29].
Overall, the drug loading capacity of TMV rods and SNPs is comparable to other virus-based materials: for example, we previously reported drug conjugation to cowpea mosaic virus (CPMV), a 30 nm-sized icosahedron. Using the CPMV platform, a maximum of ~270 DOX per particle were conjugated [59]. In another example, Pokorski et al. [60] developed atom transfer radical polymerization chemistries using the 30 nm-sized Qß bacteriophage to increase loading capacities, nevertheless, the reaction maxed out at about 500 labels per Qß particle. Another interesting approach utilizes the red clover necrotic mottle virus (RCNMV) platform, where it was shown that ~1000 DOX molecules could be loaded through non-covalent gating mechanisms [61].
In our studies we demonstrated, efficient drug release and cell killing. Based on our previous studies on cell trafficking of TMV [56, 57], it is expected that upon cell targeting and internalization, the protein-based carriers are targeted to the endolysosomal compartment, where the proteinaceous plant virus-based carriers will be metabolically degraded through hydrolase and protease activity, therefore yielding efficient drug release inducing effective cell killing. Indeed our data confirmed that drug efficacy was maintained upon conjugation to or encapsulation into the TMV and SNP-based carriers (Fig. 5).
Data indicate that both, TMV and SNP, interact with cells. SNP showed enhanced cell interactions and this was reproducible for the two breast cancer cell lines under investigation. We have made similar observations studying TMV rods of distinct aspect ratio: low-aspect ratio rods (60 nm long) are more readily taken up by cancer cells compared to high-aspect ratio materials (300 nm long) [62]. Distinct cell uptake rates of elongated vs. spherical or low-aspect ratio materials has been reported for a number of nanomaterials [63,64], and can be explained by competition of membrane (and receptor) diffusion and wrapping kinetics [65] as well as contact angle-dependence of these fluid (cell membrane)–solid (nanoparticle) interactions [66]. How these differences in cell uptake may translate into in vivo drug delivery applications remains to be evaluated; data indicate advantageous tumor homing of high aspect ratio materials [67]; the interplay between tumor homing, penetration, and cell uptake needs to be carefully studied and will be a goal of future investigation.
While no statistical significant differences between the free drug and the nanoparticle formulations were observed in vitro, the nanoparticle carriers are expected to provide advantages for delivery of drugs in vivo. The multifunctional nature of the protein-based platform enables the incorporation of stealth coatings for improved pharmacokinetics [14] and targeting ligands to confer tissue-specific targeting [26, 62,68–71] of the therapeutics, therefore overcoming the dose-limiting toxicities of the chemotherapy.
5. Conclusion
In conclusion, we have developed bioconjugate chemistries and encapsulation protocols enabling drug delivery using rod-shaped and spherical TMV-based nanocarriers. While each nanoparticle platform technology, synthetic and natural, has its advantages and disadvantages, the bio-inspired approach harnessing the nucleoprotein components of viruses for medical cargo delivery offers unique advantages: an important benefit compared to mammalian viral vector systems is that plant viruses are not virulent in animal cells [56]. Other benefits include the relative ease of their synthesis through molecular farming in plants [72]. Since plant viruses do not infect but enter mammalian cells, they provide a unique opportunity for intracellular drug delivery. These properties in combination with the ability to shape-tune and surface-engineer the nanoparticles render plant virus-based technologies and attractive platform that requires further testing and development for application in nanomedicine.
Acknowledgments
This work was supported in part by a grant from the National Science Foundation (DMR CAREER 1452257 to NFS). MAB acknowledges the NIH T32 HL105338 Cardiovascular Research Institute training grant. We thank Case Western Reserve University Farm for support in scaled-up molecular farming.
References
- 1.Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 2.Musci M, Loebe M, Grauhan O, Weng Y, Hummel M, Lange P, et al. Heart transplantation for doxorubicin-induced congestive heart failure in children and adolescents. Transplant Proc. 1997;29:578–579. doi: 10.1016/s0041-1345(96)00310-7. [DOI] [PubMed] [Google Scholar]
- 3.Lieverse RJ, Ossenkoppele GJ. Prevention of doxorubicin-induced congestive heart failure by continuous intravenous infusion in multiple myeloma; a case report and review of the literature. Neth J Med. 1991;38:33–34. [PubMed] [Google Scholar]
- 4.Ibrahim NK, Hortobagyi GN, Ewer M, Ali MK, Asmar L, Theriault RL, et al. Doxorubicin-induced congestive heart failure in elderly patients with metastatic breast cancer, with long-term follow-up: the M.D. Anderson experience. Cancer Chemother Pharmacol. 1999;43:471–478. doi: 10.1007/s002800050926. [DOI] [PubMed] [Google Scholar]
- 5.Haq MM, Legha SS, Choksi J, Hortobagyi GN, Benjamin RS, Ewer M, et al. Doxorubicin-induced congestive heart failure in adults. Cancer. 1985;56:1361–1365. doi: 10.1002/1097-0142(19850915)56:6<1361::aid-cncr2820560624>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 6.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 7.Chitale R. Merck hopes to extend gardasil vaccine to men. J Natl Cancer Inst. 2009;101:222–223. doi: 10.1093/jnci/djp014. [DOI] [PubMed] [Google Scholar]
- 8.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 9.Shirakawa T. Clinical trial design for adenoviral gene therapy products. Drug News Perspect. 2009;22:140–145. doi: 10.1358/dnp.2009.22.3.1354090. [DOI] [PubMed] [Google Scholar]
- 10.Manchester M, Singh P. Virus-based nanoparticles (VNPs): platform technologies for diagnostic imaging. Adv Drug Deliv Rev. 2006;58:1505–1522. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Kaiser CR, Flenniken ML, Gillitzer E, Harmsen AL, Harmsen AG, Jutila MA, et al. Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo. Int J Nanomedicine. 2007;2:715–733. [PMC free article] [PubMed] [Google Scholar]
- 12.Singh P, Prasuhn D, Yeh RM, Destito G, Rae CS, Osborn K, et al. Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J Control Release. 2007;120:41–50. doi: 10.1016/j.jconrel.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruckman MA, Randolph LN, Vanmeter A, Hern S, Shoffstall AJ, Taurog RE, et al. Biodistribution, pharmacokinetics, and blood compatibility of native and PEGylated tobacco mosaic virus nano-rods and -spheres in mice. Virology. 2014;449:163–173. doi: 10.1016/j.virol.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KL, Shukla S, Wu M, Ayat NR, El Sanadi CE, Wen AM, et al. Stealth filaments: polymer chain length and conformation affect the in vivo fate of PEGylated potato virus X. Acta Biomater. 2015 doi: 10.1016/j.actbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (London) 2011;6:715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 17.Raja KS, Wang Q, Gonzalez MJ, Manchester M, Johnson JE, Finn MG. Hybrid virus-polymer materials. 1. Synthesis and properties of PEG-decorated cowpea mosaic virus. Biol Macromol. 2003;3:472–476. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- 18.D’Aoust MA, Lavoie PO, Belles-Isles J, Bechtold N, Martel M, Vezina LP. Transient expression of antibodies in plants using syringe agroinfiltration. Methods Mol Biol. 2009;483:41–50. doi: 10.1007/978-1-59745-407-0_3. [DOI] [PubMed] [Google Scholar]
- 19.D’Aoust MA, Couture MM, Charland N, Trepanier S, Landry N, Ors F, et al. The production of hemagglutinin-based virus-like particles in plants: a rapid, efficient and safe response to pandemic influenza. Plant Biotechnol J. 2010;8:607–619. doi: 10.1111/j.1467-7652.2009.00496.x. [DOI] [PubMed] [Google Scholar]
- 20.Vezina LP, Faye L, Lerouge P, D’Aoust MA, Marquet-Blouin E, Burel C, et al. Transient co-expression for fast and high-yield production of antibodies with human-like N-glycans in plants. Plant Biotechnol J. 2009;7:442–455. doi: 10.1111/j.1467-7652.2009.00414.x. [DOI] [PubMed] [Google Scholar]
- 21.D’Aoust MA, Lavoie PO, Couture MM, Trepanier S, Guay JM, Dargis M, et al. Influenza virus-like particles produced by transient expression in Nicotiana benthamiana induce a protective immune response against a lethal viral challenge in mice. Plant Biotechnol J. 2008;6:930–940. doi: 10.1111/j.1467-7652.2008.00384.x. [DOI] [PubMed] [Google Scholar]
- 22.Rossmann MG. Structure of viruses: a short history. Q Rev Biophys. 2013;46:133–180. doi: 10.1017/S0033583513000012. [DOI] [PubMed] [Google Scholar]
- 23.Miller RA, Presley AD, Francis MB. Self-assembling light-harvesting systems from synthetically modified tobacco mosaic virus coat proteins. J Am Chem Soc. 2007;129:3104–3109. doi: 10.1021/ja063887t. [DOI] [PubMed] [Google Scholar]
- 24.Royston E, Ghosh A, Kofinas P, Harris MT, Culver JN. Self-assembly of virus-structured high surface area nanomaterials and their application as battery electrodes. Langmuir. 2008;24:906–912. doi: 10.1021/la7016424. [DOI] [PubMed] [Google Scholar]
- 25.Zhao X, Lin Y, Wang Q. Virus-based scaffolds for tissue engineering applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7:534–547. doi: 10.1002/wnan.1327. [DOI] [PubMed] [Google Scholar]
- 26.Bruckman M, Jiang K, Simpson EJ, Randolph L, Luyt LG, Yu X, et al. Dual-modal magnetic resonance and fluorescence imaging of atherosclerotic plaques in vivo using VCAM-1 targeted tobacco mosaic virus. Nano Lett. 2014;14:1551–1558. doi: 10.1021/nl404816m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plummer EM, Manchester M. Viral nanoparticles and virus-like particles: platforms for contemporary vaccine design. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010 doi: 10.1002/wnan.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruckman MA, Steinmetz NF. Chemical modification of the inner and outer surfaces of tobacco mosaic virus (TMV) Methods Mol Biol. 2014;1108:173–185. doi: 10.1007/978-1-62703-751-8_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruckman MA, VanMeter A, Steinmetz NF. Nanomanufacturing of tobacco mosaic virus-based spherical biomaterials using a continuous flow method. ACS Biomater Sci Eng. 2015;1:13–18. doi: 10.1021/ab500059s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnida, Janát-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. European journal of pharmaceutics and biopharmaceutics, off J Arboric für Pharmazeutische Verfahrenstechnik eV. 2011;77:417–423. doi: 10.1016/j.ejpb.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnida, Malugin A, Ghandehari H. Cellular uptake and toxicity of gold nanoparticles in prostate cancer cells: a comparative study of rods and spheres. J Appl Toxicol. 2010;30:212–217. doi: 10.1002/jat.1486. [DOI] [PubMed] [Google Scholar]
- 32.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66:2873–2896. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoyama Y, Kanamori T, Nakai T, Sasaki T, Horiuchi S, Sando S, et al. Artificial viruses and their application to gene delivery. Size-controlled gene coating with glycocluster nanoparticles. J Am Chem Soc. 2003;125:3455–3457. doi: 10.1021/ja029608t. [DOI] [PubMed] [Google Scholar]
- 35.Desai MP, Labhasetwar V, Walter E, Levy RJ, Amidon GL. The mechanism of uptake of biodegradable microparticles in caco-2 cells is size dependent. Pharm Res. 1997;14:1568–1573. doi: 10.1023/a:1012126301290. [DOI] [PubMed] [Google Scholar]
- 36.Gao H, Shi W, Freund LB. Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci U S A. 2005;102:9469–9474. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 38.Nakai T, Kanamori T, Sando S, Aoyama Y. Remarkably size-regulated cell invasion by artificial viruses. Saccharide-dependent self-aggregation of glycoviruses and its consequences in glycoviral gene delivery. J Am Chem Soc. 2003;125:8465–8475. doi: 10.1021/ja035636f. [DOI] [PubMed] [Google Scholar]
- 39.Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. A quantum dot conjugated sugar ball and its cellular uptake. On the size effects of endocytosis in the subviral region. J Am Chem Soc. 2004;126:6520–6521. doi: 10.1021/ja048792a. [DOI] [PubMed] [Google Scholar]
- 40.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244:105–115. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 41.Nan A, Bai X, Son SJ, Lee SB, Ghandehari H. Cellular uptake and cytotoxicity of silica nanotubes. Nano Lett. 2008;8:2150–2154. doi: 10.1021/nl0802741. [DOI] [PubMed] [Google Scholar]
- 42.Chithrani BD, Chan WCW. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 43.Chithrani BD, Ghazani AA, Chan WCW. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 44.Schaeublin NM, Braydich-Stolle LK, Maurer EI, Park K, MacCuspie RI, Afrooz AR, et al. Does shape matter? Bioeffects of gold nanomaterials in a human skin cell model. Langmuir. 2012;28:3248–3258. doi: 10.1021/la204081m. [DOI] [PubMed] [Google Scholar]
- 45.Bruckman MA, Jiang K, Simpson EJ, Randolph LN, Luyt LG, Yu X, et al. Dual-modal magnetic resonance and fluorescence imaging of atherosclerotic plaques in vivo using vcam-1 targeted tobacco mosaic virus. Nano Lett. 2014;14:1551–1558. doi: 10.1021/nl404816m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruckman MA, Yu X, Steinmetz NF. Engineering Gd-loaded nanoparticles to enhance MRI sensitivity via T(1) shortening. Nanotechnology. 2013;24:462001. doi: 10.1088/0957-4484/24/46/462001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karpova O, Nikitin N, Chirkov S, Trifonova E, Sheveleva A, Lazareva E, et al. Immunogenic compositions assembled from tobacco mosaic virus-generated spherical particle platforms and foreign antigens. J Gen Virol. 2012;93:400–407. doi: 10.1099/vir.0.036293-0. [DOI] [PubMed] [Google Scholar]
- 48.Bruckman MA, Hern S, Jang K, Flask CA, Yu X, Steinmetz NF. Tobacco mosaic virus rods and spheres as supramolecular high-relaxivity MRI contrast agents. J Mater Chem B. 2013 doi: 10.1039/C3TB00461A. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atabekov J, Nikitin N, Arkhipenko M, Chirkov S, Karpova O. Thermal transition of native tobacco mosaic virus and RNA-free viral proteins into spherical nanoparticles. J Gen Virol. 2011;92:453–456. doi: 10.1099/vir.0.024356-0. [DOI] [PubMed] [Google Scholar]
- 50.Namba K, Stubbs G. Structure of tobacco mosaic virus at 3.6 A resolution: implications for assembly. Science. 1986;231:1401–1406. doi: 10.1126/science.3952490. [DOI] [PubMed] [Google Scholar]
- 51.Hermanson GT. Bioconjugate Techniques. Academic Press, Elsevier; London: 1996. [Google Scholar]
- 52.Geiger FC, Eber FJ, Eiben S, Mueller A, Jeske H, Spatz JP, et al. TMV nanorods with programmed longitudinal domains of differently addressable coat proteins. Nanoscale. 2013;5:3808–3816. doi: 10.1039/c3nr33724c. [DOI] [PubMed] [Google Scholar]
- 53.Tegze B, Szallasi Z, Haltrich I, Penzvalto Z, Toth Z, Liko I, et al. Parallel evolution under chemotherapy pressure in 29 breast cancer cell lines results in dissimilar mechanisms of resistance. PLoS ONE. 2012;7:e30804. doi: 10.1371/journal.pone.0030804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yun H, Shi R, Yang Q, Zhang X, Wang Y, Zhou X, et al. Over expression of hRad9 protein correlates with reduced chemosensitivity in breast cancer with administration of neoadjuvant chemotherapy. Sci Rep. 2014;4:7548. doi: 10.1038/srep07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tassone P, Tagliaferri P, Perricelli A, Blotta S, Quaresima B, Martelli ML, et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br J Cancer. 2003;88:1285–1291. doi: 10.1038/sj.bjc.6600859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV-DOX delivers. Mol Pharm. 2013;10:3–10. doi: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wen AM, Infusino M, De Luca A, Kernan DL, Czapar AE, Strangi G, et al. Interface of physics and biology: engineering virus-based nanoparticles for biophotonics. Bioconjug Chem. 2015;26:51–62. doi: 10.1021/bc500524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobrov EN, Nikitin NA, Trifonova EA, Parshina EY, Makarov VV, Maksimov GV, et al. Beta-structure of the coat protein subunits in spherical particles generated by tobacco mosaic virus thermal denaturation. J Biomol Struct Dyn. 2014;32:701–708. doi: 10.1080/07391102.2013.788983. [DOI] [PubMed] [Google Scholar]
- 59.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV-DOX delivers. Mol Pharm. 2013;10:3–10. doi: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pokorski JK, Breitenkamp K, Liepold LO, Qazi S, Finn MG. Functional virus-based polymer-protein nanoparticles by atom transfer radical polymerization. J Am Chem Soc. 2011;133:9242–9245. doi: 10.1021/ja203286n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao J, Guenther RH, Sit TL, Opperman CH, Lommel SA, Willoughby JA. Loading and release mechanism of red clover necrotic mosaic virus derived plant viral nanoparticles for drug delivery of doxorubicin. Small. 2014 doi: 10.1002/smll.201400558. [DOI] [PubMed] [Google Scholar]
- 62.Shukla S, Eber FJ, Nagarajan AS, DiFranco NA, Schmidt N, Wen AM, et al. The impact of aspect ratio on the biodistribution and tumor homing of rigid soft-matter nanorods. Adv Healthcare Mater. 2015;4(874–2) doi: 10.1002/adhm.201400641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 64.Schaeublin NM, Braydich-Stolle LK, Maurer EI, Park K, MacCuspie RI, Afrooz AR, et al. Does shape matter? Bioeffects of gold nanomaterials in a human skin cell model. Langmuir. 2012;28:3248–3258. doi: 10.1021/la204081m. [DOI] [PubMed] [Google Scholar]
- 65.Gao H, Shi W, Freund LB. Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci U S A. 2005;102:9469–9474. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brunel FM, Lewis JD, Destito G, Steinmetz NF, Manchester M, Stuhlmann H, et al. Hydrazone ligation strategy to assemble multifunctional viral nanoparticles for cell imaging and tumor targeting. Nano Lett. 2010;10:1093–1097. doi: 10.1021/nl1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chariou PL, Lee KL, Wen AM, Gulati NM, Stewart PL, Steinmetz NF. Detection and imaging of aggressive cancer cells using an epidermal growth factor receptor (EGFR)-targeted filamentous plant virus-based nanoparticle. Bioconjug Chem. 2015;26:262–269. doi: 10.1021/bc500545z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hovlid ML, Steinmetz NF, Laufer B, Lau JL, Kuzelka J, Wang Q, et al. Guiding plant virus particles to integrin-displaying cells. Nanoscale. 2012;4:3698–3705. doi: 10.1039/c2nr30571b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang RK, Steinmetz NF, Fu CY, Manchester M, Johnson JE. Transferrin-mediated targeting of bacteriophage HK97 nanoparticles into tumor cells. Nanomedicine (Lond) 2011;6:55–68. doi: 10.2217/nnm.10.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ren Y, Wong S, Lim L. Application of plant viruses as nano drug delivery systems. Pharm Res. 2010;27:2509–2513. doi: 10.1007/s11095-010-0251-2. [DOI] [PubMed] [Google Scholar]