

Abstract
Liver receptor homolog 1 (NR5A2, LRH-1) is an orphan nuclear hormone receptor that regulates diverse biological processes, including metabolism, proliferation, and the resolution of endoplasmic reticulum stress. Although preclinical and cellular studies demonstrate that LRH-1 has great potential as a therapeutic target for metabolic diseases and cancer, development of LRH-1 modulators has been difficult. Recently, systematic modifications to one of the few known chemical scaffolds capable of activating LRH-1 failed to improve efficacy substantially. Moreover, mechanisms through which LRH-1 is activated by synthetic ligands are entirely unknown. Here, we use x-ray crystallography and other structural methods to explore conformational changes and receptor-ligand interactions associated with LRH-1 activation by a set of related agonists. Unlike phospholipid LRH-1 ligands, these agonists bind deep in the pocket and do not interact with residues near the mouth nor do they expand the pocket like phospholipids. Unexpectedly, two closely related agonists with similar efficacies (GSK8470 and RJW100) exhibit completely different binding modes. The dramatic repositioning is influenced by a differential ability to establish stable face-to-face π-π-stacking with the LRH-1 residue His-390, as well as by a novel polar interaction mediated by the RJW100 hydroxyl group. The differing binding modes result in distinct mechanisms of action for the two agonists. Finally, we identify a network of conserved water molecules near the ligand-binding site that are important for activation by both agonists. This work reveals a previously unappreciated complexity associated with LRH-1 agonist development and offers insights into rational design strategies.
Keywords: allosteric regulation, diabetes, molecular dynamics, nuclear receptor, phosphatidylcholine, lipids
Introduction
Liver receptor homolog 1 (LRH-1; NR5A2) is a nuclear hormone receptor (NR)3 that controls expression of a diverse set of genes important both in normal physiology and disease. In addition to a vital role during development (1, 2), LRH-1 regulates many genes related to metabolism, proliferation, and cell survival. In the liver, LRH-1 regulates bile acid biosynthesis (3) and reverse cholesterol transport (4, 5), affecting hepatic and circulating cholesterol levels. Glucose metabolism is also regulated by LRH-1 at several points, including GLUT-4-mediated transport (6) and glucose phosphorylation, the latter of which is essential for proper postprandial glucose sensing, flux through glycolysis and glycogenesis pathways, and de novo lipogenesis (7). LRH-1 is a key mediator of the cell stress response through control of genes involved in the hepatic acute phase response (8) and in the cytoprotective resolution of endoplasmic reticulum stress (9). Additionally, LRH-1 can be aberrantly overexpressed in certain cancers and can promote tumor growth through estrogen receptor and β-catenin signaling (10–16).
Considering the breadth and significance of these physiological effects, LRH-1 modulators are highly desired as potential therapeutic agents. Chemical modulators would also be extremely useful as tools to dissect complex or temporal aspects of LRH-1 biology. However, development of LRH-1-targeted compounds has been challenging, due in part to a lipophilic binding pocket that becomes occupied with bacterial phospholipids (PL) in recombinant protein. Very few small molecules are able to displace these PL in library screens. Moreover, ligand-mediated regulation of LRH-1 is poorly understood. Endogenous ligands for LRH-1 are unknown, but exogenous administration of dilauroylphosphatidylcholine (DLPC) (phosphatidylcholine 12:0/12:0) activates LRH-1 and has profound anti-diabetic effects in vivo, which are absent in a liver-specific LRH-1 knock-out mouse (17). In addition to phosphatidylcholines, the signaling PL phosphatidylinositol 3,4,5-triphosphate (PIP3) binds LRH-1 (18, 19), although downstream effects of this interaction have yet to be determined.
Typically, NRs are activated by a ligand-induced conformational change, which promotes recruitment of co-activator proteins to the activation function surface (AFS) in the ligand binding domain (LBD) to drive transcription. Our structural studies with DLPC have shown that, contrary to the canonical model of NR activation, LRH-1 relies on small conformational fluctuations to recruit co-activator or co-repressor proteins. These occur mainly in the AFS (composed of portions of H3, H4, and the AF-H in the LBD) as well as in the H6/β-sheet region at a distal portion of the LBD (20, 21). Flexibility in the H6/β-sheet region is required for activation by PLs (20). Mechanisms through which LRH-1 is activated by synthetic ligands have not been explored but are likely quite different, given the differing structural composition of synthetic versus PL ligands.
There are very few known chemical scaffolds capable of activating LRH-1 above basal levels, the best studied of which are the cis-bicyclo[3.3.0]-octenes discovered by Whitby et al. (22, 23). The first compound described with this scaffold, named GSK8470 (Fig. 1), was somewhat effective but was acid-labile (23). Substitution of the aniline group improved compound stability, and the GSK8470-LRH-1 crystal structure provided the basis for an extensive structure-activity relationship study (23). One of the major objectives of this study was to introduce functional groups near select polar residues within the predominantly hydrophobic pocket. The new lead compound produced from this study, named RJW100, contains an exo-hydroxyl group at the 1-position of the pentalene scaffold (indicated in red in Fig. 1) intended to interact with LRH-1 residues His-390 or Arg-393. In contrast, a diastereomer with endo stereochemistry (previously known as 24-endo, Fig. 1) was not predicted to be able to make these interactions due to the alternative conformation of the hydroxyl oxygen. The endo derivative was less active in biochemical assays, seeming to support this hypothesis. However, RJW100 was not much more potent or effective than GSK8470, and the study did not illuminate strategies for further improvement.
FIGURE 1.
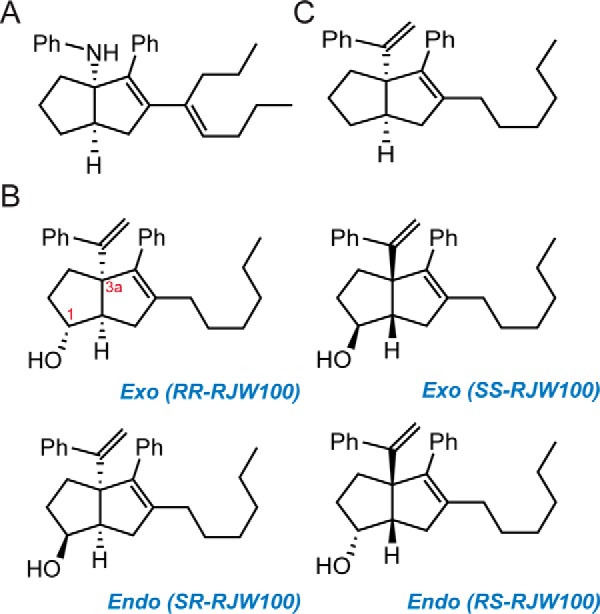
Chemical structures of LRH-1 agonists. A, GSK8470, the parent compound. B, RJW100 enantiomers. C, RJW100 analog lacking the hydroxyl group (named 18a), assayed in Fig. 8.
In this work, we present crystal structures of RJW100 and its endo-diastereomer bound to LRH-1. We demonstrate that these compounds bind quite differently than PLs and have distinct effects on protein dynamics compared with DLPC. Unexpectedly, these agonists also bind quite differently from the very closely related compound, GSK8470. We identify receptor-ligand interactions driving the repositioning and show that particular interactions are important for LRH-1 activation. These findings provide the first description of mechanisms involved in LRH-1 activation by synthetic molecules.
Results
Crystal Structure of RJW100 Bound to LRH-1
To understand how RJW100 interacts with LRH-1 and affects receptor conformation, we determined the x-ray crystal structure of LRH-1 LBD bound to the agonist and to a fragment of the co-activator, Tif2, to a resolution of 1.85 Å (Table 1 and Fig. 2A). Although the RJW100 used for crystallization was a racemic mixture of two exo stereoisomers (Fig. 1), the electron density in the structure unambiguously indicates that a single enantiomer is bound (Fig. 2B). The bound isomer has R stereochemistry at both the 1-position (hydroxyl-substituted) and 3a-position (styrene-substituted) (hereafter RR-RJW100, Fig. 1). The ligand is bound at a single site deep in the binding pocket and is fully engulfed within it. This binding mode is markedly different from that of the PL ligands, DLPC and PIP3, which extend lower in the pocket with the headgroups protruding into the solvent (seen by superposition with PDBs 4DOS and 4RWV, respectively, Fig. 2, C and D). PL ligands also increase the pocket volume and width compared with RJW100. For example, the mouth of the pocket is ∼3 Å wider and nearly 40% larger in volume when DLPC is bound versus RJW100 (Fig. 2E and Table 2). This effect appears to be mainly due to a shift of H6, which swings away from the mouth of the pocket in the DLPC structure by ∼3 Å (Fig. 2E). The direction and magnitude of the H6 movement are similar in other LRH-1-PL structures; comparison of four published human LRH-1-PL structures shows an average H6 shift of 3.0 ± 0.2 Å relative to LRH-1 in the apo-state or when synthetic ligands are bound (mean ± S.E., Fig. 2F). Although these structures exhibit diverse types of crystal packing, the movement of H6 appears to be related to whether the ligand is a PL or small molecule and not to crystal form or packing contacts. It likely occurs to avoid stearic clashes with the PL headgroup. Notably, the H6/β-sheet region has been recently identified as a site through which PL ligands allosterically communicate with the AFS to modulate LRH-1 activity (20, 21). The fact that the synthetic agonists do not displace H6 relative to the apo-receptor suggests that they utilize a different mechanism for receptor activation.
TABLE 1.
X-ray data collection and refinement statistics
Values in parentheses indicate highest resolution shell.
| Data collection | LRH-1 + RR-RJW100 +Tif2 | LRH-1 + SR-RJW100 + Tif2 |
|---|---|---|
| Space group | P422a | P43212 |
| Cell dimensions | ||
| a, b, c (Å) | 46.4 46.4, 220.5 | 46.3, 46.3, 220.0 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50−1.85 (1.92−1.85) | 50−1.93 (2.00−1.93) |
| Rpim | 0.03 (0.22) | 0.02 (0.231) |
| I / σI | 28.5 (1.98) | 33.5 (2.06) |
| CC1/2 in highest shell | 0.878 | 0.861 |
| Completeness (%) | 96.8 (86.1) | 99.7 (97.9) |
| Redundancy | 8.9 (6.8) | 11.1 (8.2) |
| Refinement | ||
| Resolution (Å) | 1.85 | 1.93 |
| No. of reflections | 21026 | 19073 |
| Rwork/Rfree (%) | 19.8/24.1 | 20.5/22.8 |
| No. of atoms | ||
| Protein | 4038 | 4062 |
| Water | 92 | 51 |
| B-factors | ||
| Protein | 38.1 | 46.9 |
| Ligand | 34.8 | 59.2 |
| Water | 36.8 | 43.5 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.004 | 0.004 |
| Bond angles (°) | 0.578 | 0.588 |
| Ramachandran favored (%) | 98 | 98 |
| Ramachandran outliers (%) | 0 | 0 |
| PDB accession code | 5L11 | 5SYZ |
a Data were processed in P422 but refined in P43212.
FIGURE 2.
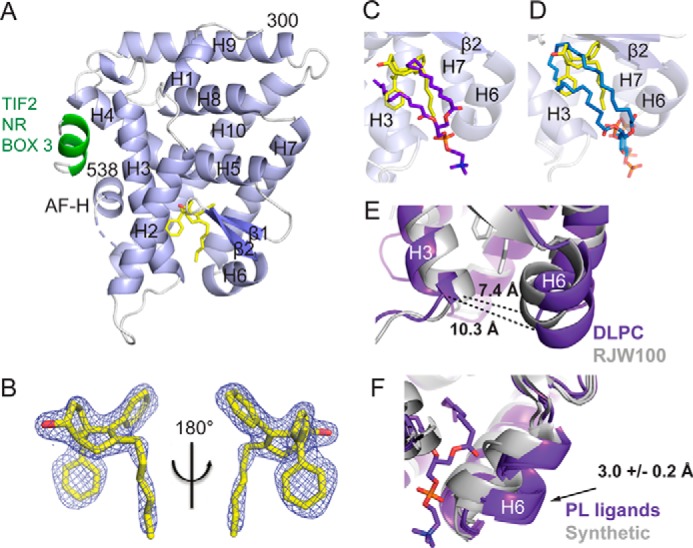
Crystal structure of LRH-1 ligand binding domain bound to RR-RJW100 and a fragment of the Tif2 co-activator. A, overall structure, with α-helices shown in light blue and β-sheets in slate. The Tif2 peptide (green) is bound at the AFS. The ligand (yellow) is bound at a single site in the binding pocket. Dashed line, region of disorder in the protein backbone that could not be modeled. B, omit map (FO − FC, contoured at 2.5 σ) showing that a single enantiomer of RJW100 is bound in the structure. C and D, superposition of RR-RJW100 (yellow) with the ligand coordinates from DLPC (purple, PDB 4DOS) (C) or PIP3 (blue, PDB 4RWV) (D) shows the very different binding mode of RR-RJW100 compared with the PL ligands. E, DLPC expands the width at the mouth of the pocket by ∼3 Å compared with RR-RJW100. The width was measured from Thr-341 to Asn-419 (α-carbons). F, superposition of four PL-bound LRH-1 crystal structures (PDBs 4DOS (20), 1YUC (46), 4RWV (18), and 4PLE (21), purple) and three structures of LRH-1 bound to synthetic agonists (PDB 3PLZ (23) and the two structures from this paper, gray) showing the shift of helix 6 by the PL ligands. One PL ligand is shown to illustrate that the shift of H6 is likely due to a stearic clash with the PL headgroup. The number in F indicates the average distance that H6 shifts when PLs are bound relative to LRH-1 in the apo-state or when synthetic ligands are bound (± S.E., determined by measuring distances between Ser-418 α-carbons). The apo-LRH-1 structure used for measurements was PDB 4PLD (21).
TABLE 2.
Binding pocket dimensions of LRH-1 in various liganded states
| PDB code | Ligand | Width | Volume |
|---|---|---|---|
| Å | Å3 | ||
| 5SYZ | SR-RJW100 | 7.2 | 1083.0 |
| 3PLZ | GSK8470 | 7.3 | 957.6 |
| 5L11 | RR-RJW100 | 7.4 | 1061.1 |
| 4PLD | Apo | 8.0 | 965.8 |
| 4PLE | E. coli lipids | 10.0 | 1368.7 |
| 4DOS | DLPC | 10.3 | 1553.9 |
| 4RWV | PIP3 | 12.0 | 1233.3 |
RJW100 Selectively Destabilizes Components of the AFS Relative to DLPC
The overall LRH-1-RJW100 structure depicts the AF-H in the active conformation and the Tif2 co-activator peptide bound at the AFS, as expected. However, there are a few indications that the active state may not be fully stabilized. There is substantial disorder in the loop connecting helix 10 to the AF-H, and three residues within this loop cannot be modeled (dashed line in Fig. 2A). This loop is not disordered in published structures of LRH-1 bound to DLPC or GSK8470, and this does not appear to be related to favorable crystal packing in those structures (data not shown). A second site of disorder occurs in the side chain of residue Glu-534 (data not shown), which plays a critical role in securing co-activators via charge clamp (24).
The disorder in the vicinity of the AFS in our structure suggests that this region is mobile when RJW100 is bound. To test this explicitly, we utilized solution-based hydrogen-deuterium exchange-mass spectrometry (HDX-MS) to compare dynamics of purified LRH-1 LBD bound to enantiomerically pure RR-RJW100 or to DLPC. As shown in Fig. 3A, RR-RJW100 destabilizes the bottom of H10 and the pre-AF-H loop versus DLPC, although the AF-H itself is slightly stabilized. Additional destabilization by RR-RJW100 occurs at the top part of H3 (part of the AFS) and in β2 (part of the alternate activation function region responsible for allosteric communication with the AFS (20, 21)). Interestingly, this localized destabilization occurs even though RJW100 increases overall thermostability of LRH-1 compared with DLPC (Fig. 3B). This finding may indicate room for improvement in agonist design; compounds that provide a stable surface for co-activator recruitment would likely be more potent activators.
FIGURE 3.
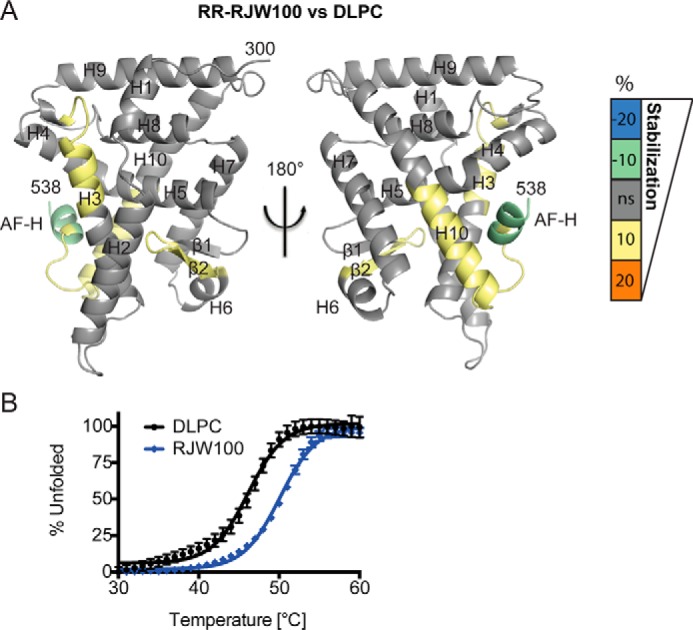
Differential effects on LRH-1 dynamics when RR-RJW100 is bound versus DLPC. A, HDX was used to probe differential effects on protein dynamics by the two ligands. The scale refers to the difference in percent deuterium incorporation for RR-RJW100-bound LRH-1 minus DLPC-bound LRH-1. The scale reflects the difference in average percent deuterium incorporation for RR-RJW100-bound versus DLPC-bound LRH-1 LBD. For example, negative numbers reflect slower deuterium incorporation (less motion) for RR-RJW100 versus DLPC. Results are mapped onto PDB 4DOS (20). B, DSF curves showing that RJW100 increases overall LRH-1 thermostability compared with DLPC. Each point represents the mean ± S.E. of values for three independent experiments, each conducted in triplicate.
Dramatic Repositioning of RJW100 Compared with a Closely Related Synthetic Agonist
Perhaps the most striking observation from our structure comes from comparison with GSK8470-bound LRH-1 (PBD 3PLZ). Overall, protein conformation is highly similar; the largest movement occurs in the bottom of H3, which moves in the direction of H6 (by 2 Å in the RR-RJW100 structure and by 4 Å in the GSK8470 structure relative to apo-LRH-1, Fig. 4A). However, there is a substantial difference in the positioning of these agonists within the binding pocket. Although GSK8470 and RR-RJW100 bind in the same vicinity, they are rotated nearly 180° from one another. The bicyclic rings at the cores of each molecule are perpendicular to each other, causing the tails to be pointed in opposite directions (Fig. 4B). Notably, the rationale for adding a hydroxyl group in the 1-position on this scaffold was to promote an interaction with a “polar patch,” consisting of residues Arg-393 and is-390 in an otherwise hydrophobic pocket (23). This interaction was predicted based on the position of the ligand in the LRH-1-GSK8470 structure; however, the actual position of RR-RJW100 in the pocket places the hydroxyl group over 6 Å away from these residues (Fig. 4C). Such a radically different binding mode for closely related molecules was unexpected, and a propensity to rotate within the pocket may contribute to difficulties improving agonist activity by modification of the GSK8470 scaffold.
FIGURE 4.
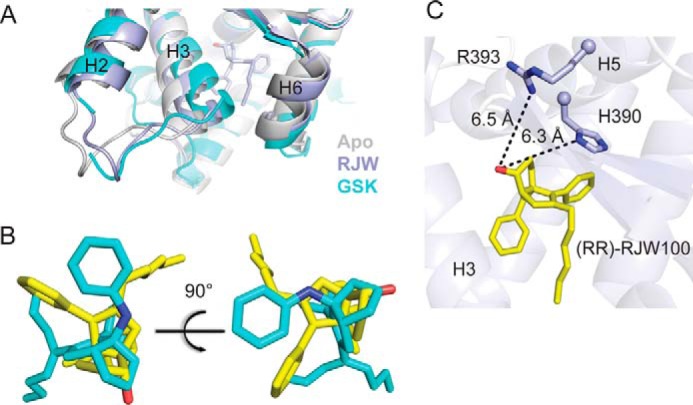
Very different binding mode of RR-RJW100 compared with a closely related synthetic agonist. A, close-up view of the bottom of the receptor, showing the shift in H3 induced by both synthetic agonists compared with apo-LRH-1. B, superposition of coordinates for GSK8470 (cyan, from PBD 3PLZ) and RR-RJW100 (yellow). C, RR-RJW100 hydroxyl group was predicted to interact with residues His-390 and Arg-393, but it is over 6 Å away from these residues in our structure.
Crystal Structure of LRH-1 Bound to an RJW100 Diastereomer
To further explore the effect of agonist structure on ligand-binding mode, we determined the 1.93 Å crystal structure of LRH-1 bound to endo-RJW100, also in complex with the Tif2 co-activator peptide (Table 1 and Fig. 5A). Overall, protein conformation is highly similar to that of the RR-RJW100 (exo) structure, and there is disorder at the same portion of the protein backbone within the pre-AFH loop (Fig. 5A).
FIGURE 5.
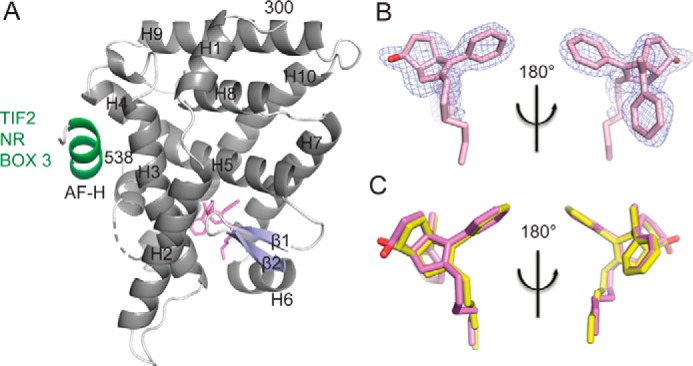
Crystal structure of LRH-1 bound to an RJW100 isomer with endo stereochemistry. A, overall structure of the LRH-1 ligand binding domain with SR-RJW100 (violet) and a peptide derived from the Tif2 co-activator (green). Dashed line, region of disorder in the protein backbone that could not be modeled. B, omit map (FO − FC, contoured at 2.5 σ) showing that a single enantiomer is bound in the structure. C, superposition of SR-RJW100 with RR-RJW100 (yellow) showing a very similar position in the binding pocket.
Like exo-RJW100, endo-RJW100 consists of a mixture of two enantiomers, only one of which is bound in the crystal structure. The presence of a single enantiomer is quite clear from the electron density surrounding the ligand, although the density for the ligand tail is much weaker than in the RR-RJW100 structure (Fig. 5B). The bound enantiomer has R stereochemistry at the styrene moiety, as in the exo-RJW100 structure, but the hydroxyl group is pointed in the opposite direction (designated SR-RJW100, Fig. 1). Superposition of the ligand coordinates from the RR-RJW100 and SR-RJW100 structures reveals nearly identical positioning, with the exception of the hydroxyl group (Fig. 5C). However, a key difference is seen in the ligand B-factors. For RR-RJW100, the average ligand B-factor is 34.8, which is slightly less than the average protein B (38.1, Table 1). In contrast, the ligand B-factor for SR-RJW100 is much higher compared with the protein B-factor (59.2 versus 46.9, Table 1). The higher average ligand B-factors do not arise solely from the disordered atoms in the ligand tail; for example, the B-factor for the hydroxyl oxygen of SR-RJW100 is 59. This is an indication of more atomic motion for SR-RJW100 than RR-RJW100, suggesting that the ligand has a reduced ability to engage in stabilizing intermolecular interactions in the pocket.
Discovery of a Novel LRH-1 Interaction Mediated by the RJW100 Hydroxyl Group
Protein-ligand interactions made by GSK8470 and the RJW100 stereoisomers were examined to gain insight into factors influencing the ligand-binding mode. A close view of the LRH-1-binding pocket reveals that RR-RJW100 makes several hydrophobic contacts, many of which are also made by GSK8470 (shown in cyan, Fig. 6A). Additionally, RR-RJW100 makes several unique contacts (shown in gray, Fig. 6A). Many of these unique contacts are also hydrophobic; however, the RR-RJW100 hydroxyl group forms an indirect polar contact with residue Thr-352 via a water molecule. A portion of the electron density map is shown in Fig. 6A to emphasize the strong evidence for this interaction. SR-RJW100 also interacts with Thr-352 through the same water molecule, despite the differing conformations of the hydroxyl group (Fig. 6B). The position of the SR-RJW100 hydroxyl group also permits a second water-mediated hydrogen-bonding interaction with the backbone nitrogen of residue Val-406 (Fig. 6B).
FIGURE 6.
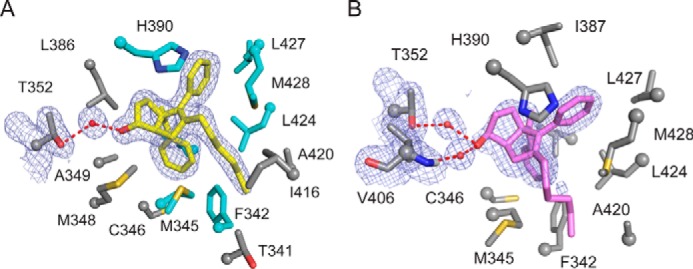
Residues interacting with LRH-1 agonists. Close-up views of the binding pockets from the structures of LRH-1 bound to RR-RJW100 (A) or SR-RJW100 (B), depicting side chains of amino acid residues that interact with each ligand. A, residues that also interact with GSK8470 are shown in cyan, whereas unique interactions made by RR-RJW100 are shown in gray. Portions of the electron density maps are shown to highlight the interactions with Thr-352 through water (FO − FC, contoured to 1σ).
Although the interaction with Thr-352 is indirect, we observed that the water molecule involved is part of a network of waters found in every LRH-1 crystal structure in the same location (for examples see PBDs 4DOS, 1YUC, 3PLZ, and 4DOR, as well as Fig. 7). Additional support for modeling water at these sites in the pocket comes from B-factors of ligating atoms (Table 3).Thus, this water network appears to be a conserved feature of the binding pocket and may play a role in receptor function or stability. To test the hypothesis that the OH-water-Thr-352 interaction was influencing ligand positioning, we analyzed the stability of this bond using molecular dynamics simulations (MDS). Throughout each simulation (200 ns), the four conserved networked water molecules remained in the same positions (if a particular water molecule occasionally left, it was immediately replaced with another in the same location). Residue Thr-352 maintained a hydrogen bond with the water molecule for 100% of each simulation, regardless of which ligand was bound. Additionally, both RR-RJW100 and SR-RJW100 maintained hydrogen bonding with the water molecule for the majority of the simulations (53.7% of the time for RR-RJW100 and 64.4% of the time for SR-RJW100). When residue Thr-352 was mutated to valine in MDS, the time spent interacting with the Thr-352-coordinated water molecule was drastically reduced (22.9 and 0.5% when RR-RJW100 and SR-RJW100 were bound, respectively), demonstrating that this mutation likely disrupts this water-mediated interaction made by these ligands.
FIGURE 7.
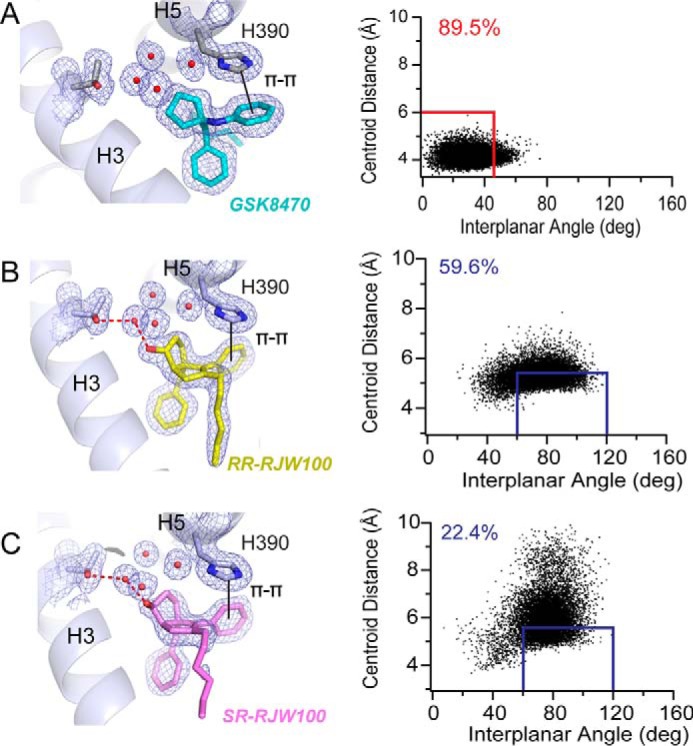
π-π-stacking with residue His-390 differs among agonists. A–C, left, views of the different types of π-π-stacking utilized by GSK8470 (A), RR-RJW100 (B), and SR-RJW100 (C). Right, MDS monitoring the distances between ring centroids (x axis) and angle between the ring planes (y axis) for the ligand phenyl group and His-390 at each time increment of the 200-ns MDS. The red and blue boxes indicate when face-to-face and edge-to-face π-π-stacking occurred, respectively. The numbers in the top left corners indicate the percentage of time spent π-π-stacking during the MDS.
TABLE 3.
B-factors for waters near the ligand and their ligating atoms
W indicates water; O indicates oxygen; N indicates nitrogen; NE indicates the ϵ nitrogen; and OG1 indicates the γ1 oxygen.
| Water | Ligating atom 1 | Ligating atom 2 | Ligating atom 3 | Ligating atom 4a |
|---|---|---|---|---|
| PDB 5L11 | ||||
| W1 (22.5) | RJW,O (30.8) | Thr-352, OG1 (22.9) | W2 (23.3) | W5 (22.8) |
| W2 (23.2) | Val-406,N (24.2) | W1 (22.5) | W9 (24.3) | |
| W5 (22.8) | Leu-386,O (25.2) | Asp-389, OD2 (28.6) | W1 (22.5) | |
| W9 (24.3) | His-390, ND1 (23.1) | Arg-393, NH1 (28.6) | W2 (23.3) | |
| PDB 5SYZ | ||||
| W17 (34.5) | ENO,O (58.8) | Thr-352, OG1 (28.9) | W16 (25.9) | W4 (32.5) |
| W16 (25.9) | Leu-386,O (26.3) | Asp-389, OD2 (34.3) | W17 | |
| W3 (32.0) | His-390, ND1 (30.7) | Arg-393, NH1 (33.0) | Phe-404,O (31.9) | W4 |
| W4 (32.5) | ENO,O (58.8) | Val-406,N (32.8) | W3 | |
| PDB 3PLZ, chain A | ||||
| W41 (19.5) | Thr-352, OG1 (14.4) | Arg-393, NH2 (21.5) | W16 (18.1) | W13 |
| W16 (18.1) | Asp-389, OD2 (19.4) | Arg-393, NH2 (21.5) | W41 (19.4) | |
| W13(21.2) | Val-406,N (20.4) | W41 | W31 | |
| W31 (21.1) | Phe-404,O (19.2) | Arg-393, NE (22.0) | His-390, ND1 (15.6) | W13 |
| PDB 3PLZ, chain B | ||||
| W25 (15.9) | Thr-352, OG1 (11.7) | W14 (13.8) | W129 (14.2) | |
| W14 (13.8) | Leu-386,O (13.8) | Asp-389, OD2 (18.7) | W25 (15.9) | W52 (18.1) |
| W129 (14.2) | Val-406,N (20.4) | W25 (15.9) | W52 (18.1) | |
| W52 (18.1) | Phe-404,O (13.6) | Arg-393, NH2 (22.7) | His-390, ND1 (12.1) | |
a When a fourth ligating atom is identified, it is likely that the waters intermittently interact with the four atoms.
Differences in π-π Stacking with Residue His-390 among LRH-1 Agonists
The π-π-stacking of GSK8470 with residue His-390 has been previously described and is hypothesized to be critical for activation of LRH-1 by synthetic compounds (23, 25). The RJW100 diastereomers also engage in π-π-stacking with His-390, but with some key differences. The π-π-stacking is face-to-face for GSK8470 and edge-to-face for the RJW100 isomers (Fig. 7). Additionally, by virtue of the very different orientations in the binding pocket, the agonists do not use analogous phenyl rings for π-π-stacking; GSK8470 uses the aniline group, whereas the RJW100 isomers use the adjacent phenyl substituent. Moreover, MDS demonstrate ligand-dependent differences in the stability of this interaction. For this latter analysis, time spend in π-π-stacking was quantified over the course of the 200-ns MDS. Face-to-face π-π-stacking was defined as a distance between ring centroids of <6 Å, an angle between ring planes of <45°, and an angle between the centroid-centroid vector and one plane of <60° (26). Edge-to-face stacking was defined as a distance of <5.5 Å between ring centroids and an angle between the ring planes between 60 and 120° (26). Applying these criteria as appropriate for the type of π-π-stacking made by each ligand revealed that GSK8470 maintained π-π-stacking with His-390 for most of the simulation (89.5% of the time, Fig. 7A). The edge-to-face π-π-stacking made by RR-RJW100 was also fairly stable, maintained for 59.6% of the MDS time (Fig. 7B). In contrast, SR-RJW100 made this interaction much less frequently (22.4% of the time) and exhibited much more motion than RR-RJW100 (Fig. 7C). These observations are consistent with the relatively high B-factors seen for SR-RJW100 in the crystal structure.
Role of Thr-352 and His-390 in LRH-1 Activation by Synthetic Agonists
The importance of the Thr-352 and His-390 interactions for binding and activation of LRH-1 by the agonists was investigated using mutagenesis. Binding and stabilization of LRH-1 were detected using DSF. Although the T352V mutation (designed to remove the water-mediated hydrogen bond with bound ligands) had little effect on the overall thermostability of DLPC-bound LRH-1, it completely abrogated the stabilizing effect of RR-RJW100 and SR-RJW100 (Fig. 8A). Likewise, disrupting this interaction by using an RJW100 analog lacking the hydroxyl group (named 18a) prevented the positive Tm shift in wild-type (WT) LRH-1 (Fig. 8B). GSK8470 did not affect the melting profile of LRH-1 in WT or T352V protein, supporting the notion that the hydroxyl group is important for stabilizing the protein-ligand complex.
FIGURE 8.
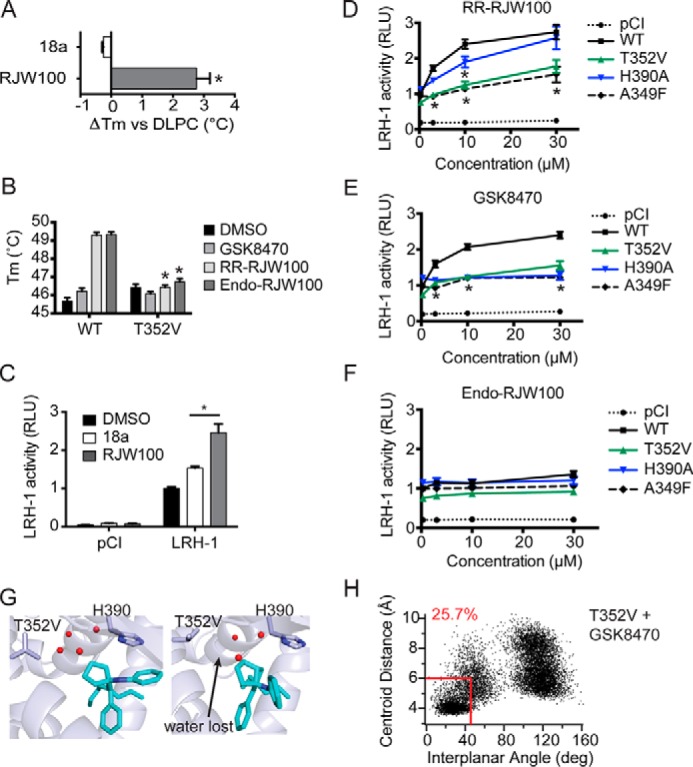
Importance of protein-ligand interactions on ligand binding and activity. A, analog 18a, lacking the hydroxyl group, does not stabilize wild-type LRH-1 in DSF assays. B, introduction of the T352V mutation to LRH-1 ablates the stabilizing effects of RR-RJW00 and endo-RJW100. Purified LRH-1 LBD, initially bound to DLPC for homogeneity, was incubated with either DMSO (control) or synthetic agonist dissolved in DMSO. C, compound 18a is a significantly weaker agonist in luciferase reporter assays. D–F, luciferase reporter assays measuring LRH-1 activity, using the SHP-luc reporter. Values have been normalized to constitutive Renilla luciferase signal and are presented as fold change versus wild-type LRH-1 + DMSO. The A349F mutation introduces a bulky aromatic side chain, which blocks the binding pocket and prevents binding of synthetic ligands (27). This was used as a negative control. G, snapshots from MDS using T352V LRH-1 with GSK8470 bound. H, plot of distances between ring centroids (x axis) and angle between the ring planes (y axis) for the GSK8470 phenyl group and His-390 at each time increment of the 200 ns with T352V LRH-1 (as described for Fig. 7). A–F, each bar (or point, for D–F) represents the mean ± S.E. for three independent experiments, each conducted in triplicate. *, p < 0.05 (significance was determined by two-way analysis of variance followed by Tukey's multiple comparisons test).
The Thr-352 interaction was also found to be important for LRH-1 activation by small molecule agonists. Compound 18a, lacking the hydroxyl group and unable to make this interaction, was an extremely poor LRH-1 activator in luciferase reporter assays (Fig. 8C). The endo-RJW100 was also a weak agonist, as reported previously (23), although statistically significant activation was achieved at the highest dose with WT LRH-1 (∼1.4-fold over DMSO, Fig. 8F). RR-RJW100 and GSK8470 were equally effective toward WT LRH-1, and both increased activity by ∼2.5-fold compared with DMSO at the highest dose, and both had EC50 values of around 4 μm (Fig. 8, D and E). Notably, the T352V mutation greatly reduced the ability of RR-RJW100 to activate LRH-1 compared with WT protein, while not significantly affecting baseline activity (Fig. 8D). Unexpectedly, this mutation similarly attenuated activation by GSK8470, perhaps suggesting a broader role for this residue (or perhaps for the water network it coordinates) in ligand-mediated activation. Indeed, introduction of a T352V mutation to GSK8470-bound LRH-1 in MDS disrupts the water network, causing complete displacement of the water molecule typically coordinated by Thr-352 (Fig. 8G). The T352V mutation also significantly reduces the amount of time GSK8470 spends π-π-stacking with His-390 (25.7% versus 89.5% of the simulation, Figs. 8H and 7A). Both the destabilization of the water network and the disruption of stable His-390 π-π-stacking by the T352V mutation could contribute to the observed loss of activity for GSK8470 in the context of this mutation.
Although the T352V mutation resulted in a loss of activity for both RR-RJW100 and GSK8470, mutating His-390 to alanine had a different effect on LRH-1 activation depending on the agonist involved. GSK8470 was completely unable to activate H390A-LRH-1, but this mutation had little to no effect on RR-RJW100-mediated activation (Fig. 8, D and E). This differential reliance on His-390 for activation is consistent with the observation that GSK8470 interacts with His-390 more stably than RR-RJW100 in MDS. This also provides evidence that RR-RJW100 must utilize a different mechanism of action than GSK8470 for LRH-1 activation.
Capacity for Productive π-π-Stacking with Residue His-390 Influences Agonist Positioning
Considering the stable nature of the π-π-stacking GSK8470 with residue His-390, the reasons why RJW100 would abandon this strong interaction for an entirely different binding mode were unclear. To answer this question, we artificially re-oriented RR-RJW100 in our structure to be aligned with GSK8470, inspected the fit, and looked at predicted interactions using MDS. Although this repositioning did not produce any obvious clashes, the planarity of the styrene moiety restricted its mobility. This constraint prevents the rotation necessary for ideal face-to-face π-π-stacking with His-390 (Fig. 9A). Although MDS using this re-oriented ligand demonstrated fairly stable face-to-face π-π-stacking for re-oriented RR-RJW100, the interaction was less stable than GSK8470 (present 71.1% of the time versus 89.5% for GSK8470, p < 0.0001, Fig. 9B). There was increased variability in both ring centroid distances and angle between the rings involved in this interaction for repositioned RR-RJW100 (mean centroid distances ± S.D. were 5.1 ± 0.6 Å versus 4.2 ± 0.3 Å for GSK8470, and mean angles were 40 ± 20° versus 30 ± 10° for GSK8470). This variability indicates more motion of the rings involved in the π-π-stacking for the repositioned ligand. Another interesting observation with this MDS pertained to the water network in the ligand-binding pocket. The hydroxyl group of the repositioned ligand was initially within hydrogen-bonding distance of the Thr-352-coordinated water molecule. Unlike native RR-RJW100, however, the hydroxyl group in the repositioned ligand was also within hydrogen-bonding distance of the three other water molecules in the immediate vicinity (Fig. 9A, right panel). During the MDS, the water network was very quickly disrupted, with a complete loss of one of the four conserved water molecules near the ligand hydroxyl group. One of the remaining three waters moved to coordinate Thr-352, whereas the ligand hydroxyl group alternated between interactions with each of the three waters without stably interacting with a single water molecule (data not shown). Thus, the water-mediated contact of the RR-RJW100 hydroxyl group with Thr-352 is not maintained for the reoriented ligand. Moreover, the loss of a typically conserved water molecule supports the idea that the ligand-binding pocket is more dynamic with the artificially repositioned ligand. Therefore, it appears that an impaired ability of the styrene phenyl ring to interact with His-390, combined with a favorable interaction mediated by the hydroxyl group with the Thr-352-coordinated water, is responsible for the dramatically different position adopted by RJW100 compared with a structurally very similar agonist.
FIGURE 9.
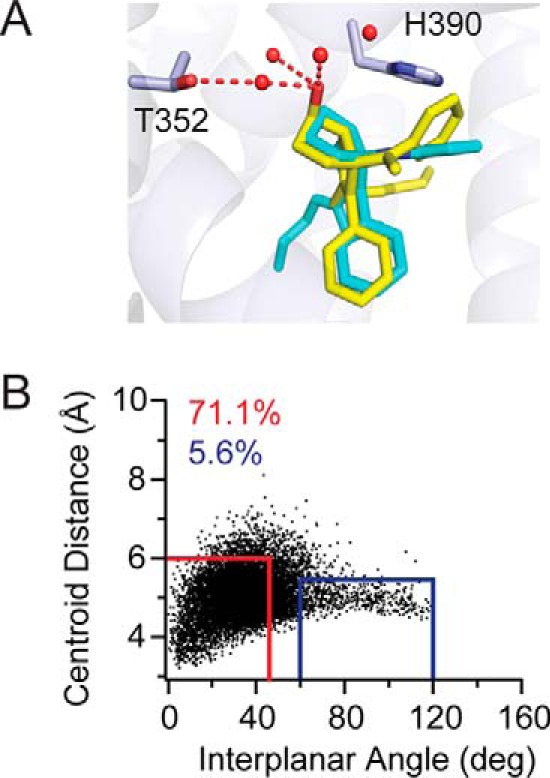
Impaired π-π-stacking of the RR-RJW100 styrene group in MDS. A, RR-RJW100 (yellow) was artificially aligned with GSK8470 (cyan) for MDS to study its ability to π-π-stack with residue His-390. Red dotted lines indicate water molecules that are within hydrogen bonding distances of the ligand or residue Thr-352. B, plot of distances between ring centroids (x axis) and angle between the ring planes (y axis) for the ligand phenyl group and His-390 at each time increment of the 200-ns MDS for the styrene phenyl ring of repositioned RR-RJW100 and residue His-390 over the course of the simulation. The red and blue boxes indicate when face-to-face and edge-to-face π-π-stacking occurred, respectively. The numbers in the top left corners indicate the percentage of time π-π-stacking during the MDS.
Discussion
Although LRH-1 synthetic modulators are highly sought as pharmacological tools and as potential therapeutic agents, a limited understanding of ligand characteristics important for binding and activating LRH-1 has impeded agonist development. This work represents the first detailed exploration of structural mechanisms governing regulation of LRH-1 by synthetic ligands. Relative to the PL LRH-1 agonist, DLPC, the current best agonist (RR-RJW100) constricts the binding pocket and destabilizes portions of the AFS (Figs. 1 and 2). In future studies, it will be interesting to investigate the causes of this latter effect, because stabilization of the AFS may facilitate co-activator binding, leading to greater potency or efficacy. Alternatively, analogs designed to enhance the AFS destabilization may be effective antagonists or inverse agonists.
In a previous study, RJW100 was the most effective of a large series of GSK8470 derivatives but still only modestly increased LRH-1 activation (23). Indeed, we find that these two agonists are statistically indistinguishable in luciferase reporter assays measuring LRH-1 activity (Fig. 8D). Given the similarities in structures and efficacies for these ligands, we expected them to utilize similar mechanisms of action; however, this is not the case. Our crystal structure reveals a dramatically different binding mode for RR-RJW100 compared with GSK8470 (Fig. 4). Although this was surprising, it is not unreasonable, considering that LRH-1 has a very large hydrophobic binding pocket and that these agonists are also quite hydrophobic, filling only 37% of the available space (excluding waters). It is possible that many of the GSK8470 analogs investigated in the previous structure-activity relationship study adopt a variety of different conformations. This seems to be the case in our docking studies with these ligands; multiple very different binding modes with similar energies are predicted (data not shown). Importantly, however, the repositioning of RR-RJW100 in our structure appears to be driven by particular interactions, because SR-RJW100 assumes a very similar pose (Fig. 5). This occurs despite the fact that the SR derivative exhibits signs of motion in our crystal structure, with significant disorder in the tail of the ligand and higher relative B-factors than RR-RJW100.
A major factor driving repositioning of the RJW100 isomers was the hydrogen bonding interaction made by the hydroxyl group. Although the contact with residue Thr-352 is indirect, it is mediated by a water molecule that is part of a network of waters found in every published LRH-1 crystal structure (with the exception of PDB 4DOR, in which a major portion of the ligand-binding pocket is disordered (20)). The existence of conserved water molecules, as well as their participation in ligand binding, has been described (28–30). Thus, this interaction could serve as an anchor point to secure the compound in a predictable orientation, enabling the targeting of desired parts of the binding pocket via strategic addition of substituents to the ligand's scaffold. Moreover, replacing the RJW100 hydroxyl group with a larger polar moiety may allow direct contact with Thr-352, leading to a stronger interaction. This strategy is being actively explored in our laboratory.
The role of the Thr-352 interaction in LRH-1 activation by RR-RJW100 was demonstrated through the marked loss of activation by this compound when this residue was mutated (Fig. 8). In addition, an RJW100 analog lacking a hydroxyl group and thus unable to make this was a poor activator. Unexpectedly, the T352V mutation also resulted in a loss of activity for GSK8470, although this compound does not interact with the Thr-352-coordinated water molecule. However, we show that the T352V mutation weakens GSK8470's interaction with His-390, perhaps via destabilization of the conserved water network (Fig. 8, G and H). This could be responsible for the loss of activity of GSK8470 when Thr-352 is mutated.
It has been hypothesized in in silico studies that π-π-stacking with residue His-390 is critical for activation of LRH-1 by this ligand class (25); however, this had not been explicitly tested. We find that this is the case for GSK8470, which stably interacts with His-390 via face-to-face π-π-stacking. Interestingly, although mutation of His-390 to alanine ablated LRH-1 activation by GSK8470, it had no effect on RR-RJW100-mediated activation (Fig. 8). We show that the RR-RJW100 interaction with His-390 is much less stable than that of GSK8470 and is mediated by a different phenyl ring (Fig. 7). Substitution of the GSK8470 aniline group with the styrene appears to have had the unexpected effect of making face-to-face stacking with His-390 less favorable (Fig. 9). This, combined with the favorable water-mediated interaction with Thr-352, influences the positioning of RJW100.
Together, these findings reveal that the interaction of small molecule agonists with LRH-1 is more complex than originally supposed. Not only do these agonists affect receptor conformation differently from PL ligands, but they also exhibit an unexpected variability in binding modes. This work has uncovered some of the molecular interactions responsible for both positioning and activation of two very similar agonists, which provide insights into strategies to improve the design of LRH-1-targeted compounds.
Experimental Procedures
Materials and Reagents
GSK8470, RJW100, and analogs were synthesized as described previously (22, 23). RR-RJW100 was separated from SS-RJW100 by chiral preparative chromatography (Diacel OD-H column (31)). endo-RJW100 is a diastereoisomer of RJW100, previously referred to as “24-endo” (23). In this paper, we use “SR-RJW100” and “RS-RJW100” to refer to the enantiomers of endo-RJW100 (Fig. 1). pCI empty vector was purchased from Promega. The SHP-luc and Renilla reporters, as well as pCI LRH-1, have been previously described (20). The vector for His-tagged tobacco etch virus (TEV) was a gift from John Tesmer (University of Texas at Austin). The pMSC7 (LIC_HIS) vector was provided by John Sondek (University of North Carolina at Chapel Hill). The Tif2 NR Box 3 peptide was purchased from RS Synthesis (Louisville, KY). DNA oligonucleotide primers were synthesized by IDT (Coralville, IA).
Protein Expression and Purification
LRH-1 LBD (residues 299–541) in the pMSC7 vector was expressed and purified as described previously (21). Briefly, protein was expressed in BL21(DE3) pLysS E. coli by induction with isopropyl 1-thio-β-d-galactopyranoside (1 mm) for 4 h at 30 °C. Protein was purified by nickel affinity chromatography. Protein used for DSF experiments was incubated with DLPC (5-fold molar excess) for 4 h at room temperature and then repurified by size exclusion into an assay buffer of 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 5% glycerol. Protein used for crystallization was incubated with TEV protease to cleave the His tag. The cleaved protein was then separated from the His tag and TEV by a second round of nickel affinity chromatography. To make protein-ligand complexes, protein was incubated with ligands overnight (10-fold molar excess) and repurified by size-exclusion, using a final buffer of 100 mm ammonium acetate, pH 7.4, 150 mm sodium chloride, 1 mm DTT, 1 mm EDTA, and 2 mm CHAPS.
Crystallization
Protein-ligand complexes were incubated with a peptide derived from human Tif2 NR box 3 (+H3N-KENALLRYLLDKDDT-CO2−) at 4-fold molar excess for 2 h at room temperature and then concentrated to 6.5 mg/ml. A crystallant of 0.05 m sodium acetate, pH 4.6, 5–11% PEG 4000, and 0–10% glycerol was used. Crystals were grown by hanging drop vapor diffusion in drops containing 1 μl of protein and 1 μl of crystallant, at a temperature of 18–20 °C.
Structure Determination
Crystals were flash-frozen in liquid nitrogen, using a cryopreservative consisting of crystallant plus 30% glycerol. Data were collected remotely from the SouthEast Regional Collaborative Access Team at the Advanced Photon Source, 22ID beamline (Argonne National Laboratories, Chicago). Data were processed and scaled using HKL2000 (32) and phased by molecular replacement using Phaser-MR (Phenix (33)). For the RR-RJW100 structure, PDB 3PLZ (23) was used as the search model, with the ligand and a portion of the bottom of the receptor omitted. For the SR-RJW100 structure, the search model was the RR-RJW100 structure with the ligand omitted. Model building and refinement were conducted with Coot (34) and phenix.refine (33), respectively. Figures were constructed using PyMOL (Schrödinger, LLC) (35).
Structure Analysis
Dimensions of the binding pocket in the presence of various ligands were calculated using CastP software (36). Ligplot+ was used to identify residues interacting with the ligands (37).
Mutagenesis
Mutations were introduced to pMSC7 and pCI LRH-1 constructs using the QuikChange site-directed mutagenesis kit (Stratagene). Constructs were sequence-verified prior to use.
Cell Culture
HeLa cells were purchased from the American Type Culture Collection and grown in phenol red-free α-minimal essential medium (CellGro) supplemented with 10% charcoal-stripped fetal bovine serum (Atlanta Biologicals). Cells were maintained using standard culture conditions.
Reporter Gene Assays
HeLa cells were seeded at a density of 10,000 cells per well in white-walled, clear-bottomed 96-well culture plates. The next day, cells were transfected with LRH-1 and reporters, using FuGENE HD (Roche Applied Science) at a ratio of 5:2 FuGENE/DNA. The transfected plasmids included full-length LRH-1 in a pCI vector (5 ng/well), and an SHP-luc reporter, encoding the LRH-1 response element and surrounding sequence from the SHP promoter cloned upstream of firefly luciferase in the pGL3 basic vector (50 ng/well). Cells were also co-transfected with a constitutive Renilla luciferase reporter (utilizing the CMV promoter), which was used for normalization of firefly signal (1 ng/well). Control cells received pCI empty vector at 5 ng/well in place of LRH-1-pCI. Following an overnight transfection, cells were treated with agonists for 24 h at the concentrations indicated in the figure legends. Agonists were dissolved in DMSO and then diluted into media, with a final concentration of 0.3% DMSO in all wells. Luciferase signal was quantified using the DualGlo kit (Promega). Experiments were conducted at least three times in triplicate.
Hydrogen-Deuterium Exchange Mass Spectrometry
Purified LRH-1 LBD protein (His tag removed) was incubated with a 5-fold molar excess DLPC or synthetic agonist overnight at 4 °C. Protein-ligand complexes were then re-purified by size exclusion to remove displaced phospholipids and unbound ligands. An additional bolus of agonist or DLPC (5-fold molar excess) was added to the complexes prior to analysis by HDX. The assay buffer consisted of 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 5% glycerol. Solution phase amide HDX was carried out with a fully automated system as described previously (38) Briefly, 5 μl of protein was diluted to 25 μl with D2O-containing HDX buffer and incubated at 25 °C for 10, 30, 60, 900, or 3600 s. Following exchange, back exchange was minimized, and the protein was denatured by dilution to 50 μl in a low pH and low temperature buffer containing 0.1% (v/v) trifluoroacetic acid in 5 m urea (held at 1 °C). Samples were then passed across an immobilized pepsin column (prepared in-house) at 50 μl min−1 (0.1% v/v TFA, 15 °C); the resulting peptides were trapped on a C8 trap cartridge (Hypersil Gold, Thermo Fisher). Peptides were then gradient-eluted (4% (w/v) CH3CN to 40% (w/v) CH3CN to 0.3% (w/v) formic acid over 5 min, 2 °C) across a 1 × 50-mm C18 HPLC column (Hypersil Gold, Thermo Fisher) and electrosprayed directly into an Orbitrap mass spectrometer (Q Exactive, Thermo Fisher). Data were processed with in-house software (39) and visualized with PyMOL (Schrödinger, LLC (35)). To measure the difference in exchange rates, we calculated the average percent deuterium uptake for hLRH-1 LBD-RR-RJW100 complexes following 10, 30, 60, 900, and 3600 s of exchange. From this value, we subtracted the average percent deuterium uptake measured for the DLPC-hLRH-1 LBD complex. Negative perturbation values indicate exchange rates are slower for these regions within the RR-RJW100-LRH-1 complex relative to DLPC-bound LRH-1.
DSF
Purified LRH-1 LBD-His protein (0.2 mg/ml) was incubated overnight with 50 μm of each compound at 4 °C. The final DMSO concentration in the reactions was 1%. SYPRO Orange dye (Invitrogen) was then added at a 1:1000 dilution. Reactions were heated at a rate of 0.5 °C per min, using a StepOne Plus Real Time PCR system (ThermoFisher). Fluorescence was recorded at every degree using the ROX filter (602 nm). Data were analyzed by first subtracting baseline fluorescence (ligands + SYPRO with no protein) and then fitting the curves using the Boltzmann equation (GraphPad Prism, version 6) to determine the Tm.
Model Construction for Molecular Dynamics Simulations
Three crystal structures of LRH-1 LBD in complex with Tif2 were used to construct models for the simulations. These were: 1) PDB 3PLZ, chains B and D (GSK8470 ligand); 2) PDB 5L11 (RR-RJW100 ligand); and 3) PDB 5SYZ (SR-RJW100 ligand). For consistency, the structures were modified at the N and C termini so that all contained residues 300–540 of LRH-1 and residues 742–751 of the Tif2 peptide. Missing residues within this protein sequence were added, as well as missing protein side chains (PDB 3PLZ). The T352V mutation was introduced by mutating the sequence of WT LRH-1 in each of these structures. Finally, in a separate simulation, RR-RJW100 ligand was artificially reoriented in WT LRH-1 to be aligned with GSK8470, allowing a face-to-face π-π-stacking interaction of LRH-1 residue His-390 with the aniline ring of RR-RJW100. Seven complexes in total were used in molecular dynamics simulations.
Molecular Dynamics Simulations
The complexes were solvated in an octahedral box of TIP3P water with a 10-Å buffer around the protein complex. Na+ and Cl− ions were added to neutralize the protein and achieve physiological conditions. All systems were set up using Xleap in AmberTools (40) with the parm99-bsc0 force field (41). Parameters for all ligands (GSK8470, RR-RJW100, and SR-RJW100) were obtained using Antechamber (42) in AmberTools. All minimizations and simulations were performed with Amber14 (43). Systems were minimized with 5000 steps of steepest decent followed by 5000 steps of conjugate gradient minimization with 500 kcal/mol·Å2 restraints on all atoms. Restraints were removed from all atoms excluding the atoms in both the ligand and the Tif2 peptide, and the previous minimization was repeated. The systems were heated from 0 to 300 K using a 100-ps run with constant volume periodic boundaries and 5 kcal/mol·Å2 restraints on all protein and ligand atoms. Twelve ns of molecular dynamics equilibration was performed with 10 kcal/mol·Å2 restraints on protein and ligand atoms using the NPT ensemble. Restraints were reduced to 1 kcal/mol·Å2 for an additional 10 ns of molecular dynamics equilibration. Then restraints were removed, and 200-ns production simulations were performed for each system in the NPT ensemble. A 2-fs time step was used, and all bonds between heavy atoms and hydrogens were fixed with the SHAKE algorithm (44). A cutoff distance of 10 Å was used to evaluate long-range electrostatics with Particle Mesh Ewald (PME) and for van der Waals forces. 10,000 evenly spaced frames were taken from each simulation for analysis. Analysis was performed with the CPPTRAJ module (45) of AmberTools. Statistical significance of differences in time π-π-stacking with residue His-390 for GSK8470 versus the re-oriented RR-RJW100 was assessed using a χ2 test with Yate's correction (GraphPad Prism version 6).
Author Contributions
S. G. M. and E. A. O. conceived the studies. S. G. M. purified protein-ligand complexes, determined crystal structures, and conducted DSF and cellular assays. C. D. O. performed and analyzed molecular dynamics simulations. D. G. and P. R. G. conducted HDX and analyzed results. R. J. W., J. S., M. C. D., A. R. F., and N. T. J. synthesized GSK8470, RJW100, and analogs. S. G. M., C. D. O., R. J. W., N. T. J., D. G., P. R. G., and E. A. O. wrote the manuscript.
Acknowledgments
We thank Sally Bloodworth (University of Southampton, United Kingdom) for assistance with synthetic chemistry, Bradley Kossmann and Dr. Ivaylo Ivanov (Georgia State University) for helpful discussions about MDS, and Dr. Kay Diederichs (Universität Konstanz) for advice in model refinement.
This work was supported in part by National Institutes of Health Grants T32-GM008602 (to S. G. M.), F31DK111171 (to S. G. M.), and R01DK095750 (to E. A. O.) and an Emory Catalyst Grant (to E. A. O. and N. T. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (codes 5SYZ and 5L11) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NR
- nuclear hormone receptor
- PDB
- Protein Data Bank
- PL
- phospholipid
- HDX
- hydrogen-deuterium exchange
- MDS
- molecular dynamics simulation
- LBD
- ligand binding domain
- DLPC
- dilauroylphosphatidylcholine
- PIP3
- phosphatidylinositol 3,4,5-triphosphate
- AFS
- activation function surface
- TEV
- tobacco etch virus
- DSF
- differential scanning fluorimetry.
References
- 1. Gu P., Goodwin B., Chung A. C., Xu X., Wheeler D. A., Price R. R., Galardi C., Peng L., Latour A. M., Koller B. H., Gossen J., Kliewer S. A., and Cooney A. J. (2005) Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol. Cell. Biol. 25, 3492–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wagner R. T., Xu X., Yi F., Merrill B. J., and Cooney A. J. (2010) Canonical Wnt/β-catenin regulation of liver receptor homolog-1 mediates pluripotency gene expression. Stem Cells 28, 1794–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., and Mangelsdorf D. J. (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 6, 507–515 [DOI] [PubMed] [Google Scholar]
- 4. Stein S., Oosterveer M. H., Mataki C., Xu P., Lemos V., Havinga R., Dittner C., Ryu D., Menzies K. J., Wang X., Perino A., Houten S. M., Melchior F., and Schoonjans K. (2014) SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab. 20, 603–613 [DOI] [PubMed] [Google Scholar]
- 5. Schoonjans K., Annicotte J.-S., Huby T., Botrugno O. A., Fayard E., Ueda Y., Chapman J., and Auwerx J. (2002) Liver receptor homolog 1 controls the expression of the scavenger receptor class B type I. EMBO Rep. 3, 1181–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolado-Carrancio A., Riancho J. A., Sainz J., and Rodríguez-Rey J. C. (2014) Activation of nuclear receptor NR5A2 increases Glut4 expression and glucose metabolism in muscle cells. Biochem. Biophys. Res. Commun. 446, 614–619 [DOI] [PubMed] [Google Scholar]
- 7. Oosterveer M. H., Mataki C., Yamamoto H., Harach T., Moullan N., van Dijk T. H., Ayuso E., Bosch F., Postic C., Groen A. K., Auwerx J., and Schoonjans K. (2012) LRH-1-dependent glucose sensing determines intermediary metabolism in liver. J. Clin. Invest. 122, 2817–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venteclef N., Smith J. C., Goodwin B., and Delerive P. (2006) Liver receptor homolog 1 is a negative regulator of the hepatic acute-phase response. Mol. Cell. Biol. 26, 6799–6807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mamrosh J. L., Lee J. M., Wagner M., Stambrook P. J., Whitby R. J., Sifers R. N., Wu S. P., Tsai M. J., Demayo F. J., and Moore D. D. (2014) Nuclear receptor LRH-1/NR5A2 is required and targetable for liver endoplasmic reticulum stress resolution. eLife 3, e01694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bayrer J. R., Mukkamala S., Sablin E. P., Webb P., and Fletterick R. J. (2015) Silencing LRH-1 in colon cancer cell lines impairs proliferation and alters gene expression programs. Proc. Natl. Acad. Sci. U.S.A. 112, 2467–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bianco S., Brunelle M., Jangal M., Magnani L., and Gévry N. (2014) LRH-1 governs vital transcriptional programs in endocrine-sensitive and -resistant breast cancer cells. Cancer Res. 74, 2015–2025 [DOI] [PubMed] [Google Scholar]
- 12. Chand A. L., Herridge K. A., Thompson E. W., and Clyne C. D. (2010) The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion. Endocr. Relat. Cancer 17, 965–975 [DOI] [PubMed] [Google Scholar]
- 13. Clyne C. D., Kovacic A., Speed C. J., Zhou J., Pezzi V., and Simpson E. R. (2004) Regulation of aromatase expression by the nuclear receptor LRH-1 in adipose tissue. Mol. Cell. Endocrinol. 215, 39–44 [DOI] [PubMed] [Google Scholar]
- 14. Lai C. F., Flach K. D., Alexi X., Fox S. P., Ottaviani S., Thiruchelvam P. T., Kyle F. J., Thomas R. S., Launchbury R., Hua H., Callaghan H. B., Carroll J. S., Charles Coombes R., Zwart W., Buluwela L., and Ali S. (2013) Co-regulated gene expression by oestrogen receptor α and liver receptor homolog-1 is a feature of the oestrogen response in breast cancer cells. Nucleic Acids Res. 41, 10228–10240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thiruchelvam P. T., Lai C. F., Hua H., Thomas R. S., Hurtado A., Hudson W., Bayly A. R., Kyle F. J., Periyasamy M., Photiou A., Spivey A. C., Ortlund E. A., Whitby R. J., Carroll J. S., Coombes R. C., et al. (2011) The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res. Treat. 127, 385–396 [DOI] [PubMed] [Google Scholar]
- 16. Lin Q., Aihara A., Chung W., Li Y., Huang Z., Chen X., Weng S., Carlson R. I., Wands J. R., and Dong X. (2014) LRH1 as a driving factor in pancreatic cancer growth. Cancer Lett. 345, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee J. M., Lee Y. K., Mamrosh J. L., Busby S. A., Griffin P. R., Pathak M. C., Ortlund E. A., and Moore D. D. (2011) A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature 474, 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sablin E. P., Blind R. D., Uthayaruban R., Chiu H. J., Deacon A. M., Das D., Ingraham H. A., and Fletterick R. J. (2015) Structure of liver receptor homolog-1 (NR5A2) with PIP hormone bound in the ligand binding pocket. J. Struct. Biol. 192, 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krylova I. N., Sablin E. P., Moore J., Xu R. X., Waitt G. M., MacKay J. A., Juzumiene D., Bynum J. M., Madauss K., Montana V., Lebedeva L., Suzawa M., Williams J. D., Williams S. P., Guy R. K., et al. (2005) Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell 120, 343–355 [DOI] [PubMed] [Google Scholar]
- 20. Musille P. M., Pathak M. C., Lauer J. L., Hudson W. H., Griffin P. R., and Ortlund E. A. (2012) Antidiabetic phospholipid-nuclear receptor complex reveals the mechanism for phospholipid-driven gene regulation. Nat. Struct. Mol. Biol. 19, 532–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Musille P. M., Kossmann B. R., Kohn J. A., Ivanov I., and Ortlund E. A. (2016) Unexpected allosteric network contributes to LRH-1 coregulator selectivity. J. Biol. Chem. 291, 1411–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Whitby R. J., Dixon S., Maloney P. R., Delerive P., Goodwin B. J., Parks D. J., and Willson T. M. (2006) Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. J. Med. Chem. 49, 6652–6655 [DOI] [PubMed] [Google Scholar]
- 23. Whitby R. J., Stec J., Blind R. D., Dixon S., Leesnitzer L. M., Orband-Miller L. A., Williams S. P., Willson T. M., Xu R., Zuercher W. J., Cai F., and Ingraham H. A. (2011) Small molecule agonists of the orphan nuclear receptors steroidogenic factor-1 (SF-1, NR5A1) and liver receptor homologue-1 (LRH-1, NR5A2). J. Med. Chem. 54, 2266–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y., Lambert M. H., and Xu H. E. (2003) Activation of nuclear receptors. Structure 11, 741–746 [DOI] [PubMed] [Google Scholar]
- 25. Lalit M., Gangwal R. P., Dhoke G. V., Damre M. V., Khandelwal K., and Sangamwar A. T. (2013) A combined pharmacophore modeling, 3D-QSAR and molecular docking study of substituted bicyclo-[3.3.0]oct-2-enes as liver receptor homolog-1 (LRH-1) agonists. J. Mol. Struct. 1049, 315–325 [Google Scholar]
- 26. Zhang Z., Chen H., Bai H., and Lai L. (2007) Molecular dynamics simulations on the oligomer-formation process of the GNNQQNY peptide from yeast prion protein Sup35. Biophys. J. 93, 1484–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benod C., Carlsson J., Uthayaruban R., Hwang P., Irwin J. J., Doak A. K., Shoichet B. K., Sablin E. P., and Fletterick R. J. (2013) Structure-based discovery of antagonists of nuclear receptor LRH-1. J. Biol. Chem. 288, 19830–19844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ogata K., and Wodak S. J. (2002) Conserved water molecules in MHC class-I molecules and their structural and functional roles. Protein Eng. 15, 697–705 [DOI] [PubMed] [Google Scholar]
- 29. Barillari C., Taylor J., Viner R., and Essex J. W. (2007) Classification of water molecules in protein binding sites. J. Am. Chem. Soc. 129, 2577–2587 [DOI] [PubMed] [Google Scholar]
- 30. Klebe G. (2015) Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discov. 14, 95–110 [DOI] [PubMed] [Google Scholar]
- 31. Stec J. (2010) Tandem Reaction Sequences on a Zirconocene Template. Ph.D. thesis, University of Southampton, United Kingdom [Google Scholar]
- 32. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 33. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 35. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 36. Binkowski T. A., Naghibzadeh S., and Liang J. (2003) CASTp: computed atlas of surface topography of proteins. Nucleic Acids Res. 31, 3352–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 38. Goswami D., Devarakonda S., Chalmers M. J., Pascal B. D., Spiegelman B. M., and Griffin P. R. (2013) Time window expansion for HDX analysis of an intrinsically disordered protein. J. Am. Soc. Mass Spectrom. 24, 1584–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pascal B. D., Willis S., Lauer J. L., Landgraf R. R., West G. M., Marciano D., Novick S., Goswami D., Chalmers M. J., and Griffin P. R. (2012) HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Case D., and Kollman P. (2012) AmberTools, Version 12, University of California, San Francisco [Google Scholar]
- 41. Pérez A., Marchán I., Svozil D., Sponer J., Cheatham T. E. 3rd., Laughton C. A., and Orozco M. (2007) Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 92, 3817–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J., Wang W., Kollman P. A., and Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 43. Case D., Babin V., Berryman J., Betz R., Cai Q., Cerutti D., Cheatham Iii T., Darden T., Duke R., and Gohlke H. (2014) Amber Tools, Version 14, University of California, San Francisco [Google Scholar]
- 44. Ryckaert J.-P., Ciccotti G., and Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 45. Roe D. R., and Cheatham T. E. 3rd. (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 [DOI] [PubMed] [Google Scholar]
- 46. Ortlund E. A., Lee Y., Solomon I. H., Hager J. M., Safi R., Choi Y., Guan Z., Tripathy A., Raetz C. R., McDonnell D. P., Moore D. D., and Redinbo M. R. (2005) Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat. Struct. Mol. Biol. 12, 357–363 [DOI] [PubMed] [Google Scholar]