

Abstract
Shroom-mediated remodeling of the actomyosin cytoskeleton is a critical driver of cellular shape and tissue morphology that underlies the development of many tissues including the neural tube, eye, intestines, and vasculature. Shroom uses a conserved SD2 domain to direct the subcellular localization of Rho-associated kinase (Rock), which in turn drives changes in the cytoskeleton and cellular morphology through its ability to phosphorylate and activate non-muscle myosin II. Here, we present the structure of the human Shroom-Rock binding module, revealing an unexpected stoichiometry for Shroom in which two Shroom SD2 domains bind independent surfaces on Rock. Mutation of interfacial residues impaired Shroom-Rock binding in vitro and resulted in altered remodeling of the cytoskeleton and loss of Shroom-mediated changes in cellular morphology. Additionally, we provide the first direct evidence that Shroom can function as a Rock activator. These data provide molecular insight into the Shroom-Rock interface and demonstrate that Shroom directly participates in regulating cytoskeletal dynamics, adding to its known role in Rock localization.
Keywords: cytoskeleton; Ras homolog gene family, member A (RhoA); serine/threonine protein kinase; structural biology; X-ray crystallography; Rho-kinase; RhoA; Shroom; coiled-coil
Introduction
The actomyosin cytoskeleton is a dynamic network that assembles, disassembles, and generates contractile forces to change the shape of a cell in response to stimuli. Dramatic rearrangement of cellular architecture is required for essential processes including migration, division, and changes in cell shape, events that must be executed correctly and efficiently to ensure the proper development and survival of every organism. Misregulation of the cytoskeleton can result in a variety of birth defects including exencephaly, spina bifida (1), and congenital myopathy (2); it also plays a role in other human disease states including cancer (3), immune function (4), synaptic function (5), and neural degeneration (6).
Cells utilize the actin cytoskeleton and myosin II to generate the force needed to control cell shape, motility, and behavior. The contractile activity of myosin II is largely regulated by the phosphorylation status of its associated myosin regulatory light chain (MRLC).2 Activity of non-muscle myosin II is stimulated by several serine/threonine kinases, which phosphorylate MRLC at Thr-18 and Ser-19 (7, 8) and drive contraction of F-actin bundles (9, 10). The phosphorylation of MRLC is opposed by the activity of myosin phosphatase (MYPT), which decreases phosphorylated MRLC levels and reduces the contractile activity of non-muscle myosin II (11).
Rho-associated kinase (Rock) is a serine/threonine kinase that both directly and indirectly activates non-muscle myosin II. Rock has been shown to positively regulate contractility by phosphorylating MRLC at position Ser-19 (12). Additionally, Rock phosphorylates MYPT at positions Thr-697 and Ser-854 (13), resulting in inactivation of MYPT and effectively augmenting myosin II activity by preventing its dephosphorylation (13–15).
Rock activity has been shown to drive cytoskeletal remodeling associated with cell migration (16), membrane blebbing (17–19), vasculogenesis (20), and apical constriction (21–23). The Rock protein is composed of an N-terminal kinase domain, a large central coiled coil domain, and C-terminal pleckstrin homology (PH) and cysteine-rich C1 domains (Fig. 1A). Autoinhibitory interactions between the kinase and PH/C1 domains provide a mechanism by which Rock activity is tightly controlled (24–26). Rock can be activated through several mechanisms, the best studied of which is the binding of RhoA-GTP to the Rho-binding domain within the coiled coil domain of Rock (27, 28) (see Fig. 1A). Additional mechanisms exist that affect Rock autoinhibitory interactions including lipid binding to the PH domain (29) and protease cleavage of the C terminus by caspase-3 (19) or granzyme B (18). In all of these scenarios, activation is thought to be achieved by displacement of the C-terminal PH/C1 motifs from the catalytic domain.
FIGURE 1.

Architecture of the human Shrm SD2-Rock SBD complex. A, signaling cascade and domain diagram of human Shrm2 and human Rock1 proteins. B, positions of peptides identified by mass spectrometry from isolated Shrm-Rock complexes digested with trypsin. The observed Shrm-binding site on Rock is indicated in green. C, ribbon diagram of the hShrm2 SD2-hRock1 SBD binding module. Two views are shown with a 90° rotation in between as indicated. Two SD2 monomers (orange) and one SBD dimer (blue) are shown with important regions within each indicated. RBD, Rho-binding domain; nm-myosin, non-muscle myosin.
Members of the Shroom (Shrm) family of cytoskeletal proteins bind to the coiled coil domain of Rock and are required for epithelial cell shape change (23, 30). Humans and many other vertebrates have four Shrm isoforms (Shrm1–4), and each isoform associates with a distinct population of F-actin, driving its subcellular localization and subsequent function (31, 32). Shrm family members share similar domain architecture with two highly conserved domains called Shrm domain 1 (SD1) and Shrm domain 2 (SD2) that bind F-actin and Rock, respectively (22, 23, 31, 33) (see Fig. 1A). Sequences at the N terminus are more diverse but, in combination with SD1, appear to modulate subcellular localization (31, 33–35).
The Shrm-Rock interaction module is conserved from Drosophila to humans and has been shown to play a role in the development of multiple tissues including the neural tube (1, 22), gut (36, 37), eye (38, 39), and vasculature (40). Shrm-Rock interactions recruit Rock to Shrm-associated regions of the cytoskeleton where Rock activity can drive specific and localized changes to the cytoskeletal architecture and cellular morphology (23, 30, 33). Finally, the importance of the Shrm-Rock interaction module has been established in mice where a single point mutation in the Shrm3 SD2 has been shown to disrupt Rock binding and result in neural tube defects (21).
Structural data exist for many domains within Shrm and Rock including the Shrm-binding domain (SBD) of human Rock1 (41), which forms a parallel coiled coil, consistent with previous structural data from the RhoA-binding and central regions of Rock (42, 43). Biochemical analysis identified residues important for Shrm binding and found them to be positioned on opposing faces of the Rock coiled coil (41). Surprisingly, the structure of the SD2 domain from Drosophila Shrm revealed that this conserved domain adopted a three-segmented, antiparallel coiled coil with mutational analysis indicating two Rock-binding sites located ∼80 Å apart (44). The two observed structures therefore appeared inadequate to describe a molecular mechanism for the formation of the Shrm-Rock binding module.
Here, we present crystal structures of human Shrm2 SD2 determined independently and in a complex with human Rock1 SBD. The structure of the Shrm-Rock module reveals two two-segmented, antiparallel coiled coil human Shrm2 SD2 monomers binding to opposing faces of a single parallel coiled coil dimer of human Rock1 SBD. Our models reveal striking oligomeric differences between Drosophila and human Shrm proteins while retaining a similar core structure. We have used this model to identify critical residues responsible for Shrm-Rock recognition both in vitro and in vivo. Lastly, we also demonstrate, using purified components, that the Shrm SD2 can stimulate Rock kinase activity independently of RhoA. These findings indicate that Shrm proteins can both simultaneously recruit and activate Rock in vivo to control cell morphology.
Results
Architecture of the Shrm-Rock Binding Module
To understand the molecular mechanism driving formation of the Shrm-Rock interaction module, we first sought to crystallize and determine the structure of a Shrm-Rock complex. Previous binding data indicated that Rock proteins containing additional coiled coil sequence N-terminal to the SBD bind to Shrm more tightly than the minimal SBD. Therefore, we reasoned that additional contacts between these regions of Rock might be revealed if we utilized larger fragments of Rock. In an effort to identify these potential contacts, we expressed and purified a large portion of human Rock1 (residues 707–946) containing the SBD core (residues 834–913) as well as the SD2 from human Shrm2. The resulting complex was subjected to limited proteolysis, and the progress of digestion was monitored by native and denaturing PAGE. This resulted in the formation of a species that migrated farther when analyzed by native PAGE. This species was extracted from the gel and trypsinized, and the resulting peptides were identified by mass spectrometry (Fig. 1B). In this analysis, we observed enrichment in Rock peptides between amino acid positions ∼750 and 890. Guided by these data, we attempted to crystallize a variety of Shrm-Rock complexes, generating crystals with several variants of Rock. We only obtained suitable diffraction data from crystals of the smallest Shrm-Rock module tested, which contained human Shrm2 SD2 (residues 1427–1610) and Rock1 SBD (residues 834–913). Diffraction data were weak and highly anisotropic as was observed for Rock1 (41). The SD2-SBD structure was refined to 3.57-Å resolution with an Rwork/free of 27.3/28.7% (see “Experimental Procedures” and Table 1 for a complete description of the structure determination process).
TABLE 1.
Data collection and refinement statistics
| Human Shrm2 SD2 | Human Shrm-Rock complex | |
|---|---|---|
| Protein Data Bank code | 5F4Y | 5F5P |
| Data collection | ||
| Space group | C2221 | P212121 |
| Cell dimensions a, b, c (Å) | 90.89, 110.00, 118.86 | 97.59, 133.80, 135.87 |
| Reflections | ||
| Total | 280,990 | 1,327,044 |
| Unique | 8,552 | 21,121 |
| Resolution (Å) | 50.00–3.29 (3.36–3.29)a | 50.00–3.57 (3.63–3.57) |
| Rmerge (%)b | 8.0 | 8.0 |
| I/σI | 23.7 (2.26) | 24.5 (1.72) |
| Completeness (%) | 92.7 (91.8) | 97.8 (97.9) |
| Redundancy | 5.6 | 5.3 |
| Wilson B-factor | 115.4 | 130.0 |
| Refinement | ||
| Resolution (Å) | 14.93–3.29 (3.46–3.29) | 20–3.57 (3.74–3.57) |
| Rworkc/Rfreed (%) | 24.6/29.9 (30.8/39.0) | 27.3/28.7 (51.1/47.9) |
| Number of atoms | ||
| Protein | 5,198 | 15,406 |
| B-factors (Å2) | 125.1 | 149.8 |
| r.m.s.e deviations | ||
| Bond lengths (Å) | 0.003 | 0.002 |
| Bond angles (°) | 0.45 | 0.54 |
| Ramachandran | ||
| Favored (%) | 98.1 | 98.68 |
| Allowed (%) | 1.9 | 1.32 |
| Clash score | 1.54 | 1.49 |
a Values in parentheses are for highest resolution shell.
b Rmerge = (|(ΣI − 〈I〉)|)/(ΣI) where 〈I〉 is the average intensity of multiple measurements.
c Rwork = Σhkl‖Fobs(hkl)| − |Fcalc(hkl)‖/Σhkl|Fobs(hkl)|.
d Rfree represents the cross-validation R factor for 10.1% (Shrm SD2) or 5.3% (Shrm-Rock complex) of the reflections against which the model was not refined.
e Root mean square.
The asymmetric unit contains four copies of each protein, comprising two complete Shrm-Rock modules (module A and module B) (Fig. 1C). Overall, electron density and the resulting model were best for module A (chains A–D), and thus all of our analysis and figures are derived from that module unless noted. The two modules are similar to each other (r.m.s.d. of 0.7 Å over 310 Cα atoms) with each module containing one dimer of Rock SBD with two Shrm SD2 monomers bound on opposing faces (Fig. 1C). Nearly all of the Shrm SD2 residues were observed in our structure with the exception of the hinge region where the quality of the electron density was variable. The dimeric, parallel coiled coil arrangement observed for the Rock SBD is in general agreement with other structures of the central region of Rock (42, 43). However, unlike our previous structure of the Rock SBD (41), residues 895–913 were disordered.
Human Shrm2 SD2 Adopts an Unanticipated Fold
Prior to this study, the only structural data for a Shrm SD2 domain was that of the SD2 domain from Drosophila Shrm (44). In this structure, an extensive array of protein-protein interactions between Drosophila SD2 molecules supported the formation of a novel three-segmented, antiparallel coiled coil dimer with internal symmetry (44) (Fig. 2A). The structure of human Shrm2 SD2 observed in the SD2-SBD complex, however, is clearly monomeric (Figs. 1C and 2A), forming four helices (A–D) that come together to form two segments of coiled coil, one containing helices A and D with helices B and C forming the second coiled coil segment. A region called the “elbow” forms a bend between the two segments, and an eight-residue stretch in the middle of the SD2, called the “hinge,” forms a turn that allows the protein to fold back on itself, forming the B/C segment. The resulting fold is similar to both halves of the Drosophila SD2 structure (average r.m.s.d. of 1.7 Å over 168 Cα atoms) (Fig. 2A) but lacks significant structural similarity to any protein in the structural database. The positions of previously identified mutants that disrupted homodimerization in Drosophila Shrm SD2 are conserved in human Shrm2 SD2 where they now form interactions that stabilize the A/D and B/C segments. Electron density in the hinge region is of variable quality; however, density for backbone residues can be observed for several of the SD2 molecules (Fig. 2B). The hinge spans human Shrm2 SD2 residues Leu-1519 to Pro-1525, a region previously termed the “symmetry point” as it was located directly in between the two structurally identical halves of Drosophila SD2 (44) (Fig. 2A). The hinge sequence is absent in Drosophila Shrm and may restrict its ability to form a B/C segment in the manner that human Shrm SD2 does.
FIGURE 2.
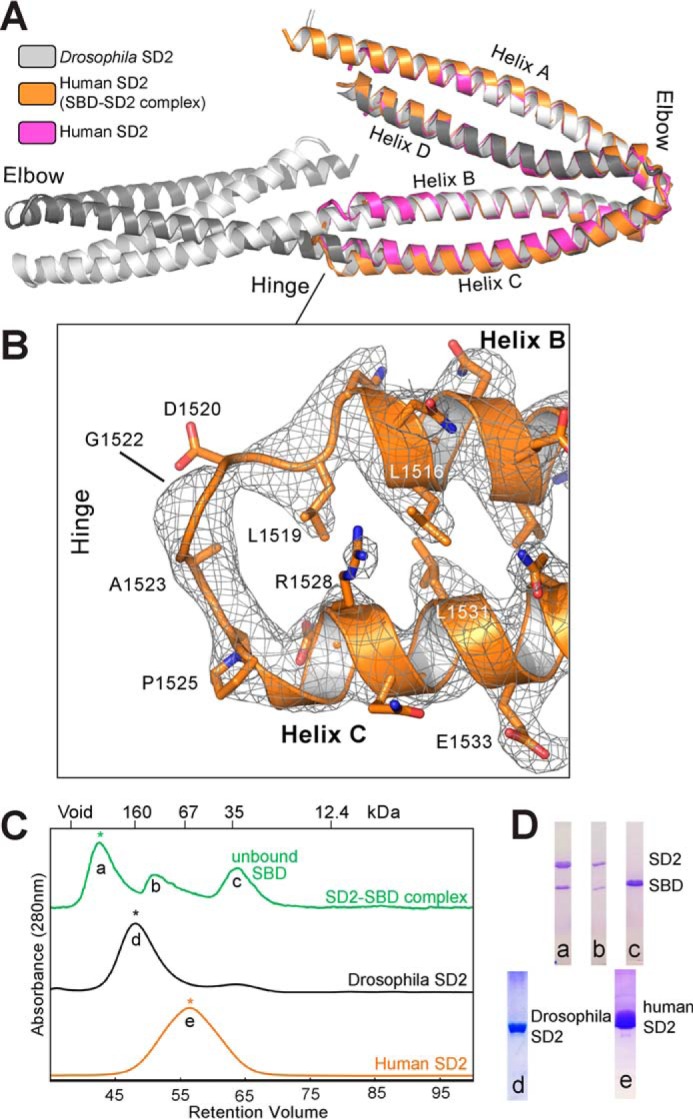
Structural similarities and oligomeric differences between the Drosophila Shrm and Human Shrm2 SD2 domains. A, crystal structures of Drosophila and human Shrm SD2 domains superposed over the SD2 domain from the Shrm-Rock complex. Both human SD2 domains are highly similar to either half of the Drosophila SD2 structure. B, electron density (2Fo − Fc contoured at 1.0σ) of the hinge region in human Shrm2 SD2 from the Shrm-Rock complex. C, indicated species of human Shrm SD2, Drosophila Shrm SD2, and the human Shrm-Rock complex were resolved by size exclusion chromatography using a Sephacryl S200 column. The elution of globular molecular mass standards are indicated for comparison. Homogenous species with Gaussian profiles indicated by an asterisk were used in structural studies. D, SDS-PAGE showing the composition of the species a–e.
One significant difference between the two Shrm structures is the presence of Rock SBD in the structure of our complex. We therefore sought to compare human Shrm2 and Drosophila Shrm SD2 proteins in the absence of Rock. First, we subjected purified SD2 domain from Drosophila Shrm, human Shrm2, and the Shrm-Rock complex to size exclusion chromatography (Fig. 2C). As described previously, we observed two species when performing size exclusion chromatography with Drosophila SD2, a major species that we observed to be a dimer in our crystals (44) and a second minor peak, which was presumed to be monomeric. We also observed two species for the Shrm-Rock complex with the major species being significantly larger than the SD2 domain from either human or Drosophila. We observed only a single species for human Shrm2 SD2 with a retention volume that indicates a smaller radius of gyration under the same conditions, consistent with a smaller monomeric species rather than the extended, dimeric conformation of Drosophila SD2. In all cases, the apparent molecular weights were significantly higher than expected presumably due to the extended nature of these proteins.
To ascertain whether there was a conformational change that resulted upon Rock binding and might explain the difference in SD2 conformation, we crystallized and determined the structure of human Shrm2 SD2 in the absence of Rock, refining against native data to 3.3-Å resolution with an Rwork/free of 24.6/29.9% (Fig. 2A and Table 1). We observed that, in the absence of Rock, human Shrm2 SD2 adopts a monomeric fold, which is very similar to human SD2 in complex with Rock (r.m.s.d. of 0.9 Å over 147 Cα). Although the electron density for the hinge region was once again of variable quality, we were able to fit the hinge sequence into the electron density in one of the two molecules in the asymmetric unit, demonstrating that this feature is characteristic of the human Shrm2 SD2 fold. From these data, we conclude that, in contrast to Drosophila Shrm, the SD2 from human Shrm2 is a monomer and that formation of the 1:2:1 (Shrm-Rock2-Shrm) arrangement we observed for the human Shrm-Rock complex does not require any dramatic conformational changes within the SD2.
The Molecular Basis for Shrm-Rock Recognition in Vitro
The four Shrm-Rock interfaces within the asymmetric unit bury an average of 887.2 Å2 of surface area. Calculating shape complementarity for each interface yielded values ranging from 0.54 to 0.61, which is consistent with values for the Rock SBD dimer interface (0.56) as well many other protein-protein interfaces, particularly those that have multiple binding interfaces (45). Given that the Shrm-Rock interaction is conserved throughout metazoans, we hypothesized that the interface would be enriched in highly conserved and invariant residues. To test this, we generated a multiple sequence alignment of the Shrm2 SD2 (23 sequences) and the Rock1 SBD (33 sequences). Mapping sequence conservation onto the surface of both proteins revealed a large patch of residues with >95% identity within our alignment that cluster near the N terminus of the Rock SBD. This conserved patch also correlates with the location of the Shrm-Rock binding interface observed in our structure (Fig. 3). Sequence conservation within the Shrm2 SD2 is more extended with smaller patches existing on both faces of the molecule. The largest patch of residues with >95% identity in our alignment nevertheless corresponds with the observed binding interface. It also includes residue Arg-1508, a position that is conserved in all SD2 and when mutated in Shrm3 results in neural tube defects in a mouse model (Fig. 3) (21).
FIGURE 3.

The conserved SD2-SBD interface. Conservation from a multiple sequence alignment of 23 Shrm or 33 Rock1 proteins was mapped onto a surface representation of each protein. Residues with over 95% identity within this alignment are colored orange for Shrm (left box), and residues over 85% identical (light blue) and over 95% identical (blue) are indicated on the Rock1 surface (lower right box). The boundaries of the observed binding interface are outlines for both proteins. A ribbon diagram of the complex is shown for reference.
To demonstrate that the Shrm-Rock recognition we observed in our crystal is the biologically relevant interface, we designed several single point mutations to probe the SD2-SBD interface and identify residues critical to binding. We generated mutant proteins in the context of fluorescent fusions of Shrm SD2 (His10-mRuby2-1427–1610) and Rock SBD (His10-Clover-707–946) (46). We assayed the ability of purified Shrm2 SD2 variants (L1501A, L1548A, and K1487A) to bind Rock1 SBD by native PAGE. Leu-1501 and Leu-1548 are both buried within the binding interface (Fig. 4, A and B), and alanine substitutions at those positions abolish binding (Fig. 4E). Lys-1487 is predicted to interact with Rock Glu-862 at the periphery of the binding interface where it makes one of a relatively small number of hydrogen bonds observed within the interface (Fig. 4A). Despite its peripheral location, the K1487A mutant also abolishes binding (Fig. 4E).
FIGURE 4.

The molecular basis for Shroom-Rock recognition. A and B, ribbon diagram of the Shrm-Rock interface featuring important residues as sticks. Residues previously shown to mediate Shrm-Rock interactions are indicated in green, additional residues shown to mediate interactions as a part of this study are colored yellow, and the position Arg-1508 shown to mediate neural tube defects in mice is shown in magenta. Selected hydrogen bonding interactions within the interface are indicated with black dotted lines. C and E, mutants in the Shrm-Rock interface block complex formation. Wild-type or mutant Shrm SD2 (mRuby-SD2) or Rock1 SBD (residues 707–946 fused to Clover) were mixed as indicated, and their ability to form a complex was assayed by native PAGE, imaged at 460 or 630 nm, and presented as a false colored overlay. D, WT and mutant Rock1 (residues 707–946) proteins were subjected to limited proteolysis using 0.025% trypsin, and samples were taken at the indicated time points. In this assay, the mutant proteins behaved similarly to wild type. F, limited proteolysis for Shrm SD2 (residues 1427–1610) mutants was performed as in D.
We generated Rock interface mutations (Y851A, F852A, L855A, Q859A, and E862A) in a similar manner, once again testing them for complex formation by native PAGE. Here, we observed that F852A and L855A substitutions in the hydrophobic center of the interface cannot support complex formation, whereas substitutions of residues in the periphery (Y851A, Q859A, and E862A) had little to no effect on binding in this assay (Fig. 4C). The effect of substituting Glu-862 in Rock was surprising as it was observed hydrogen bonding with the critical Shrm residue Lys-1487 in our structure. Previous studies have demonstrated that an alanine triple mutant in Rock1, K857A/T858A/Q859A, was unable to bind Shrm3. These results indicate that either the hydrogen bond between Rock Gln-859 and Shrm Asn-1551 is, on its own, not essential for the Shrm-Rock interaction in vitro; that Lys-857 and Thr-858 contribute significantly to binding; or that these three residues contribute more to Shrm3 binding than Shrm2. The purified proteins used in this assay were subjected to limited proteolysis using 0.025% trypsin (Fig. 4, D and F) and in all cases were found to behave like wild type, indicating that the indicated substitutions had no effect on overall structure. Together, these data are consistent with our previous biochemical analysis performed in the absence of structural information on the binding interface (21, 30, 41, 44) and pinpoint residues within both Shrm and Rock that are critical for complex formation in vitro.
Changes in the Shrm-Rock Interface Block Shrm-mediated Regulation of the Cytoskeleton
We next sought to determine whether single point mutants that did not support binding in vitro would be functional within a cellular context. To do this, we substituted human Shrm2 SD2 or the SD2 with selected point mutants into a modified pCS2 vector containing an endolyn targeting sequence (amino acids 1–187) (47) fused to amino acids 1372–1572 of mShrm3. This system allows us to target the SD2 to the apical surface of cells where we have previously shown that mouse Shrm2 and Shrm3 SD2 motifs are both necessary and sufficient to cause apical constriction in polarized epithelial cells (23, 31). Using this system, we demonstrate that human Shrm2 SD2 can also elicit apical constriction similarly to mouse Shrm3 SD2 (Fig. 5, B and C) and consistent with apical constriction assays using mouse Shrm2 SD2 (31). Introducing the K1487A, L1501A, or L1548A variant into this assay, we found that all three proteins are stably expressed and targeted to the apical surface but are unable to trigger apical constriction to any appreciable degree (Fig. 5, D–F). Thus, we conclude that the Shrm-Rock interface that we observed in our crystal structure is critical for both binding to Rock in vitro and for SD2-mediated changes in cellular morphology in vivo.
FIGURE 5.

Shrm SD2 and Rock SBD point mutations disrupt binding in vitro. A–F, MDCK cells were transiently transfected on Transwell filters with the indicated constructs and stained to detect Shrm (green) and ZO-1 (red). Scale bars represent 10 μm. Images are projections of 0.5-μm confocal sections. G–L, MDCK cells were transiently co-transfected on Transwell filters with Myc-Rock (residues 681–942) and the indicated constructs and then stained to detect Shrm (green) or Myc (red). Scale bars represent 10 μm. Images are projections of 0.5-μm confocal sections. ap., apical surface; ba., basolateral surface; hSD2, human SD2.
To ensure that the loss of apical constriction observed in our mutants is due to a defect in Shrm-mediated recruitment of Rock, we co-transfected MDCK cells with both the endolyn-tagged Shrm variants and a Myc-tagged Rock SBD (residues 681–942). We stained these transfected cells to determine the localization of both Shrm and Rock proteins. Although wild-type mShrm3 SD2 and human Shrm2 SD2 can recruit Rock to the apical surface of MDCK cells (Fig. 5, H and I), Shrm mutants K1487A, L1501A, and L1548A as well as a mShrm3 construct lacking the SD2 domain cannot (Fig. 5, G and J–L). We conclude that the phenotype observed in Fig. 5, D–F, is due to the loss of Shrm-mediated Rock recruitment to the apical plasma membrane.
Shrm SD2 Can Directly and Independently Activate Rock Kinase
Intramolecular autoinhibitory interactions between the N-terminal kinase domain and C-terminal regions of Rock are believed to hold Rock in an “inactive” state in which only basal levels of activity can be measured (24, 25). It has been established that binding of Shrm localizes Rock to various subcellular locales where Rock activity drives cytoskeletal rearrangement (23, 31). However, it has remained unclear whether the primary role of Shrm-Rock interactions was to govern Rock localization or whether Shrm also possesses the ability to relieve Rock autoinhibition directly. To address whether human Shrm2 SD2 can function as a direct Rock activator, we performed an on-bead kinase assay (48) using purified SD2 domains and His10-tagged full-length Rock1 protein expressed in and purified from COS7 cells. Rock1 kinase was immobilized onto beads and washed thoroughly to remove any potential activating proteins (48). We then assayed the ability of Rock1, in the presence and absence of human Shrm2 SD2, to phosphorylate a purified substrate consisting of amino acids 658–714 of MYPT, which contains the phosphorylation site at Thr-696 (13). Rock activity was monitored via Western blotting using an α-pMYPT antibody and normalized to Rock1 signal. As shown in Fig. 6, Rock1 kinase has basal activity for the MYPT substrate in the absence of activators and is stimulated by the addition of the canonical Rock activator arachidonic acid (50 μm). This activity is nearly abolished in the presence of the Rock-specific inhibitor Y-27632 as reported previously (29, 49) (Fig. 6A).
FIGURE 6.

Shrm SD2 can directly activate Rock1 kinase. A, magnetic nickel-nitrilotriacetic acid resin coated with full-length (FL) Rock1 were incubated in a kinase assay containing MYPT. Samples were analyzed by Western blotting using antibodies to pMYPT and Rock1. B, the effect of the Shrm2 SD2 variant L1501A on Rock activation. pMYPT levels were normalized to Rock1 enzyme signal and Rock1 activity was compared with the Rock1 alone basal level and reported as the mean with error bars representing the standard deviation from five independent replicates. A student t-test was used to calculate p-values with statistically significant values indicated with an asterisk.
We then asked whether wild-type and mutant Shrm2 SD2 domains could also activate Rock. In this assay, the addition of wild-type Shrm2 SD2 at a concentration of 1.5 μm resulted in a 3.5-fold stimulation of Rock activity over basal levels, consistent with RhoA-mediated stimulation of Rock activity (28). Addition of L1501A mutant Shrm2 SD2 reduced the levels of stimulation to 1.6-fold, indicating that the Shrm-mediated stimulation we observed is specific for the Shrm-Rock binding interface visualized crystallographically. These data demonstrate that Shrm is a direct activator of Rock, and thus its role in regulating changes to the actomyosin network extends beyond localization and recruitment of Rock. Together with the cellular assays described earlier, we conclude that the Shrm SD2, through the Shrm-Rock binding module, elevates pMYPT levels and thereby levels of active non-muscle myosin at the apical surface. Through this mechanism, the Shrm SD2 domain both regulates Rock activity and positions it properly within the cell, generating localized forces that lead to changes in cellular morphology.
Discussion
Structure of the Shroom-Rock Complex
The interaction between Shrm family proteins and Rock is critical for the development of many tissues, but the molecular basis for this interaction is not fully understood. We have crystallized and determined the structure of the Shrm-Rock binding module in an effort to identify significant interactions driving complex formation. Consistent with previous results, the SBD of Rock is a parallel coiled coil dimer when bound to Shrm2 SD2 (41, 42). The human Shroom2 SD2, however, is monomeric both when crystallized independently and in the context of a Shrm-Rock complex. Interestingly, our model indicates that two SD2 proteins are capable of interacting with the SBD coiled coil at the same time, and we did not observe any potentially cooperative interactions between SD2 molecules. Previous quantitative measurements of binding affinity have shown that longer versions of the Rock coiled coil region (amino acids 707–946) bind Shrm SD2 with higher affinity than the minimal SBD described here (41), suggesting that additional Rock residues not contained within our structure may mediate contacts within the Shrm SD2 domain. We expect these additional contacts to be made through conserved surfaces on the SD2. We note that our structure reveals three such surfaces on the SD2 domain that are not occupied by the Rock SBD, although the function of these is currently unknown (Fig. 3). One, containing residue Lys-1582, is located in a groove between the A/D and B/C segments adjacent to the observed Rock-binding site. The second site, containing Glu-1513, is located on the opposite face of the B/C segment from Arg-1508 that houses several highly conserved and surface-exposed residues, suggesting that it might mediate a protein-protein interaction. The third site is the conserved patch containing Leu-1440 and Gln-1602, located on the A/D segment. Substitutions in this patch did not block Shrm-Rock complex formation or the ability to support apical constriction (44), suggesting that they do not play a primary role; however, these assays are qualitative, and thus we cannot exclude the possibility that this surface mediates a minor Shrm-Rock contact that improves Shrm-Rock affinity. Another possibility is that these surfaces on Shrm mediate interactions with other proteins. To date, besides Rock, only Scribble has been suggested to bind the SD2 motif of Shrm2. Interestingly, this interaction appears to be mediated by the phosphorylation of Scribble by Rock (50), implying that the Scribble recognition surface on the SD2 domain should both accommodate and specifically recognize this post-translational modification.
Oligomeric Differences between Drosophila and Human Shrm SD2
Previous structural data revealed that the SD2 domain of Drosophila Shroom adopts a unique three-segmented, antiparallel coiled coil dimer fold (44). This is a striking difference from the two crystal structures presented here that both capture human Shrm2 SD2 domain in a monomeric state. It is possible that the fold captured in the Drosophila Shrm SD2 structure is that of an inactive, unbound conformation of Shrm SD2 that changes when the domain associates with its binding partner. The Drosophila Shrm SD2 conformation could also be the result of unique stresses present in crystallization reactions, or the precise nature of the protein construct used facilitated an extreme domain swapping event. Finally, an evolutionary difference between the two proteins might explain these differences. Importantly, although the domains differ in their oligomeric state, the locations of key residues (Leu-1467, Leu-1509, and Lys-1453 in Drosophila Shrm SD2) within the Rock-binding site are conserved in both SD2 proteins. It remains to be determined whether human Shrm is capable of dimerization, but we propose that it interacts with the SBD of Rock as a monomer.
The Role of the Shrm-Rock Signaling Module in Rock Regulation
When its activity is not needed, Rock is believed to reside in an autoinhibited state in which its C-terminal PH/C1 domains directly bind to its N-terminal kinase domain (24, 51). Recent electron microscopy data on full-length Rock2 have sought to challenge this view, suggesting that instead of autoinhibition Rock regulation is spatially organized within the cell at a distance governed by the length of its coiled coil region (52). This is in contrast to existing data (24, 51, 53) demonstrating protein-protein interactions between C-terminal regulatory sequences and the kinase domain. Furthermore, binding events within the regulatory regions of Rock have been demonstrated to alter Rock activity. It is possible that the inability to observe autoinhibition or stimulation of Rock2 activity reflects a difference in the regulatory mechanisms between Rock1 and Rock2. Mutational data suggest that phosphorylation of the activation loop is unlikely to be relieving a regulatory barrier for activity (52). Alternatively, it is possible that the Sf9-purified Rock2 used in that study was trapped in an activated state by the presence of a co-purifying activator or that the phosphorylation events observed within the C-terminal sequences of Rock2 at positions 1352–1353 prevent autoinhibitory interactions. It will be interesting to see whether Rock2 activity can be stimulated in a Shrm-mediated manner. We predict that it can be as important residues within the Rock1-Shrm interface are conserved in Rock2. However, Rock2 may use an entirely different regulatory mechanism.
We have previously shown that Rock binding to RhoA is not essential for Shrm-induced apical constriction of MDCK cells (41). However, this does not eliminate the possibility of cooperation between Shrm and RhoA for regulating Rock in other biological process. The binding sites in Rock for Shrm and RhoA are adjacent but do not overlap. It will be interesting to test whether there are cooperative interactions between Shrm and RhoA that influence binding of either of these partners to Rock as the available crystal structures suggest that simultaneous binding is reasonable (41, 42). It remains to be determined what effect this might have on kinase activation.
Conservation of critical residues within the already highly conserved SD2 domain makes it an attractive prediction that all Shrm proteins retain the ability to activate Rock directly as we have shown here. Internal cleavage by caspase-3 and granzyme B in Rock1 and Rock2, respectively, also activate Rock (18, 19) but through a different mechanism. It is interesting to note that these cleavage sites are located C-terminal to the Shrm-binding site on Rock. This would suggest that Shrm proteins could still play a role in regulating Rock distribution even under conditions in which the kinases themselves are activated by cleavage. This would add an additional layer of Shrm-mediated regulation of Rock activity during certain in vivo processes.
Many studies have aimed to understand the mechanisms of Rock activation and autoinhibition in an effort to design therapeutic drugs to down-regulate Rock activity. The widespread role Rock plays as a cytoskeletal regulator makes it both an attractive and challenging target for drug design, so information on the specific molecular mechanisms of Rock regulation is immensely valuable. One such study has already shown the usefulness of an inhibitor of the Shroom3-Rock2 interaction in stimulating axon outgrowth during repair of neural tissue (54). We anticipate that the structural information provided by our investigation will inform on more targeted drug development for this highly conserved interaction.
Experimental Procedures
Protein Expression and Purification
Coding sequences for human Rock1 (residues 834–913) and human Shrm2 (residues 1427–1610) were amplified by PCR and cloned into pET151-D/Topo (Invitrogen). Human Shrm2 SD2 (residues 1427–1610) was expressed in BL21(DE3)-Gold cells (Agilent) using autoinduction media (55). Cells were harvested by centrifugation and lysed by homogenization. Lysates were cleared by centrifugation at 30,000 × g, and proteins were purified using nickel affinity purification (Qiagen) followed by TEV protease digestion and a second round of nickel affinity purification to remove the His10 tag. The protein was then further purified via anion exchange and size exclusion chromatography. The same strategy was also used to purify human Rock1 (residues 834–913).
Shrm SD2 and Rock SBD proteins used in the native PAGE binding assays were expressed as fusions containing N-terminal His10-Ruby (Shrm SD2) or His10-Clover (Rock) tag using a modified pET28a vector. The proteins were purified as described above, omitting the TEV cleavage and second nickel column steps to preserve the fluorescent tag as needed.
Generation of SBD-SD2 Complex for Crystallography
Human Shrm2 SD2 and Rock1 SBD were purified, then mixed at a 2:1 Rock:Shrm ratio, and dialyzed into buffer containing 50 mm NaCl and 20 mm Tris at pH 8.0 overnight. Following dialysis, size exclusion chromatography was performed, and peak fractions containing homogenous SBD-SD2 complex were identified by both SDS- and native PAGE and concentrated to 5.3 mg/ml prior to crystallization.
Crystallization
Crystals of the SD2-SBD complex were generated via the vapor diffusion method using a well solution containing 20.5% PEG 3350 and 233 mm ammonium chloride. Rectangular prism-shaped crystals were cryoprotected in mother liquor supplemented with 30% glycerol and then flash frozen in liquid nitrogen prior to data collection. Human Shrm2 SD2 crystals were generated using a well solution containing 20% PEG 3350, 200 mm magnesium chloride, and 100 mm Tris, pH 8.5, and cryoprotected in a similar manner.
Structure Determination
Diffraction data were collected at the National Synchrotron Light Source at Brookhaven National Laboratory on Beamline X25 using a Pilatus 6M detector. Integration, merging, and scaling of the diffraction data were performed using HKL2000 (56).
Crystals of the Shrm-Rock complex belong to the space group P212121. Diffraction was weak and anisotropic, extending to only 5.3 Å along the a* axis (UCLA Anisotropy server (58)). Based on this analysis as well as values for completeness and CC* (57), we chose to truncate the data at 3.57-Å resolution. Phases were determined through molecular replacement using search models generated from the structure of the SBD as well as fragments of Drosophila SD2 (Protein Data Bank codes 4L2W and 3THF, respectively). Refinement was carried out in PHENIX (59) using rigid body and group B-factor refinement with non-crystallographic symmetry restraints. Model building and positional refinement were performed in Coot (60). Crystals of human Shrm2 SD2 belong to space group C2221 and were only mildly anisotropic. Phases were determined using molecular replacement using a monomer of SD2 from the Shrm-Rock binding module as a search model. Rigid body and B-factor refinement were performed in PHENIX (59), and manual model building was performed within Coot (61, 62). MolProbity (62) was used to assess model validity, and images were generated using PyMOL (63).
Native PAGE Binding Assays
Binding reactions containing 25 μm SD2 and 15 μm SBD were carried out in 8% glycerol, 125 mm NaCl, 1 mm β-mercaptoethanol, and 25 mm Tris, pH 8.0, and incubated at room temperature for 5 min before being resolved by 10% native PAGE and quantified using ImageJ (64).
Limited Proteolysis and Mass Spectrometry
Complexes of Shrm and Rock were isolated and truncated by limited proteolysis using trypsin. After isolation of gel slices representing the truncated complexes from native gels, the slices were subject to in-gel trypsin digestion to generate tryptic peptides, which were identified by tandem MS using a ThermoFisher Orbitrap mass spectrometer (Fred Hutchinson Cancer Research Center Proteomics Facility). Peptide Prophet (65) was used to evaluate the validity of peptide identifications, and we eliminated peptides with a score <0.85 (<2.5% false discovery rate).
Cell Culture and in Vivo Cytoskeletal Assay
COS7 cells were grown in DMEM supplemented with 10% FBS, l-glutamine, and penicillin/streptomycin. Transient transfection was performed in 6-well plates using 300,000 cells, 2 μg of DNA, and 5 μl of Lipofectamine/well. Transfected cells were incubated for 24 h before processing. MDCK cells were grown in a similar manner using Eagle's minimum essential medium.
pCS2 endolyn-Shrm chimeric constructs were generated as reported previously (23). For analysis of the cytoskeleton, cells were washed three times in PBS, fixed in 100% MeOH for 5 min, washed three times with PBST, and then incubated with primary antibody in PBST for 1 h. Antibodies used were UPT132 (1:200), anti-ZO-1 (1:200) (Promega), and 9E10 (1:100) (ATCC). Cells were washed three times in PBST and then incubated for 1 h with secondary antibody (Alexa Fluor 488- and 568-conjugated antibodies; 1:400). Cells were washed three times with PBST and then finally mounted on slides using Immuno-Fluore mounting medium (MP Biomedicals).
Kinase Activity Assay
Full-length human Rock1 coding sequences were cloned into a modified pCAG-myc vector containing a His10 tag and TEV protease cleavage site. This plasmid was then transfected into COS7 cells using Lipofectamine. 24 h post-transfection, cells were rinsed with PBS and then resuspended in lysis buffer containing 50 mm HEPES, 300 mm NaCl, 5 mm β-mercaptoethanol, 10% glycerol, 20 mm imidazole, and 0.05% Nonidet P-40. Lysates were cleared by centrifugation, and the soluble fraction was subjected to nickel affinity chromatography using magnetic nickel beads (Thermo Scientific). Full-length Rock1 protein was retained on the nickel resin and washed with lysis buffer and with lysis buffer containing 1 m NaCl prior to washing into reaction buffer before being used in activity assays.
Kinase activity assays were carried out by incubating 1 μl of magnetic nickel beads coated with full-length Rock1 in a 25-μl reaction using a reaction buffer consisting of 20 mm Tris-HCl, pH 7.0, 1 mm EDTA, 1 mm EGTA, 5 mm MgCl2, and 5 μm MYPT with and without SD2, Y-27632, or 50 μm arachidonic acid. Reactions were started with the addition of 10 μm ATP and then incubated at 30 °C for 10 min. Substrate phosphorylation was evaluated by Western blotting using a primary antibody against pMYPT (Millipore). Quantification was performed using ImageJ (64).
Author Contributions
J. K. Z., J. D. H., and A. P. V. conceived and designed the experiments. J. K. Z., J. H. M., S. H., and R. G. G. performed the experiments. A. H. collected diffraction data for the crystallographic studies, which were processed and refined by J. K. Z. and A. P. V. J. K. Z., J. H. M., R. G. G., J. D. H., and A. P. V. analyzed the biochemical and cell-based data. J. K. Z., J. D. H., and A. P. V. wrote the paper.
Acknowledgments
We thank Dr. Andrea Berman, Dr. Joel Rosenbaum, and Ryan Corbo for helpful discussions and advice and Doowon Lee for technical assistance. Operations at the National Synchrotron Light Source are supported by the Department of Energy, Office of Basic Energy Sciences, and by the National Institutes of Health.
This work was supported by National Institutes of Health Grant GM097204 (to J. D. H. and A. P. V.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5F4Y and 5F5P) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MRLC
- myosin regulatory light chain
- MYPT
- myosin phosphatase
- Rock
- Rho-associated kinase
- Shrm
- Shroom
- PH
- pleckstrin homology
- SD
- Shrm domain
- SBD
- Shrm-binding domain
- r.m.s.d.
- root mean square deviation
- mShrm
- mouse Shrm
- MDCK
- Madin-Darby canine kidney
- TEV
- tobacco etch virus
- PBST
- PBS with Tween 20
- pMYPT
- phosphorylated MYPT.
References
- 1. Hildebrand J. D., and Soriano P. (1999) Shroom, a PDZ domain-containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell 99, 485–497 [DOI] [PubMed] [Google Scholar]
- 2. Clarkson E., Costa C. F., and Machesky L. M. (2004) Congenital myopathies: diseases of the actin cytoskeleton. J. Pathol. 204, 407–417 [DOI] [PubMed] [Google Scholar]
- 3. Hall A. (2009) The cytoskeleton and cancer. Cancer Metastasis Rev. 28, 5–14 [DOI] [PubMed] [Google Scholar]
- 4. Malinova D., Fritzsche M., Nowosad C. R., Armer H., Munro P. M., Blundell M. P., Charras G., Tolar P., Bouma G., and Thrasher A. J. (2016) WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts. J. Leukoc. Biol. 99, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spence E. F., and Soderling S. H. (2015) Actin out: regulation of the synaptic cytoskeleton. J. Biol. Chem. 290, 28613–28622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brandt R. (2001) Cytoskeletal mechanisms of neuronal degeneration. Cell Tissue Res. 305, 255–265 [DOI] [PubMed] [Google Scholar]
- 7. Ikebe M., Koretz J., and Hartshorne D. J. (1988) Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J. Biol. Chem. 263, 6432–6437 [PubMed] [Google Scholar]
- 8. Moussavi R. S., Kelley C. A., and Adelstein R. S. (1993) Phosphorylation of vertebrate nonmuscle and smooth muscle myosin heavy chains and light chains. Mol. Cell. Biochem. 127–128, 219–227 [DOI] [PubMed] [Google Scholar]
- 9. Sellers J. R., Eisenberg E., and Adelstein R. S. (1982) The binding of smooth muscle heavy meromyosin to actin in the presence of ATP. Effect of phosphorylation. J. Biol. Chem. 257, 13880–13883 [PubMed] [Google Scholar]
- 10. Somlyo A. P., and Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 11. Dent P., MacDougall L. K., MacKintosh C., Campbell D. G., and Cohen P. (1992) A myofibrillar protein phosphatase from rabbit skeletal muscle contains the beta isoform of protein phosphatase-1 complexed to a regulatory subunit which greatly enhances the dephosphorylation of myosin. Eur. J. Biochem. 210, 1037–1044 [DOI] [PubMed] [Google Scholar]
- 12. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., and Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 13. Kawano Y., Fukata Y., Oshiro N., Amano M., Nakamura T., Ito M., Matsumura F., Inagaki M., and Kaibuchi K. (1999) Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 147, 1023–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., and Kaibuchi K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 15. Fukata Y., Amano M., and Kaibuchi K. (2001) Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 22, 32–39 [DOI] [PubMed] [Google Scholar]
- 16. Somlyo A. V., Bradshaw D., Ramos S., Murphy C., Myers C. E., and Somlyo A. P. (2000) Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem. Biophys. Res. Commun. 269, 652–659 [DOI] [PubMed] [Google Scholar]
- 17. Coleman M. L., Sahai E. A., Yeo M., Bosch M., Dewar A., and Olson M. F. (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3, 339–345 [DOI] [PubMed] [Google Scholar]
- 18. Sebbagh M., Hamelin J., Bertoglio J., Solary E., and Bréard J. (2005) Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J. Exp. Med. 201, 465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sebbagh M., Renvoizé C., Hamelin J., Riché N., Bertoglio J., and Bréard J. (2001) Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3, 346–352 [DOI] [PubMed] [Google Scholar]
- 20. Renault M. A., Roncalli J., Tongers J., Thorne T., Klyachko E., Misener S., Volpert O. V., Mehta S., Burg A., Luedemann C., Qin G., Kishore R., and Losordo D. W. (2010) Sonic hedgehog induces angiogenesis via Rho kinase-dependent signaling in endothelial cells. J. Mol. Cell. Cardiol. 49, 490–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Das D., Zalewski J. K., Mohan S., Plageman T. F., VanDemark A. P., and Hildebrand J. D. (2014) The interaction between Shroom3 and Rho-kinase is required for neural tube morphogenesis in mice. Biol. Open 3, 850–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haigo S. L., Hildebrand J. D., Harland R. M., and Wallingford J. B. (2003) Shroom induces apical constriction and is required for hingepoint formation during neural tube closure. Curr. Biol. 13, 2125–2137 [DOI] [PubMed] [Google Scholar]
- 23. Hildebrand J. D. (2005) Shroom regulates epithelial cell shape via the apical positioning of an actomyosin network. J. Cell Sci. 118, 5191–5203 [DOI] [PubMed] [Google Scholar]
- 24. Amano M., Chihara K., Nakamura N., Kaneko T., Matsuura Y., and Kaibuchi K. (1999) The COOH terminus of Rho-kinase negatively regulates rho-kinase activity. J. Biol. Chem. 274, 32418–32424 [DOI] [PubMed] [Google Scholar]
- 25. Chen X. Q., Tan I., Ng C. H., Hall C., Lim L., and Leung T. (2002) Characterization of RhoA-binding kinase ROKα implication of the pleckstrin homology domain in ROKα function using region-specific antibodies. J. Biol. Chem. 277, 12680–12688 [DOI] [PubMed] [Google Scholar]
- 26. Feng J., Ito M., Kureishi Y., Ichikawa K., Amano M., Isaka N., Okawa K., Iwamatsu A., Kaibuchi K., Hartshorne D. J., and Nakano T. (1999) Rho-associated kinase of chicken gizzard smooth muscle. J. Biol. Chem. 274, 3744–3752 [DOI] [PubMed] [Google Scholar]
- 27. Ishizaki T., Maekawa M., Fujisawa K., Okawa K., Iwamatsu A., Fujita A., Watanabe N., Saito Y., Kakizuka A., Morii N., and Narumiya S. (1996) The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 15, 1885–1893 [PMC free article] [PubMed] [Google Scholar]
- 28. Matsui T., Amano M., Yamamoto T., Chihara K., Nakafuku M., Ito M., Nakano T., Okawa K., Iwamatsu A., and Kaibuchi K. (1996) Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 15, 2208–2216 [PMC free article] [PubMed] [Google Scholar]
- 29. Araki S., Ito M., Kureishi Y., Feng J., Machida H., Isaka N., Amano M., Kaibuchi K., Hartshorne D. J., and Nakano T. (2001) Arachidonic acid-induced Ca2+ sensitization of smooth muscle contraction through activation of Rho-kinase. Pflugers Arch. 441, 596–603 [DOI] [PubMed] [Google Scholar]
- 30. Nishimura T., and Takeichi M. (2008) Shroom3-mediated recruitment of Rho kinases to the apical cell junctions regulates epithelial and neuroepithelial planar remodeling. Development 135, 1493–1502 [DOI] [PubMed] [Google Scholar]
- 31. Dietz M. L., Bernaciak T. M., Vendetti F., Kielec J. M., and Hildebrand J. D. (2006) Differential actin-dependent localization modulates the evolutionarily conserved activity of Shroom family proteins. J. Biol. Chem. 281, 20542–20554 [DOI] [PubMed] [Google Scholar]
- 32. Hagens O., Ballabio A., Kalscheuer V., Kraehenbuhl J. P., Schiaffino M. V., Smith P., Staub O., Hildebrand J., and Wallingford J. B. (2006) A new standard nomenclature for proteins related to Apx and Shroom. BMC Cell Biol. 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bolinger C., Zasadil L., Rizaldy R., and Hildebrand J. D. (2010) Specific isoforms of drosophila shroom define spatial requirements for the induction of apical constriction. Dev. Dyn. 239, 2078–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dye D. E., Karlen S., Rohrbach B., Staub O., Braathen L. R., Eidne K. A., and Coombe D. R. (2009) hShroom1 links a membrane bound protein to the actin cytoskeleton. Cell. Mol. Life Sci. 66, 681–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoder M., and Hildebrand J. D. (2007) Shroom4 (Kiaa1202) is an actin-associated protein implicated in cytoskeletal organization. Cell Motil. Cytoskeleton 64, 49–63 [DOI] [PubMed] [Google Scholar]
- 36. Grosse A. S., Pressprich M. F., Curley L. B., Hamilton K. L., Margolis B., Hildebrand J. D., and Gumucio D. L. (2011) Cell dynamics in fetal intestinal epithelium: implications for intestinal growth and morphogenesis. Development 138, 4423–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Plageman T. F. Jr, Zacharias A. L., Gage P. J., and Lang R. A. (2011) Shroom3 and a Pitx2-N-cadherin pathway function cooperatively to generate asymmetric cell shape changes during gut morphogenesis. Dev. Biol. 357, 227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fairbank P. D., Lee C., Ellis A., Hildebrand J. D., Gross J. M., and Wallingford J. B. (2006) Shroom2 (APXL) regulates melanosome biogenesis and localization in the retinal pigment epithelium. Development 133, 4109–4118 [DOI] [PubMed] [Google Scholar]
- 39. Plageman T. F. Jr., Chung M. I., Lou M., Smith A. N., Hildebrand J. D., Wallingford J. B., and Lang R. A. (2010) Pax6-dependent Shroom3 expression regulates apical constriction during lens placode invagination. Development 137, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farber M. J., Rizaldy R., and Hildebrand J. D. (2011) Shroom2 regulates contractility to control endothelial morphogenesis. Mol. Biol. Cell 22, 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mohan S., Das D., Bauer R. J., Heroux A., Zalewski J. K., Heber S., Dosunmu-Ogunbi A. M., Trakselis M. A., Hildebrand J. D., and Vandemark A. P. (2013) Structure of a highly conserved domain of Rock1 required for Shroom-mediated regulation of cell morphology. PLoS One 8, e81075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dvorsky R., Blumenstein L., Vetter I. R., and Ahmadian M. R. (2004) Structural insights into the interaction of ROCKI with the switch regions of RhoA. J. Biol. Chem. 279, 7098–7104 [DOI] [PubMed] [Google Scholar]
- 43. Tu D., Li Y., Song H. K., Toms A. V., Gould C. J., Ficarro S. B., Marto J. A., Goode B. L., and Eck M. J. (2011) Crystal structure of a coiled-coil domain from human ROCK I. PLoS One 6, e18080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohan S., Rizaldy R., Das D., Bauer R. J., Heroux A., Trakselis M. A., Hildebrand J. D., and VanDemark A. P. (2012) Structure of Shroom domain 2 reveals a three-segmented coiled-coil required for dimerization, Rock binding, and apical constriction. Mol. Biol. Cell 23, 2131–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lawrence M. C., and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 46. Lam A. J., St-Pierre F., Gong Y., Marshall J. D., Cranfill P. J., Baird M. A., McKeown M. R., Wiedenmann J., Davidson M. W., Schnitzer M. J., Tsien R. Y., and Lin M. Z. (2012) Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 9, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ihrke G., Bruns J. R., Luzio J. P., and Weisz O. A. (2001) Competing sorting signals guide endolyn along a novel route to lysosomes in MDCK cells. EMBO J. 20, 6256–6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riento K., and Ridley A. J. (2006) Inhibition of ROCK by RhoE. Methods Enzymol. 406, 533–541 [DOI] [PubMed] [Google Scholar]
- 49. Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Tamakawa H., Yamagami K., Inui J., Maekawa M., and Narumiya S. (1997) Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994 [DOI] [PubMed] [Google Scholar]
- 50. Amano M., Hamaguchi T., Shohag M. H., Kozawa K., Kato K., Zhang X., Yura Y., Matsuura Y., Kataoka C., Nishioka T., and Kaibuchi K. (2015) Kinase-interacting substrate screening is a novel method to identify kinase substrates. J. Cell Biol. 209, 895–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Craft J. W. Jr, Zhang H., Charendoff M. N., Mindrebo J. T., Schwartz R. J., and Briggs J. M. (2013) Associations between the Rho kinase-1 catalytic and PH domain regulatory unit. J. Mol. Graph. Model. 46, 74–82 [DOI] [PubMed] [Google Scholar]
- 52. Truebestein L., Elsner D. J., Fuchs E., and Leonard T. A. (2015) A molecular ruler regulates cytoskeletal remodelling by the Rho kinases. Nat. Commun. 6, 10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leung T., Chen X. Q., Manser E., and Lim L. (1996) The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 16, 5313–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dickson H. M., Wilbur A., Reinke A. A., Young M. A., and Vojtek A. B. (2015) Targeted inhibition of the Shroom3-Rho kinase protein-protein interaction circumvents Nogo66 to promote axon outgrowth. BMC Neurosci. 16, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 56. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 57. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Strong M., Sawaya M. R., Wang S., Phillips M., Cascio D., and Eisenberg D. (2006) Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 8060–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 62. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.8 Schrödinger, LLC, New York [Google Scholar]
- 64. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]