

Abstract
The ubiquitous second messenger cAMP mediates signal transduction processes in the malarial parasite that regulate host erythrocyte invasion and the proliferation of merozoites. In Plasmodium falciparum, the central receptor for cAMP is the single regulatory subunit (R) of protein kinase A (PKA). To aid the development of compounds that can selectively dysregulate parasite PKA signaling, we solved the structure of the PKA regulatory subunit in complex with cAMP and a related analogue that displays antimalarial activity, (Sp)-2-Cl-cAMPS. Prior to signaling, PKA-R holds the kinase's catalytic subunit (C) in an inactive state by exerting an allosteric inhibitory effect. When two cAMP molecules bind to PKA-R, they stabilize a structural conformation that facilitates its dissociation, freeing PKA-C to phosphorylate downstream substrates such as apical membrane antigen 1. Although PKA activity was known to be necessary for erythrocytic proliferation, we show that uncontrolled induction of PKA activity using membrane-permeable agonists is equally disruptive to growth.
Keywords: cyclic AMP (cAMP), malaria, plasmodium, protein kinase A (PKA), protein structure
Introduction
Despite significant inroads in mitigating their toll on human health, Plasmodium parasites, the causative agents of malaria, remain deadly human pathogens (1, 2). Their complex parasitic life cycle alternates between mosquito and vertebrate hosts; however, it is the asexual intraerythrocytic stage with which disease is associated. To ensure timely and successful transmission and propagation, the parasite must constantly monitor its surroundings to coordinate its development. At the molecular level, signal transduction pathways orthologous to those found in other eukaryotes are responsible for dynamic post-transcriptional phosphorylation reactions that control key developmental proteins (3). The phylum Apicomplexa to which Plasmodium belongs is evolutionarily distant to most commonly studied organisms (4), and significant phylogenetic diversity thus separates the Plasmodium kinome from other systems studied. This diversity can be exploited however if inhibitory compounds can be developed that selectively block parasite regulatory enzymes while leaving orthologous human variants unaffected (5).
As in other eukaryotic species, Plasmodium developmental pathways utilize the diffusible second messenger 3′-5′-cyclic adenosine monophosphate (cAMP), the main effector for which is the cAMP-dependent kinase, protein kinase A (6). cAMP-dependent signaling is generally activated when ligands bind to membrane-bound receptors that go on to activate intracellular adenylate cyclases, yielding a localized rise in the cAMP concentration and hence activation of PKA. Plasmodium falciparum PKA activity is instrumental to a number of molecular mechanisms, including merozoite egress, motility and red blood cell invasion (7, 8), schizogony (7), and the progression from schizonts to invasive merozoites (9). Specifically, during merozoite invasion PfPKA has been implicated in regulating microneme secretion of erythrocyte host-recognition receptors (10), and once inside the host cell, PKA signaling pathways influence parasite growth and differentiation (11) possibly by initiating the formation of new permeation pathways (12). Following differentiation into gametocytes, cAMP-dependent kinase activity has also been found to regulate the host cell membrane stiffness to prevent splenic clearance (13).
PKA orthologues are ubiquitous among eukaryotes and are composed of a catalytic kinase subunit (PKA-C) in association with a regulatory subunit (PKA-R) containing two cAMP-binding sites. Compounds that specifically inhibit the Plasmodium PKA-C kinase domain are known to block parasite development (14). Less is known about the action of the PfPKA-R subunit and whether this can also be therapeutically targeted. Mammalian PKA has been extensively studied and serves as a useful frame of reference from which to understand the distinct P. falciparum PKA system (15). In mammals, the regulatory PKA-R subunit is a highly dynamic molecular switch (16, 17) that serves as a competitive inhibitor of the catalytic subunit, holding it inactive in the absence of cAMP but rapidly releasing its inhibitory check as the cAMP concentration rises. Mammals have four R-subunit isoforms within their PKA systems, which are split into related type I and type II pairs (RIα and Iβ, IIα, and IIβ, respectively) (18). Each of these isoforms are functionally distinct and influence their respective holoenzyme cellular localization, sensitivity to activation, and regulatory feedback mechanisms (19). In contrast, the unicellular Plasmodium parasites contain a single PfPKA-R subunit whose features appear to be a hybrid of the mammalian subtypes (15). Primary structure analysis indicates that Plasmodium PKA-R has its own loosely structured N-terminal sequence, but as in mammalian R subunits, the C terminus contains two consecutive cyclic nucleotide-binding domains (CBDs).3 These CBDs share ∼35% sequence identity with their mammalian equivalents and ∼54% identity with non-plasmodium Apicomplexans.
The CBDs themselves are an ancient signaling module often found in enzymes regulated by cAMP or cGMP (20), and their fold consists of a conserved 8-stranded β-barrel along with three or more accessory α-helices. Cyclic nucleotides typically insert phosphate first into the barrel's core and in doing so induce structural rearrangements of the α-helices to induce the second messenger response. In human PKA holoenzymes, the catalytic kinase subunit is held inactive through its association with the apo dumbbell-shaped PKA-R subunits, a shape derived from the two CBDs lying on either side of a rigid connecting helix (21). This dumbbell shape exposes two inhibitory regions as follows: a pseudosubstrate sequence N-terminal to CBD1 that locks into the kinase domain's active site, and the helix between the two CBDs that binds to the large lobe of the kinase domain and prevents catalytically necessary “breathing” motions. These mutual inhibitory effects are lost when cyclic nucleotide binding causes the regulatory domain's dumbbell shape to collapse into a more compact form that masks the kinase domain-interacting regions facilitating subunit dissociation (17). The catalytic domain is thus freed from its allosteric restraint allowing it to transmit and amplify the cAMP signal to multiple downstream targets via its kinase activity. We sought to assess to what extent this molecular activation mechanism is conserved in PfPKA-R. Furthermore, we examine the effects on parasite growth of membrane-permeable compounds that cause PKA dysregulation through premature activation of the holoenzyme.
Results
Expression of the P. falciparum PKA Regulatory Subunit
The P. falciparum 3D7 genome contains a single 441 amino acid PfPKA-R subunit (PlasmoDB PF3D7_1223100) known to associate with a cAMP-inducible kinase activity (12). The PfPKA-R sequence encompasses two predicted cyclic nucleotide binding domains, CBD1 (residues 166–294) and CBD2 (residues 310–441) (Fig. 1). We cloned, expressed, and purified four PfPKA-R constructs spanning either one or both CBDs (residues 2–441, 69–441, 141–441 and 297–441). The constructs PfPKA-R.141.441 and PfPKA-R.297.441 expressed with 5–10 mg/liter yields and were thus favored for further study (Fig. 1).
FIGURE 1.

Domain structure of Plasmodium PKA regulatory subunit. Diagram of P. falciparum PKA regulatory (PfPKA-R) gene showing regions expressed for functional and structural analysis. Left, PfPKA-R has two cAMP binding regions CBD1 and CBD2, inhibitory sequence (IS), dimerization domain (DD), and predicted N-myristoylation (Gly-2) and palmitoylation (Cys-5) sites for membrane anchorage. Four regions of PfPKA-R were expressed of which two generated high yield (right, 141–441/297–441).
Following purification of the PfPKA-R constructs, UV-visible light absorbance spectra indicated the presence of a 260-nm absorbance peak presumed to be bound nucleotide scavenged from the Escherichia coli lysate. To liberate these PfPKA-R-bound nucleotides, we performed a standard soluble expression protocol but retained PfPKA-R.141.441 on the immobilized metal affinity column, washed with progressively higher concentrations of urea-containing buffers, and slowly returned to a standard buffer prior to protein elution and further purification. Absorbance measurements indicated that ∼50 column volumes of 8 m urea were required to liberate all bound nucleotide. This refolded apo-PfPKA-R.141.441 sample represents a conformation of PfPKA-R with two empty CBD-binding sites primed for re-association with the PKA-C (22). This on column refolding reduced total protein yield (∼1 mg/liter) from that of a standard soluble purification (∼5 mg/liter). We append the prefix “apo-” to distinguish between PfPKA-R purifications that required nucleotide dissociation (kinase and affinity assays and drug-bound structures).
Recombinant PfPKA-R Is Functionally Active
To verify that the recombinant apo-PfPKA-R.141.441 was active, we aimed to investigate whether it could block PKA-C activity and thus inhibit PfPKA-driven phosphorylation. Unfortunately, we were unable to bacterially express soluble recombinant PfPKA-C and instead utilized late-stage P. falciparum schizont lysates as our kinase source. The cytoplasmic tail of apical membrane antigen 1 (AMA1), which is a highly validated target of PfPKA (8, 23, 24), was used as our kinase substrate. The AMA1-tail is phosphorylated by PfPKA at amino acid Ser-610, and this event is required for merozoite invasion of erythrocytes by P. falciparum (8). For our assays, the 56-amino acid AMA1-tail was expressed as a fusion with glutathione S-transferase (GST) and immobilized in the wells of an ELISA microplate (Fig. 2A). Schizont lysate was added to each well with varying concentrations of cAMP to stimulate PfPKA phosphorylation and/or recombinant PfPKA-R to inhibit phosphorylation. Following the reaction, phosphorylation of Ser-610 was detected with a highly specific antibody raised to a synthetic Ser(P)-610 phosphopeptide (Fig. 2A) (23). In the absence of supplementary apo-PfPKA-R or cAMP, phosphorylation of AMA1 was quite low, indicating minimal levels of PfPKA-C activity in the parasite lysates (Fig. 2B). This activity was, however, strongly boosted by addition of supplementary cAMP. Furthermore, addition of our apo-PfPKA-R fragment strongly suppressed AMA1 phosphorylation in a concentration-dependent manner presumably because it allosterically inhibited native PfPKA-C (Fig. 2B). Importantly, this inhibitory activity could be reversed in a concentration-dependent fashion by increasing amounts of supplementary cAMP (Fig. 2B).
FIGURE 2.
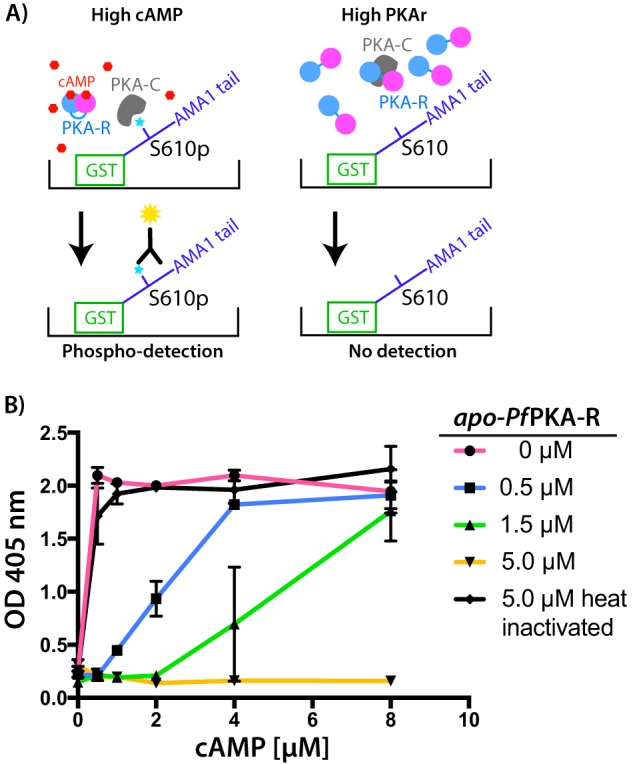
Recombinant apo-PfPKA-R inhibits PKA-mediated phosphorylation. A, illustration of the ELISA-based phosphorylation assay using the recombinant tail of AMA1, a validated PfPKA target. B, native phosphorylation activity of PfPKA-C (indicated by A405 nm) can be efficiently inhibited by addition of recombinant apo-PfPKA-R in a concentration-dependent manner. This effect can be efficiently reversed by addition of supplementary cAMP and is due specifically to the apo-PfPKA-R as the inhibitory effect of the fragment is lost upon heat denaturation (heat-inactivated). Each data point and whiskers are the mean/standard deviation of two technical replicates. Graph shown is representative of three independent biological replicates.
To ensure that this inhibitory effect was specifically due to the action of the recombinant protein and not a nonspecific contaminant, we heated the recombinant protein and repeated the assay. The heat-denatured protein was poorly efficient at inhibiting AMA1 phosphorylation when compared with the untreated protein validating that inhibition was due to recombinant apo-PfPKA-R (Fig. 2B). Our recombinantly produced regulatory subunit thus associates with the parasite's catalytic subunit influencing its activity.
Structure of the PfPKA-R Subunit
Broad screens identified conditions facilitating protein crystal growth for the PfPKA-R.141.441 and 297.441 constructs in complex with cAMP, with X-ray diffraction data then obtained to 2.0 and 1.1 Å resolution, respectively. Initial electron density maps were calculated using phases obtained via molecular replacement (see under “Materials and Methods” for details and Table 1 for data collection and refinement statistics).
TABLE 1.
Data collection and refinement statistics
Values in parentheses refer to the highest resolution bin. r.m.s.d. is root mean square deviation.
| PKAR.141.441, cAMP-bound | PKAR.297.441, cAMP-bound | PKAR.297.441, (Sp)-2Cl-cAMPS-bound | |
|---|---|---|---|
| Data collection | |||
| Space group | P21212 | P212121 | P43212 |
| Wavelength | 0.9537 | 0.9537 | 0.9537 |
| Cell dimensions | |||
| a, b, c (Å) | 78.7, 103.8, 104.2 | 39.3, 71.8, 106.5 | 64.2, 64.2, 195.7 |
| Resolution (Å) | 46.6–2.00 (2.11–2.00) | 42.8–1.15 (1.21–1.15) | 37.3–2.40 (2.53–2.40) |
| Rpima | 3.1 (55.5) | 4.1 (44.0) | 3.8 (31.6) |
| I/σ1 | 12.4 (1.6) | 9.7 (1.8) | 11.2 (2.2) |
| Completeness (%) | 100.0 (100.0) | 97.2 (89.5) | 99.9 (100.0) |
| No. of unique observations | 58,452 (8419) | 104,403 (13799) | 16,863 (2389) |
| Multiplicity | 9.0 (9.1) | 7.9 (5.9) | 11.2 (11.6) |
| Refinement statistics | |||
| Rfactor b (%) | 24.5 (36.0) | 19.8 (29.0) | 25.1 (32.3) |
| Rfreec (%) | 28.5 (37.8) | 21.8 (33.2) | 30.7 (38.1) |
| No. of atoms | |||
| Protein | 4534 | 2469 | 2392 |
| Water (cAMP) | 36 (4) | 356 (2) | 10 (2 cAMPS; 5I−) |
| Ramachandran plot (%) | |||
| Most favored | 97.1 | 98.7 | 96.1 |
| Allowed region | 2.9 | 1.2 | 3.9 |
| Outlier | 0.0 | 0.0 | 0.0 |
| B-Factors (Å2) | |||
| Protein | 66.1 | 22.0 | 93.3 |
| r.m.s.d. bonds (Å) | 0.009 | 0.012 | 0.014 |
| r.m.s.d. angles (°) | 1.26 | 1.31 | 2.2 |
| Protein Data Bank code | 5KBF | 5K8S | 5T3N |
a Rpim = Σhkl (1/(N − 1))1/2 Σi|Ihkl, i − 〈Ihkl〉|/Σhkl 〈Ihkl〉.
b Rfactor = (Σ‖Fo| − |Fc‖)/(Σ|Fo|), for all data except as indicated in Footnote c.
c 5% of data was used for the Rfree calculation.
Our final model of PfPKA-R.141.441 has clear electron density for all structural elements of the two CBD modules (Fig. 3, A and B). These are accorded standard nomenclature for the fold, which comprises eight β-strands (β1–8), three major helices (αA, αB, and αC), a short helical turn overlaying the phosphate moiety (αP), and an N-terminal helical element (αN). Fig. 3C delineates the residues within each structural element, with those from CBD2 referenced using a suffixing quotation mark (e.g. αA′). The asymmetric unit of the PfPKA-R.141.441 crystals contained two protomers related by non-crystallographic symmetry whose structural differences are limited to different side chain conformations within surface residues. During the refinement of PfPKA-R, the unbiased omit maps showed cAMP molecules clearly occupying each domain's nucleotide-binding site (Fig. 4, A and B). Thus, our structure represents the dissociated nucleotide-bound state of the regulatory subunit.
FIGURE 3.

Structure of PfPKA-R. A, schematic representation of the mammalian PKA holoenzyme and its activation mechanism. Catalytic domain depicted in gray and CBD1 and CBD2 domains in blue and purple. B, schematic representation of the nucleotide-bound PfPKA-R structure. β-Strands are colored tan, CBD1 α-helices blue, and CBD2 α-helices purple. All secondary structure elements are labeled within each CBD. The two bound cAMP molecules are shown in red. C, sequence alignment of the residues within the respective CBD domains for the PKA-R subunits from P. falciparum, Plasmodium vinckei vinckei, Toxoplasma gondii, and Homo sapiens RIα and RIIβ. The conserved PBC arginines are highlighted in black, the αP glutamates and the tyrosine with which they interact in red, and the inhibitory sequence in pink.
FIGURE 4.

Structure of the cAMP-binding sites within PfPKA-R. A, residues within the cAMP-binding site of CBD2 as observed within the 1.1 Å PfPKAR.297.441 structure. The nucleotide is clearly bound as seen in the Fo − Fc omit map contoured at 3.4σ. The main residues contributing to the binding site are displayed in stick representation. B, CBD1 cAMP-binding site as observed within the 2.0 Å PfPKAR.141.441 structure. Tyr-319 is highlighted. C, backbone alignment of PfPKA-R with the orthologous cAMP-bound structure of B. taurus PKA-R (green). The bound nucleotides of BtPKA-R are colored light brown, and the PfPKA-R is depicted using the color scheme of Fig. 2B. D, potential mobility of the helical elements of the CBD2 domain can be seen when comparing an alignment of its structure ascertained from the different crystals.
In CBD1 (Fig. 4B), a conserved phosphate binding cassette (PBC) pincers the cyclophosphate moiety of cAMP between the N terminus of the αP helix and a conserved arginine (Arg-268), a position that also facilitates the formation of an H-bond between the cAMP hydroxyl group and Glu-259. The residues making up these PBC interactions are highly conserved across different CBD modules with equivalent residues present in CBD2 (αP′, Arg-386′, and Glu-377′, respectively). The cyclic nucleotide adenosine base projects outward where it is clamped on one side by hydrophobic residues originating from the β4-β5 loop (Ile-236 and Leu-248 in CBD1 and Ile-366′ in CBD2). On the other side, the base is clamped by a hydrophobic capping residue, the origin of which is an important point of distinction between the CBD1- and CBD2-binding sites; within CBD2, the cAMP′ molecule is extensively buried by the subunit's C-terminal helix that lies across it. This αC′ helix is positioned such that Tyr-424′ forms a critical base-stacking interaction (Fig. 4A), which along with Val-427′ and Leu-428′ creates an enclosed hydrophobic cAMP-binding site within this domain. C-terminal to αC′ a kink in the backbone structure brings Cys-437′ and Ile-438′ back into proximity where they interact with cAMP near its N1-C2-N3 edge. The CBD2-binding site is thus hydrophobic, masks the nucleotide from solvent, and is entirely self-contained (the structure of PfPKA-R.297.441 is essentially CBD2 by itself in complex with cAMP, Fig. 4D). In contrast the CBD1-binding site is far more open and solvent-exposed; Tyr-319′ provides an analogous base-stacking hydrophobic cap (Fig. 4B), but importantly this residue actually originates from the N terminus of the CBD2 domain's αA′ helix. The αA′ helix also contributes a second charged H-bond interaction between Asp-317′ and the N6 amide. Direct allosteric inter-domain interactions are thus required to form the nucleotide-binding site of CBD1.
Comparison with Other Known PKA Regulatory Subunits
We compared the malarial PfPKA regulatory subunit structure with those of mammalian and yeast orthologues as follows: Bos taurus PKA type RIα (16, 25), Rattus norvegicus type RIIβ (26), and Saccharomyces cerevisiae Bcy1 (27). Nucleotide-free holoenzyme structures of the mammalian orthologues are also known (17, 21) and were instrumental in elucidating the molecular basis of PKA signaling. When RIα and RIIβ are in complex with their shared kinase domain, their structures align with their tandem CBDs lying separated on either end of their respective straight conjoined αB/C helices. This rigid helix, along with their αA′ and αB′ helices, forms a binding site for the large lobe of the catalytic domain (21). cAMP binding collapses this dumbbell shape, with the previously co-linear αB/C helix bending to reduce its length by up to 20%. This moves the CBD modules into contact with each other masking their αA′ and αB′ KD-interacting surfaces, creating the active cAMP-bound form of their regulatory subunit, a form also reported for yeast Bcy1 (27). Although the two mammalian orthologues adopt similar structures in their kinase-associated state (17, 21), the CBD modules collapse together utilizing different orientations upon cAMP binding. In RIα, cAMP induces a bend in the previously rigid helix between αB and αC, although in RIIβ the bend occurs midway along αC. This distinction is important as it influences how their respective CBD2 modules rotate in the cAMP-collapsed state. In RIα the N terminus of αA′ moves to become proximal to the cAMP base of CBD1 (Fig. 5A) (17), providing a hydrophobic capping tryptophan that plays a role similar to that of Tyr-319′ in PfPKA-R (Fig. 5B). In contrast, the bending mechanism of RIIβ moves the N terminus of its αB′ helix next to the adenine ring, capping it with an underlying arginine (21) in an orientation perpendicular to that of RIα (Fig. 5C). Our cAMP-bound structure of PfPKA-R mimics RIα most closely (see Fig. 4C). Plasmodium and RI regulatory subunits both interact with the adenine ring of cAMP via aromatic N-terminal αA′ residues, whereas the RII isoforms utilize an αB′ arginine (Fig. 5C).
FIGURE 5.

Comparison of the cAMP-binding domains in the known structures of PKA orthologues. A, schematic representation of the A- or CBD1 cAMP-binding site from B. taurus-type RIα (25). Helices within CBD1 are colored aquamarine, and those within CBD2 teal, and the bound cAMP is shown in red. The interdomain interfacial hydrophobic capping tryptophan contributed by αA′ is shown in stick representation. Similar representations are presented for the CBD1 domains of PfPKA-R (B) and RnPKA-RIIβ (C) (26). Similar representations of the CBD2 domains are presented for BtPKA-RIα (D). E, PfPKA-R; F, RnPKA-RIIβ. Yellow arrows indicate the orthologue structures to which each malarial PfPKA-R binding domain is most similar.
PfPKA-R also has RIIβ-like features as each have a long αC′ helix that extensively buries their respective CBD2 nucleotides. In PfPKA-R and RIIβ, the αC′ helix is twice the length of that of RIα. In PfPKA-R, the first cAMP-binding site could thus be considered more similar to that of mammalian RI isoforms, and the second site could be considered more reminiscent of RII (Fig. 5). Both nucleotide-binding sites are important for signaling within the vertebrate PKA regulatory subunits (22). In their holoenzyme state, the CBD2 or “B” domain acts as a “gatekeeper,” lying exposed and ready for binding due to residues within the nucleotide-clamping αC′ helix being disordered (21, 28). In PfPKA-R the αC′ helix displays a degree of flexibility even in its cAMP-bound state, and if we align the CBD2 domains from all protomers of our structures, there are slight relative movements within αC′ residues (Fig. 4D). This appears to arise from slight deviations in backbone hydrogen bond distances from those of an ideal α helix for the clamping residue Tyr-424′.
PfPKA-R shares ∼39% sequence identity with mammalian type I and II isoforms but also displays a similar level of identity with the yeast regulatory subunit Bcy1 (27). Despite sharing similar levels of sequence identity with Bcy1 and both being unicellular, PfPKA-R demonstrates closer structural homology to the mammalian regulatory subunits than that of yeast.
Kinase Domain Interaction Site
Overall, the PfPKA-C subunit shares ∼50% sequence identity with the mammalian catalytic subunits. However, residues orthologous to those at the intersubunit docking interface of the mammalian holoenzymes are almost stringently conserved. The cAMP-bound structures of RI and RII subunits are distinct, yet their holoenzyme forms overlay. As the cAMP-bound forms of PfPKA-R and mammalian RIα also overlay (root mean square deviation of 2.7 Å over 227 Cα), it appears likely that the more stringently conserved holoenzyme docking interface will also closely resemble that of the mammalian isoforms.
In mammalian PKA's an inhibitory pseudosubstrate region extends away from the CBDs and docks into the kinase domain (17, 21). Upon cAMP binding and dissociation, these residues contract against CBD1 creating a “parked” conformation stabilized via an arginine within the preceding linker interacting with negatively charged surface presented by αB, an interaction that facilitates PKA activation (29). cAMP-bound PfPKA-R adopts an equivalent parked intermolecular linker, but the interactions stabilizing its association with CBD1 are distinct, with αB being primarily contacted by hydrophobic linker residues (Pro-167 and Val-169).
The sequence of the PfPKA-R pseudosubstrate residues (145KKRLSVS151, see Fig. 3C) is most similar to the equivalent region of mammalian RI; they lack the flexible glycines of the RII sequence, and Ser-149 corresponds to the γ-phosphate-interacting serine of RI. Consistent with this assumption, phosphorylation of Ser-149 by PfPKA-C and Ser-151 by PfPKG is known to mitigate the inhibitory action of the PfPKA-R pseudosubstrate region (15). Although present in our construct, we were not able to observe what form these residues take, perhaps indicating that they may be only partially ordered in the dissociated cAMP-bound state.
Cyclic-AMP Affinity Measurements
We developed the purification protocol described for apo-PfPKA-R.141.441 to eliminate scavenging of cyclic nucleotides from the bacterial lysate, representing a conformation of the subunit primed for binding to the catalytic subunit. We next ascertained the binding kinetics of cAMP to this form of the PfPKA-R. Protein sample at ∼25 μm within the cell of a Microcal isothermal titration calorimeter was titrated against sequential injections of cAMP (Fig. 6A).
FIGURE 6.

Isothermal titration calorimetry affinity measurements. A, binding isotherm measured at 295 K for the interaction between cAMP and apo-PfPKA-R.141.441; below is the resulting thermogram obtained from the integrated heats using a one-site binding model. Equivalent experiments for binding to the CBD2 site of apo-PfPKA-R.297.441 are shown for cAMP (B), (Sp)-2-Cl-cAMPS (C), and(Sp)-8-Br-cAMPS (D).
Approximate equilibrium thermodynamics for cAMP binding to PfPKA-R can be determined from the calorimetry data by fitting to the appropriate binding model. cAMP binding resulted in an isotherm that was clearly uniphasic and was able to be fit with a single-site interaction model having ligand stoichiometry of 2.1 ± 0.1. If two binding sites are known to be structurally non-overlapping, a uniphasic isotherm typically results from positive cooperativity between them. This can cause a tendency for the sites within each molecule to saturate together with a single enthalpic change of ΔH1 + ΔH2 (30). In such circumstances, fitting to a single-site interaction model is more robust and provides the binding constant for only the lowest affinity site, in our case yielding a Kd of ∼1 nm (see Table 2) for the tandem CBDs of PfPKA-R. Repeating the experiment for the CBD2 domain in isolation (Fig. 6B and Table 2) indicated the presence of only a single cAMP-binding site with a nearly equivalent Kd of ∼6 nm and is consistent with it being the binding site first occupied by cAMP.
TABLE 2.
Isothermal calorimetry binding constants
| Construct | Ligand | N | Kda | ΔH | ΔS |
|---|---|---|---|---|---|
| nm | kcal mol−1 | cal mol−1 K−1 | |||
| PfPKAR.141.441 | cAMP | 2.1 ± 0.1 | 1.2 ± 0.2 | −18.9 ± 0.6 | −22 ± 2 |
| PfPKAR.141.441 | (Sp)-8-Br-cAMPS | 1.2 ± 0.1 | 1300 ± 850 | −10.5 ± 2 | −8.1 ± 1.1 |
| PfPKAR.141.441 | (Sp)-2-Cl-cAMPS | 2.1 ± 0.1 | 8.9 ± 4 | −11.9 ± 0.4 | −3 ± 2 |
| PfPKAR.297.441 | cAMP | 0.9 ± 0.1 | 6.0 ± 0.6 | −11.1 ± 0.1 | −0.2 ± 0.3 |
| PfPKAR.297.441 | (Sp)-8-Br-cAMPS | ND | ND | ND | |
| PfPKAR.297.441 | (Sp)-2-Cl-cAMPS | 0.9 ± 0.1 | 9.1 ± 11 | −7.1 ± 0.1 | 11.7 ± 0.9 |
a Data were determined at 295 K; ND, none determined.
Inhibition of PfPKA-R Blocks Parasite Proliferation
Differences between the cAMP-binding sites of PfPKA-R and other eukaryotes raised the possibility that the subunit could be chemically exploited to dysregulate PKA signaling and arrest parasite proliferation. To evaluate this, P. falciparum chloroquine-sensitive and -resistant strains 3D7 and W2mef, respectively, were grown in the presence of four membrane-permeable cyclic nucleotide phosphodiesterase-resistant cAMP analogues for 72 h. As a marker for parasite proliferation, the levels of lactate dehydrogenase activity were measured at different drug concentrations to calculate an EC50 for growth. The most potent compound with an EC50 of 3.71 μm was (Sp)-2-Cl-cAMPS followed by (Sp)-8-Br-cAMPS with an EC50 of 11.30 μm for 3D7 parasites (Fig. 7A). The cAMP analogue 6-Bnz-cAMP was only weakly inhibitory at the highest concentrations (Fig. 7A). The generic PKA-C inhibitor H89 was included as a positive control as it had already been demonstrated to inhibit parasite growth (14) and with an EC50 5.76 μm was of similar potency as (Sp)-2-Cl-cAMPS (Fig. 7A). H89, (Sp)-2-Cl-cAMPS, and (Sp)-8-Br-cAMPS blocked the proliferation of W2mef parasites nearly as well as 3D7 demonstrating the compounds are effective against multiple parasite strains (data not shown).
FIGURE 7.

Compounds binding PfPKA-R block parasite proliferation and cause PKA dysregulation. A, four cAMP analogues and the PKA-C inhibitor H89 were assessed for their capacity to block proliferation of the asexual blood stage of P. falciparum 3D7 parasite strain. After 72 h of growth in serial dilutions of the compounds, the levels of the parasite enzyme lactate dehydrogenase were measured at A650 nm, and growth curves are presented. Mean EC50 values and S.E. for three replicates are shown on the right. B–D, cyclic AMP and its analogues can stimulate PKA phosphorylation. The level of phosphorylation of an immobilized AMA1 substrate was detected using an anti-phospho-AMA1 Ser-610 protein and measured using an anti-rabbit HRP conjugate producing a colorimetric change quantified at 405 nm. Parasite lysate provided the source of PKA-C whose activity was stimulated with the compounds indicated. Supplementary recombinant apo-PfPKA-R was added to verify whether this protein was the target of the cAMP analogues whose structures are shown. Each data point and whiskers are the mean and standard deviations of duplicate measurements. Representative graphs of three independent experiments are shown. E, structure of (Sp)-2-Cl-cAMPS-bound PfPKA-R.297.441 is shown; residues capable of forming van der Waals interactions with the 2′-Cl are highlighted with black lines. Coloring and labeling are as used in Fig. 4A, and the Fo − Fc omit map generated during refinement contoured at 3σ is displayed with a green surface representation. For clarity, the 1st half of αC′ is not displayed.
(Sp)-2-Cl-cAMPS Relieves the Allosteric Inhibitory Activity of Pf-PKA-R
To provide evidence that the mechanism of action of the cAMP analogues was through dysregulation of PfPKA-R function instead of through inhibition (31), we sought to determine whether the analogues could reverse the inhibitory effects of supplementary apo-PfPKA-R in in vitro PKA-dependent phosphorylation assays. The phosphorylation assays were performed as in Fig. 2 with parasite lysate providing the kinase source, the tail of AMA1 being the protein substrate and a phospho-Ser-610 antibody being the detector. The growth inhibitory (Sp)-2-Cl-cAMPS and (Sp)-8-Br-cAMPS compounds were tested alongside a cAMP control, and PfPKA-dependent phosphorylation of the AMA1 substrate was reconstituted in their presence (Fig. 7, B–D). If the cAMP analogues act on PfPKA-R directly then they would be expected to alleviate the addition of supplementary apo-PfPKA-R.141.441. Following the addition of 0.5 or 1.5 μm PfPKA-R, high concentrations of (Sp)-2-Cl-cAMPS and cAMP were effective at alleviating the subunit's allosteric inhibitory action with the analogue being as effective as cAMP (Fig. 7, B and D). Interestingly, the second analogue (Sp)-8-Br-cAMPS had difficulty overcoming apo-PfPKA-R supplementation (Fig. 7C). We repeated the isothermal titration calorimetry experiments with both analogues and found that (Sp)-2-Cl-cAMPS bound to the CBD2 domain of PfPKA-R with a Kd of ∼9 nm (Fig. 6C and Table 2) equivalent to that of cAMP. Moreover, it bound effectively to both sites within the tandem CBD construct (Table 2). In contrast, (Sp)-8-Br-cAMPS had no detectable affinity for the CBD2 site of PfPKA-R (Fig. 6D) and only bound the longer construct at a single site, presumably CBD1, with a Kd of 1.2 μm.
(Sp)-2-Cl-cAMPS Binding to CBD2
Finally, we examined the mechanism of (Sp)-2-Cl-cAMPS binding to the CBD2 domain of PfPKA-R. Ligand-bound CBD2 crystals gave the best diffraction but still displayed significant anisotropy (2.4 Å on two axes and 3.8 Å resolution on the c axis). This was sufficient to allow a tentative model to be built and indicated (Sp)-2-Cl-cAMPS binding occurs in an almost identical manner as cAMP (Fig. 7E). The thiophosphate modification is accommodated without obvious changes (Fig. 7E). The additional chlorine on the adenosine's C2 carbon potentially requires a slight shift of the proteins extreme C-terminal residues (437′–441′) to be accommodated (Fig. 7, D and E). Our 1.1 Å cAMP-bound structure would indicate a close contact if the C terminus was to remain entirely stationary, but this movement is relatively minor. The remaining interactions of the binding site, including the base-stacking interaction with Tyr-424′, appear largely unchanged. Residues 437′–441′ are not directly associated with any crystal packing interface.
If we overlay the structures of the other cAMP analogues tested onto our cAMP-bound structures, it indicates that unlike (Sp)-2-Cl-cAMPS they could not be accommodated without more serious structural changes. The hydroxyl group of Tyr-369′ makes a direct van der Waals contact with the C8 carbon and may be the cause for the reduced affinity for 8-bromoadenosine analogues within CBD2. In contrast, there is no obvious hindrance to (Sp)-8-Br-cAMPS binding within the PfPKA-R CBD1 site. During the more extended growth assays, this may be sufficient to effect a reasonable growth arrest (Fig. 7A) yet is insufficient to strongly alleviate the addition of recombinant PfPKA-R within the phosphorylation assay (Fig. 7C). Our structures suggest (Sp)-2-Cl-cAMPS could be accommodated in both CBD sites.
In vertebrate PKA's, 6-Bnz-cAMP is selective for the CBD1 or “A” site, yet our assays suggest it is ineffective at inducing growth arrest via interaction with PfPKA-R (Fig. 7A). It would clearly not be accommodated within CBD2, and a charged H-bond between Asp-317′ and the N6-amide of cAMP may also limit its ability to interact within CBD1.
Discussion
The conserved cAMP-dependent protein kinases are a major regulator of signal transduction that arose prior to the origin of multicellularity in eukaryotes. Modern day unicellular eukaryotes often contain only a single catalytic/regulatory subunit pair, but the increased requirement for signaling complexity in multicellular organisms such as vertebrates appears to have developed hand-in-hand with multicomponent PKA signaling systems forged from gene duplications and subsequent isoform specialization (32). The structures of PKA regulatory subunits from vertebrates and S. cerevisiae have previously been compared (27), with the latter seen to have a novel fungus-specific quaternary structure and allosteric activation mechanism. The structure of the P. falciparum PKA regulatory subunit represents the first from a human pathogen as well as that of a species evolutionary distant from previously studied vertebrate and yeast orthologues (16, 17, 21, 25, 26, 33).
In mammalian PKAs, the two binding sites of the R subunits display positive cooperativity upon cAMP binding. The C-terminal CBD2, or B site, is always exposed and immediately available for nucleotide binding. When this site is occupied, it stabilizes structural changes within CBD1 that drastically increase its affinity for cAMP and promote subunit dissociation and hence activation (22). Our isothermal titration calorimetry data are consistent with PfPKA-R utilizing a similar two-state cooperative binding mechanism that provides an enthalpically driven interaction with nanomolar affinity for cAMP, as in vertebrates (34). Our structure of cAMP-bound PfPKA-R.141.441 is consistent with this and details the interdomain interactions between the CBD modules.
The compact cAMP-bound conformation of PfPKA-R has features reminiscent of both mammalian regulatory subtypes. The allosteric interface that creates the CBD1 nucleotide-binding site resembles that of RI (27). This may be indicative of it being the more ancestral allosteric regulatory architecture, with the orthogonal CBD interaction of RII isoforms and that of Bcy1 perhaps later evolutionary developments. Meanwhile, the self-contained CBD2-binding site of PfPKA-R extensively buries its adenosine base using a large hydrophobic C-terminal helix in a manner similar to that of the RII isoforms yielding a hybrid set of vertebrate features. The differences between the cAMP-binding sites of RI and RII have led others to identify compounds that selectively activate the different mammalian isoforms; cAMP analogues containing N6-substitutions preferentially bind RII, whereas C8-substitutions target type RI (35). The CBD1 domain of PfPKA-R is relatively open within the plane of the base and accordingly may accommodate a number of adenosine-modified cAMP analogues. In contrast, the adenosine base within the CBD2 domain is extensively buried in our structures with C2-modified bases appearing to be most easily accommodated.
We then showed that some membrane-permeable cAMP analogues disrupt P. falciparum growth; thus, although PKA activity is necessary for parasite growth, the timing of that activity is also critical. It would be appealing to identify ligands that more selectively target PfPKA-R than those of their human host. Our (Sp)-2-Cl-cAMPS bound structure highlights one of the unique features of the CBD2 cAMP-binding site in Plasmodium sp., the proximity of Cys-437′ and its interactions near C2 of the adenosine base. Because of the conservation of Cys-437′ among Plasmodium species, it may stand as a useful point-of-difference that could be productively targeted when attempting to create Plasmodium-specific PKA activators. The action of (Sp)-2-Cl-cAMPS on PKA phosphorylation demonstrates that C2 modifications near Cys-437′ are well tolerated and disrupt erythrocytic stage parasite growth at low micromolar concentrations.
Erythrocytic stage P. falciparum growth was effectively inhibited by (Sp)-2-Cl-cAMPS; however, the therapeutic potential of this compound is reduced by its potential to influence human R isoforms. Given the low similarity of PfPKA-R to orthologous human proteins, more specific allosteric agonists could, in theory, be developed. Our current work focuses on direct interference of cAMP sensing, yet the phosphate binding cassettes are a highly conserved part of the fold. Greater specificity may be obtained by targeting compounds to other sites within PfPKA-R such as near the inter-CBD interface or the intermolecular linker N-terminal to CBD1. Moreover, the function of the PfPKA-R N terminus and its essentiality to the parasite remain to be ascertained.
Despite their divergent evolutionary history, the regulatory architecture of PKA signaling in Plasmodium parasites and their hosts bears recognizable similarities. We provide a first glimpse at the molecular details of cAMP signaling in a major human pathogen, and we detail the residues involved in providing allosteric cooperativity between its two CBD domains. Moreover, although compounds blocking the kinase activity of PfPKA-C are known to limit parasite growth (14), we herein demonstrate that uncontrolled premature activation of PfPKA-R is equally disruptive to parasite survival.
Materials and Methods
Protein Cloning and Expression
Synthetic cDNA encoding the P. falciparum 3D7 PKA regulator subunit (PF3D7_1436600) was cloned into pET-28b-based expression vector. Five different constructs were made consisting of PKAR residues 1–441, 69–441, 141–441, 297–441, and 141–297. Recombinant protein was expressed in E. coli BL21(DE3) cells grown for 4 h at 37 °C in Luria Broth containing 34 μm kanamycin before being induced for 4 h with 0.1 mm isopropyl thiogalactopyranoside. Cells were harvested and resuspended in buffer A (20 mm HEPES, pH 7.5, 250 mm NaCl, 1 mm TCEP) with 20 mm imidazole prior to lysis with 1 mg/ml lysozyme and DNase along with 5 min of sonication on ice; cell lysate was clarified by centrifugation and loaded onto immobilized metal affinity column with the bound material and then washed extensively with buffer A before being eluted in buffer supplemented with 400 mm imidazole. After elution from the immobilized metal affinity chromatography column, the hexahistidine tag was removed by incubating at 4 °C overnight with 0.5 mg of human rhinovirus-3c protease. After exchanging into 20 mm HEPES, pH 7.5, 50 mm NaCl, 1 mm TCEP, PfPKA-R was loaded onto a 1-ml HiTrap-SP cation exchange column and eluted under a 25-ml gradient to buffer A with 1 m NaCl. Protein eluted as a single peak and was subsequently loaded onto a size exclusion chromatography column equilibrated with buffer A.
When apo-PfPKA-R was required, the protein was purified as per normal, but after loading onto the immobilized metal affinity chromatography column the bound material was washed extensively with 20 mm HEPES, pH 7.5, 8 m urea, 250 mm NaCl, 1 mm TCEP before slowly exchanging back into buffer A.
Culture of P. falciparum
P. falciparum asexual stage parasites were cultured in human erythrocytes (blood group O+) supplied by the Australian Red Cross Blood Bank at 4% hematocrit in RPMI 1640/HEPES medium supplemented with 0.43% AlbuMAX II lipid-rich bovine serum (Gibco) and 0.18% NaHCO3 as described previously (36). Lysates for phosphorylation were made from late stage schizonts 42–46 h post-invasion at 2% parasitemia. The schizonts were treated with 0.09% saponin in PBS and Complete protease inhibitors (Roche Applied Science) to lyse the membranes of their erythrocytes. The erythrocyte contents, including hemoglobin and human PKA, were removed by washing the parasite cells multiple times in PBS and frozen at −80 °C. Immediately prior to performing phosphorylation reactions, the parasite pellets were thawed in 20 mm Tris, pH 7.4, 1% Triton X-100, 150 mm NaCl, 20 mm MgCl2, 1 mm ATP, 1 mm DTT with PhosSTOP phosphatase inhibitors and Complete protease inhibitors (Roche Applied Science). Before use, the lysates were cleared by centrifugation, and a volume equivalent to 0.2 μl of pelleted saponin-lysed schizonts was used per reaction (see below).
Functional Validation of Recombinant PfPKA-R
Nunc MaxiSorp 96-well ELISA plates were coated with 1 μg/ml GST-PfAMA1 (8) substrate overnight at 4 °C. Coated plates were subsequently blocked in 1% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 1 h prior to initiation of phosphorylation reactions in kinase activity buffer (20 mm Tris, 20 mm MgCl2, 2 mm MnCl2, 1 mm DTT, PhosSTOP (Roche Applied Science)) supplemented with lysates made from P. falciparum late stage schizonts and 100 μm ATP and 0–8 μm cAMP. Recombinant PfPKA-R (5, 1.5, 0.5, or 0 μm) was titrated into the phosphorylation reactions in either an active or heat-inactivated state (70 °C for 15 min prior to placement on ice for 10 min) at 37 °C for 30 min. Following the reaction, the wells were washed three times with TBS and probed with rabbit anti-phospho-PfAMA1Ser-610 antibodies raised to the synthetic peptide SFWGEEKRASpHTTPV phosphorylated on the Ser-610 residue (Genscript). The resulting antisera had been depleted of non-phospho-specific antibodies by depletion against a non-phosphorylated version of the peptide (Genscript). The ELISA plates were washed as before and subsequently probed with goat anti-rabbit-HRP (Abcam) at room temperature/shaking. Following washing, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) was added (30 min/shaking) to develop the assay, and the reaction was stopped by addition of 1% SDS before reading at 405 nm on a spectrophotometer. Absorbance values were plotted in Prism 6 (GraphPad).
Parasite Proliferation Assays
Asexual blood stage P. falciparum parasites were cultured for 72 h in a dilution series of cAMP analogues (Sp)-8-Br-cAMPS, (Sp)-isomer (Calbiochem), 6-Bnz-cAMP (BioLog), and (Sp)-2-Cl-cAMPS, (Sp)-isomer (BioLog). H89 dihydrochloride hydrate was from Sigma. After culturing, lactate dehydrogenase activity was measured using a modified form of the Malstat assay (23, 37).
Protein Crystallization
PfPKA-R.141.441 at 12 mg/ml with 2 mm cAMP was crystallized at 4 °C within a hanging drop vapor diffusion experiment. Protein drops were set in a 1:1 ratio against a reservoir consisting of 20% w/v polyethylene glycol 3350, 0.1 m NaF, and 0.1 m 1,3-bispropane, pH 7.5, with crystals appearing after 7–10 days. Crystals were transferred into a cryoprotectant consisting of reservoir and 250 mg/ml sucrose before being plunged into liquid nitrogen.
PfPKA-R.297.441 at 4 mg/ml with 2 mm cAMP also crystallized at 4 °C in a similar experiment using a reservoir consisting of 22% w/v polyethylene glycol 6000, 0.1 m LiCl, and 0.1 m CH3COONa, pH 4. Apo-PfPKA-R.297.441 without removal of the His tag at 6 mg/ml with 2 mm (Sp)-2-Cl-cAMPS was crystallized at 22 °C over a reservoir consisting of 20% w/v polyethylene glycol 3350, 0.2 m NH4I. Prior to being plunged into liquid nitrogen crystals were transferred into a solution containing 250 mg/ml sucrose as cryoprotectant.
Data Collection and Structure Solution
X-ray diffraction data were collected at the Australian Synchrotron MX1 and MX2 beamlines. Data were integrated and scaled with the programs iMOSFLM and Scala. A trimmed version of the bovine PKA-R subunit (16) was used as a molecular replacement probe in the program Phaser (38). The resulting model was then improved by simulated annealing and rounds of manual building and refinement using the programs COOT (39) and PHENIX (40). The final PfPKA-R.297.441 model has two protomers in the asymmetric unit, with all residues observed along with two molecules of cAMP. The final PfPKA-R.141.441 model consists of two molecules within the asymmetric unit with clear electron density for residues 167–441, four molecules of cAMP, and four tentatively identified N-terminal residues.
Isothermal Titration Calorimetry
Apo-PfPKA-R.141.441 or PfPKA-R.297.441 was purified, concentrated, and finally dialyzed overnight against buffer containing 20 mm HEPES, pH 7.0, 150 mm NaCl, 0.5 mm TCEP. After dialysis, the protein concentration was adjusted to ∼0.026 mm. Cyclic nucleotides used for titration were prepared using the dialysis buffer to a final concentration of ∼0.5 mm. Isothermal titration calorimetry measurements were performed with a MicroCal VP-ITC calorimeter (GE Healthcare) at 25 °C. Nucleotides were injected 50–70 times (1 μl for the first two injections and 2 μl for subsequent injections), with 240-s intervals between injections. Background data obtained from the buffer sample were subtracted before data analysis. All data were fitted using the Origin 7.0 software package (MicroCal, Northampton, MA). Two independent measurements were performed with errors representing the standard deviation of fitted constants.
Author Contributions
D. R. L. performed recombinant protein expression, crystallization, data collection, and solved the X-ray crystal structures. H. E. B. and K. L. H. established and performed the growth and kinase assays. J. R., T. B., B. S. C., D. R. L., and P. R. G. analyzed the data and wrote the manuscript.
Acknowledgments
This work was performed with the help of the staff of the Australian Synchrotron MX1 and MX2 beamlines. We thank the Australian Red Cross Blood Bank for providing human blood.
This work was supported by National Health and Medical Research Council of Australia Project Grant APP1068287 and funding from the Victorian Operational Infrastructure Support Program received by the Burnet Institute. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5KBF, 5K8S, and 5T3N) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CBD
- cyclic nucleotide-binding domain
- TCEP
- tris(2-carboxyethyl)phosphine
- (Sp)-8-Br-cAMPS
- 8-bromoadenosine-3′,5′-cyclic monophosphorothioate
- 6-Bnz-cAMP
- N6-benzoyladenosine-3′,5′-cyclic monophosphate
- (Sp)-2-Cl-cAMPS
- 2-chloroadenosine-3′,5′-cyclic monophosphorothioate
- PBC
- phosphate binding cassette.
References
- 1. World Health Organization (2014) World Malaria Report 2014, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. WHO Malaria Policy Advisory Committee and Secretariat (2016) Malaria Policy Advisory Committee to the WHO: conclusions and recommendations of eighth biannual meeting (September 2015). Malaria Journal 15, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Solyakov L., Halbert J., Alam M. M., Semblat J. P., Dorin-Semblat D., Reininger L., Bottrill A. R., Mistry S., Abdi A., Fennell C., Holland Z., Demarta C., Bouza Y., Sicard A., Nivez M. P., et al. (2011) Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2, 565. [DOI] [PubMed] [Google Scholar]
- 4. van Dooren G. G., and Striepen B. (2013) The algal past and parasite present of the apicoplast. Annu. Rev. Microbiol. 67, 271–289 [DOI] [PubMed] [Google Scholar]
- 5. Lucet I. S., Tobin A., Drewry D., Wilks A. F., and Doerig C. (2012) Plasmodium kinases as targets for new-generation antimalarials. Future Med. Chem. 4, 2295–2310 [DOI] [PubMed] [Google Scholar]
- 6. Hopp C. S., Bowyer P. W., and Baker D. A. (2012) The role of cGMP signalling in regulating life cycle progression of Plasmodium. Microbes Infect. 14, 831–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lasonder E., Green J. L., Camarda G., Talabani H., Holder A. A., Langsley G., and Alano P. (2012) The Plasmodium falciparum schizont phosphoproteome reveals extensive phosphatidylinositol and cAMP-protein kinase A signaling. J. Proteome Res. 11, 5323–5337 [DOI] [PubMed] [Google Scholar]
- 8. Leykauf K., Treeck M., Gilson P. R., Nebl T., Braulke T., Cowman A. F., Gilberger T. W., and Crabb B. S. (2010) Protein kinase A dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathog. 6, e1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lasonder E., Green J. L., Grainger M., Langsley G., and Holder A. A. (2015) Extensive differential protein phosphorylation as intraerythrocytic Plasmodium falciparum schizonts develop into extracellular invasive merozoites. Proteomics 15, 2716–2729 [DOI] [PubMed] [Google Scholar]
- 10. Dawn A., Singh S., More K. R., Siddiqui F. A., Pachikara N., Ramdani G., Langsley G., and Chitnis C. E. (2014) The central role of cAMP in regulating Plasmodium falciparum merozoite invasion of human erythrocytes. PLoS Pathog. 10, e1004520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beraldo F. H., Almeida F. M., da Silva A. M., and Garcia C. R. (2005) Cyclic AMP and calcium interplay as second messengers in melatonin-dependent regulation of Plasmodium falciparum cell cycle. J. Cell Biol. 170, 551–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merckx A., Nivez M. P., Bouyer G., Alano P., Langsley G., Deitsch K., Thomas S., Doerig C., and Egée S. (2008) Plasmodium falciparum regulatory subunit of cAMP-dependent PKA and anion channel conductance. PLoS Pathog. 4, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramdani G., Naissant B., Thompson E., Breil F., Lorthiois A., Dupuy F., Cummings R., Duffier Y., Corbett Y., Mercereau-Puijalon O., Vernick K., Taramelli D., Baker D. A., Langsley G., and Lavazec C. (2015) cAMP-signalling regulates gametocyte-infected erythrocyte deformability required for malaria parasite transmission. PLoS Pathog. 11, e1004815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Syin C., Parzy D., Traincard F., Boccaccio I., Joshi M. B., Lin D. T., Yang X. M., Assemat K., Doerig C., and Langsley G. (2001) The H89 cAMP-dependent protein kinase inhibitor blocks Plasmodium falciparum development in infected erythrocytes. Eur. J. Biochem. 268, 4842–4849 [DOI] [PubMed] [Google Scholar]
- 15. Haste N. M., Talabani H., Doo A., Merckx A., Langsley G., and Taylor S. S. (2012) Exploring the Plasmodium falciparum cyclic-adenosine monophosphate (cAMP)-dependent protein kinase (PfPKA) as a therapeutic target. Microbes Infect. 14, 838–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., and Varughese K. I. (1995) Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science 269, 807–813 [DOI] [PubMed] [Google Scholar]
- 17. Kim C., Cheng C. Y., Saldanha S. A., and Taylor S. S. (2007) PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 [DOI] [PubMed] [Google Scholar]
- 18. Reimann E. M., Walsh D. A., and Krebs E. G. (1971) Purification and properties of rabbit skeletal muscle adenosine 3′,5′-monophosphate-dependent protein kinases. J. Biol. Chem. 246, 1986–1995 [PubMed] [Google Scholar]
- 19. Amieux P. S., and McKnight G. S. (2002) The essential role of RIα in the maintenance of regulated PKA activity. Ann. N.Y. Acad. Sci. 968, 75–95 [DOI] [PubMed] [Google Scholar]
- 20. Berman H. M., Ten Eyck L. F., Goodsell D. S., Haste N. M., Kornev A., and Taylor S. S. (2005) The cAMP binding domain: an ancient signaling module. Proc. Natl. Acad. Sci. U.S.A. 102, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang P., Smith-Nguyen E. V., Keshwani M. M., Deal M. S., Kornev A. P., and Taylor S. S. (2012) Structure and allostery of the PKA RIIβ tetrameric holoenzyme. Science 335, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akimoto M., McNicholl E. T., Ramkissoon A., Moleschi K., Taylor S. S., and Melacini G. (2015) Mapping the free energy landscape of PKA inhibition and activation: a double-conformational selection model for the tandem cAMP-binding domains of PKA RIα. PLoS Biol. 13, e1002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buskes M. J., Harvey K. L., Richards B. J., Kalhor R., Christoff R. M., Gardhi C. K., Littler D. R., Cope E. D., Prinz B., Weiss G. E., O'Brien N. J., Crabb B. S., Deady L. W., Gilson P. R., and Abbott B. M. (2016) Antimalarial activity of novel 4-cyano-3-methylisoquinoline inhibitors against Plasmodium falciparum: design, synthesis and biological evaluation. Org. Biomol. Chem. 14, 4617–4639 [DOI] [PubMed] [Google Scholar]
- 24. Howard B. L., Harvey K. L., Stewart R. J., Azevedo M. F., Crabb B. S., Jennings I. G., Sanders P. R., Manallack D. T., Thompson P. E., Tonkin C. J., and Gilson P. R. (2015) Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem. Biol. 10, 1145–1154 [DOI] [PubMed] [Google Scholar]
- 25. Bruystens J. G., Wu J., Fortezzo A., Kornev A. P., Blumenthal D. K., and Taylor S. S. (2014) PKA RIα homodimer structure reveals an intermolecular interface with implications for cooperative cAMP binding and Carney complex disease. Structure 22, 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diller T. C., Madhusudan, Xuong N. H., and Taylor S. S. (2001) Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II β regulatory subunit. Structure 9, 73–82 [DOI] [PubMed] [Google Scholar]
- 27. Rinaldi J., Wu J., Yang J., Ralston C. Y., Sankaran B., Moreno S., and Taylor S. S. (2010) Structure of yeast regulatory subunit: a glimpse into the evolution of PKA signaling. Structure 18, 1471–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ilouz R., Bubis J., Wu J., Yim Y. Y., Deal M. S., Kornev A. P., Ma Y., Blumenthal D. K., and Taylor S. S. (2012) Localization and quaternary structure of the PKA RIbeta holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 12443–12448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akimoto M., Selvaratnam R., McNicholl E. T., Verma G., Taylor S. S., and Melacini G. (2013) Signaling through dynamic linkers as revealed by PKA. Proc. Natl. Acad. Sci. U.S.A. 110, 14231–14236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brautigam C. A. (2015) Fitting two- and three-site binding models to isothermal titration calorimetric data. Methods 76, 124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Badireddy S., Yunfeng G., Ritchie M., Akamine P., Wu J., Kim C. W., Taylor S. S., Qingsong L., Swaminathan K., and Anand G. S. (2011) Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: conformational selection highlights a new concept in allosteric inhibitor design. Mol. Cell. Proteomics 10, M110.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Søberg K., Jahnsen T., Rognes T., Skålhegg B. S., and Laerdahl J. K. (2013) Evolutionary paths of the cAMP-dependent protein kinase (PKA) catalytic subunits. PLoS One 8, e60935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu J., Jones J. M., Nguyen-Huu X., Ten Eyck L. F., and Taylor S. S. (2004) Crystal structures of RIα subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry 43, 6620–6629 [DOI] [PubMed] [Google Scholar]
- 34. Moll D., Schweinsberg S., Hammann C., and Herberg F. W. (2007) Comparative thermodynamic analysis of cyclic nucleotide binding to protein kinase A. Biol. Chem. 388, 163–172 [DOI] [PubMed] [Google Scholar]
- 35. Brown S. H., Cheng C. Y., Saldanha S. A., Wu J., Cottam H. B., Sankaran B., and Taylor S. S. (2013) Implementing fluorescence anisotropy screening and crystallographic analysis to define PKA isoform-selective activation by cAMP analogs. ACS Chem. Biol. 8, 2164–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trager W., and Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193, 673–675 [DOI] [PubMed] [Google Scholar]
- 37. Makler M. T., and Hinrichs D. J. (1993) Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 48, 205–210 [DOI] [PubMed] [Google Scholar]
- 38. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]