

Abstract
The sirtuin family of proteins catalyze the NAD+-dependent deacylation of acyl-lysine residues. Humans encode seven sirtuins (Sirt1–7), and recent studies have suggested that post-translational modification of Sirt1 by cysteine S-nitrosation correlates with increased acetylation of Sirt1 deacetylase substrates. However, the mechanism of Sirt1 inhibition by S-nitrosation was unknown. Here, we show that Sirt1 is transnitrosated and inhibited by the physiologically relevant nitrosothiol S-nitrosoglutathione. Steady-state kinetic analyses and binding assays were consistent with Sirt1 S-nitrosation inhibiting binding of both the NAD+ and acetyl-lysine substrates. Sirt1 S-nitrosation correlated with Zn2+ release from the conserved sirtuin Zn2+-tetrathiolate and a loss of α-helical structure without overall thermal destabilization of the enzyme. Molecular dynamics simulations suggested that Zn2+ loss due to Sirt1 S-nitrosation results in repositioning of the tetrathiolate subdomain away from the rest of the catalytic domain, thereby disrupting the NAD+ and acetyl-lysine-binding sites. Sirt1 S-nitrosation was reversed upon exposure to the thiol-based reducing agents, including physiologically relevant concentrations of the cellular reducing agent glutathione. Reversal of S-nitrosation resulted in full restoration of Sirt1 activity only in the presence of Zn2+, consistent with S-nitrosation of the Zn2+-tetrathiolate as the primary source of Sirt1 inhibition upon S-nitrosoglutathione treatment.
Keywords: acetylation, allosteric regulation, enzyme inactivation, inhibition mechanism, nicotinamide adenine dinucleotide (NAD), nitric oxide, nitrosylation, sirtuin 1 (SIRT1), zinc, nitrosation
Introduction
Sirtuins are a class of NAD+-dependent enzymes that catalyze the deacylation of acyl-lysine residues, producing O-acyl-ADP-ribose and nicotinamide in the process (1). The seven human sirtuin protein family members (Sirt1–7) have diverse subcellular localization and diverse deacylase targets (2). Collectively, sirtuins are known for their role in lifespan extension, as a decrease in sirtuin activity is implicated in early aging and the development of aging-related diseases, including neurodegeneration, cardiovascular disease, and type II diabetes (3).
Sirt1 is localized to both the nucleus and cytoplasm where it deacetylates histones, transcription factors, and many other proteins (2). Recent work has implicated oxidative post-translational modification of cysteine residues within Sirt1 as a mechanism of physiological inhibition (4–11). In particular, S-nitrosation of Sirt1 was recently suggested to inhibit Sirt1 deacetylase activity, resulting in increased acetylation of Sirt1 deacetylase substrates PGC1-α, p53, and the p65 subunit of NF-κB (4, 5). An increase in S-nitrosation of Sirt1 was also suggested to occur in the context of aging and to correspond to decreased Sirt1 deacetylation of cellular targets (5). However, in this study (5) endogenous nitric oxide (NO) was generated through lipopolysaccharide and cytokine treatment to induce inducible nitric-oxide synthase (iNOS)3 expression, conditions known to also stimulate production of hydrogen peroxide by NADPH oxidases (12). Therefore, cysteine sulfenylation could also have contributed to the observed Sirt1 cysteine oxidation. It has also been demonstrated that Sirt1 can be S-glutathionylated by S-nitrosoglutathione (GSNO) (6). More recently, Sirt1 glutathionylation was shown to correlate with increased acetylation of cellular Sirt1 substrates and induction of apoptosis (7).
The primary Sirt1 sequence is composed of the following three major domains: the N-terminal domain that binds and positions acetylated protein substrates (13, 14) and pharmaceutical activators (15–18); the catalytic domain that binds acetyl-lysine and NAD+ and catalyzes lysine deacetylation; and the C-terminal domain that extends the Rossmann-fold motif in the catalytic core (Fig. 1a) (14). The catalytic domain is further subdivided into the primarily α-helical Zn2+-tetrathiolate subdomain and the Rossmann-fold composed of α-helices and parallel β-strands. The Zn2+-tetrathiolate consists of four surface-exposed cysteine residues that are conserved among all seven human sirtuins and have been suggested to be the site of Sirt1 S-nitrosation (4, 5). However, the mechanism of inhibition by S-nitrosation remains unclear. Here, we demonstrate that Sirt1 S-nitrosation results in a conformational change that allosterically inhibits acetyl-lysine substrate and NAD+ binding through reversible modification of the Sirt1 Zn2+-tetrathiolate.
FIGURE 1.

Sirt1 is inhibited by S-nitrosation. a, crystal structure (PDB code 4ZZJ (17)) of the Sirt1 catalytic domain with carba-NAD+ and an acetyl-lysine peptide bound in the Rossmann-fold subdomain, located proximal to the conserved Zn2+-tetrathiolate. b, comparison of Sirt1 inhibition by cysteine oxidants. Sirt1 (0.5–1 μm) deacetylase activity was assessed via an enzyme-coupled assay following treatment with varying concentrations of GSNO (red squares), DEA-NONOate (gray diamonds), H2O2 (blue circles), H2O2/GSH (purple triangles), and GSSG (green triangles) for 1 h at 37 °C. Percentage of deacetylase activity in the absence of oxidants was plotted versus log10 of the oxidant concentration (n = 3 ± S.E.). Relative Sirt1 S-nitrosation (c), glutathionylation (d), and sulfenylation (e) levels were assessed for Sirt1 WT treated with the indicated concentrations of GSNO for 1 h at 37 °C (representative blots, n = 3 for biotin switch S-nitrosation detection, n = 4 for anti-glutathionylation, and n = 3 for DYn-2 sulfenylation detection). GAPDH treated with 100 μm H2O2 and 100 μm GSH was used as a positive control for glutathionylation. GAPDH treated with 100 μm H2O2 was used as a positive control for sulfenylation. f, relative Sirt1 S-nitrosation levels for Sirt1 WT treated with varying concentrations of GSNO and DEA-NONOate for 1 h at 37 °C were assessed via the biotin switch assay (representative blot, n = 3). Full blots and loading control gels are shown in supplemental Fig. S1.
Results
Sirt1 Activity Is Inhibited by Transnitrosation
Previous reports have demonstrated a decrease in cellular Sirt1 deacetylase activity in response to cysteine S-nitrosation (4, 5) and glutathionylation (6, 7). However, a systematic study directly comparing the relative susceptibility of Sirt1 to physiological cysteine oxidants has not been performed. Therefore, Sirt1 activity was monitored following treatment with varying concentrations of GSNO, NO (released from DEA-NONOate), hydrogen peroxide, hydrogen peroxide in the presence of reduced glutathione (GSH), and oxidized glutathione (GSSG). A concentration-dependent decrease in Sirt1 deacetylase activity as measured by an enzyme-coupled assay (19) was only observed in response to treatment with GSNO and DEA-NONOate, where GSNO (IC50 = 17 ± 5 μm) exhibited greater potency than DEA-NONOate (IC50 >100 μm) (Fig. 1b). To determine whether the observed Sirt1 inhibition by GSNO was due to transnitrosation (4, 5) or other potential oxidative modifications that may occur upon GSNO treatment such as glutathionylation (6, 7) or sulfenylation, Sirt1 was treated with increasing concentrations of GSNO, and cysteine oxidation was probed using the biotin switch assay (Fig. 1c). In the biotin switch assay, protein is treated with a cysteine-selective alkylating agent (e.g. iodoacetamide) to block all unmodified cysteine residues. The alkylating agent is removed by acetone precipitation of the protein, and oxidized cysteines (e.g. nitrosothiols) are then reduced with ascorbate followed by labeling of the newly reduced thiols with biotin-iodoacetamide. Proteins are resolved via SDS-PAGE, and biotin labeling is detected by blotting with a streptavidin-based chemiluminescent probe. The observed Sirt1 biotin switch signal was completely dependent on the use of ascorbate and correlated with the degree of Sirt1 inhibition. As ascorbate is not completely selective for nitrosothiols and under certain conditions can also reduce disulfides (20) and sulfenic acids (21), GSNO-treated Sirt1 was probed using anti-glutathionylation immunoblotting and the DYn-2 probe for protein sulfenylation (22). Neither glutathionylation (Fig. 1d) nor sulfenylation (Fig. 1e) of Sirt1 was observed above background levels compared with the robust glutathionylation and sulfenylation detected for the positive control, GAPDH (23, 24). To determine whether, like GSNO, the observed Sirt1 inhibition by NO (released from DEA-NONOate) correlated with the degree of Sirt1 S-nitrosation, biotin switch assays were performed in parallel to compare the level of Sirt1 S-nitrosation in response to GSNO and DEA-NONOate. A concentration-dependent increase in Sirt1 S-nitrosation was observed with both GSNO and DEA-NONOate. However, although maximal S-nitrosation was observed at 60 μm GSNO, the level of S-nitrosation was still increasing at 1 mm DEA-NONOate (Fig. 1f). Therefore, Sirt1 displayed higher sensitivity toward S-nitrosation by GSNO compared with DEA-NONOate (Fig. 1f), consistent with the more potent Sirt1 inhibition by GSNO compared with DEA-NONOate (Fig. 1b). Taken together, these results are consistent with S-nitrosation and not glutathionylation or sulfenylation being primarily responsible for the observed Sirt1 inhibition.
Sirt1 S-Nitrosation of the Zn2+-Tetrathiolate Results in Zn2+ Release
Previous mutagenesis studies have localized the sites of Sirt1 S-nitrosation to two of the Zn2+-tetrathiolate cysteine residues, Cys-395 and Cys-398 (4, 5). To further confirm that the Zn2+-tetrathiolate is the target of Sirt1 S-nitrosation, four individual tetrathiolate cysteine to serine mutant constructs were generated (C371S, C374S, C395S, and C398S), as well as a cysteine to serine double mutant (C395S/C398S). Biotin switch assays were performed to assess transnitrosation of each of these Zn2+-tetrathiolate mutants by GSNO (Fig. 2a). Robust S-nitrosation was observed for Sirt1 WT, whereas S-nitrosation was reduced to background levels for the C395S/C398S double mutant or any of the individual tetrathiolate mutants (C371S, C374S, C395S, and C398S). To correlate loss of deacetylase activity upon GSNO treatment of Sirt1 (Fig. 1b) to modification of the Zn2+-tetrathiolate, the deacetylase activity of each Zn2+-tetrathiolate mutant was measured via an enzyme-coupled assay (19), which demonstrated near background levels of deacetylase activity for the C395S/C398S double mutant consistent with previous work (5), as well as each of the individual tetrathiolate mutants (C371S, C374S, C395S, and C398S) (Fig. 2b). To determine whether S-nitrosation of the Sirt1 Zn2+-tetrathiolate resulted in Zn2+ release, Sirt1 was treated with varying concentrations of GSNO in the presence of the colorimetric Zn2+-chelating agent Zincon, and the rate of Zn2+ release was monitored spectrophotometrically. A concentration-dependent increase in the rate of Zn2+ release was observed in response to GSNO treatment of Sirt1 (Fig. 2c), confirming that Zn2+ is released from the Zn2+-tetrathiolate upon modification by S-nitrosation consistent with previous work (5). Based on the rate of Zn2+ release from Sirt1 (1.4 ± 0.3 nm/s; determined at 7.5 μm Sirt1) treated with 100 μm GSNO, ∼67% of Zn2+ would be lost after 1 h of GSNO treatment, corresponding to the observed 73 ± 9% inhibition of Sirt1 deacetylase activity at 100 μm GSNO (Fig. 1b). In contrast, background levels of Zn2+ release were observed for the Sirt1 C395S/C398S double mutant as well as the individual tetrathiolate mutants (C371S, C374S, C395S, and C398S) indicating that the tetrathiolate mutants have lost the ability to stably bind Zn2+. Based on these results, all the Sirt1 tetrathiolate mutants are predicted to behave similarly, but previous studies have suggested that Cys-395 and/or Cys-398 are the site(s) of Sirt1 S-nitrosation (4, 5). Therefore, for the sake of simplicity Sirt1 C395S/C398S was used as a representative S-nitrosation-deficient Sirt1 mutant.
FIGURE 2.
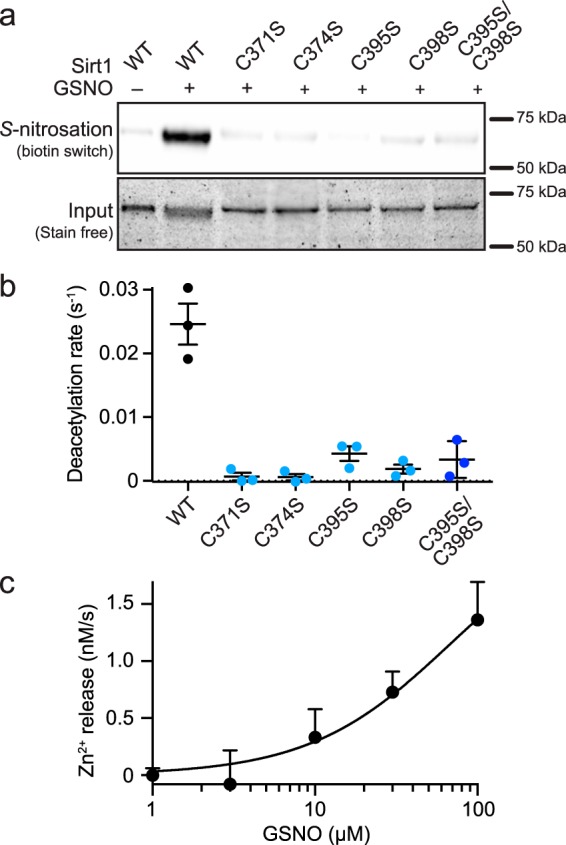
Sirt1 S-nitrosation of the Zn2+-tetrathiolate results in Zn2+ release. a, relative Sirt1 S-nitrosation levels were assessed for Sirt1 WT and Sirt1 tetrathiolate mutants (C371S, C374S, C395S, C398S, and C395S/C398S) treated with 100 μm GSNO for 1 h at 37 °C. Full blot and loading control gel are shown in supplemental Fig. S2. b, Sirt1 tetrathiolate mutants do not display deacetylase activity significantly above background. Deacetylase activity of Sirt1 WT and tetrathiolate mutants (1 μm) was assessed via continuous enzyme-coupled assay (n = 3 ± S.E.). c, Sirt1 S-nitrosation results in Zn2+ release. Sirt1 (7.5 μm) was treated with varying concentrations of GSNO in the presence of Zincon (40 μm). Rates of Zn2+ release were measured by following changes in absorbance at 529 and 620 nm for 30 min at 37 °C. Data were plotted as rate of Zn2+ release versus log10 of the GSNO concentration (n = 3 ± S.E.).
Inhibition of Sirt1 Deacetylase Activity by S-Nitrosation Is Reversible
In cellular contexts, transnitrosation is a reversible reaction in equilibrium with other low molecular weight (e.g. GSH) and protein thiols (25). To determine whether the activity of S-nitrosated Sirt1 could be recovered by subsequent denitrosation by thiol-based reducing agents as reported previously (5), Sirt1 was transnitrosated by GSNO and then incubated with the thiol-based reducing agent, DTT, in the presence or absence of Zn2+. Although the biotin switch assay demonstrated clear reversal of Sirt1 S-nitrosation by DTT (Fig. 3c), Sirt1 deacetylase activity was not recovered by DTT or Zn2+ treatment alone; instead, deacetylase activity was only restored following treatment with both DTT and Zn2+ (Fig. 3a). Sirt1 C395S/C398S was also treated with GSNO followed by treatment with DTT, Zn2+, or both; however, deacetylase activity was not increased for the C395S/C398S mutant, which suggests that DTT and Zn2+ treatment restore GSNO-treated Sirt1 WT deacetylase activity solely through the Zn2+-tetrathiolate. Inhibition of GSNO-treated Sirt1 and recovery of Sirt1 activity following DTT and Zn2+ treatment were confirmed via an HPLC assay (Fig. 3b) that quantitates acetylated and deacetylated peptides instead of nicotinamide formation (the basis of the enzyme-coupled sirtuin assay (19)). To assess the concentration dependence of the primary cellular low molecular weight thiol (i.e. GSH), S-nitrosated Sirt1 was treated with varying concentrations of GSH in the presence of Zn2+. A concentration-dependent recovery of Sirt1 activity was observed (Fig. 3d) that correlated with a concentration-dependent reversal of S-nitrosation (Fig. 3e). In contrast, deacetylase activity was not restored when Sirt1 C395S/C398S was treated with GSH and Zn2+.
FIGURE 3.
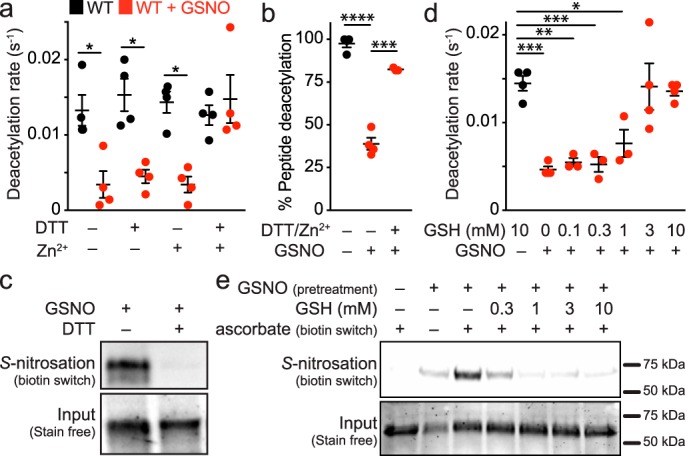
Sirt1 deacetylase inhibition by S-nitrosation is reversible in the presence of reductants and Zn2+. a, Sirt1 (2 μm) was treated with buffer or GSNO (100 μm) for 1 h at 37 °C, followed by incubation with DTT (10 mm) and/or ZnCl2 (1 μm) for 5 min at 37 °C. Sirt1 activity (0.5 μm) was assessed via the enzyme-coupled assay. Data were plotted as the mean deacetylation rate for each condition, and significance was determined using a one-way ANOVA and Tukey post-test (n = 4 ± S.E.). b, Sirt1 (2.5 μm) was treated with buffer or GSNO (60 μm) for 30 min at 37 °C followed by incubation with DTT (5 mm) and ZnCl2 (1–10 μm) for 5 min at 37 °C. Sirt1 activity (1 μm) was assessed via HPLC as detailed under “Experimental Procedures.” Data were plotted as the mean percent peptide deacetylation for each condition, and significance was determined via Student's t test (n = 3 ± S.E.). c, Sirt1 S-nitrosation is reversed by DTT. Sirt1 was treated with GSNO (100 μm) for 1 h at 37 °C, followed by immediate treatment with DTT (10 mm). The relative level of S-nitrosation was assessed via the biotin switch assay (n = 3, representative blot). d, Sirt1 inhibition by S-nitrosation is recovered by GSH. Sirt1 (2 μm) was treated with buffer or GSNO (100 μm) for 1 h at 37 °C, followed by incubation with varying concentrations of GSH and ZnCl2 (1 μm) for 5 min at 37 °C. Sirt1 activity (0.5 μm) was assessed via the enzyme-coupled assay, and data were plotted as the mean deacetylation rate versus GSH concentration (n = 4 ± S.E.). e, Sirt1 S-nitrosation is reversed by GSH. Sirt1 was treated with GSNO (100 μm) for 1 h at 37 °C followed by immediate treatment with varying concentrations of GSH for 5 min at 37 °C. The relative level of S-nitrosation was assessed via the biotin switch assay. Full blot and loading control gel are shown in supplemental Fig. S3 (n = 3, representative blot). Values that are significantly different are indicated by bars and asterisks as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Sirt1 S-Nitrosation Inhibits Binding to Acetyl-lysine and NAD+-binding Pockets
The acetyl-lysine and NAD+ substrate-binding sites are located near the interface of the Zn2+-tetrathiolate and Rossmann-fold subdomains (Fig. 1a). Therefore, Sirt1 S-nitrosation may alter positioning of the Zn2+-tetrathiolate subdomain relative to the Rossmann-fold subdomain and allosterically inhibit substrate binding. To determine whether inhibition by S-nitrosation was due to a decreased overall rate of catalysis or a decrease in substrate binding affinity, the deacetylase rates for control and GSNO-treated Sirt1 WT as well as Sirt1 C395S/C398S were determined at varying concentrations of acetylated H3 peptide (H3K14ac; Fig. 4a) and NAD+ (Fig. 4b) substrates. For buffer control-treated Sirt1 WT, the kcat and Km values varying H3K14ac were 0.083 ± 0.007 s−1 and 130 ± 33 μm, and the kcat and Km values varying NAD+ were 0.087 ± 0.005 s−1 and 668 ± 82 μm. For GSNO-treated Sirt1 WT, the kcat and Km values varying H3K14ac were 0.026 ± 0.007 s−1 and 129 ± 104 μm, and the kcat and Km values varying NAD+ were 0.02 ± 0.05 s−1 and 712 ± 413 μm. Thus, for both Sirt1 substrates, the maximum catalytic activity (kcat) of GSNO-treated Sirt1 WT was significantly decreased compared with the control (H3K14ac, p = 0.046; NAD+, p = 0.0003); however, the Km values for each substrate were unaffected (p > 0.05). In addition, Sirt1 C395S/C398S exhibited only background levels of deacetylase activity (Fig. 4a).
FIGURE 4.
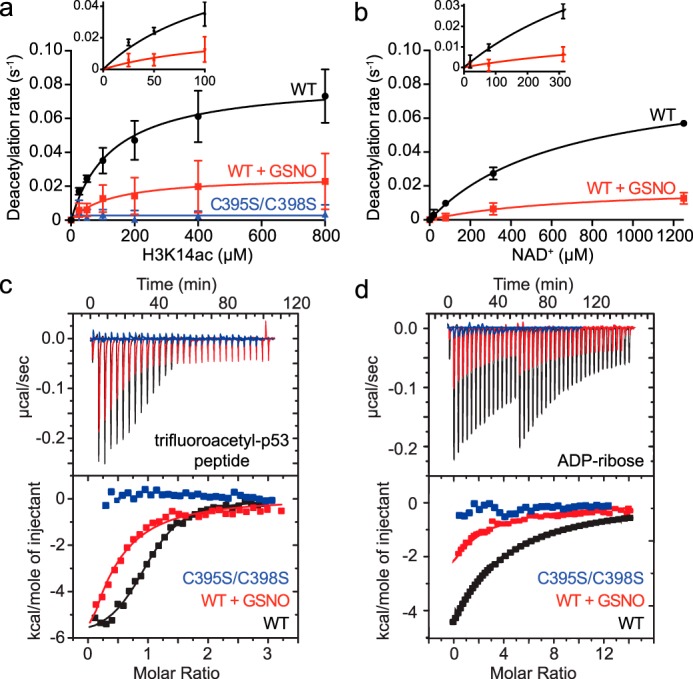
Effect of Sirt1 S-nitrosation on substrate binding and catalysis. a, titrations of H3K14ac peptide substrate were performed under saturating NAD+ concentrations (2 mm) following treatment of Sirt1 WT or Sirt1 C395S/C398S (2 μm) with buffer or GSNO (100 μm) for 1 h at 37 °C. Sirt1 (0.5 μm) deacetylase activity was assessed via the enzyme-coupled assay. Deacetylation rates were plotted versus H3K14ac concentration and fit to the Michaelis-Menten equation to obtain steady-state kinetic parameters (WT n = 3 ± S.D.; C395S/C398S n = 2 ± S.D.). Inset displays quality of nonlinear fit at low substrate concentrations. b, titrations of NAD+ were performed under saturating concentrations of H3K14ac peptide (1.4 mm) following treatment of Sirt1 (2 μm) with buffer or GSNO (100 μm) for 1 h at 37 °C. Sirt1 (1 μm) deacetylase activity was assessed via the enzyme-coupled assay. Deacetylation rates were plotted versus H3K14ac concentration and fit to the Michaelis-Menten equation to obtain steady-state kinetic parameters (n ≥3 ± S.D.). Inset displays quality of nonlinear fit at low substrate concentrations. c, determining the effect of Sirt1 S-nitrosation on binding to the acetyl-lysine-binding site by ITC. Sirt1 WT or C395S/C398S was pretreated with buffer or GSNO (100 μm) for 1 h at 37 °C. p53tfa peptide (200 μm) was titrated into Sirt1. The area under each peak was integrated and plotted as kcal/mol of p53tfa versus the molar ratio of p53tfa/Sirt1 (representative data shown, n ≥2 for each condition). d, determining the effect of Sirt1 S-nitrosation on binding to the NAD+-binding site by ITC. Sirt1 WT or C395S/C398S was pretreated with buffer or GSNO (100 μm) for 1 h at 37 °C. ADPr (400 μm) was titrated into Sirt1. The area under each peak was integrated and plotted as kcal/mol of ADPr versus the molar ratio of ADPr/Sirt1 (representative data shown, n ≥2 for each condition).
These data could be explained by two possible models. In one model, the entire Sirt1 population is modified by GSNO, and S-nitrosated Sirt1 possesses a lower overall rate of catalysis (kcat) with no effect on substrate binding, positioning, and other steps reflected in the Km value. In an alternative model, only a fraction of the Sirt1 is transnitrosated by GSNO, and the S-nitrosated fraction is completely inactive due to an inability of S-nitrosated Sirt1 to bind or position the acetyl-lysine and/or NAD+ substrates for catalysis. In this case, the observed kcat would decrease, reflecting a decrease in the concentration of active Sirt1; however, the Km value would remain the same, as the remaining unmodified Sirt1 WT could properly bind substrates and catalyze deacetylation, and the Km value is independent of active enzyme concentration. To distinguish between these two models, substrate binding affinities (Kd) and stoichiometry were determined using isothermal titration calorimetry. If the first model is operative, the Kd value of each substrate is predicted to increase, whereas the stoichiometry of Sirt1/substrate remains 1:1. If the second model is operative, the Kd value of each substrate is predicted to remain the same, although the stoichiometry of the Sirt1/substrate will decrease to <1 and reflect the fraction of Sirt1 that remains unmodified. To measure binding to the acetyl-lysine-binding site, a tight-binding acetyl-lysine analogue peptide was used (trifluoroacetylated p53 peptide; p53tfa) (26–29). The NAD+ analogue ADP-ribose (ADPr) was used to measure binding to the NAD+-binding site (30, 31). These substrate analogues were titrated with control and GSNO-treated Sirt1 WT as well as Sirt1 C395S/C398S to determine the Kd values and binding stoichiometry (n). Consistent with the second model, the p53tfa peptide Kd values for GSNO and control-treated Sirt1 WT were not altered within error (4.0 ± 3.2 μm versus 1.2 ± 1.0 μm) (Fig. 4c). In contrast, the apparent stoichiometry of binding for GSNO-treated Sirt1 WT was decreased compared with control-treated Sirt1 WT (0.44 ± 0.15 versus 1.00 ± 0.02). Further consistent with the second model, the Kd values for ADPr binding to GSNO and control-treated Sirt1 WT were unchanged within error (38 ± 2 μm versus 42 ± 5 μm), whereas the apparent stoichiometry of binding for GSNO-treated Sirt1 WT was decreased compared with control-treated Sirt1 WT (0.4 ± 0.1 versus 1.0 ± 0.1) (Fig. 4d). In contrast, Sirt1 C395S/C398S did not bind either p53tfa or ADPr (Fig. 4, c and d) suggesting that quantitatively nitrosated Sirt1 is unable to bind both the acetyl-lysine and NAD+ substrates.
S-Nitrosation of Sirt1 Results in Selective Alteration of Secondary Structure without Overall Thermal Destabilization
As the Sirt1 Zn2+-tetrathiolate subdomain is rich in α-helical secondary structure (Fig. 1a), S-nitrosation of the Sirt1 Zn2+-tetrathiolate was hypothesized to result in loss of α-helical character. Circular dichroism was performed to assess changes in secondary structure of Sirt1 following GSNO treatment. Compared with control-treated Sirt1 (17.3 ± 0.3% α-helix), GSNO-treated Sirt1 (14.8 ± 0.9% α-helix) exhibited a significant (p = 0.04) loss in α-helical character (Fig. 5, a and b), accompanied by a significant gain (p = 0.007) in β-sheet character (control, 27.8 ± 0.3% β-sheet; GSNO-treated, 31 ± 1.2% β-sheet) with no change in turn (control, 24.7 ± 0.5% turn; GSNO-treated, 24.5 ± 0.9% turn) or random coil (control, 30 ± 0.4% coil; GSNO-treated, 29.8 ± 0.9% coil) composition. Sirt1 C395S/C398S demonstrated an even greater significant loss in α-helicity (13.3 ± 0.6%; p = 0.0008) and gain in β-sheet composition (32.5 ± 0.9%; p < 0.0001) as compared with Sirt1 WT, with no accompanying change in turn (22.8 ± 0.3%) or random coil (31 ± 0.7%) character (Fig. 5, a and b). Therefore, Sirt1 C395S/C398S likely represents an upper limit to the effect of Sirt1 S-nitrosation on Sirt1 secondary structure. To determine whether the observed loss in α-helical and gain in β-sheet secondary structure upon GSNO treatment altered the overall thermal stability of Sirt1, the melting temperature of control-treated Sirt1 WT (Tm = 48 ± 0.5 °C), GSNO-treated Sirt1 WT (Tm = 52 ± 0.2 °C), DEA-NONOate-treated Sirt1 WT (Tm = 49 ± 2 °C), and Sirt1 C395S/C398S (Tm = 52 ± 0.1 °C) was assessed using a thermal shift assay (Fig. 5c). In the thermal shift assay, thermal denaturation of the protein results in exposure of the hydrophobic protein core, resulting in a fluorescence increase of the SYPRO Orange dye upon binding to these exposed hydrophobic regions. The decrease in signal following the fluorescence maximum after the melting transition is typically attributed to the loss of hydrophobic patches that bind SYPRO Orange upon protein aggregation at high temperature (32). Disruption of the Sirt1 Zn2+-tetrathiolate region through S-nitrosation or mutation resulted in significant stabilization of Sirt1 (Sirt1-SNO, p = 0.002; Sirt1 C395S/C398S, p = 0.0009) suggesting the changes in secondary structure upon Sirt1 S-nitrosation are localized to the Zn2+-tetrathiolate subdomain and surrounding regions rather than global Sirt1 unfolding.
FIGURE 5.

Sirt1 S-nitrosation results in selective alteration of Sirt1 secondary structure without a decrease in thermal stability. a, circular dichroism analyses of Sirt1 WT or C395S/C398S (14 μm) treated with buffer or GSNO (100 μm) for 1 h at 37 °C. CD spectra of desalted Sirt1 (2 mg/ml or 3.5 μm) were obtained by scanning from 190 to 280 nm in 0.1-nm intervals. b, percentages of α-helix, β-sheet, turn, and random coil were calculated by analyzing CD spectra with DICHROWEB software. Data were plotted as the percentage of each secondary structure observed under each condition, and significance was determined by two-way ANOVA and Tukey post-test (n = 4 ± S.E.). c, disruption of the Sirt1 Zn2+-tetrathiolate by S-nitrosation or mutation increases thermal stability. Sirt1 WT or C395S/C398S (14 μm) was treated with buffer, GSNO (100 μm), or DEA-NONOate (100 μm) for 1 h at 37 °C (n = 3 for each condition). Melting curves for desalted Sirt1 (7.5 μm) were obtained by following SYPRO Orange fluorescence as a function of temperature using a temperature gradient of 20–95 °C. Values that are significantly different are indicated by bars and asterisks as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Molecular Dynamics Support Allosteric Inhibition of Sirt1 Substrate Binding
To further elucidate the mechanism of Sirt1 inhibition by S-nitrosation of the Sirt1 Zn2+-tetrathiolate, molecular dynamics simulations were performed using a crystal structure of Sirt1 (PDB code 4KXQ (33)). To compare the conformational dynamics of unmodified and S-nitrosated Sirt1, docked substrates and Zn2+ were removed from the crystal structure, and cysteines 395 and 398 were left unmodified, modified by S-nitrosation (Sirt1-SNO), or mutated to serine. The relative conformational dynamics of each residue was determined using root mean square fluctuation (RMSF) values calculated during each 100-ns simulation trajectory (Fig. 6a). RMSF calculations derived from each simulation demonstrated significant differences (>1 S.D.) in localized residue dynamics of Sirt1-SNO and Sirt1 C395S/C398S compared with Sirt1 WT (Fig. 6, b and c). Specifically, increased dynamics of the Zn2+-tetrathiolate subdomain (residues 288–291, 367, 371–379, and 395–405) was observed when Cys-395 and Cys-398 were either S-nitrosated or mutated to serine (Fig. 6, b and c). In contrast, residues 264–280, 324–325, and 480–483 that bind the adenosine portion of NAD+ but are located distal to the Zn2+-tetrathiolate displayed decreased dynamics when Cys-395 and Cys-398 were either S-nitrosated or mutated to serine (Fig. 6, b and c). Examination of the final state of each 100-ns simulation trajectory revealed a tilting of the Zn2+-tetrathiolate subdomain away from the Rossmann-fold subdomain of the catalytic core (Fig. 6d).
FIGURE 6.
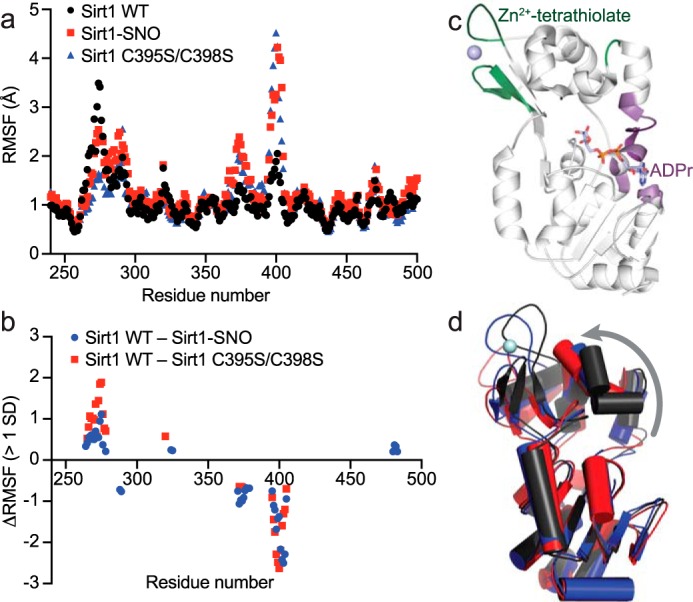
Molecular dynamics simulations support an allosteric mechanism of Sirt1 inhibition by S-nitrosation. 100-ns molecular dynamics simulations of Sirt1 WT, Sirt1-SNO, and Sirt1 C395S/C398S were performed using the Schrödinger Maestro molecular dynamics modeling platform. a, RMSF of each residue was calculated using Schrödinger Maestro and plotted against residue number. b, RMSF values of Sirt1-SNO and Sirt1 C395S/C398S were subtracted from Sirt1 WT RMSF values. ΔRMSF values within 1 S.D. of the average difference in RMSF were excluded, revealing localized regions of significant dynamic change between wild-type and modified Sirt1. c, differences in Sirt1-SNO or Sirt1 C395S/C398S RMSF outside of 1 S.D. plotted in b were mapped to the crystal structure of Sirt1 with ADP-ribose bound (PDB code 4KXQ (33)). Residues colored green indicate predicted regions of increased dynamics in Sirt1-SNO and Sirt1 C395S/C398S. Residues colored purple indicate predicted regions of increased rigidity in Sirt1-SNO and Sirt1 C395S/C398S. d, aligned end point structures from the Sirt1 WT (black), Sirt1-SNO (blue), and Sirt1 C395S/C398S (red) molecular dynamics simulations. The gray arrow represents the observed tilting away of the Zn2+-tetrathiolate subdomain relative to the Rossmann-fold subdomain in Sirt1-SNO and Sirt1 C395S/C398S compared with Sirt1 WT.
Discussion
Several recent studies have demonstrated a correlation between S-nitrosation of Sirt1 and increased acetylation of the Sirt1 deacetylase substrates PGC-1α, p53, and p65 (4, 5). Although these studies revealed a potential link between S-nitrosation and inhibition of cellular sirtuin activity, the precise mechanism of Sirt1 inhibition was unknown. Titration of GSNO, NO, H2O2, H2O2/GSH, and GSSG revealed selective inhibition by GSNO and to a lesser extent by NO (Fig. 1b). Inhibition by GSNO and NO correlated with the extent of S-nitrosation but not glutathionylation or sulfenylation (Fig. 1, d–f) suggesting Sirt1 deacetylase activity can be directly inhibited by S-nitrosation. The ability of Sirt1 to be inhibited by glutathionylation cannot be completely excluded as the Sirt1 construct for recombinant expression and purification utilized in these studies lacks Cys-67, a previously identified site of glutathionylation (6). However, the physiological relevance of the Cys-67 glutathionylation site is unclear as this site was identified upon treatment of Sirt1 with 2 mm GSNO (6), several orders of magnitude higher than the low micromolar GSNO concentrations measured in cells (34). Furthermore, Sirt1 deacetylation was inhibited more potently by GSNO than NO, consistent with the extent of S-nitrosation (Fig. 1, b and f). GSNO can directly transnitrosate protein thiols, whereas NO must undergo a net one-electron oxidation to form a nitrosothiol (25). The greater inhibitory potency of GSNO suggests transnitrosation rather than chemical S-nitrosation by NO may be the more relevant mechanism of Sirt1 S-nitrosation and subsequent inhibition under physiological conditions. Although GSNO is a known cellular nitrosothiol, GSNO is not the only potential cellular Sirt1 transnitrosating agent. Recent work has pointed to the presence of numerous cellular protein and low molecular weight nitrosothiols, including S-nitrosocysteine (35), S-nitrosocoenzyme A (36), S-nitrosothioredoxin (37), and S-nitroso-GAPDH (25). GAPDH in particular has been implicated as a Sirt1-transnitrosating agent (4).
Previous mutagenesis studies localized the sites of Sirt1 S-nitrosation to two of the Sirt1 Zn2+-tetrathiolate cysteine residues, Cys-395 and Cys-398 (4, 5). Similarly, an increase in cellular sirtuin deacetylase activity has been observed when the other two tetrathiolate cysteine residues, Cys-371 and Cys-374, are maintained in a reduced state (38). When Sirt1 was treated with GSNO, a concentration-dependent increase in the rate of Zn2+ release was observed (Fig. 2c) that correlated with both decreased deacetylase activity (Fig. 1b) and increased S-nitrosation (Fig. 1, c and f) consistent with a previous study (5). Furthermore, the C395S/C398S double mutant as well as the individual tetrathiolate cysteine mutants were not transnitrosated by GSNO, exhibited deacetylase activity near background levels, and did not bind Zn2+ (Fig. 2, a and b). Moreover, reversal of S-nitrosation with reductants was insufficient to recover Sirt1 deacetylase activity, and full recovery of deacetylase activity required both nitrosothiol reduction and the presence of Zn2+ to reform the Zn2+-tetrathiolate (Fig. 3) (5). Taken together, these data indicate that Zn2+-tetrathiolate S-nitrosation is primarily responsible for the observed Sirt1 inhibition and that S-nitrosation of any one of the four Zn2+-tetrathiolate cysteine residues is sufficient to release Zn2+ and inhibit Sirt1 deacetylase activity (Fig. 7).
FIGURE 7.

Proposed mechanism of Sirt1 inhibition by S-nitrosation. S-Nitrosation of Sirt1 occurs at one or more cysteines within the Zn2+-tetrathiolate, resulting in loss of Zn2+ coordination and loss of α-helical secondary structure. Local conformational changes are allosterically translated to the Sirt1 substrate-binding sites where acetyl-lysine and NAD+ binding and deacetylation are inhibited through rotation of the α-helical Zn2+-binding subdomain (green) away from the substrate-binding domain (blue) as indicated by the gray arrow. S-Nitrosation is reversible in the presence of reductants (e.g. GSH), and deacetylase activity is recoverable upon restoration of the Zn2+-tetrathiolate in the presence of Zn2+ and reductants.
Several steps along the sirtuin-catalyzed deacetylation mechanism may lead to inhibition, including binding or positioning of the NAD+ or acetyl-lysine substrates, a subsequent chemical step (27), or product release (39). As the Sirt1 Zn2+-tetrathiolate subdomain is structurally adjacent to the NAD+ and acetyl-lysine substrate-binding sites (Fig. 1a), loss of Zn2+ coordination upon S-nitrosation of the tetrathiolate may result in structural alterations that allosterically inhibit Sirt1 substrate binding and subsequent catalysis. Comparison of the steady-state kinetics of S-nitrosated and wild-type Sirt1 revealed an overall decrease in the kcat value with no significant effect on the Km values for either the acetyl-lysine or NAD+ substrates (Fig. 4, a and b). The ITC binding data (Fig. 4, c and d) supports a model where S-nitrosated Sirt1 is unable to bind either the acetyl-lysine or NAD+ substrate and therefore unable to catalyze deacetylation. This model is further supported by the observation that Sirt1 C395S/C398S was completely unable to bind acetyl-lysine and NAD+ substrate analogues (Fig. 4, c and d) and unable to catalyze deacetylation significantly above background levels (Fig. 4a).
S-Nitrosation of Sirt1 allosterically inhibits substrate binding likely through alterations in secondary and tertiary structure upon loss of Zn2+ from the Zn2+-tetrathiolate that are translated to the substrate-binding pockets. Indeed, a significant loss in overall α-helical content of S-nitrosated Sirt1 and Sirt1 C395S/C398S was observed (Fig. 5, a and b) consistent with local structural unraveling of the α-helical Zn2+-tetrathiolate subdomain leading to an allosteric mechanism of Sirt1 inactivation. Interestingly, the loss in α-helical character did not result in a decrease in overall thermal stability of S-nitrosated Sirt1 or Sirt1 C395S/C398S (Fig. 5c). Instead, disruption of the Zn2+-tetrathiolate stabilized the protein (Fig. 5c), potentially due to an increase in β-sheet secondary structure (Fig. 5, a and b). Two regions of Sirt1 exhibited changes in dynamics upon Zn2+-tetrathiolate S-nitrosation (Fig. 6). In particular, the residues surrounding the Zn2+-tetrathiolate exhibited increased dynamics likely due to release of the conformational constraints of Zn2+ ligation. In contrast, residues surrounding the binding site of the adenosine portion of NAD+, distal to the Zn2+-tetrathiolate, exhibited decreased dynamics, consistent with the inability of Sirt1 to undergo the necessary conformational changes to bind or position substrates when S-nitrosated.
Several lines of evidence suggest that S-nitrosation may be a reversible signaling mechanism that responds to cellular nitrosative stress. Intriguingly, physiologically relevant GSH concentrations were able to reduce S-nitrosation of Sirt1 in a concentration-dependent manner (Fig. 3e), and Sirt1 deacetylase activity was completely recovered in the presence of Zn2+ (Fig. 3d). These data support S-nitrosation as a reversible mechanism to regulate cellular Sirt1 deacetylase activity. Zn2+-tetrathiolate reconstitution likely requires an uncharacterized process of Zn2+ insertion similar to initial Zn2+ insertion after Sirt1 protein synthesis. The ability of GSH to reverse S-nitrosation in vivo will likely also be highly dependent on the local GSH concentration. Under normal conditions cellular GSH concentrations range from 1 to 5 mm (40). As GSH concentrations in excess of 1 mm were required to fully reverse Sirt1 S-nitrosation and recover Sirt1 deacetylase activity (Fig. 3, d and e), cellular GSH concentrations would likely need to fall into the micromolar range for significant Sirt1 S-nitrosation and inhibition to occur. This drop in GSH concentrations occurs under conditions of oxidative or nitrosative stress such as macrophage activation (41), ischemia/reperfusion (42), and endotoxemia (43, 44). Under these and other related nitrosative/oxidative stress conditions, it may be of benefit to the cell to inhibit Sirt1 deacetylation of Sirt1 substrates such as p53. For instance, if genomic damage exceeds the ability of the cell to repair its DNA, then inhibition of Sirt1 deacetylase activity by S-nitrosation would increase p53 acetylation and promote apoptosis (45). Interestingly, we recently observed a similar dependence on physiological GSH concentrations for iNOS inhibition by S-nitrosation of the iNOS Zn2+-tetrathiolate (46).
In summary, we have demonstrated that Sirt1 is directly modified and inhibited by cysteine S-nitrosation. S-Nitrosation of a single cysteine with the Sirt1 Zn2+-tetrathiolate is likely sufficient to result in release of Zn2+ and local alteration of secondary and tertiary structure to allosterically inhibit binding to both the acetyl-lysine and NAD+-binding pockets (Fig. 7). The loss of substrate binding results in complete inhibition of Sirt1 deacetylase activity upon S-nitrosation. Treatment of Sirt1 with physiologically relevant concentrations of the primary cellular reductant GSH in the presence of Zn2+ results in nitrosothiol reduction and full recovery of Sirt1 deacetylase activity. Although our data are consistent with S-nitrosation as the relevant modification for GSNO-mediated inhibition, further work is necessary to directly compare the relative abundance and importance of Sirt1 S-nitrosation and other oxidative cysteine modifications such as glutathionylation in cellular contexts. Furthermore, pharmacological prevention of the accumulation of Sirt1 S-nitrosation during aging (5) may restore Sirt1 activity and protect against aging-related diseases, including neurodegeneration, cardiovascular disease, and type II diabetes.
Experimental Procedures
Materials
Acetonitrile, ADP-ribose, type II (l)-glutamic dehydrogenase (from bovine liver), dithiothreitol, hydrogen peroxide, NAD+, sodium hydrosulfide, and trifluoroacetic acid were purchased from Sigma. DEA-NONOate and GSNO were purchased from Cayman Chemical (Ann Arbor, MI). Fmoc amino acids, reduced glutathione, oxidized glutathione, α-ketoglutaric acid, NADH, and Zincon monosodium were purchased from Chem-Impex (Wood Dale, IL). Rink-amide 4-methylbenzhydrylamine resin was purchased from Novabiochem. Ni-NTA superflow resin was purchased from 5 PRIME (Hilden, Germany). Micro BioSpin-6 and ENrich SEC 650 10 × 300-mm columns were purchased from Bio-Rad. GSNO stocks were made in HDN (200 mm HEPES, pH 7.7, 1 mm diethylenetriaminepentaacetic acid, 0.1 mm neocuproine) or HEN (200 mm HEPES, pH 7.7, 1 mm EDTA, 0.1 mm neocuproine).
Solid-phase Peptide Synthesis
An 11-mer acetyl-lysine H3 peptide (H3K14ac: H2N-KSTGGK(acetyl)APRKQ-NH2), a 7-mer trifluoroacetyl-lysine p53 peptide (p53tfa: H2N-RHKK(trifluoroacetyl)LMF-NH2), and a 5-mer p53-based peptide (p53W: acetyl-RHKK(acetyl)W-NH2) were synthesized using standard tert-butyl/Fmoc solid-phase peptide synthesis techniques (47) on an Applied Biosystems ABI 433A peptide synthesizer (FastMoc 0.10 mmol). Fmoc amino acids were coupled to 100–200-mesh Rink-amide 4-methylbenzhydrylamine resin. The protecting groups used were as follows: Boc for lysine and tryptophan; for histidine; tert-butyl for serine and threonine; and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl for arginine. Each amino acid was coupled using ∼10 eq of activated amino acid (9 eq of 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, 9 eq of hydroxybenzotriazole, and 20 eq of N,N-diisopropylethylamine). The acetyl- and trifluoroacetyl-lysine derivatives were synthesized from their corresponding commercially available Fmoc-Lys (Ac)-OH and Fmoc-Lys(trifluoroacetyl)-OH building blocks. After completion of the synthesis, the resin was rinsed with dichloromethane and dried. The full-length peptide was then deprotected and cleaved from the resin with 95% v/v TFA, 2.5% v/v H2O, and 2.5% v/v triisopropylsilane. The peptide was precipitated with cold (−20 °C) diethyl ether and pelleted by centrifugation washing twice with cold diethyl ether. The precipitate was dried, redissolved in water, and lyophilized. Crude peptides were purified by semipreparative HPLC on a μBondapak C18 column (Waters, 3.9 × 300 mm) using an Agilent 1100 series HPLC eluting with a gradient of 0–80% v/v acetonitrile in water with 0.1% v/v TFA. Fractions collected were lyophilized to yield final peptides as dry white powders. Masses of the cleaved peptides were confirmed by direct injection electrospray ionization-mass spectrometry (QExactive, Thermo Scientific). The observed mass for each peptide matched the predicted mass (H3K14ac, calculated for C50H93N19O15 [M + 2H]2+: 599.85438, found 599.85461; p53tfa, calculated for C46H75F3N15O8S [M + H]+: 1054.55904, found 1054.56058; p53Wac, calculated for C39H61N14O7 [M + H]+: 837.48422, found 837.48619). Peptide concentrations were determined from the weight of the peptide TFA salt assuming 1 eq of TFA per cationic group.
Sirt1 Site-directed Mutagenesis
Plasmids encoding Sirt1 C371S, C374S, C395S, C398S, and C395S/C398S were generated by site-directed mutagenesis on a pET28a-LIC vector coding for wild-type Sirt1, amino acids 156–664. Primers were purchased from Integrated DNA Technologies (Coralville, IA) (supplemental Table 1). Mutants were confirmed by DNA sequencing (Retrogen, San Diego, or Functional Biosciences, Madison, WI).
Expression and Purification of Nicotinamidase
MBP-tagged nicotinamidase (pTEV6 vector) was purified from BL21(DE3) Escherichia coli by nickel affinity chromatography as described previously (19). Transformed cells were grown at 37 °C in 2× YT media supplemented with 50 mg/liter ampicillin to an absorbance of 0.7 at 600 nm. Protein expression was induced overnight with 0.5 mm IPTG at 25 °C. Cells were harvested by centrifugation at 5,000 × g and frozen at −80 °C. Frozen cell pellets were thawed on ice and resuspended in 20 mm potassium phosphate, pH 7.5, containing 500 mm NaCl and 5 mm imidazole. Cells were lysed via sonication and insoluble debris pelleted by centrifugation at 30,000 × g. Cleared lysate was applied to Ni-NTA resin (0.5 ml of Ni-NTA resin/liter culture) and rocked at 4 °C for 1 h. Bound nickel resin was applied to a column and washed with 10 column volumes of lysis buffer, followed by 10 column volumes of 20 mm potassium phosphate, pH 7.5, containing 500 mm NaCl and 25 mm imidazole. Purified MBP-PncA was eluted from the column in 5 column volumes of 20 mm potassium phosphate, pH 7.5, containing 500 mm NaCl, and 300 mm imidazole. Nicotinamidase was further purified by size exclusion chromatography on an ENrich SEC 650 10 × 300-mm column eluting with 50 mm potassium phosphate, pH 7.5, containing 100 mm NaCl and 10% v/v glycerol. Purified nicotinamidase was concentrated, aliquoted, flash-frozen in liquid N2, and stored at −80 °C until use.
Expression and Purification of GAPDH
GST-tagged GAPDH (pGEX 6P-3 vector) was purified from BL21(DE3) E. coli. Transformed cells were grown at 37 °C in TB media supplemented with 50 mg/liter ampicillin to an absorbance of 0.6 at 600 nm. Protein expression was induced overnight by 1 mm IPTG at 20 °C. Cells were harvested by centrifugation at 5,000 × g and frozen at −80 °C. Frozen cell pellets were thawed at room temperature and resuspended into PBS, pH 7.4, supplemented with 1 mg/ml lysozyme and 1 μg/ml DNase I. Cells were lysed via sonication, and insoluble debris was pelleted by centrifugation at 30,000 × g. Cleared lysate was applied to glutathione-agarose resin (Pierce) and rocked at 4 °C for 1 h. Protein-bound resin was washed with 30 bead bed volumes of PBS, pH 7.4. The resin-bound protein was resuspended in PreScission protease cleavage buffer (50 mm Tris, pH 8, 100 mm NaCl, 1 mm EDTA, 1 mm DTT), and PreScission protease (GE Healthcare) was added at 50 units/ml buffer volume. The mixture was rocked at 4 °C for 2.5 h to cleave GAPDH from the resin. Cleaved GAPDH was further purified by size exclusion chromatography on an S-75 size exclusion column (GE Healthcare), eluting with 50 mm HEPES, pH 7.7, 100 mm NaCl, 5% v/v glycerol. Purified protein was concentrated with a 10,000 molecular weight cutoff spin concentrator (Sartorius), aliquoted, flash-frozen in liquid N2, and stored at −80 °C until use.
Expression and Purification of Sirt1
Recombinant Sirt1 WT(156–664) (pET28a-LIC) and tetrathiolate mutants were purified from BL21(DE3) E. coli by nickel affinity chromatography. Transformed cells were grown at 37 °C in 2× YT media supplemented with 50 mg/liter kanamycin to an absorbance of 0.7 at 600 nm. Protein expression was induced overnight with 0.5 mm IPTG at 16 °C. Cells were harvested by centrifugation at 5,000 × g and frozen at −80 °C. Frozen cell pellets were thawed on ice and resuspended in 20 mm Tris, pH 8.0, containing 500 mm NaCl, 10% v/v glycerol, 5 mm β-mercaptoethanol, and 2.5 mm imidazole. Cells were lysed via sonication, and insoluble debris was pelleted by centrifugation at 30,000 × g. Cleared lysate was applied to Ni-NTA resin and rocked at 4 °C for 1 h. Bound nickel resin was applied to a column and washed with 10 column volumes of lysis buffer, followed by 10 column volumes of 20 mm potassium phosphate, pH 7.5, containing 500 mm NaCl and 25 mm imidazole. Purified Sirt1 was eluted from the column with 5 column volumes of 20 mm Tris, pH 8.0, containing 500 mm NaCl, 10% v/v glycerol, 5 mm β-mercaptoethanol, and 300 mm imidazole. Sirt1 was further purified by size exclusion chromatography on an ENrich SEC 650 10 × 300-mm column eluting with 10 mm HEPES, pH 7.5, containing 150 mm NaCl, 10% v/v glycerol, and 1 mm DTT. Purified Sirt1 was concentrated, aliquoted, flash-frozen in liquid N2, and stored at −80 °C until use.
Sirt1 Enzymatic Activity Assays
Sirt1 activity was monitored under initial rate conditions using a continuous coupled assay for nicotinamide-producing enzymes as described previously (19). In general, the assay was performed in 20 mm potassium phosphate, pH 7.5, at 25 °C in a reaction mixture containing NAD+, H3K14ac, 0.5 μm Sirt1, 2 μm MBP-PncA, 3.3 mm α-ketoglutarate, 200 μm NADH, and 2.5 units of (l)-glutamic dehydrogenase. Rates were monitored continuously at 340 nm for 10 min in a clear flat-bottom 96-well plate (Greiner Bio-One) on a BioTek Synergy Mx microplate reader (Winooski, VT).
For comparison of kinetic parameters between wild-type and S-nitrosated Sirt1, 2 μm Sirt1 was treated with 20 mm potassium phosphate, pH 7.5, or 100 μm GSNO diluted in phosphate buffer in the dark for 1 h at 37 °C. Michaelis-Menten titrations were performed by varying the concentration of either H3K14ac or NAD+ at saturating concentrations of the second substrate (1.4 mm H3K14ac or 2 mm NAD+). Reactions were initiated by the addition of NAD+. The resulting curves were fit to the Michaelis-Menten equation, and kcat and Km values were calculated using GraphPad Prism (La Jolla, CA).
Concentration-dependent oxidant responses were performed by treating 2 μm Sirt1 with varying concentrations of each oxidant (GSNO, DEA-NONOate, and GSSG) for 1 h at 37 °C. Deacetylation rates were determined at subsaturating concentrations of both H3K14ac (100 μm) and NAD+ (500 μm). Reactions were initiated by the addition of oxidant-treated Sirt1. The resulting rates were fit to Equation 1 using GraphPad Prism (La Jolla, CA),
| (Eq. 1) |
where [Oxidant] is the concentration of GSNO, DEA-NONOate, or GSSG in the assay.
For DTT/Zn2+ oxidation reversal experiments, 2 μm Sirt1 was treated with 20 mm potassium phosphate, pH 7.5, or 100 μm GSNO diluted in phosphate buffer for 1 h at 37 °C in the dark. Buffer or GSNO-treated Sirt1 (diluted to 0.5 μm) was then added to the reaction mixture and treated with or without 10 mm DTT and 1 μm ZnCl2 for 5 min at 37 °C. Reactions were observed at subsaturating concentrations of H3K14ac (100 μm) and NAD+ (500 μm) and initiated by the addition of NAD+.
Reversal of inhibition with DTT and Zn2+ was also confirmed using an HPLC assay. 2.5 μm Sirt1 was treated with 20 mm potassium phosphate, pH 7.5, or 60 μm GSNO diluted in phosphate buffer for 30 min at 37 °C in the dark. Buffer- or GSNO-treated Sirt1 was then treated with 1–10 μm ZnCl2 and 5 mm DTT. Treated Sirt1 was added to a reaction mixture containing 20 mm potassium phosphate, pH 7.5, 100 μm p53Wac, and 500 μm NAD+. After 30 min, reactions were quenched with 1% v/v final concentration of TFA. Samples were pelleted at 20,800 × g prior to HPLC analysis to remove particulates. Acetylated and deacetylated p53W peptides were separated on a C18 column (Thermo Scientific 25005–254630) using an Agilent 1100 series HPLC. A gradient of 0–40% v/v acetonitrile in water with 0.1% v/v TFA was applied over 20 min where deacetylated p53W eluted at 15 min and p53Wac eluted at 16.4 min (confirmed using acetylated and deacetylated p53W standards). The area under the curve at 280 nm was integrated and utilized to calculate percent conversion of the acetylated peptide.
For the GSH S-nitrosation reversal experiments, 2 μm Sirt1 was treated with 20 mm potassium phosphate, pH 7.5, or 100 μm GSNO diluted in phosphate buffer for 1 h at 37 °C in the dark. GSNO or buffer-treated Sirt1 was then incubated with varying concentrations of GSH in the presence of 1 μm ZnCl2 at 37 °C for 5 min. Rates were observed at subsaturating conditions of both H3K14ac (100 μm) and NAD+ (500 μm) substrates and initiated with the addition of NAD+.
Isothermal Titration Calorimetry
Equilibrium binding constants were determined in the presence and absence of GSNO using a MicroCal VP-ITC isothermal titration calorimeter. For all ITC experiments, Sirt1 was pre-treated with 20 mm potassium phosphate, pH 7.5, or 100 μm GSNO at 37 °C for 1 h in the dark. Peptide Kd values were determined by titrating 200 μm p53tfa peptide (syringe) into ∼12 μm Sirt1 (cell) in 10 mm HEPES, pH 7.5, containing 150 mm NaCl and 10% v/v glycerol. ADP-ribose Kd values were determined by titrating 400 μm (determined spectrophotometrically using an extinction coefficient at 260 nm of 13.5 mm−1 cm−1 (48)) ADP-ribose (syringe) into ∼6 μm Sirt1 (cell), 10 mm HEPES, pH 7.5, containing 150 mm NaCl and 10% v/v glycerol. For p53tfa and the ADP-ribose Sirt1 C395S/C398S titrations, an initial 2-μl injection followed by 30 injections of 8 μl was used. For ADP-ribose control-treated and GSNO-treated Sirt1 WT titrations, an initial 2-μl injection was followed by 15 injections of 4 μl and then 26 injections of 8 μl. Data were transformed and fit to one set of sites using MicroCal LLC ITC software. Protein concentration was normalized to generate an n-value of 1 for control-treated Sirt1.
Circular Dichroism
Sirt1 (14 μm) in 20 mm potassium phosphate, pH 7.5, was treated with 20 mm potassium phosphate, pH 7.5, or 100 μm GSNO diluted in phosphate buffer for 1 h at 37 °C in a total volume of 100 μl. Treated protein was then desalted using a Micro BioSpin-6 column pre-equilibrated with 20 mm potassium phosphate, pH 7.5. Desalted Sirt1 was diluted to a final volume of 400 μl (3.5 μm Sirt1) in 20 mm potassium phosphate, pH 7.5, and pelleted at 16,100 × g for 5 min at 4 °C to remove particulates. Circular dichroism spectra were measured using a Jasco J-710 spectropolarimeter in a 0.1-cm pathlength quartz cuvette scanning from 190 to 280 nm at 0.1 nm resolution. CD spectra were analyzed using Dichroweb analysis software (49, 50), applying the CDSSTR algorithm using data set 4.
Thermal Shift Assay
Sirt1 (8 μm) in 20 mm potassium phosphate, pH 7.5, was treated with 100 μm GSNO, DEA-NONOate, or 20 mm potassium phosphate for 1 h at 37 °C in a total volume of 75 μl. Treated protein was then desalted using a Micro BioSpin-6 column pre-equilibrated with 20 mm potassium phosphate, pH 7.5. SYPRO Orange dye was added to a final concentration of 5× in 90-μl total volume. 30 μl of each reaction was added to a PCR plate, and melting curves were generated by monitoring SYPRO Orange fluorescence in FRET mode over a temperature gradient of 20–95 °C over 50 min using a CFX96 real time system (Bio-Rad). Fluorescence values before the minimum and after the maximum fluorescence were excluded from curve fitting, and the resulting curve was fit to Boltzmann Equation 2 using GraphPad Prism (La Jolla, CA),
| (Eq. 2) |
where UL and LL are the maximum and minimum fluorescence values; a is the slope of the curve within the melting region; and Tm is the melting temperature. Differences in melting temperature were analyzed for significance by one-way ANOVA and Tukey post-test.
Zinc Release Assay
Zinc release assays for Sirt1 WT were performed by treating Sirt1 (7.5 μm) with increasing concentrations of GSNO in the presence of 40 μm Zincon in 100 mm HEPES, pH 7.7, at 37 °C. Zn2+ release from Sirt1 was measured by continuously monitoring changes in absorbance at 529 and 620 nm for 30 min using a BioTek Synergy Mx microplate reader. Rates were determined by plotting the difference in absorbance between 620 and 529 nm versus time. The slope of each line was transformed into a rate (nm Zn2+/s) using a standard curve generated with ZnCl2 in the presence of BSA and GSNO. Relative Zn2+ content of Sirt1 WT, C371S, C374S, C395S, C398S, and C395S/C398S was assessed by treating 40 μm Sirt1 with 1.5 mm iodoacetamide for 10 min at 100 °C. Samples were spun at 20,800 × g to remove precipitated protein. Zn2+-containing supernatant was added to 100 mm HEPES, pH 7.7, containing 40 μm Zincon. Zn2+ release from Sirt1 was detected by measuring absorbance at 529 and 620 nm using a BioTek Synergy Mx microplate reader.
Biotin Switch Assay
Biotin switch assays were performed under low ambient light conditions. Sirt1 (20 μm) was incubated with 100 μm TCEP for 15 min at room temperature and then desalted with a Micro BioSpin-6 column. Desalted protein was incubated with 6–100 μm GSNO, 100 μm GSH, or the molar equivalent of DEA-NONOate for 60 min at 37 °C in HDN (250 mm HEPES, pH 7.7, 1 mm diethylenetriaminepentaacetic acid, 0.1 mm neocuproine) in a 100-μl total volume. In oxidation reversal experiments, this step was followed by immediate addition of 0.3–100 mm GSH or DTT and incubation for 5 min at 37 °C. Free thiols were alkylated with an equal volume of blocking buffer (HDN with 1% w/v SDS and 200 mm iodoacetamide) for 1 h at 37 °C. Proteins were precipitated with 1.2 ml of cold (−20 °C) high purity acetone and incubated at −80 °C overnight. Precipitated protein was pelleted at >12,000 × g at 4 °C, and the supernatant was discarded. Pellets were washed once more with 1 ml of ice-cold acetone and pelleted by centrifugation, and the supernatant was removed. Pellets were air-dried for 15 min at room temperature and then resuspended into label buffer (PBS with 1% w/v SDS, 30 mm ascorbate, 200 μm EZ-link biotin iodoacetamide). In a control condition, ascorbate was omitted from the reduction and labeling step. After 1 h at 37 °C, the reactions were quenched with 6× Laemmli Sample Buffer (LSB) containing 10% v/v β-mercaptoethanol. 30 μl of each sample was loaded onto a Bio-Rad Any kDa TGX Stain-Free gel. Equal protein loading was verified by imaging under UV light on a ChemiDoc MP gel imager (Bio-Rad). Proteins were transferred to a nitrocellulose membrane using a TransBlot Turbo semi-dry transfer system (Bio-Rad). The membrane was blocked for 1 h at room temperature in PBS containing 1% v/v Tween 20 (PBST) and 2.5% w/v dry milk. The membrane was washed three times for 5 min with PBST and then rocked for 1 h at room temperature with the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) where reagents A and B were diluted into 15 ml of PBST. The membrane was again washed three times for 5 min in PBST. The membrane was imaged after a 2-min incubation in chemiluminescence buffer (50 mm Na2HPO4, 50 mm NaHCO3, 150 mm NaCl, 10 mm NaBO3, 225 μm p-coumaric acid, 1.5 mm luminol, and 0.6% v/v H2O2).
Anti-glutathionylation Blots
Purified Sirt1 WT protein was incubated with 100 μm TCEP for 15 min at room temperature and then desalted with a Micro BioSpin-6 column (Bio-Rad). Desalted 2.5 μm Sirt1 was incubated with 0–1000 μm freshly dissolved GSNO (concentration confirmed by A336 nm (ϵ = 0.9 mm−1 cm−1) at 37 °C for 1 h in HDN buffer. Purified GAPDH was used as a positive control for glutathionylation. GAPDH (2.5 μm) was treated with 100 μm GSH and 100 μm H2O2 as described previously (23). The Sirt1 samples were subsequently split into 2 aliquots. One aliquot was analyzed for S-nitrosation by the biotin switch assay. The other aliquot was alkylated with 20 mm N-ethylmaleimide for 5 min at 37 °C. Non-reducing Laemmli sample buffer was added, and samples were resolved by SDS-PAGE, transferred to nitrocellulose, and blocked in PBST with 2.5% w/v milk. The membrane was incubated with mouse anti-glutathione antibody (Virogen catalogue no. 101-A, lot 58) diluted 1:1,000 in PBST with 2.5% w/v milk for 16 h at 4 °C, washed three times with PBST, and then incubated with anti-mouse HRP secondary antibody diluted 1:5,000 in PBST with 2.5% w/v milk for 1 h at room temperature. After three PBST washes, the membrane was imaged with chemiluminescence, and a Stain-Free gel image of samples treated with reducing Laemmli sample buffer was used as a loading control.
In Vitro Sulfenylation Assays
The in vitro sulfenylation protocol was adapted from Truong and Carroll (24). Purified Sirt1 and GAPDH proteins (75 μm) were reduced with 1 mm TCEP for 15 min at room temperature and then desalted with Micro BioSpin-6 columns pre-equilibrated with Tris labeling buffer (50 mm Tris, pH 7.4, 150 mm NaCl). Desalted GAPDH protein (25 μm) was treated with 0 or 100 μm H2O2 and 0 or 1 mm DYn-2 for 1 h at 37 °C in Tris labeling buffer. Desalted Sirt1 was treated with 0 or 100 μm GSNO and 0 or 1 mm DYn-2 for 1 h at 37 °C in Tris labeling buffer. The proteins were desalted a second time with Micro BioSpin-6 columns pre-equilibrated with click label buffer (50 mm triethanolamine, pH 7.4, 1% w/v SDS). Desalted proteins were treated with 100 μm tetramethylrhodamine azide (Thermo Fisher catalogue no. T10182), 1 mm TCEP, 100 μm tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, and 1 mm CuSO4 for 1 h at room temperature, protected from light and under constant agitation. Reactions were quenched and proteins precipitated with the addition of 1.3 ml of ice-cold MeOH and incubated for 18 h at −80 °C. Proteins were pelleted at 16,000 × g for 30 min at 4 °C; the supernatant was discarded and pellets were washed with 1.3 ml of ice-cold MeOH and pelleted by centrifugation. The supernatant was discarded. The resulting pellets were dried by leaving the tubes open for 30 min at room temperature and covered to protect nitrosothiols from light. Pellets were resuspended into 1× Laemmli sample buffer with 10% v/v β-mercaptoethanol. Samples were boiled and resolved on a 10% SDS-polyacrylamide gel. The gel was incubated twice for 10 min in destain solution (40% v/v MeOH, 10% v/v acetic acid, 50% v/v water) and then equilibrated in water for 10 min before imaging on a Typhoon Trio Visible Mode Imager using an excitation wavelength of 532 nm and an emission wavelength of 580 nm for tetramethylrhodamine. Following in-gel fluorescence detection, the gel was incubated for 30 min at room temperature in destain solution and then incubated for 18 h at room temperature with SYPRO Ruby stain. The gel was washed twice for 15 min in SYPRO Ruby wash solution (10% v/v MeOH, 7% v/v acetic acid, 83% v/v water). The gel was washed once for 10 min in water, and SYPRO Ruby signal was imaged on a Typhoon Trio Visible Mode Imager using an excitation wavelength of 532 nm and an emission wavelength of 610 nm for SYPRO Ruby.
Molecular Dynamics Simulations
Conformational dynamics of Sirt1 WT, S-nitrosated Sirt1 (Sirt1-SNO), and Sirt1 C395S/C398S were simulated using the Schrödinger Maestro molecular dynamics modeling suite using the OPSL3 force field. The Sirt1 crystal structure (PDB code 4KXQ (33)) was preprocessed using the protein preparation window to remove ligands and add hydrogens. For Sirt1-SNO and Sirt1 C395S/C398S, cysteines 395 and 398 were modified by S-nitrosation or mutated to serine, and Zn2+ was removed using the build tool following preprocessing. The most likely protonation state for each cysteine residue at pH 7.0 ± 2.0 was calculated by the ligand preparation tool using Epik empirical pKa prediction (51, 52). The protein was placed in a neutral simple point charge environment containing 0.15 m sodium chloride using the molecular dynamics system setup window. After adding an aqueous shell, a 100-ns molecular dynamics simulation was performed following initial relaxation (energy minimization) of the system. Data summaries were generated using the molecular dynamics simulation interactions diagram function.
Author Contributions
K. S. K. expressed and purified Sirt1 and nicotinamidase, performed site-directed mutagenesis, and conducted deacetylase, Zn2+ release, isothermal titration calorimetry, thermofluor, and circular dichroism assays as well as the molecular dynamics simulations and related analyses. S. L. W.-S. purified GAPDH and performed site-directed mutagenesis, biotin switch assays, DYn-2 sulfenylation assays, and anti-glutathionylation immunoblots. M. D. O. synthesized and purified peptides, conducted isothermal titration calorimetry, and assisted with the thermofluor and molecular dynamics analyses. B. C. S., S. L. W.-S., K. S. K., and M. D. O. conceived and designed the experiments, prepared figures, and wrote the manuscript. B. C. S. acquired funding and coordinated the study. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Anthony Brandt (Medical College of Wisconsin) for assistance in purifying Sirt1 and optimization of Zincon assays. We thank Trudy Holyst in the Protein Chemistry Core Lab at the BloodCenter of Wisconsin for assistance with peptide synthesis. We thank Dr. Kirkwood Pritchard (Medical College of Wisconsin) for the gift of ABI 433A peptide synthesizer. We thank the Office of Research and the Research Computing Center of Medical College of Wisconsin for help with Schrödinger and computational server resources.
This work was supported in part by American Heart Association Scientist Development Grant 15SDG25830057 (to B. C. S.), the Medical College of Wisconsin Research Affairs Committee (to B. C. S.), Institutional Research Grants 14-247-29-IRG and 86-004-26-IRG from the American Cancer Society (to B. C. S.), and the Advancing a Healthier Wisconsin Endowment (to B. C. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S3 and Table 1.
- iNOS
- inducible nitric-oxide synthase
- GSNO
- S-nitrosoglutathione
- Ni-NTA
- nickel-nitrilotriacetic acid
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- TCEP
- tris(2-carboxyethyl)phosphine
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- ADPr
- ADP-ribose
- RMSF
- root mean square fluctuation
- PDB
- Protein Data Bank
- ITC
- isothermal titration calorimetry
- ANOVA
- analysis of variance
- DEA
- diethylamine
- MBP
- maltose-binding protein.
References
- 1. Feldman J. L., Dittenhafer-Reed K. E., and Denu J. M. (2012) Sirtuin catalysis and regulation. J. Biol. Chem. 287, 42419–42427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith B. C., Hallows W. C., and Denu J. M. (2008) Mechanisms and molecular probes of sirtuins. Chem. Biol. 15, 1002–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sebastián C., Satterstrom F. K., Haigis M. C., and Mostoslavsky R. (2012) From sirtuin biology to human diseases: an update. J. Biol. Chem. 287, 42444–42452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kornberg M. D., Sen N., Hara M. R., Juluri K. R., Nguyen J. V., Snowman A. M., Law L., Hester L. D., and Snyder S. H. (2010) GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shinozaki S., Chang K., Sakai M., Shimizu N., Yamada M., Tanaka T., Nakazawa H., Ichinose F., Yamada Y., Ishigami A., Ito H., Ouchi Y., Starr M. E., Saito H., Shimokado K., et al. (2014) Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci. Signal. 7, ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zee R. S., Yoo C. B., Pimentel D. R., Perlman D. H., Burgoyne J. R., Hou X., McComb M. E., Costello C. E., Cohen R. A., and Bachschmid M. M. (2010) Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 13, 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shao D., Fry J. L., Han J., Hou X., Pimentel D. R., Matsui R., Cohen R. A., and Bachschmid M. M. (2014) A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 289, 7293–7306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodríguez-Ortigosa C. M., Celay J., Olivas I., Juanarena N., Arcelus S., Uriarte I., Marín J. J., Avila M. A., Medina J. F., and Prieto J. (2014) A GAPDH-mediated trans-nitrosylation pathway is required for feedback inhibition of bile salt synthesis in rat liver. Gastroenterology 147, 1084–1093 [DOI] [PubMed] [Google Scholar]
- 9. Caito S., Rajendrasozhan S., Cook S., Chung S., Yao H., Friedman A. E., Brookes P. S., and Rahman I. (2010) SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 24, 3145–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hwang J.-W., Yao H., Caito S., Sundar I. K., and Rahman I. (2013) Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 61, 95–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santos L., Escande C., and Denicola A. (2016) Potential modulation of sirtuins by oxidative stress. Oxid. Med. Cell. Longev. 2016, 9831825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ginnan R., Guikema B. J., Halligan K. E., Singer H. A., and Jourd'heuil D. (2008) Regulation of smooth muscle by inducible nitric-oxide synthase and NADPH oxidase in vascular proliferative diseases. Free Radic. Biol. Med. 44, 1232–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghisays F., Brace C. S., Yackly S. M., Kwon H. J., Mills K. F., Kashentseva E., Dmitriev I. P., Curiel D. T., Imai S.-I., and Ellenberger T. (2015) The N-terminal domain of SIRT1 is a positive regulator of endogenous SIRT1-dependent deacetylation and transcriptional outputs. Cell Rep. 10, 1665–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pan M., Yuan H., Brent M., Ding E. C., and Marmorstein R. (2012) SIRT1 contains N- and C-terminal regions that potentiate deacetylase activity. J. Biol. Chem. 287, 2468–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borra M. T., Smith B. C., and Denu J. M. (2005) Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 280, 17187–17195 [DOI] [PubMed] [Google Scholar]
- 16. Cao D., Wang M., Qiu X., Liu D., Jiang H., Yang N., and Xu R.-M. (2015) Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 29, 1316–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dai H., Case A. W., Riera T. V., Considine T., Lee J. E., Hamuro Y., Zhao H., Jiang Y., Sweitzer S. M., Pietrak B., Schwartz B., Blum C. A., Disch J. S., Caldwell R., Szczepankiewicz B., et al. (2015) Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 6, 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hubbard B. P., Gomes A. P., Dai H., Li J., Case A. W., Considine T., Riera T. V., Lee J. E., E SY, Lamming D. W., Pentelute B. L., Schuman E. R., Stevens L. A., Ling A. J., Armour S. M., et al. (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith B. C., Hallows W. C., and Denu J. M. (2009) A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 394, 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giustarini D., Dalle-Donne I., Colombo R., Milzani A., and Rossi R. (2008) Is ascorbate able to reduce disulfide bridges? A cautionary note. Nitric Oxide 19, 252–258 [DOI] [PubMed] [Google Scholar]
- 21. Monteiro G., Horta B. B., Pimenta D. C., Augusto O., and Netto L. E. (2007) Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc. Natl. Acad. Sci. U.S.A. 104, 4886–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paulsen C. E., Truong T. H., Garcia F. J., Homann A., Gupta V., Leonard S. E., and Carroll K. S. (2011) Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peralta D., Bronowska A. K., Morgan B., Dóka É., Van Laer K., Nagy P., Gräter F., and Dick T. P. (2015) A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 11, 156–163 [DOI] [PubMed] [Google Scholar]
- 24. Truong T. H., and Carroll K. S. (2012) Bioorthogonal chemical reporters for analyzing protein sulfenylation in cells. Curr. Protoc. Chem. Biol. 10.1002/9780470559277.ch110219 [DOI] [Google Scholar]
- 25. Smith B. C., and Marletta M. A. (2012) Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr. Opin Chem. Biol. 16, 498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith B. C., Settles B., Hallows W. C., Craven M. W., and Denu J. M. (2011) SIRT3 substrate specificity determined by peptide arrays and machine learning. ACS Chem. Biol. 6, 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith B. C., and Denu J. M. (2007) Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 46, 14478–14486 [DOI] [PubMed] [Google Scholar]
- 28. Smith B. C., and Denu J. M. (2007) Sir2 deacetylases exhibit nucleophilic participation of acetyl-lysine in NAD+ cleavage. J. Am. Chem. Soc. 129, 5802–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith B. C., and Denu J. M. (2007) Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J. Biol. Chem. 282, 37256–37265 [DOI] [PubMed] [Google Scholar]
- 30. Borra M. T., Langer M. R., Slama J. T., and Denu J. M. (2004) Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry 43, 9877–9887 [DOI] [PubMed] [Google Scholar]
- 31. Pan P. W., Feldman J. L., Devries M. K., Dong A., Edwards A. M., and Denu J. M. (2011) Structure and biochemical functions of SIRT6. J. Biol. Chem. 286, 14575–14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huynh K., and Partch C. L. (2015) Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 79, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davenport A. M., Huber F. M., and Hoelz A. (2014) Structural and functional analysis of human SIRT1. J. Mol. Biol. 426, 526–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seneviratne U., Godoy L. C., Wishnok J. S., Wogan G. N., and Tannenbaum S. R. (2013) Mechanism-based triarylphosphine-ester probes for capture of endogenous RSNOs. J. Am. Chem. Soc. 135, 7693–7704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Broniowska K. A., Zhang Y., and Hogg N. (2006) Requirement of transmembrane transport for S-nitrosocysteine-dependent modification of intracellular thiols. J. Biol. Chem. 281, 33835–33841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anand P., Hausladen A., Wang Y.-J., Zhang G.-F., Stomberski C., Brunengraber H., Hess D. T., and Stamler J. S. (2014) Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 111, 18572–18577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mitchell D. A., and Marletta M. A. (2005) Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat. Chem. Biol. 1, 154–158 [DOI] [PubMed] [Google Scholar]
- 38. Jung S.-B., Kim C.-S., Kim Y.-R., Naqvi A., Yamamori T., Kumar S., Kumar A., and Irani K. (2013) Redox factor-1 activates endothelial SIRTUIN1 through reduction of conserved cysteine sulfhydryls in its deacetylase domain. PLoS ONE 8, e65415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith B. C., and Denu J. M. (2006) Sir2 protein deacetylases: evidence for chemical intermediates and functions of a conserved histidine. Biochemistry 45, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meister A., and Anderson M. E. (1983) Glutathione. Annu. Rev. Biochem. 52, 711–760 [DOI] [PubMed] [Google Scholar]
- 41. Hothersall J. S., Cunha F. Q., Neild G. H., and Norohna-Dutra A. A. (1997) Induction of nitric oxide synthesis in J774 cells lowers intracellular glutathione: effect of modulated glutathione redox status on nitric-oxide synthase induction. Biochem. J. 322, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McKelvey T. G., Höllwarth M. E., Granger D. N., Engerson T. D., Landler U., and Jones H. P. (1988) Mechanisms of conversion of xanthine dehydrogenase to xanthine oxidase in ischemic rat liver and kidney. Am. J. Physiol. 254, G753–G760 [DOI] [PubMed] [Google Scholar]
- 43. Keller G. A., Barke R., Harty J. T., Humphrey E., and Simmons R. L. (1985) Decreased hepatic glutathione levels in septic shock. Predisposition of hepatocytes to oxidative stress: an experimental approach. Arch. Surg. 120, 941–945 [DOI] [PubMed] [Google Scholar]
- 44. Okabe H., Irita K., Taniguchi S., Kurosawa K., Tagawa K., Yoshitake J.-I., and Takahashi S. (1994) Endotoxin causes early changes in glutathione concentrations in rabbit plasma and liver. J. Surg. Res. 57, 416–419 [DOI] [PubMed] [Google Scholar]
- 45. Yi J., and Luo J. (2010) SIRT1 and p53, effect on cancer, senescence and beyond. BBA-Proteins Proteomics 1804, 1684–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith B. C., Fernhoff N. B., and Marletta M. A. (2012) Mechanism and kinetics of inducible nitric-oxide synthase auto-S-nitrosation and inactivation. Biochemistry 51, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen W. C., and White P. D. (eds) (2000) Fmoc Solid Phase Peptide Synthesis, pp. 9–76 Oxford University Press, New York [Google Scholar]
- 48. Schultheisz H. L., Szymczyna B. R., and Williamson J. R. (2009) Enzymatic synthesis and structural characterization of 13C,15N-poly(ADP-ribose). J. Am. Chem. Soc. 131, 14571–14578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whitmore L., and Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 51. Greenwood J. R., Calkins D., Sullivan A. P., and Shelley J. C. (2010) Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24, 591–604 [DOI] [PubMed] [Google Scholar]
- 52. Shelley J. C., Cholleti A., Frye L. L., Greenwood J. R., Timlin M. R., and Uchimaya M. (2007) Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.