

Abstract
A large percentage of redox-responsive gene promoters contain evolutionarily conserved guanine-rich clusters; guanines are the bases most susceptible to oxidative modification(s). Consequently, 7,8-dihydro-8-oxoguanine (8-oxoG) is one of the most abundant base lesions in promoters and is primarily repaired via the 8-oxoguanine DNA glycosylase-1 (OOG1)-initiated base excision repair pathway. In view of a prompt cellular response to oxidative challenge, we hypothesized that the 8-oxoG lesion and the cognate repair protein OGG1 are utilized in transcriptional gene activation. Here, we document TNFα-induced enrichment of both 8-oxoG and OGG1 in promoters of pro-inflammatory genes, which precedes interaction of NF-κB with its DNA-binding motif. OGG1 bound to 8-oxoG upstream from the NF-κB motif increased its DNA occupancy by promoting an on-rate of both homodimeric and heterodimeric forms of NF-κB. OGG1 depletion decreased both NF-κB binding and gene expression, whereas Nei-like glycosylase-1 and -2 had a marginal effect. These results are the first to document a novel paradigm wherein the DNA repair protein OGG1 bound to its substrate is coupled to DNA occupancy of NF-κB and functions in epigenetic regulation of gene expression.
Keywords: 8-oxoguanine (8-oxoG), 8-oxoguanine glycosylase (OGG1), NF-κB transcription factor, promoter, reactive oxygen species (ROS)
Introduction
Oxidatively modified DNA base lesions generated by reactive oxygen species (ROS)6 are genotoxic and mutagenic and implicated in various human pathologies (1). Although the levels of the base lesions in DNA vary according to sequence context, chromatin accessibility, and the nature of the oxidants, 7, 8-dihydro-8-oxoguanine (8-oxoG) is the most abundant lesion due to guanine's lowest redox potential among the four nucleobases (2, 3). 8-OxoG and its open-ringed product 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) are primarily repaired by 8-oxoguanine DNA glycosylase-1 (OGG1) during the DNA base excision repair pathway (BER), a well studied process (4, 5).
When OGG1-initiated BER is overwhelmed and/or repair efficiency is decreased, 8-oxoG accumulates in the genome, which has been associated with heritable mutations and altered cellular/tissue homeostasis, diseases, and aging processes (1, 6). Indeed, supra-physiological levels of 8-oxoG in the genome of Ogg1-null (Ogg1−/−) mice (7, 8) are associated with mis-regulation of gene expression leading to aberrant immune responses (9–11) and metabolic disorders (12). These conditions may be explained by a lack of OGG1, whose role in activation of small GTPases and downstream signaling has been documented (13–16). Studies have also shown that localized demethylation produces ROS, which generates 8-oxoG and the recruited OGG1 and topoisomerase II β that are essential for estrogen-induced gene expression (17, 18). A temporal association of OGG1 with the introduction of DNA single strand breaks (ssbs) in sequences adjacent to hypoxia-response elements and gene expression was also documented (19). A recent study showed OGG1 to function as a coactivator for signal transducer and activator of transcription 1 (STAT1) (20).
Regulatory regions (>70%) of RNA polymerase II-dependent genes (e.g. proto-oncogenes, growth factors, chemokines, and cytokines) are guanine-rich (21, 22). Moreover, binding sites of transacting factors (TFs; e.g. specificity protein 1 (SP1), nuclear factor κB (NF-κB), activator protein 1, transcription factor E2F)) contain runs of guanines. For example, NF-κB, a master regulator of gene expression necessary for inflammatory response, cell proliferation, and differentiation (23–25), classically binds to DNA sequences that contain three or more guanines (5′-GGGRNYYYCC-3′; where R is purine; Y is pyrimidine, and N can be any nucleotide) (26–28). It has also been shown that guanine lesions (particularly 8-oxoG) can either change the sequence context (due to its mutagenicity) of the TF-binding site or directly modulate DNA-protein interactions and thus gene expression (28).
Our recent studies show that in OGG1-expressing cells there was enhanced association of NF-κB, SP1, transcription initiation factor II-D (TFIID), and phosphorylated-RNA polymerase II with promoter compared with those in OGG1-depleted cells (29). Here, using chromatin immunoprecipitation (ChIP), ChIP-sequencing, Flare-PCR, RT-PCR, and electromobility shift as well as reporter assays, we tested whether OGG1 bound to its genomic substrate is utilized for gene expression in oxidatively stressed cells. As an example, NF-κB's DNA occupancy was studied. We showed that TNFα-induced ROS increased levels of oxidatively modified guanines and OGG1's association preferentially with proximal promoter regions, which precede the association of NF-κB with its motif. OGG1 binding to 8-oxoG, 8–11 bp 5′ from the NF-κB motif robustly increased its DNA occupancy. These intriguing observations along with other data shown here suggest that base lesion 8-oxoG and its cognate repair enzyme OGG1 act cooperatively with NF-κB binding to promote prompt gene expression upon oxidative exposure.
Results
TNFα Induces an Association of Both OGG1 and NF-κB with Promoter Sequences
Our previous studies have documented an increased association of transacting factors (TFIID, SP1, and NF-κB) with promoter in OGG1-expressing cells (29). To further explore these observations, we examined the stimulus-driven engagement of OGG1 with gene regulatory sequences and its potential role in NF-κB binding. FLAG-OGG1-expressing cells were TNFα-exposed and ChIP-ed using anti-FLAG (OGG1) and anti-RelA(NF-κB) antibodies (Ab). qPCR amplification of the proximal promoter segment of the TNF (where TNF indicates the human promoter and gene) in anti-FLAG (OGG1)-Ab-ChIP-ed DNA was increased by an average of 3.2-, 16.5-, and 10.6-fold at 15, 30, and 60 min after TNFα addition, respectively (Fig. 1A). In the DNA ChIP-ed with anti-RelA(NF-κB) Ab, the level of an identical segment of the TNF promoter was increased by an average of 1.8-, 4.4-, and 4.9-fold at 15, 30, and 60 min post-exposure, respectively (Fig. 1A). TNFα exposure also enriched levels of TNF, CCL20, and CXCL1 promoter (where CCL20 is CC chemokine ligand 20 gene/promoter, and CXCL1 is chemokine (CXC motif) ligand-1 (GRO α) gene/promoter) segments in (OGG1)-Ab ChIP-ed DNA by an average of 16.8-, 14.2-, and 15.9-fold (30 min), respectively (Fig. 1B). The Ab to RelA(NF-κB)-enriched levels of promoter sequences by an average of 5.1-, 5.8-, and 6.2-fold for TNF, CCL20, and CXCL1, respectively (Fig. 1B, 30-min time point is shown). The Ab to OGG1-enriched levels of the β2-microglobulin (where is B2M human β2-microglobulin gene) promoter segment by 12.1-fold, whereas RelA(NF-κB) Ab did not do so. The latter is in line with the lack of an NF-κB-binding site in the B2M proximal promoter region (30). These data strongly imply a stimulus-dependent association of both OGG1 and NF-κB with promoter sequences.
FIGURE 1.

Binding of NF-κB to promoters is OGG1-dependent. A, TNFα-induced enrichment of OGG1 and NF-κB on TNF promoter. Cells expressing FLAG-OGG1 were mock- or TNFα-exposed, and ChIP was performed at 0, 15, 30, and 60 min using anti-FLAG(OGG1) or anti-RelA(NF-κB) Ab. Uncross-linked DNA was subjected to PCR amplification as described under “Experimental Procedures” (n = 4). B, TNFα-induced enrichment of OGG1 and NF-κB on CCL20, CXCL1, and B2M promoters. Cells expressing FLAG-OGG1 were mock- or TNFα-exposed, and ChIP was performed at 30 min using anti-FLAG(OGG1) or anti-RelA(NF-κB) Ab. Uncross-linked DNA was subjected to PCR amplification of TNF, CCL20, CXCL1, and B2M promoters as described under “Experimental Procedures.” C, OGG1 expression-dependent enrichment of NF-κB on TNF promoter. OGG1 expression was down-regulated by siRNA (scrambled siRNA was used as a control, see under “Experimental Procedures”), and cells were TNFα-exposed. ChIP was performed at 30 min using anti-RelA(NF-κB) Ab (in controls, IgG). The levels of the TNF promoter in ChIP-ed DNA were determined by qPCR. The upper panel is a graphical depiction of changes in NF-κB binding to the TNF promoter in the presence and absence of OGG1 (n = 3). Lower panel, ethidium bromide-stained agarose gel from a representative experiment. Inset, cells were transfected with control and target-specific (OGG1) siRNA. At 48 h thereafter, cells were lysed to prepare protein extracts, and levels of OGG1 were determined by Western blotting. D, OGG1 depletion, but not that of NEIL1 or NEIL2, decreases NF-κB's association with promoters. NEIL1, NEIL2, or OGG1 expression was down-regulated by siRNA as described under “Experimental Procedures,” and cells were TNFα-exposed. ChIP was performed using Ab to RelA(NF-κB). The levels of TNF promoter in ChIP-ed DNA were determined by qPCR (30-min time point is shown). E, enrichment of OGG1 and NF-κB on the TNF promoter. FLAG-OGG1-expressing cells were TNFα-exposed and ChIP-ed at 0, 15, 30, and 60 min using Abs against RelA(NF-κB) or FLAG(OGG1). ChIP-ed DNA was subjected to sequencing, and sequence data were analyzed as described under “Experimental Procedures.” Images are directly taken from the Integrative Genomics Viewer. Enrichment levels (fold) of OGG1 and NF-κB for each time points are shown (left-hand side). Blue, green, and red bars are positions of NF-κB-binding motifs in promoter. Inset, distribution of OGG1 and NF-κB on TSS-adjacent sequences ±2000 bp at whole genome level. F, enrichment of OGG1 at the CXCL1 and CCL20 promoters. FLAG-OGG1-expressing cells were TNFα-exposed and ChIP-ed using Ab FLAG(OGG1), and ChIP-ed DNA was sequenced (see under “Experimental Procedures”). Images show enrichment peaks of OGG1 at 30 min after TNFα exposure of cells. Images are directly taken from the Integrative Genomics Viewer. Red bars within CXCL1 and CCL20 promoter regions are the locations of NF-κB-binding motifs. G, OGG1 digestion decreases qPCR product levels of TNF, CCL20, CXCL1, and B2M promoters. Cells were exposed to TNFα (20 ng/ml) for various lengths of time, and then the isolated DNAs were OGG1-digested and subjected to qPCR as under “Experimental Procedures” (n = 3). H, TNFα exposure of cells induces oxidative modifications of OGG1 and decreases its enzymatic activity. FLAG-OGG1-expressing cells were TNFα-exposed, and nuclear extracts were made in the presence or absence of DCP-Bio1 (a cysteine sulfenic acid-reacting agent). Levels of DCP-Bio1-tagged OGG1 were determined by streptavidin-coupled chemiluminescence. Total OGG1 levels were determined by immunoblotting using Ab to FLAG (OGG1). Lower panel: enzymatic activity of OGG1 is decreased in TNFα-exposed cells. NE was isolated at the 30-min post-exposure, and OGG1's excision activity was determined in the presence or absence of DTT in the reaction buffer, as described under “Experimental Procedures.” * = p < 0.05; ** = p < 0.01; *** = p < 0.001; Cells used are as follows: HEK293; CCL20, promoter of that chemokine (C-C motif) ligand 20 gene; CXCL1, promoter of chemokine (CXC motif) ligand 1 gene; B2M, promoter of β2-microglobulin gene; NEIL1, gene human Nei-like DNA glycosylase-1; NEIL2, gene human Nei-like DNA glycosylase 2; OGG1, gene of human 8-oxoguanine DNA glycosylase.
To test for a possible association between OGG1 and NF-κB in enrichment on promoter sequences, OGG1-depleted and OGG1-expressing cells (where OGG1 is human 8-oxoguanine DNA glycosylase-1 gene/mRNA and Ogg1 is mouse 8-oxoguanine DNA glycosylase-1 gene/mRNA (as indicated)) were TNFα-exposed, and ChIPs were performed using Ab to RelA(NF-κB). In OGG1-depleted cells, enrichment of the TNF promoter in ChIP-ed DNA was nearly identical to that of pre-TNFα exposure levels (Fig. 1C). In OGG1-expressing cells, promoter enrichment in ChIP-ed DNA was significantly increased (4.7 ± 0.9; 30-min time point is shown). Depletion of NEIL1 and NEIL2 DNA glycosylases (mammalian orthologs of Escherichia coli Nei (31), Fig. 1D, right panel) had some, but not a significant, effect on NF-κB binding to the TNF promoter (Fig. 1D) implying that the association of NF-κB with the proximal promoter of TNF is primarily linked to OGG1 in TNFα-exposed cells.
To further investigate these unexpected observations, we utilized ChIP sequencing. FLAG-OGG1-expressing cells were TNFα-exposed for 0, 15, 30, and 60 min, and ChIPs were performed using Abs to RelA(NF-κB) or OGG1 (anti-FLAG Ab). ChIP-ed DNA was sequenced and analyzed as described under “Experimental Procedures.” As found with ChIP-coupled PCR (Fig. 1, A–D), fold increases in enrichment (Integrative Genomics Viewer; IGV) revealed that at time 0 there was a low level association of OGG1 with the TNF promoter; however, at 15 min OGG1 occupancy increased (∼7-fold), but NF-κB was only 1.8-fold. After 30 min of exposure, OGG1 was enriched by ∼14-fold, whereas NF-κB showed an ∼5-fold increase. At 60 min, OGG1 level decreased, whereas NF-κB showed an ∼3-fold enrichment on the promoter (Fig. 1E). IGV also showed that NF-κB enrichment peaks were aligned with its putative binding sites (Fig. 1E). Sequence alignment predicted that the enrichment peaks of OGG1 were primarily aligned with guanine runs in close proximity to the NF-κB motifs on the TNF promoter (Fig. 1E). Similarly, NF-κB and OGG1 were enriched on regulatory sequences of CXCL1 and CCL20 (Fig. 1F, OGG1 is shown). We note that against our expectations there were no significant levels of engagement of OGG1 with exons, introns, or untranslated regions of TNF, CXCL1, and CCL20, and genome-wide distributions of OGG1 and the NF-κB enrichment sites were similar. As an example, we show the distribution of OGG1 and NF-κB at TSS-adjacent sequences (±2000) (Fig. 1E, inset; 30 min) at the whole genome level, the significance of which is being further investigated.
As putative DNA-binding motifs for OGG1 cannot be found, we examined levels of OGG1 substrate(s) 8-oxoG in the TNF promoter surrounding NF-κB-binding motifs (as example), using “Flare”-qPCR (29, 32). DNAs were isolated at 0, 10, 20, 30, 60, and 120 min after TNFα exposure, and a promoter region (−350 to −21 bp) was PCR-amplified prior to and after OGG1 digestion. Compared with time 0, OGG1 digestion of DNA significantly decreased the levels of PCR product between 20 and 30 min of TNFα exposure (Fig. 1G, left panel). OGG1 digestion also decreased levels of similar size PCR products of CCL20, CXCL1, and B2M promoters (Fig. 1G, right panel, 30-min time point is shown), suggesting an accumulation of OGG1's substrate in regions. Kinetic changes in levels of Flare-PCR promoter products are in line with changes in TNFα-induced intracellular ROS levels, which are similar to those previously documented (29, 33).
To address whether 8-oxoG accumulation in promoter(s) was due to both ROS and decreases in OGG1's enzymatic activity, FLAG-OGG1-expressing cells were exposed to TNFα, and nuclear extracts (NEs) were prepared in the presence or absence of a cysteine sulfenic acid-reacting agent DCP-Bio1 (29, 34). The levels of DCP-Bio1-linked FLAG-OGG1 protein were determined using IP-coupled Western blotting (see under “Experimental Procedures”). Fig. 1H (left panel) shows a transient increase in cysteine sulfenic acid-containing OGG1, which temporally correlated with increases in 8-oxoG levels in TNF promoter regions (Fig. 1G). In parallel experiments, 8-oxoG incision activity of OGG1 was under detectable levels (Fig. 1H, right panel, 30-min time point was analyzed) in NEs isolated from TNFα-exposed cells. However, the activity of OGG1 was regained after the addition of DTT to the NEs (Fig. 1H, lower panel). These data together imply that TNFα-induced ROS cause both accumulation OGG1 substrate and a decrease in OGG1's enzymatic activity, which explains enrichment of OGG1 on promoters (Fig. 1, A–F).
NF-κB DNA Occupancy Is Modulated by OGG1
Given our data showing OGG1-dependent enrichment of NF-κB on TNF promoter sequences (ChIP-qPCR; Fig. 1, A–C, E, and F), we first tested whether all putative NF-κB binding sequences bind homo- and heterodimeric forms of NF-κB. NEs from TNFα-exposed cells were subjected to EMSA using biotin-labeled 31-bp-long probes containing each of the three putative NF-κB motifs (located between −98 and −88 bp, 5′-GGGGCTGTCC-3′; −214 and −204 bp, 5′-GGGGTATCC-3′; and −598 and −588 bp, 5′-GGGGTTTCTCC-3′). The extensive NF-κB binding to these motifs was specific as they were competed out by excess (100-fold) unlabeled probe containing a canonical NF-κB motif (5-GGGATCATTCCC-3′) (Fig. 2A) (26). Time course studies also show that activated NF-κB fully entered the nucleus from cytoplasm, as there were no further increases in the formation of the NF-κB-DNA gel-shift complexes from 15 min onward (Fig. 2B).
FIGURE 2.

NF-κB's DNA occupancy as a function of OGG1 expression. A, putative NF-κB motifs located in TNF promoter are functional. NEs isolated from OGG1-expressing TNFα (20 ng per ml)-exposed cells were incubated with synthetic DNA (probe) containing NF-κB-binding sites derived from TSS-adjacent region of TNF promoter (5′-GGGGTTTCTCC-3′, −598 bp to −588 bp; 5′-GGGGTATCC-3′, −214 bp to −204 bp; and 5′-GGGGCTGTCC-3′, −98 bp to −88 bp). EMSA was performed using 2 μg of NE per individual probes. A cold probe containing canonical NF-κB motif (5′-GGGATCATTCCC-3′ (26)) prevented binding of NF-κB. B, nuclear accumulation of NF-κB in OGG1-expressing cells as assessed by EMSA. Cells were TNFα-exposed, and NEs were prepared at the time points indicated in the figure. EMSA was carried out (see under “Experimental Procedures”) using a probe containing an NF-κB-binding motif (5′-GGGGTATCC-3′, −214 bp to −204 bp upstream from TSS in the TNF promoter). C, OGG1 present in crude NE modulates NF-κB's DNA occupancy in an 8-oxoG position manner. NEs from TNFα-exposed OGG1-expressing cells were isolated at 30 min, and EMSAs were performed using probes lacking (WT) or containing 8-oxoG (G1 to G11) in sense (5′-TG7GGG8AGT GTG5AG1G2G3G4TAT CCG6AT GCTTG-3′) and complementary strand (3′-ACCCCTCACACTCCCCATAG9G10AACTAC G11AAC-5). Inset, graphical depiction of NF-κB binding to individual probes (n = 3). Band intensities of p50 homodimers and p50-p65 heterodimers were determined by densitometry using ImageJ software (version 1.44). D, NF-κB's occupancy of its binding site within G7 and G8 probes is specific. Crude NEs from OGG1-expressing cells were incubated with cold probe containing the IκB motif, and then G7 or G8 was added, and EMSAs were performed. E, OGG1 increases in occupancy of NF-κB's on DNA containing 8-oxoG 11 bp upstream of its binding site. Crude NEs from TNFα-exposed, OGG1-expressing cells were diluted stepwise (2, 1, 0.5, and 0.25, 0.125, and 0.006 μg per assay) and added to WT (left side) and G7 (right side) probes for 5 min, and EMSA was performed. Inset, fold increases in occupancy of homo- and heterodimeric NF-κB on the G7 probe (n = 3). Band intensities were determined by densitometry using ImageJ software, and fold changes in binding were calculated. F, binding of NF-κB to WT and 8-oxoG-containing probes. Recombinant p50 (3.75 ng) and RelA(p65) (2.75 ng) proteins were annealed in binding buffer for 60 min, and then individual probes were added for 5 min, and mixtures were subjected to EMSA. Inset, graphical depictions of changes in band intensities of p50-p50 homodimers and p50-p65 heterodimers. Band intensities were determined using ImageJ software (version 1.44), and fold changes were calculated (n = 3). G, occupancy of NF-κB on DNA, containing 8-oxoG within and adjacent to its motif. Upper panel, crude NEs (2 μg per assay) from OGG1-expressing, mock-, or TNFα-exposed cells were added to probes lacking (WT) or containing 8-oxoG within (G1) or 11 bp upstream of NF-κB's binding sequence (G7). Inset, longer exposure. Lower panel, NF-κB's DNA occupancy on G7 probe in crude NEs from Ogg1+/+ and Ogg1−/− cells. H, NF-κB's occupancy of DNA-containing 8-oxoG within its binding motif is re-established in NE from OGG1-depleted cells. OGG1 siRNA and control siRNA-transfected cells were TNFα-exposed (30 min), and NEs were prepared to perform EMSAs using the G1 probe. I, occupancy of NF-κB on 8-oxoG-containing DNA is diminished in NEs isolated from OGG1-depleted cells. OGG1 and control siRNA-transfected cells were TNFα-exposed for 30 min, NEs were prepared to perform EMSAs using the 8-oxoG-containing G7 probe (8-oxoG is 11 bp distance from its binding site). Lane 1, control; lane 2, TNFα-exposed OGG1-expressing; lane 3, mock-exposed OGG1-depleted; lane 4, TNFα-exposed, OGG1-depleted. J, OGG1 has insignificant impact on TNFα-induced nuclear translocation of RelA(NF-κB). Cells were TNFα-exposed for 30 min, and NEs were prepared. Proteins were fractionated by SDS-PAGE and immunoblotted using Abs to RelA(NF-κB) or OGG1. Quality control of NEs: 5 μg per lane, NEs were SDS-PAGE-fractionated and immunoblotted using Abs to tubulin and histone H1. A, B, and D–J, NEs were prepared from HEK293 cells; NS, nonspecific; TNF, human gene encoding for TNFα; WT, is a 31-bp DNA probe without 8-oxoG; G1 to G11 are 31-bp DNA probes containing 8-oxoG.
To explore the possibility that OGG1 facilitates NF-κB binding, we selected a 31-bp promoter segment of the TNF promoter surrounding an NF-κB-binding motif (5′-GGGGTATCC-3′) located between −214 and −204 bp from TSS. Guanines within and outside of the NF-κB-binding motifs were individually replaced with the OGG1 substrate 8-oxoG in the sense (5′-TG7GGG8AGTGTG5AG1G2G3G4TATCCTTG6ATGCTTG-3′) and complementary (3′-ACCCCTCACACTCCCCATAG9G10AACTACG11AAC-5′) strand. In designing probes, we kept in mind that charges are usually trapped at the 5′ end of guanine runs (35, 36), and thus 8-oxoG at G1, G7, and G10 positions could potentially exist in chromatin. The 8-oxoG-containing synthetic DNAs (probes) are numbered from G1 to G11. The probe lacking 8-oxoG is designated as wild type (WT).
NEs were isolated from TNFα-exposed OGG1-expressing cells, and EMSAs were performed using individual probes. Compared with the WT probe, an 8-oxoG lesion within the p50-p50 interacting sequences (probes: G1, G2, G3, and G4) of the NF-κB motif decreased DNA occupancy of both p50-p50 and p50-p65 dimers (Fig. 2C). Similar decreases in NF-κB binding were observed, when OGG1's substrate 8-oxoG was placed within a complementary strand (probe: G9 and G10). Likewise, 8-oxoG adjacent to the NF-κB motif (probes: G5 and G6) or at 5 bp distance (probe: G11) of the binding sequence (Fig. 2C) in the complementary strand DNA decreased the occupancy of NF-κB's homo- and heterodimers. In contrast with OGG1 substrate being 8 and 11 bp upstream in the sense strand (G7 and G8 probes) from the NF-κB motif, the binding of both homo- and heterodimers became significantly increased. Extensive NF-κB binding to G7 and G8 was specific because it can be prevented by excess “cold” probe (Fig. 2D) containing an IκB motif (37). Quantification analysis showed that the G7 probe occupancy by heterodimers (p50-p65) was 25.5 times (±5.1) higher than the WT probe. The p50-p50 occupancy of G7 was 16.5 ± 1.3 times higher than that of WT (Fig. 2E, inset). These results suggest a role of 8-oxoG-bound OGG1 in regulating DNA occupancy of NF-κB.
In control experiments, to test the possibility that 8-oxoG itself affects the DNA occupancy of NF-κB (38), we utilized recombinant NF-κB proteins (p50R and RelA(p65)R, for binding studies. Subunits p50R and RelA(p65)R were preincubated for annealing (see under “Experimental Procedures”) at 37 °C for 60 min, and occupancy by NF-κB of WT and 8-oxoG-containing probes was examined using EMSA. Compared with the WT probe, the DNA occupancy of p50-p50R homodimer was not affected by 8-oxoG being at positions G1, G3, G5 to G8, and G11. However, 8-oxoG in the G2, G4, G9, and G10 positions decreased binding of p50-p50R (Fig. 2F). Interestingly, compared with the WT, p50-RelA(p65)R showed a decrease in DNA occupancy on all probes except G1, G3, G2, G6 and G11 (Fig. 2F). Importantly, 8-oxoG in the G7 and G8 probes did not increase in NF-κB's DNA occupancy compared with WT probe.
To further evaluate the potential role of OGG1 in modulating NF-κB's DNA occupancy, we selected G1 and G7 probes because 8-oxoG located at the 5′ end of guanine runs within the NF-κB-binding site (G1) and 11 bp upstream (G7) representing scenarios that may occur on promoters. By using NEs from OGG1-expressing cells, NF-κB DNA occupancy on the G1 probe was low, but G7 occupancy was robust (Fig. 2G, upper panel) compared with that of the WT probe, as predicted from results shown in Fig. 2, C–E. NF-κB binding to G7 probe was decreased in NEs isolated from Ogg1−/− cells compared with its binding in NEs of Ogg1+/+ cells (Fig. 2G, lower panel). In contrast, using the G1 probe, we consistently observed increased NF-κB-G1(DNA) complex in crude NE isolated from cells lacking OGG1 (Fig. 2H, lane 4). As predicted from the above results, OGG1 depletion decreased the binding of NF-κB homo- and heterodimers to G7 (Fig. 2I, lane 2 versus lane 3). Together, these results suggest that binding of OGG1 to 8-oxoG outside its DNA footprint (39, 40) from the NF-κB sequence motif is increased, although binding within interfered with NF-κB's DNA occupancy. Because in OGG1-expressing cells both proteins are present in crude NEs, these data also mean that OGG1 has a higher affinity to 8-oxoG compared with that of NF-κB's affinity to its binding motif. To exclude the possibility that the decreased DNA occupancy of NF-κB was associated with an impairment in its nuclear translocation in OGG1-depleted cells, we showed that the levels of NF-κB in the nuclear compartment were similar between OGG1-depleted and -expressing TNFα-exposed cells (Fig. 2J).
Next, crude NE from mock- and TNFα-exposed cells were preincubated with biotin-labeled G7 probes, and DNA-protein complexes were pulled down using magnetic streptavidin beads. Levels of bound proteins were evaluated by immunoblotting. Results show that OGG1 bound to 8-oxoG-containing probe G7 in the NE of OGG1-expressing cells (Fig. 3A, upper panel). As expected, there was ∼10 times less OGG1 association with G7 probe in NE isolated from OGG1-depleted cells (Fig. 3A, upper panel). There was no detectable OGG1 binding to WT probe (Fig. 3A, middle panel). In controls, blots were probed using Abs to RelA(p65) and p50. Results show that TNFα exposure increased the association of both RelA(p65) and p50 with G7 and WT probes (Fig. 3A, upper and middle panels). Importantly, OGG1 depletion substantially decreased levels of NF-κB that were pulled down using G7, whereas no change was observed using WT probe (Fig. 3A). Because the only difference between WT and G7 is an OGG1 substrate 8-oxoG located 11 bp upstream from the NF-κB-binding site, these results strongly suggest involvement of OGG1 in the increased DNA occupancy of NF-κB. We note that NEs were isolated from cells expressing poly(ADP-ribose) polymerase-1 (PARP1) and ribosomal protein S3 (RSP3), so their concerted action with OGG1 may not be excluded. Both PARP1 and RSP3 have previously been shown to modulate DNA occupancy of NF-κB (41, 42).
FIGURE 3.

OGG1 facilitates NF-κB binding to its motif. A, OGG1 and NF-κB's occupancy of 8-oxoG-containing DNA in NE. NE (50 μg per sample) from ±TNFα-exposed, OGG1-expressing, and depleted cells were incubated with G7 (upper panel) or WT probe (middle panel) for 5 min, and protein DNA complexes were pulled down using magnetic streptavidin beads. The washed pellets were subjected to SDS-PAGE and immunoblotting using Abs to OGG1, p50, or RelA(p65). Lower panel (input): NE (3 μg per sample) was subjected SDS-PAGE and immunoblotted by using Ab to OGG1 and lamin A. B, OGG1 increases DNA occupancy of NF-κB. Annealed RelA(p65)R and p50R and OGG1R were mixed with the 8-oxoG-containing G7 probe conjugated to magnetic streptavidin beads. Five min thereafter, streptavidin bead-G7-associated proteins were collected, washed, and subjected to SDS-PAGE and immunoblotted using Abs to OGG1, p50, and RelA(p65). Lane 1, RelA(p65)R + p50R+OGG1; lane 2, p65R+p50R; lane 3, OGG1R alone; lane 4, RelA(p65)R alone; lane 5, p50R alone. Lanes 7–9 are OGG1R, RelA(p65)R, and p50R protein loaded as molecular size markers, respectively. Right panel, fold changes in p50 and p65(RelA) binding to G7 probe ± OGG1 (n = 3). Band intensities were determined by densitometry using ImageJ software (version 1.44). C, mutually positive interactions between NF-κB and OGG1 in binding to 8-oxoG containing DNA. Lanes 1–4, OGG1's binding to 8-oxoG-containing G7 probe. Increasing amounts of OGG1 were mixed with G7 probes for 5 min and subjected to EMSA. Lanes 5–8, OGG1 increases NF-κB's DNA occupancy. Annealed p50-RelA(p65) was added to the probe simultaneously with increasing concentrations of OGG1 for 5 min, and EMSAs were carried out. Results from a representative experiment (n = 3) are shown. D, physical interactions between OGG1 and NF-κB. p50R (15 ng) and p65R (20 ng) was preincubated for annealing, and GST or GST-tagged OGG1 (OGG1GST) was added in binding buffer at 37 °C for 30 min. OGG1GST·NF-κB complexes were pulled down using glutathione-Sepharose; GST served as a control (see under “Experimental Procedures”). NF-κB subunit proteins as well as OGG1GST were detected by immunoblotting using Ab to p50, RelA(p65) (upper panels) or GST (lower panels). p50R or Rel(p65)Ralone served as molecular size markers. Results of a representative experiment out of three are shown. Band intensities were determined by densitometry using ImageJ software (version 1.44), and fold changes were calculated (n = 3). * = p < 0.05; ** = p < 0.01; *** = p < 0.001; cells used are as follows: HEK293; OGG1R, recombinant human 8-oxoguanine DNA glycosylase-1; OGG1GST, glutathione S-transferase-tagged OGG1; NF-κBR, recombinant p50R and RelA(p65)R.
To confirm OGG1-facilitated NF-κB binding to G7 probe (and exclude potential roles of PARP1 as well as RPS3), recombinant proteins (RelA(p65)R and p50R) were mixed with OGG1R and added to a streptavidin bead-conjugated G7 probe. Immunoblotting of bound proteins show that RelA(p65)R and p50R as well as OGG1 extensively bound to the G7 probe (Fig. 3B, lane 1). In the absence of OGG1, binding of RelA(p65)R and p50R was notably less (Fig. 3B, lane 2). RelA(p65)R alone did not bind (Fig. 3B, lanes 2 and 4) as the G7 probe lacks a p65-binding site (5′-GGAATTTTC-3′) (26). As markers, OGG1R, RelA(p65)R, and p50R were directly loaded into SDS-PAGE (Fig. 3B, lanes 7–9). Densitometry of band intensities (in lanes 1 and 2) revealed a significantly increased association of p50R and RelA(p65)R with DNA in the presence of OGG1 compared with their levels without it (Fig. 3B, right panel). These results together support a role of OGG1 in NF-κB's DNA occupancy.
Results from pulldown assays were confirmed using EMSA. RelA(p65)R plus p50R was added to the G7 probe simultaneously with increasing amounts of OGG1R. Results show increased occupancy of both homo- and heterodimeric NF-κB in an OGG1 concentration-dependent manner (Fig. 3C, lanes 5-8). OGG1 alone bound to the 8-oxoG-containing probe (Fig. 3C, lanes 1–4) similar to that documented previously (43–45). Surprisingly, NF-κB being in the assay mixture significantly increased the levels of OGG1-DNA complexes (Fig. 3C, lanes 5–8). The NF-κB-mediated increase in DNA binding of OGG1 is a novel observation, and its significance is the focus of our investigations.
The increase in NF-κB's DNA occupancy in the presence of OGG1 (Fig. 2C) raises the possibility of protein-protein interactions. To explore this, pulldown assays were performed. OGG1 interacts with both subunits of NF-κB (Fig. 3D). Densitometry of band intensities revealed a more abundant interaction between OGG1 and p50R compared with OGG1 and RelA(p65)R (Fig. 3D, right panel).
The observed increase in NF-κB's DNA occupancy in NEs and that of recombinant NF-κB subunits in the presence of OGG1 (Figs. 2, C and E and 3, B and C), the time required for NF-κB's DNA occupancy was determined with and without OGG1. Annealed NF-κB subunits were mixed with OGG1 (or same volume of binding buffer), and G7 probe was added for increasing lengths of time. Results from EMSAs showed that OGG1 accelerated interactions between DNA and homo- and heterodimeric forms of NF-κB (Fig. 4, A and B). For example, at 1 min in the presence of OGG1, levels of DNA-associated p50-p50R and p50-RelA(p65)R were similar to that of 8 min without OGG1 (compare lane 8 to lane 10, in Fig. 4A). OGG1 had no effect on homo- or the heterodimer occupancy of WT probe (lacking 8-oxoG).
FIGURE 4.

OGG1 accelerates association of NF-κB with its DNA motif. A, kinetics of binding of homo- and heterodimeric NF-κB to DNA ± OGG1. Annealed NF-κB subunits were mixed with OGG1, and 8-oxoG-containing DNA (G7 probe) was added. Aliquots were taken at the times indicated, and the levels of NF-κB·DNA complexes were determined by EMSA. B, graphical depiction of NF-κB's binding kinetic to DNA. Band intensities of NF-κB' homo- and heterodimers were determined by using ImageJ software (version 1.44) (n = 3).
OGG1 Augments NF-κB-driven Gene Expression
OGG1-expressing and depleted cells were TNFα-exposed and mRNA levels were determined. In OGG1-depleted cells, TNFα stimulation produced significantly lower TNF mRNA levels (Fig. 5A, left panels) compared with those in OGG1-expressing HEK293 cells (Fig. 5A, right panel). Similar results were obtained using airway epithelial (MLE12) cells (Fig. 5B, left and right panels). OGG1 depletion also decreased mRNA levels of CCL20 and CXCL1. TNFα exposure had only a marginal effect on B2M mRNA levels (Fig. 5C) in line with the lack of an NF-κB motif in its promoter (30). OGG1 siRNA had no effect on B2M mRNA levels. A decrease in mRNA levels of TNF, CXCL1, and CCL20 in OGG1-depleted cells was similar to those observed in the IκB kinase inhibitor (BMS-345541)-treated (46) TNFα-exposed cells (Fig. 5E). In controls, silencing NEIL1 expression had no effect on mRNA levels, whereas NEIL2 silencing somewhat increased it (Fig. 5D), an observation being followed up in future investigations.
FIGURE 5.

OGG1-dependent gene expression in TNFα-exposed cells. A and B, OGG1-dependent gene expression from TNF/Tnf promoters. Left panels, parallel cultures of cells were transfected with control siRNA (□) or siRNA to OGG1 (■), and cells were exposed to TNFα (20 ng/ml) for 30 min. TNF mRNA levels were determined by RT-PCR. A and B, insets, expression of OGG1 at protein levels in cells after two cycles of transfections of cells with siRNA (Western blotting analysis). A and B, right panels, HEK293 and MLE-12 cells were exposed to TNFα (20 ng/ml), and mRNA levels were determined by RT-PCR at the time points indicated (n = 3). C, OGG1-dependent expression of CCL20 and CXCL1 mRNA. Cells were transfected with control siRNA (□) or siRNA to OGG1 (■) and TNFα-exposed (20 ng/ml). In controls, expression from B2M was tested. mRNA levels were determined at 30 min by RT-PCR (n = 3). D, depletion of NEIL1 and NEIL2 has no significant impact on TNF mRNA levels. Parallel cultures of cells were sequentially transfected with control siRNA (Cont) or siRNA to NEIL1, NEIL2, or OGG1. Inset, Western blotting analysis of protein levels. TNFα (20 ng/ml) was added for 30 min, and RNAs were isolated. mRNA levels of TNF were determined by RT-PCR (n = 3). E, IκB kinase inhibitor BMS-345541 decreases gene expression. Cells were pretreated with BMS-345541 (10 μm) for 3 h, and TNFα-exposed (20 ng/ml). mRNA levels were determined at 30 min by RT-PCR (n = 4). F, decreases in luciferase mRNA expression driven by TNF promoter in OGG1-depleted cells. OGG1 was depleted by siRNA, and cells were transfected with pGL4.2-Luc vector containing a promoter region (−974 + 90 bp) of human TNF (see under “Experimental Procedures”). mRNA expression levels of firefly luciferase were assessed at 30 min after mock (□) or TNFα (■) exposure (n = 4). G, NF-κB-dependent expression from the TNF promoter. Cells were co-transfected with IκB super-repressor (IκBSR) and pGL4.2-Luc, and then mock-exposed (□) or TNFα-exposed (■, 20 ng/ml). mRNA levels of firefly luciferase were assessed by RT-PCR (n = 3). ** = p < 0.01; *** = p < 0.001. HEK293 cells were utilized for experiments described in C–G. OGG1, 8-oxoguanine DNA glycosylase-1; CCL20, mRNA encoded by chemokine (C-C motif) ligand 20 gene; CXCL1, mRNA encoded by chemokine (CXC motif) ligand 1 gene; B2M, mRNA encoded by β2-microglobulin gene; IκBSR, super-repressor of κ-light-chain-enhancer of activated B cells; pGL4.2, luciferase (Luc) reporter vector 4.2; NEIL1, gene human Nei-like DNA glycosylase-1; NEIL2, gene human Nei-like DNA glycosylase2; OGG1, gene of human 8-oxoguanine DNA glycosylase-1; Tnf-Luc, human TNF promoter-containing pGL4.2-Luc expressing vector.
Next, TNF promoter containing three putative NF-κB-binding sites was cloned into a pGL4.2 vector containing luciferase (TNF-pGL4.2-Luc). The TNF-pGL4.2-Luc vector was transfected into OGG1-silenced and -expressing cells and then TNFα-exposed. Results showed that Firefly-Luc mRNA levels were significantly lower in OGG1-depleted compared with that in OGG1-expressing ones (Fig. 5F). To test whether transcriptional events were primarily NF-κB-dependent, the TNF-pGL4.2-Luc expression construct was co-transfected with an NF-κB super-repressor (IκBSR)-expressing vector leading to an >80% decrease in Firefly-Luc mRNA levels (Fig. 5G). Together, these data underline the significance of OGG1 in NF-κB-driven gene expression, and these data are in line with those showing OGG1-mediated increases in DNA occupancy of NF-κB.
Discussion
Damage to genomic DNA is an inevitable event upon an increase in ROS levels generated by ligand-receptor interactions, metabolic processes, and/or environmental exposures. One of the most prevalent reactions of ROS with DNA is oxidation of guanine to 8-oxoG. 8-OxoG can modify gene transcriptional output directly by altering DNA-transacting factor interactions or through DNA repair intermediates (e.g. AP-sites) (47). As cellular response(s) to stimuli are prompt, we hypothesized that DNA base lesions and their cognate repair protein OGG1 are utilized in transcriptional gene activation. Here, we present experimental evidence showing that NF-κB-driven gene expression requires enrichment of 8-oxoG and oxidatively modified OGG1 in the promoter sequences after TNFα exposure of cells. OGG1 bound to 8-oxoG facilitates NF-κB's DNA occupancy. OGG1 depletion decreased both NF-κB's association with promoter and cellular transcriptional response to challenge. These studies are the first to document a novel paradigm wherein OGG1 bound to its substrate functions in the epigenetic regulation of gene expression in oxidatively stressed cells.
To test the potential implication of OGG1 in NF-κB-driven gene expression, we examined a stimulus-specific OGG1 enrichment in TNF, CCL20, and CXCL1. In our detailed analyses, using the TNF gene promoter (as an example), sequence alignments consistently show that enrichment peaks of OGG1 are located in close proximity to the NF-κB motifs. Importantly, similar to NF-κB, OGG1's enrichment peaks were localized to distinct genomic region(s) (mostly at regulatory sequences) and were not distributed along the genome as expected from a genome-wide distribution of 8-oxoG and OGG1's evolutionally conserved role in executing unbiased repair of DNA (48). Enrichment of OGG1 in promoter regions co-existed with increased levels of 8-oxoG (and/or FapyG), implying that OGG1 likely binds to its substrate. We also observed oxidative modification to OGG1 at cysteine (sulfenic acid), and nearly undetectable enzymatic activity has previously been documented (29, 49, 50).
Enrichment of OGG1 on the promoter preceded NF-κB's DNA occupancy, and OGG1 depletion resulted in a decreased level of NF-κB binding. For specificity, we show that depletion of NEIL1, whose substrates include thymine glycol, 5-hydroxycytosine guanidine-hydantoin, 5-hydroxyuracil, and spiroiminodihydantoin (51), had no effect on NF-κB's DNA occupancy. Interestingly, the lack of NEIL2 (whose primary substrates are oxidized cytosine (31, 51)) had substantially increased occupancy of NF-κB on the TNF promoter in the context of chromatin. It may not be related to the observed enhancement of NF-κB binding, but a recent study showed an increased inflammatory response of Neil2−/− mice to oxidative stress, TNFα, or LPS (52).
When 8-oxoG was placed within or 2, 3, or 5 bp (5′ or 3′) distance from the NF-κB-binding motif in probes, EMSA consistently showed a decrease in DNA occupancy of homo- and heterodimeric NF-κB. As crude NEs contain both proteins, these observations suggest a physical interference of 8-oxoG-bound OGG1 with NF-κB binding. Indeed, OGG1's DNA “footprint” covers up to 8 bp around 8-oxoG (39, 40). These data also indicate that OGG1 has a higher affinity for 8-oxoG compared with the affinity of NF-κB to its binding sequence. In support of this, occupancy by NF-κB of DNA containing 8-oxoG was re-established in NEs isolated from OGG1-depleted cells. A recent study documented similar results; OGG1 inhibited binding of cyclic AMP-response element-binding protein 1 to CRE-containing probe when 8-oxoG was placed within its binding site (53).
Importantly, when 8-oxoG was placed in the synthetic probes outside of OGG1's “footprint” (39, 40) at 8–11 bp from the NF-κB-binding motif, OGG1 increased its DNA occupancy. Compared with DNA lacking 8-oxoG, the levels of DNA-associated p50-p50 and p50-RelA(p65) were higher by ∼15- and ∼25-fold, respectively. Moreover, in the presence of OGG1, the time required for DNA occupancy of homo- and heterodimeric NF-κB was significantly decreased compared with those without OGG1 in the reaction mixture. The mechanism by which OGG1 increases NF-κB binding is unknown; however, one may propose that OGG1-induced DNA architectural changes are creating a specific interface in DNA that allows for the recognition of motifs in chromatin possibly lowering the previously documented energy need of NF-κB for “twisting” DNA (54). Our studies performed in vitro show that a minimum 8-bp distance was needed between OGG1–8-oxoG and NF-κB-binding site; however, the distance could be more as facilitation of NF-κB's DNA occupancy by sequence-specific transcription factors may occur from ≥20 bp (55–58). OGG1-mediated DNA architectural changes are well documented (39, 40).
OGG1 physically interacts primarily with the heterodimeric NF-κB. These results predict OGG1's interaction with NF-κB·DNA complex; however, such a complex was not observed using EMSA (supershift). It is possible that OGG1·NF-κB ternary complex is disrupted under electrophoretic conditions or the complex is transient similar to those described, e.g. for interaction between OGG1 and special AT-rich sequence-binding protein-1 (59), allowing us to observe individual NF-κB·DNA and OGG1·DNA complexes. In support of a ternary complex, we have shown that OGG1 is present in the transcriptional complex along with NF-κB(RelA), Sp1, TFII-D, and p-RNA·polymerase II only in TNFα-exposed cells (29).
An unexpected observation was that NF-κB increased association of OGG1 with substrate-containing DNA, pointing to a novel discovery-bidirectional interaction between NF-κB and OGG1. Because the underlying molecular mechanisms and significance have yet to be elucidated, one may propose that increased (or prolonged) OGG1-DNA interaction by NF-κB could have a role in gene expression in which repair of DNA base lesions is delayed for the time period of cellular response. Nonetheless, the observed cooperative binding points to a yet to be discovered regulatory mechanism(s) that may exist in oxidatively stressed cells.
The decreased expression of NF-κB-driven genes (e.g. TNF, CXCL1, and CCL20) in OGG1-depleted TNFα-exposed cells is alarming. Both TNFα-receptor- and ROS-induced signaling are potent inducers of NF-κB activation, its nuclear translocation, and gene expression (33, 60). As OGG1 depletion did not affect the level and nuclear translocation of activated NF-κB, these data point to the importance of oxidative guanine modification(s) in promoters in cytokine-induced gene expression. Because the promoter of genes encoding for TNFα, CXCL1, and CCL20 are guanine-rich, one may extend our observations to those genes with guanine-rich regulatory regions, including RNA pol II-dependent ones (21, 22).
OGG1 is an authentic DNA repair protein, which is continuously searching for damage while migrating/diffusing along the DNA strands (48). When damage is detected, OGG1 diffusion significantly decreases in parallel with its actions (39) involving the following: 1) intrusion of its amino acid residues into the DNA helix; 2) extraction of the damaged base from the DNA; 3) its insertion into OGG1's active-site pocket; and 4) un-stocking cytosine opposite to 8-oxoG from the DNA helix. These events result in a 5′ sharp (∼70°) bending of the DNA duplex and architectural changes at the site of action and in adjacent sequences as well (39, 61), which could facilitate NF-κB binding to its DNA motif. In support of this idea, OGG1 1) increased/accelerated DNA occupancy of NF-κB, and 2) a lack of OGG1 in assay buffer decreased NF-κB's DNA binding. Importantly, OGG1-depleted cells showed 3) pretreatment levels of NF-κB binding; 4) OGG1 binding to promoter preceded DNA occupancy of NF-κB; and 5) OGG1 depletion decreased NF-κB-driven gene expression. Although these results may be predicted from cooperative associations among partner transcription factors (28, 55, 58, 62), they also point to a new paradigm utilization of OGG1-mediated DNA architectural changes in transcription factor binding.
The role of OGG1 in gene expression is not entirely novel. For example, demethylation of histone 3 at lysine generates focal ROS, which oxidize guanine to 8-oxoG in the surrounding DNA, and the OGG1-generated DNA single strand gap recruit topoisomerase IIβ to induce chromatin remodeling to ensure efficient expression of genes, including c-Myc (17, 18). Follow-up studies found that DNA base modifications and ssbs adjacent to hypoxia-response elements were accompanied by recruitment of BER enzymes (OGG1 and AP-endonuclease1) and gene expression (19, 32). In our studies, generation of gaps in DNA strands (AP sites and DNA ssbs) after TNFα exposure was under the detectable level. These deviations among data can be explained by differences in gene regulation-pro-inflammatory genes versus hypoxia and estrogen-response genes or the way OGG1–8-oxoG is utilized by NF-κB versus estrogen receptor or the hypoxia-inducible factor.
Overall, it is evident that not only the release of NF-κB from inhibitory complexes in the cytoplasm and its nuclear translocation but also access of NF-κB to the binding motifs in the chromatin are also actively controlled, the mechanism of which is poorly understood (28, 63). In our model, binding of the authentic DNA repair protein OGG1 to its substrate induces architectural changes in DNA and modulates the transcription factor (NF-κB)-DNA interface (Fig. 6). These repair-associated changes within the chromatin may be considered an unexplored epigenetic regulatory mechanism of gene expression where an oxidatively modified base and its cognate repair protein are utilized by transcriptional machinery to provide timely cellular response upon oxidative exposure.
FIGURE 6.
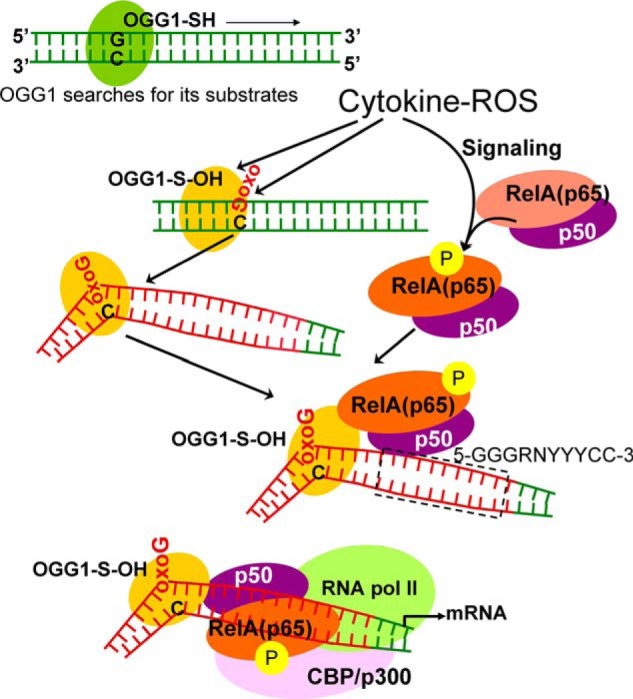
Model is shown for OGG1-mediated “homing” of NF-κB. OGG1 is a prototypic base excision repair protein, which searches for its substrates oxoG (8-oxoG, FapyG) in DNA (48). ROS-induced damage to DNA and OGG1's oxidative modification at cysteine (OGG1-SH → OGG1-S-OH) are inevitable events. OGG1-S-OH has a compromised base excision activity, although it is capable of base extrusion from DNA helix and interaction with the opposite cytosine and structural DNA modifications adjacent to its DNA footprint (39). OGG1-driven architectural DNA modification is utilized by NF-κB subunits for DNA occupancy. OGG1, 8-oxoguanine DNA glycosylase-1; oxoG, 8-oxoguanine; CBP/p300, cAMP-response element-binding protein (transcriptional co-activator); RNA pol II, RNA polymerase II; RelA(p65), 65-kDa regulatory subunit of NF-κB; p50, 50-kDa protein A DNA-binding subunit of NF-κB; 5′-GGGRNYYYCC-3′, NF-κB binding motif; G, guanine; R, purine; Y, pyrimidine; N can be any nucleotide (26).
Experimental Procedures
Cell Culture and Reagents
Human embryonic kidney 293 (HEK293) cells were maintained in DMEM/high glucose medium, and MLE-12 (an immortalized type 2 mouse lung epithelial cell line) was cultured in RPMI 1640 medium, per the instructions of the American Type Culture Collection (ATCC). Ogg1−/− and Ogg1+/+ mouse embryo fibroblasts (MEFs) (7) were kindly provided by Dr. Deborah E. Barnes (Imperial Cancer Research Fund, Clare Hall Labs, UK) and cultured in DMEM/Ham's F-12 medium (64). All media were supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, NJ), penicillin (100 units/ml; Gibco, Life Technologies, Inc.), and streptomycin (100 μg/ml; Gibco, Life Technologies, Inc.). Cells were regularly tested for mycoplasma contamination.
Recombinant human TNFα was purchased from Sigma (catalog no. H8916). Human OGG1 recombinant protein was purchased from GenWay (catalog no. GWB-P1370E, San Diego). Human NEIL1 and NEIL2 recombinant (His) proteins (catalog no. LS-G1742 and LS-G1567) were from LifeSpan BioSciences, Inc. (Seattle); human NF-κB/p50 (catalog no. AG-40T-0021-C002, San Diego) and NF-κB/p65 (catalog no. AG-40T-0020-C002, San Diego) recombinant proteins were from Adipogen. ChIP-quality Abs to NF-κB/RelA (catalog no. sc-372X), and control IgG (catalog no. sc-2027) were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-OGG1 (catalog no. ab135940) and anti-FLAG Abs (catalog no. F1804) were from Abcam (Cambridge, MA) and Sigma, respectively.
Real Time PCR (RT-PCR)
RNA was isolated (Qiagen, Valencia, CA), and 1 μg was reverse-transcribed using a SuperScript® III first-strand synthesis system (Invitrogen, Life Technologies, Inc.). RT-PCRs were performed in triplicate with primers purchased from Integrated DNA Technologies, using SYBR Green PCR Master Mix (Qiagen) in an ABI7000 thermal cycler (Applied Biosystem). The relative transcription level was calculated using the ΔΔCt method we described previously (29, 65). The validated primers were obtained from OriGene Technologies, Inc. (Rockville, MD). The following were used: human OGG1 (catalog no. HP231384), human NEIL1 (catalog no. HP223287), NEIL2 (catalog no. HP217364), human CCL20 (catalog no. HP207883), human CXCL1 (catalog no. HP205376), human TNF (catalog no. HP200561), mouse Tnf (catalog no. MP217748), and human B2M (catalog no. HP207421).
Assessment of Oxidatively Modified Guanine Levels
Changes in oxidatively modified guanines in promoter regions were determined by OGG1 digestion-coupled qPCR (Flare-qPCR) of DNA (29, 32). Briefly, cells were exposed to TNFα (20 ng per ml), and DNA was extracted at 0, 10, 20, 30, 60, 90 and 120 min using QIAamp DNA kits (Qiagen, Stanford Valencia, CA). Genomic DNA (2 μg) from each sample was incubated with or without recombinant OGG1 (5 ng) in a 100-μl reaction buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 2 mm DTT, 1 mg/ml bovine serum albumin (BSA), at 37 °C for 20 min, and DNA was subjected to phenol/chloroform extraction. Twenty ng of DNA from each sample was used as a template to amplify the proximal regions of promoters by real time qPCR. TSS-adjacent promoter regions of genes were identified using the “Transcriptional Regulatory Element Database.” Primers were designed using Blast (www.ncbi.nlm.nih.gov), and products were validated by sequencing. Validated primers (synthesized by Integrated DNA Technologies (IDT, Coralville, IA) were used: TNF (−350 to −21 bp): F, 5′-GGTCCCCAAAAGAAATGGAGG-3′, and R, 5′-TTTATATGTCCCTGGGGCGA-3′; chemokine (C-C motif) ligand 20, CCL20 (−366 to −70 bp): F, 5′-TGTTCCTGTGTGGGGCTG-3′, and R, 5′-TTGCCACATGGGGTTTTCC-3′; chemokine (CXC motif) ligand 1 (CXCL1; −432 to −73 bp): F, 5′-ACAAATCCGAGACACAACGC-3′, and R, 5′-ATTCCCGGAGTTCCAGATCG-3′; β2-microglobulin (B2M; −351 to −27): F, 5′-GGGGCACCATTAGCAAGTCA-3′, and R, 5′-ATATTAAACGCGTGCCCAGC-3′.
siRNA-mediated Depletion of Gene Expression
Cells were transfected with small interfering (si) RNAs using the N-TER Nanoparticle Transfection System (Sigma) per the manufacturer's instructions. The siRNA sequences for human RPS3 (catalog no. M-013607-01-0005), OGG1 (catalog no. M-005147-03-0005), mouse Ogg1 (catalog no. M-048121-01-0005), human NEIL1 (catalog no. M-005147-03-0005), and human NEIL2 (catalog no. M-005147-03-0005) were purchased from Dharmacon (GE Healthcare and Dharmacon Inc. (Lafayette, CO). The extent of depletion was determined by RT-PCR (29) using primers (human OGG1, catalog no. HP231384; mouse OGG1, catalog no. MP200099; NEIL1, catalog no. HP223287; NEIL2, catalog no. HP217364; and RPS3, catalog no. HP201499) purchased from OriGene Technologies, Inc. (Rockville, MD). Depletion of the target proteins was determined by Western blotting.
Luciferase Reporter Assays
Transient transfection of DNA constructs was described previously (29). Briefly, cells were cotransfected at a ratio of 10:1 with firefly luciferase constructs driven by the human TNF promoter (−974 to +90), together with the Renilla luciferase plasmid (Promega). To assess expression at mRNA levels, cells were exposed to TNFα (20 ng/ml) for 30 min and harvested, and RNA was extracted for RT-PCR. In parallel experiments, IκBα super-repressor (SR) or control pcDNA 3 vectors were cotransfected with reporter plasmids. After mock or TNFα exposure, cells were harvested; RNA was extracted, and RT-PCR was performed. The following primers were used: firefly luciferase: F, 5′-GGTGGACATCACTTACGC-3′, and R, 5′-CTCACGCAGGCAGTTCTA-3′, and Renilla luciferase: F, 5′-AGCCAGTAGCGCGGTGTATT-3′, and R, 5′-TC AAGTAACCTATAAGAACCATTACCAGATT-3′.
Assessment of Cysteine-modified OGG1
Cells expressing FLAG-OGG1 were exposed to TNFα for 0, 10, 20, 30, 60, and 120 min, and nuclei were isolated and then lysed in an ice-cold de-oxygenized buffer containing 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Nonidet P-40, 2.5 mm sodium pyrophosphate, protease inhibitor mixture (Sigma), 100 μm diethylenetriaminepentaacetic acid, 5 mm iodoacetamide, 200 units/ml catalase, and 0.1 mm 3-(2,4-dioxocyclohexyl)propyl 5-((3aR,6S,6aS)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-6-yl)pentanoate (DCP-Bio1; KeraFAST, Inc., Boston) as documented previously (34). NEs were clarified, and the supernatants were incubated with 30 μl of protein G-Sepharose (Millipore Corporation Billerica, MA) at 4 °C for 1 h. The precleared supernatants were then incubated with Ab against FLAG for 3 h and added to protein G-Sepharose for 3 h. The immunoprecipitates were resolved by SDS-PAGE, and OGG1-DCP-Bio1 was detected by streptavidin-conjugated horseradish peroxidase (HRP)-coupled chemiluminescence.
EMSA
NEs were prepared using CelLyticTMNuCLEARTM extraction kit (NXTRACT, Sigma), and protein concentration was determined with the DCTM protein assay kit (Bio-Rad). Biotinylated wild-type and 8-oxoG and NF-κB-binding site containing double-stranded oligonucleotide probes were used for EMSA. Probes (10 fmol) were mixed with NE (2 μg) in buffer containing 2 mm HEPES (pH 7.5), 4 mm KCl, 1 mm dithiothreitol (DTT), 0.25 mm MgCl2, 0.001% Nonidet P-40, 50 ng/μl of poly(dI-dC), and 1 mg/ml BSA. EMSAs for recombinant NF-κB subunits (RelA(p65R) and/or p50R) were first annealed in 10 mm Tris (pH 7.5), 5 mm NaCl, 1 mm DTT, and 1 mm EDTA at 37 °C for 1 h. The mixture was incubated with synthetic DNA probes for 5 min at room temperature or 10 min at 4 °C. Unlabeled probe was used for competition experiments. Samples were resolved on a 6% DNA retardation gel (Invitrogen) in 0.25× Tris/borate/EDTA (TBE) buffer. Visualization of NF-κB·DNA complexes was carried out with LightShift chemiluminescent EMSA kit (Thermo Scientific) with modifications.
Streptavidin Pulldown Assay
Streptavidin-Dynabeads (catalog no. 11205D, Invitrogen) were blocked with 1 mg/ml BSA, washed, and incubated for 30 min with biotin-labeled probe(s) containing the NF-κB-binding motif. Unbound probes were removed by washing, and NE (20 μg per assay) (or recombinant p50, p65, or OGG1) was added in binding buffer at room temperature for 10 min. The streptavidin·probe·protein complexes were washed three times with binding buffer, and gel-loading buffer was added. Proteins were separated using SDS-10% polyacrylamide gel. Protein were transferred to nitrocellulose membranes, blocked in 5% nonfat dry milk containing TBST (20 mm Tris base, 50 mm NaCl, 0.05% Tween 20 (pH 7.5)), and then incubated for 3 h at 4 °C with primary Ab(s) to p50, RelA(NF-κB), or OGG1, washed with TBST, and subsequently incubated with HRP-conjugated secondary Ab(s) at a 1:4000 dilution (Southern Biotech, Birmingham, AL). The chemiluminescent signals were detected using the ECL Plus detection system (GE Healthcare, Buckinghamshire, UK).
GST Pulldown Assay
A GST-OGG1 construct was obtained by inserting the coding sequence of human OGG1 into vector pGEX 4T-2 via its EcoRI and XhoI restriction enzyme sites. GST-OGG1 and GST were expressed in Escherichia coli strain BL-21 (DE3). GST-OGG1 was purified on glutathione-Sepharose 4B beads per the manufacturer's instructions (Amersham Biosciences). To test whether OGG1 interacts with recombinant NF-κB complex (p50 and p65), GST-OGG1 pulldown assays were conducted. Briefly, recombinant NF-κB subunit proteins (p50R, 20 ng of p65R, 15 ng per assay) were annealed in 200 μl of binding buffer at 37 °C for 1 h, and glutathione-Sepharose beads (20 μl) coated with GST or GST-OGG1 were then introduced into the reaction buffer. After 1 h of incubation, the beads were collected by centrifugation and washed three times with ice-cold binding buffer. Bound proteins were then subjected to SDS-PAGE and detected by immunoblotting using Abs against p50, p65, and GST.
DNA Cleavage Assays Using OGG1
OGG1's base excision activity was determined using a 40-mer oligonucleotide containing an 8-oxoG at position 19 and labeled at the 3′ end with Cy5 (5′-AGAGAAGAAGAAGAAGAA(8oxoG)AGATGGGTTATTCGAACTAGC/3Cy5Sp/-3′) (substrate-Cy5), as we previously described (66). In a standard excision reaction, recombinant OGG1 was added to a 10-μl reaction mixture containing 200 fmol of the substrate Cy5-labeled duplex in 20 mm Tris-HCl (pH 7.1), 1 mm EDTA, 100 mm NaCl, 1 mg/ml bovine serum albumin (BSA), and 5% glycerol. After 15 min at 37 °C, the excision reaction was stopped by adding 4 μl of formamide dye and heated for 5 min at 95 °C. The cleaved product was separated from the intact substrate in a 20% polyacrylamide gel containing 8 m urea in TBE buffer. Fluorescence in the separated DNA bands was visualized using a LI-COR Odyssey CLx system (LI-COR Biosciences, Lincoln, NE).
ChIP
ChIP assays were performed as described by “ChIP-Sequencing guidelines and Practices” of the ENCODE and modENCODE consortia (67) using slight modifications (68). Briefly, FLAG-OGG1-transfected HEK 293 cells (107) were stimulated with TNFα (20 ng/ml) for 0, 15, 30, and 60 min, and DNA·protein were cross-linked with 1% paraformaldehyde and sheared (average 300 bp) with 10-s pulses using Cole-Parmer's GEX 130 Ultrasonic processor (Vernon Hills, IL) set to 30% of maximum power. DNA·protein complexes were immunoprecipitated with ChIP quality Abs (NF-κB-RelA, catalog no. sc-372X; IgG, catalog no. sc-2027; Santa Cruz Biotechnology; anti-FLAG Ab, catalog no. F1804, Abcam (Cambridge, MA)), using a magna ChIPTM G kit (Millipore, Billerica, MA). The precipitates were washed three times, de-cross-linked, and subjected to qPCR. The following were used: TNF primers (−293 to −64 bp): F, 5′-CCTCCAGGGTCCTACACACA-3′, and R, 5′-AGGAAGTTTTCCGCTGGTTGA-3′; CCL20 primers (−280 to −58): F, CCCCTCCTCCTTGACTGGTT, and R, 5′-GCAACACGCCTTCTGTGTAC-3′; CXCL1 (GRO α) primers (−412 to −185 bp): F, 5′-TCTTCCTCCAAAGAGGTCGC-3′, and R, 5′-GCCTCGCCCTTCAGAGTAAC-3′; and B2M, F, 5′-TTAATGTGCCTCCAGCCTGA-3′, and R, 5′-GCAGTGCCAGGTTAGAGAGAG-3′.
ChIP-Seq
To prepare samples for ChIP-Seq, the immunoprecipitated DNA was isolated, and DNA fragments were gel-purified. Sequencing analyses (10 ng of ChIP DNA) were performed at the University of Texas Medical Branch Next-Generation Sequencing (NGS) Core Facility (Dr. Thomas G. Wood, Director) using Illumina HiSeq 1500 sequencing system (Illumina Inc., San Diego). ChIP-Seq data have been deposited in the Gene Expression Omnibus (GEO), NCBI, and is accessible through GEO series accession no. GSE75652.
CHIP-Seq Data Analysis
All sequence reads produced by the Illumina sequencing system were analyzed by BioInfoRx, Inc. (University Research Park, Madison, WI). In brief, quality of raw data (bam files) was determined by FastQC program. Sequence reads were aligned to the human genome (hg38) using bowtie2. The Homer program was used to detect peaks that are enriched by 4-fold over background and have a cumulative Poisson p value less than 0.0001. The sequence reads were further analyzed by Model-based Analysis for ChIP-Sequencing (MACS) programs, and BED and wiggle (WIG) files were created and visualized using the University of California at Santa Cruz Genome Browser. To confirm results we utilized IGV. Peak detection was performed by running model-based analysis of ChIPSeq (MACS) (69) within ArrayStar (parameters were mfold (second structure) of 10, bandwidth of 300 bp, and a p value threshold of 1 × 10−10). The DNA-binding motif was identified using the multiple motif elicitation (MEME; version 4.11.0) software package. NF-κB(RelA) ChIPSeq enrichment signals were plotted around TSS. The transcription factor-binding motifs were predicted on the basis of the highest number of occurrences with the lowest p value under the zero- or one-per-sequence option. Position-dependent letter-probability matrices generated by MEME were represented in the logo format by using the WebLogo application. The PWM was used to determine a score for any 6–20-bp sequence, and all motifs with a p value of <1 × 10−10 were examined (70).
Statistical Analysis
Statistical analysis was performed using Student's t test to analyze changes at the mRNA and protein levels. The data are presented as the means ± the S.E. of the mean. Differences were considered to be statistically significant at p < 0.05
Author Contributions
I. B. and X. B. designed the study. L. P., B. Z., W. H., M. L. H., A. B., and S. A. V. carried out the research experiments. I. B. and X. B. wrote the manuscript, and A. R. B., S. A. V., T. K. H., Z. R., and X. Z. edited and gave scientific advice. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. David Konkel (Institute of Translational Sciences), Mardelle Susman, and Dr. Lynsey A. Yeager (Department of Microbiology and Immunology) for their scientific input and for critically editing the manuscript. We appreciate the help of Dr. L. Aguilera-Aguirre for managing ChIP-Sequence analysis.
This work was supported by National Institutes of Health Grant RO1 ES018948 from NIEHS (to I. B.), Grant PO1-AI062885 from NIAID (to A. R. B. and I. B.), Grant P30 ES006676 from NIEHS (to A. R. B. and I. B.), and National Science Foundation of China Grant 31571339 (to X. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- 8-oxoG
- 8-oxo-7,8-dihydroguanine
- Ab
- antibody
- BER
- DNA base excision repair
- B2M
- human β2 microglobulin gene
- ChIP-Seq
- chromatin immunoprecipitation-coupled sequencing
- NE
- nuclear extract
- NF-κB
- nuclear factor κB (referred here as a p50-RelA heterodimer)
- ssbs
- DNA single strand breaks
- TFIID
- transcription initiation factor II-D
- TSS
- transcription start site
- F
- forward
- R
- reverse
- IGV
- Integrative Genomics Viewer
- qPCR
- quantitative PCR.
References
- 1. David S. S., O'Shea V. L., and Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dizdaroglu M. (1992) Oxidative damage to DNA in mammalian chromatin. Mutat. Res. 275, 331–342 [DOI] [PubMed] [Google Scholar]
- 3. Steenken S., and Jovanovic S. V. (1997) How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 119, 617–618 [Google Scholar]
- 4. Mitra S., Hazra T. K., Roy R., Ikeda S., Biswas T., Lock J., Boldogh I., and Izumi T. (1997) Complexities of DNA base excision repair in mammalian cells. Mol. Cells 7, 305–312 [PubMed] [Google Scholar]
- 5. Lindahl T., and Barnes D. E. (2000) Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 65, 127–133 [DOI] [PubMed] [Google Scholar]
- 6. Shigenaga M. K., Hagen T. M., and Ames B. N. (1994) Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U.S.A. 91, 10771–10778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klungland A., Rosewell I., Hollenbach S., Larsen E., Daly G., Epe B., Seeberg E., Lindahl T., and Barnes D. E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. U.S.A. 96, 13300–13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Minowa O., Arai T., Hirano M., Monden Y., Nakai S., Fukuda M., Itoh M., Takano H., Hippou Y., Aburatani H., Masumura K., Nohmi T., Nishimura S., and Noda T. (2000) Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 4156–4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mabley J. G., Pacher P., Deb A., Wallace R., Elder R. H., and Szabó C. (2005) Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 19, 290–292 [DOI] [PubMed] [Google Scholar]
- 10. Bacsi A., Aguilera-Aguirre L., Szczesny B., Radak Z., Hazra T. K., Sur S., Ba X., and Boldogh I. (2013) Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair. 12, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ba X., Aguilera-Aguirre L., Rashid Q. T., Bacsi A., Radak Z., Sur S., Hosoki K., Hegde M. L., and Boldogh I. (2014) The role of 8-oxoguanine DNA glycosylase-1 in inflammation. Int. J. Mol. Sci. 15, 16975–16997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sampath H., Vartanian V., Rollins M. R., Sakumi K., Nakabeppu Y., and Lloyd R. S. (2012) 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 7, e51697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boldogh I., Hajas G., Aguilera-Aguirre L., Hegde M. L., Radak Z., Bacsi A., Sur S., Hazra T. K., and Mitra S. (2012) Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J. Biol. Chem. 287, 20769–20773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hajas G., Bacsi A., Aguilera-Aguirre L., Hegde M. L., Tapas K. H., Sur S., Radak Z., Ba X., and Boldogh I. (2013) 8-Oxoguanine DNA glycosylase-1 links DNA repair to cellular signaling via the activation of the small GTPase Rac1. Free Radic. Biol. Med. 61, 384–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo J., Hosoki K., Bacsi A., Radak Z., Hegde M. L., Sur S., Hazra T. K., Brasier A. R., Ba X., and Boldogh I. (2014) 8-Oxoguanine DNA glycosylase-1-mediated DNA repair is associated with Rho GTPase activation and α-smooth muscle actin polymerization. Free Radic. Biol. Med. 73, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aguilera-Aguirre L., Bacsi A., Radak Z., Hazra T. K., Mitra S., Sur S., Brasier A. R., Ba X., and Boldogh I. (2014) Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway. J. Immunol. 193, 4643–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perillo B., Ombra M. N., Bertoni A., Cuozzo C., Sacchetti S., Sasso A., Chiariotti L., Malorni A., Abbondanza C., and Avvedimento E. V. (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206 [DOI] [PubMed] [Google Scholar]
- 18. Amente S., Bertoni A., Morano A., Lania L., Avvedimento E. V., and Majello B. (2010) LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 29, 3691–3702 [DOI] [PubMed] [Google Scholar]
- 19. Pastukh V., Roberts J. T., Clark D. W., Bardwell G. C., Patel M., Al-Mehdi A. B., Borchert G. M., and Gillespie M. N. (2015) An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L1367–L1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim H. S., Kim B. H., Jung J. E., Lee C. S., Lee H. G., Lee J. W., Lee K. H., You H. J., Chung M. H., and Ye S. K. (2016) Potential role of 8-oxoguanine DNA glycosylase 1 as a STAT1 coactivator in endotoxin-induced inflammatory response. Free Radic. Biol. Med. 93, 12–22 [DOI] [PubMed] [Google Scholar]
- 21. Saxonov S., Berg P., and Brutlag D. L. (2006) A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U.S.A. 103, 1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kadonaga J. T., Carner K. R., Masiarz F. R., and Tjian R. (1987) Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell 51, 1079–1090 [DOI] [PubMed] [Google Scholar]
- 23. Brasier A. R. (2010) The nuclear factor-κB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 86, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian B., and Brasier A. R. (2003) Identification of a nuclear factor κB-dependent gene network. Recent Prog. Horm. Res. 58, 95–130 [DOI] [PubMed] [Google Scholar]
- 25. Vlahopoulos S. A., Cen O., Hengen N., Agan J., Moschovi M., Critselis E., Adamaki M., Bacopoulou F., Copland J. A., Boldogh I., Karin M., and Chrousos G. P. (2015) Dynamic aberrant NF-κB spurs tumorigenesis: a new model encompassing the microenvironment. Cytokine Growth Factor Rev. 26, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen F. E., Huang D. B., Chen Y. Q., and Ghosh G. (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 391, 410–413 [DOI] [PubMed] [Google Scholar]
- 27. Ghosh R., and Mitchell D. L. (1999) Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 27, 3213–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghosh S., and Karin M. (2002) Missing pieces in the NF-κB puzzle. Cell 109, S81–S96 [DOI] [PubMed] [Google Scholar]
- 29. Ba X., Bacsi A., Luo J., Aguilera-Aguirre L., Zeng X., Radak Z., Brasier A. R., and Boldogh I. (2014) 8-Oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 192, 2384–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vraetz T., Ittel T. H., van Mackelenbergh M. G., Heinrich P. C., Sieberth H. G., and Graeve L. (1999) Regulation of β2-microglobulin expression in different human cell lines by proinflammatory cytokines. Nephrol. Dial. Transplant. 14, 2137–2143 [DOI] [PubMed] [Google Scholar]
- 31. Hazra T. K., Izumi T., Boldogh I., Imhoff B., Kow Y. W., Jaruga P., Dizdaroglu M., and Mitra S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 3523–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pastukh V., Ruchko M., Gorodnya O., Wilson G. L., and Gillespie M. N. (2007) Sequence-specific oxidative base modifications in hypoxia-inducible genes. Free Radic. Biol. Med. 43, 1616–1626 [DOI] [PubMed] [Google Scholar]
- 33. Jamaluddin M., Wang S., Boldogh I., Tian B., and Brasier A. R. (2007) TNF-α-induced NF-κB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal 19, 1419–1433 [DOI] [PubMed] [Google Scholar]
- 34. Kaplan N., Urao N., Furuta E., Kim S. J., Razvi M., Nakamura Y., McKinney R. D., Poole L. B., Fukai T., and Ushio-Fukai M. (2011) Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free Radic. Res. 45, 1124–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giese B. (2002) Long-distance electron transfer through DNA. Annu. Rev. Biochem. 71, 51–70 [DOI] [PubMed] [Google Scholar]
- 36. Margolin Y., Cloutier J. F., Shafirovich V., Geacintov N. E., and Dedon P. C. (2006) Paradoxical hotspots for guanine oxidation by a chemical mediator of inflammation. Nat. Chem. Biol. 2, 365–366 [DOI] [PubMed] [Google Scholar]
- 37. Ghosh G., van Duyne G., Ghosh S., and Sigler P. B. (1995) Structure of NF-κB p50 homodimer bound to a κB site. Nature 373, 303–310 [DOI] [PubMed] [Google Scholar]
- 38. Hailer-Morrison M. K., Kotler J. M., Martin B. D., and Sugden K. D. (2003) Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-κB promoter element. Biochemistry 42, 9761–9770 [DOI] [PubMed] [Google Scholar]
- 39. Bruner S. D., Norman D. P., and Verdine G. L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403, 859–866 [DOI] [PubMed] [Google Scholar]
- 40. Banerjee A., Yang W., Karplus M., and Verdine G. L. (2005) Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 434, 612–618 [DOI] [PubMed] [Google Scholar]
- 41. Nakajima H., Nagaso H., Kakui N., Ishikawa M., Hiranuma T., and Hoshiko S. (2004) Critical role of the automodification of poly(ADP-ribose) polymerase-1 in nuclear factor-κB-dependent gene expression in primary cultured mouse glial cells. J. Biol. Chem. 279, 42774–42786 [DOI] [PubMed] [Google Scholar]
- 42. Wan F., Anderson D. E., Barnitz R. A., Snow A., Bidere N., Zheng L., Hegde V., Lam L. T., Staudt L. M., Levens D., Deutsch W. A., and Lenardo M. J. (2007) Ribosomal protein S3: a KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 131, 927–939 [DOI] [PubMed] [Google Scholar]
- 43. Donley N., Jaruga P., Coskun E., Dizdaroglu M., McCullough A. K., and Lloyd R. S. (2015) Small molecule inhibitors of 8-oxoguanine DNA glycosylase-1 (OGG1). ACS Chem. Biol. 10, 2334–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hill J. W., and Evans M. K. (2006) Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 34, 1620–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mokkapati S. K., Wiederhold L., Hazra T. K., and Mitra S. (2004) Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry 43, 11596–11604 [DOI] [PubMed] [Google Scholar]
- 46. Tian B., Li X., Kalita M., Widen S. G., Yang J., Bhavnani S. K., Dang B., Kudlicki A., Sinha M., Kong F., Wood T. G., Luxon B. A., and Brasier A. R. (2015) Analysis of the TGFβ-induced program in primary airway epithelial cells shows essential role of NF-κB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics 16, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Allgayer J., Kitsera N., Bartelt S., Epe B., and Khobta A. (2016) Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 44, 7267–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wallace S. S. (2013) DNA glycosylases search for and remove oxidized DNA bases. Environ. Mol. Mutagen. 54, 691–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morreall J., Limpose K., Sheppard C., Kow Y. W., Werner E., and Doetsch P. W. (2015) Inactivation of a common OGG1 variant by TNF-α in mammalian cells. DNA Repair. 26, 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bravard A., Vacher M., Gouget B., Coutant A., de Boisferon F. H., Marsin S., Chevillard S., and Radicella J. P. (2006) Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol. Cell Biol. 26, 7430–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dizdaroglu M. (2005) Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat Res. 591, 45–59 [DOI] [PubMed] [Google Scholar]
- 52. Chakraborty A., Wakamiya M., Venkova-Canova T., Pandita R. K., Aguilera-Aguirre L., Sarker A. H., Singh D. K., Hosoki K., Wood T. G., Sharma G., Cardenas V., Sarkar P. S., Sur S., Pandita T. K., Boldogh I., and Hazra T. K. (2015) Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation. J. Biol. Chem. 290, 24636–24648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moore S. P., Kruchten J., Toomire K. J., and Strauss P. R. (2016) Transcription factors and DNA repair enzymes compete for damaged promoter sites. J. Biol. Chem. 291, 5452–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schreck R., Zorbas H., Winnacker E. L., and Baeuerle P. A. (1990) The NF-κB transcription factor induces DNA bending which is modulated by its 65-kD subunit. Nucleic Acids Res. 18, 6497–6502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen-Park F. E., Huang D. B., Noro B., Thanos D., and Ghosh G. (2002) The κB DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J. Biol. Chem. 277, 24701–24708 [DOI] [PubMed] [Google Scholar]
- 56. Falvo J. V., Thanos D., and Maniatis T. (1995) Reversal of intrinsic DNA bends in the IFNβ gene enhancer by transcription factors and the architectural protein HMG I(Y). Cell 83, 1101–1111 [DOI] [PubMed] [Google Scholar]
- 57. Panne D. (2008) The enhanceosome. Curr. Opin. Struct. Biol. 18, 236–242 [DOI] [PubMed] [Google Scholar]
- 58. Agresti A., Lupo R., Bianchi M. E., and Müller S. (2003) HMGB1 interacts differentially with members of the Rel family of transcription factors. Biochem. Biophys. Res. Commun. 302, 421–426 [DOI] [PubMed] [Google Scholar]
- 59. Kaur S., Coulombe Y., Ramdzan Z. M., Leduy L., Masson J. Y., and Nepveu A. (2016) SATB1 functions as an accessory factor in base excision repair. J. Biol. Chem. 291, 22769–22780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brasier A. R. (2006) The NF-κB regulatory network. Cardiovasc. Toxicol. 6, 111–130 [DOI] [PubMed] [Google Scholar]
- 61. Norman D. P., Chung S. J., and Verdine G. L. (2003) Structural and biochemical exploration of a critical amino acid in human 8-oxoguanine glycosylase. Biochemistry 42, 1564–1572 [DOI] [PubMed] [Google Scholar]
- 62. Ghosh G., Wang V. Y., Huang D. B., and Fusco A. (2012) NF-κB regulation: lessons from structures. Immunol. Rev. 246, 36–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Natoli G., Saccani S., Bosisio D., and Marazzi I. (2005) Interactions of NF-κB with chromatin: the art of being at the right place at the right time. Nat. Immunol. 6, 439–445 [DOI] [PubMed] [Google Scholar]
- 64. Bacsi A., Chodaczek G., Hazra T. K., Konkel D., and Boldogh I. (2007) Increased ROS generation in subsets of OGG1 knockout fibroblast cells. Mech. Ageing Dev. 128, 637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aguilera-Aguirre L., Bacsi A., Saavedra-Molina A., Kurosky A., Sur S., and Boldogh I. (2009) Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 183, 5379–5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. German P., Szaniszlo P., Hajas G., Radak Z., Bacsi A., Hazra T. K., Hegde M. L., Ba X., and Boldogh I. (2013) Activation of cellular signaling by 8-oxoguanine DNA glycosylase-1-initiated DNA base excision repair. DNA Repair. 12, 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Landt S. G., Marinov G. K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B. E., Bickel P., Brown J. B., Cayting P., Chen Y., DeSalvo G., Epstein C., Fisher-Aylor K. I., Euskirchen G., et al. (2012) ChIP-Seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tian B., Yang J., and Brasier A. R. (2012) Two-step cross-linking for analysis of protein-chromatin interactions. Methods Mol. Biol. 809, 105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., and Liu X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gupta S., Stamatoyannopoulos J. A., Bailey T. L., and Noble W. S. (2007) Quantifying similarity between motifs. Genome Biol. 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]