

Abstract
Protein 4.1R (4.1R) isoforms are expressed in both cardiac and skeletal muscle. 4.1R is a component of the contractile apparatus. It is also associated with dystrophin at the sarcolemma in skeletal myofibers. However, the expression and function of 4.1R during myogenesis have not been characterized. We now report that 4.1R expression increases during C2C12 myoblast differentiation into myotubes. Depletion of 4.1R impairs skeletal muscle differentiation and is accompanied by a decrease in the levels of myosin heavy and light chains and caveolin-3. Furthermore, the expression of myogenin at the protein, but not mRNA, level is drastically decreased in 4.1R knockdown myocytes. Similar results were obtained using MyoD-induced differentiation of 4.1R−/− mouse embryonic fibroblast cells. von Hippel-Lindau (VHL) protein is known to destabilize myogenin via the ubiquitin-proteasome pathway. We show that 4.1R associates with VHL and, when overexpressed, reverses myogenin ubiquitination and stability. This suggests that 4.1R may influence myogenesis by preventing VHL-mediated myogenin degradation. Together, our results define a novel biological function for 4.1R in muscle differentiation and provide a molecular mechanism by which 4.1R promotes myogenic differentiation.
Keywords: E3 ubiquitin ligase, myogenesis, protein degradation, protein stability, ubiquitylation (ubiquitination), myogenin, protein 4.1R, von Hippel Lindau
Introduction
The 80-kDa erythroid protein 4.1R (4.1R)2 is a major structural component of the erythrocyte membrane skeleton, where it links transmembrane proteins to the underlying spectrin-actin complex and enhances membrane mechanical stability. A diverse collection of 4.1R isoforms, regulated by tissue-specific alternative splicing, has been shown to participate in several cellular processes in non-erythroid cells (1–5). 4.1R isoforms are expressed in both cardiac (6) and skeletal muscle (7). We have shown that a 135-kDa isoform localizes at the A-band and interacts with the components of the contractile apparatus in skeletal myofibers (7). 4.1R epitopes are found at the sarcolemma of both slow and fast myofibers in normal tissue but are absent from dystrophin-deficient sarcolemma in muscles of individuals affected with Duchene muscular dystrophy (DMD) (8). Isoforms of 4.1R are differentially compartmentalized in the heart, where they form specific complexes with proteins critical to cardiomyocyte Ca2+ metabolism (9) and are required for normal function of several cardiac ion transporters (10). However, the expression and importance of 4.1R during myogenesis remains to be characterized. In this paper, we describe studies suggesting that 4.1R is important for the development as well as the function of skeletal muscle.
Skeletal muscle differentiation is a tightly regulated process that starts with cell cycle arrest of myoblasts. This leads to cell fusion and formation of multinucleated myotubes. The myogenesis program is orchestrated by sequential and coordinated expression of a basic helix-loop-helix family of myogenic regulatory factors (MRFs), including MyoD, Myf5, myogenin, and MRF4. MyoD and Myf5 are expressed in proliferating myoblasts and are required for initial commitment of the myogenic lineage (11). Myogenin, MRF4, and myocyte-specific enhancer factors (MEF2a and MEF2c) participate in differentiation of the committed cells (12, 13). A remarkable property of cultured muscle precursor cells is the activation of the myogenic program upon changes in cell culture conditions. The differentiated cells fuse together to form muscle fibers (14). Ectopic expression of any one of the MRFs is sufficient to induce myogenic differentiation in various non-muscle cells (15, 16). Although cultured cells only partially recapitulate the regulation of developmental myogenesis by extracellular cues, they have been instrumental for the identification and the characterization of myogenic pathways.
The expression of MRFs is controlled by both transcriptional and post-transcriptional mechanisms that increase and stabilize their mRNAs during myogenesis (17, 18). For example, HuR protects MyoD and myogenin mRNAs from mRNA decay during myogenesis (19), whereas Rbm24 interacts with the 3′-untranslated region of myogenin mRNA and regulates its stability (20). Like many transcription factors, MRFs are also short lived regulatory proteins. Pax7 (21), Pax3 (22), MyoD (23), Myf5 (24, 25), and myogenin (26, 27) have all been shown to be degraded by the ubiquitin-proteasome system. In this system, proteins subjected to degradation are covalently linked to a ubiquitin (Ub) chain by a cascade involving Ub-activating E1 enzyme, Ub-conjugating E2 enzyme, and E3 Ub ligase (28). Activated Ub is transferred to a target protein by Ub ligase, and the resultant polyubiquitinated proteins are degraded by proteasomes. Ub ligase represents either an E3-E2 holoenzyme or its E3 component. Three classes of E3 with substrate specificity have been identified: HECT (homologous to E6-AP C terminus), RING finger, and U-box domain types. The mammalian genome encodes at least 1,000 E3 Ub ligases that allow for a high degree of substrate specificity (29). All three classes of E3 are involved in the ubiquitin-mediated degradation of MRFs. MAFbx (muscle atrophy F-box protein)/AT-1 (atrogin-1) (30), and HUWE1 (31) E3 ubiquitin ligase can ubiquitinate MyoD and lead to proteasomal degradation. The von Hippel-Lindau (VHL) tumor suppressor protein is crucial to the activity of an E3 ubiquitin-protein ligase (32, 33). It has been shown that VHL interacts with myogenin and promotes both the ubiquitination and destabilization of the myogenin protein (26).
Conversely, several proteins have been demonstrated to promote myogenesis through an anti-ubiquitination ability. TBP-interacting protein 120B (TIP120B)/cullin-associated and neddylation-dissociated 2 (CAND2) inhibit SCF-dependent ubiquitination of myogenin and result in accelerated myogenic differentiation (27). EGLN3 prolyl hydroxylase binds to myogenin and antagonizes VHL-mediated ubiquitination and degradation of myogenin and also accelerates skeletal muscle differentiation (26). Understanding the factors modulating proteolysis of MRFs under physiological conditions is thus important for understanding MRF homeostasis. In the present study, we investigated the role of protein 4.1R in muscle differentiation using C2C12 and MyoD-induced mouse embryonic fibroblast (MEF) differentiation as model systems. We find that differentiation is delayed significantly in cells lacking 4.1R. Both muscle-specific myosin and caveolin-3 expressions were compromised due to increased ubiquitination and reduced myogenin protein levels. We show that 4.1R associates with VHL and, when overexpressed, reverses myogenin ubiquitination and stability. Our study suggests that 4.1R may prevent VHL-mediated myogenin degradation. Together, our results define a novel function for 4.1R in muscle differentiation and suggest a molecular mechanism by which 4.1R promotes myogenic differentiation.
Results
Protein 4.1R is Up-regulated during Myogenic Differentiation of C2C12 Cells
We have shown earlier that 4.1R is a component of the contractile apparatus and associates with myosin heavy chain (MHC) in the A-band (7). Using a C2C12 myoblast differentiation system, we investigated the role of 4.1R during myogenesis. C2C12 cells proliferate in growth medium and undergo myogenic differentiation in differentiation medium. We first characterized the developmental profiles of muscle protein markers, MHC and α-actinin, and 4.1R during a 10-day (d1–d10) differentiation period. The expression of MHC was undetectable in d1 and d2 cells. Initial expression was noted around d3 and continuously increased throughout the differentiation process (Fig. 1A). The expression of α-actinin occurred at an earlier time point in d1 cells and continuously increased during differentiation (Fig. 1A). 4.1R protein was detected in non-differentiated myoblasts and also increased at a steady rate through differentiation (Fig. 1A). 4.1R protein levels increased by 2-fold at d4-d6 when cultures exhibited contractile behavior and by 5-fold in d10 myotubes when compared with that of d0 cells (Fig. 1B).
FIGURE 1.

Expression and localization of 4.1R, myosin heavy chain, and α-actinin in C2C12 cells during myogenic differentiation. C2C12 cells were cultured in growth medium (GM) and switched to differentiation medium (DM) for a specific number of days as indicated. A, lysates from C2C12 at the indicated days of differentiation were separated by SDS-PAGE and immunoblotted with anti-MHC, anti-α-actinin, anti-4.1R, or anti-β-tubulin antibodies. B, the relative expression of 4.1R in each sample was quantified using densitometry, adjusted for protein loading with β-tubulin, and compared with 4.1R levels in the day 0 sample, which was arbitrarily assigned a value of 1.0. The results represent the means of three separate experiments. C, intracellular localization of 4.1R, MHC, and α-actinin were examined using its respective antibody and revealed with Zeiss microscopy. Day 0 and day 3 cells were also stained with DAPI to visualize the nucleus. Bar, 5 μm. D, double-stained day 10 myotubes with MHC and α-actinin or 4.1R. Bar, 5 μm. E, staining with anti-4.1R (green) and anti-MHC (red) of a longitudinal section of adult mouse skeletal muscle. Bar, 5 μm. Error bars, S.D.
We then characterized the localization of 4.1R in differentiating myotubes. In non-differentiated C2C12 cells, MHC was absent in d0, discontinuously distributed along filaments on d3, and then cross-striated into the mature A-band in d10 myotubes (Fig. 1C). A similar progression and localization of α-actinin were observed from d0 to d3 differentiation, before subsequently organizing into the Z-line (Fig. 1C). 4.1R was abundantly concentrated in the nucleus and diffusely distributed in the cytoplasm of d0 cells. A reduction in 4.1R localization to the nucleus with an increase in localization to the cytoplasm was detected in d3 cells (Fig. 1C). Day 10 myotubes exhibited A-band as well as cytoplasmic localization of 4.1R (Fig. 1C). When superimposed, MHC and α-actinin are labeled at the A-band and Z-line, respectively, whereas 4.1R partially coincides with MHC in the A-band of d10 cells (Fig. 1D). The co-localization of MHC and 4.1R is not discrete due to the strong cytoplasmic staining of 4.1R. To characterize 4.1R distribution relative to that of MHC in vivo, we stained longitudinal sections of adult mouse muscle section for the presence of 4.1R and MHC. Coincident staining between MHC and 4.1R indicated that the latter is in line with A-bands (Fig. 1E). The correlation between the increase in 4.1R expression and development of myotubes suggests that 4.1R may be involved in myogenic differentiation and/or maturation.
4.1R Expression Modulates C2C12 Myogenic Differentiation and Myogenic Protein Expression
To establish the role of 4.1R in myogenic differentiation, we characterized the impact of reduced 4.1R expression on differentiation and myogenic related protein expression in differentiating C2C12. A tet-on lentiviral vector containing either 4.1R shRNA or a control scramble shRNA sequence was transduced into C2C12 cells, and stable lines were selected. 4.1R protein levels did not change in control shRNA cells grown in the presence or absence of doxycycline (Fig. 2A). 4.1R shRNA cells exhibited an 80–90% reduction in 4.1R protein levels when grown in doxycycline-containing medium (Fig. 2A).
FIGURE 2.

4.1R depletion delays C2C12 myogenic differentiation and reduces myogenic related protein expression. A, validation of 4.1R silencing. C2C12 cells transduced with a control scramble shRNA (Ctrl) or 4.1R shRNA (sh4.1R) in pTRIPZ were stably selected with puromycin and treated with or without doxycycline for 24 h. Cell extracts were analyzed for the efficiency of 4.1R knockdown by immunoblotting with an anti-4.1R Ab. β-Actin served as a loading control. B, 4.1R silencing suppresses myogenic differentiation. Control or sh4.1R stable C2C12 cells were grown for 3 days in differentiation medium in the presence or absence of doxycycline. Differentiation was analyzed by immunofluorescence staining for MHC with an anti-MF20 Ab and FITC-conjugated secondary Ab and revealed with Zeiss microscopy (×40). Nuclei were counterstained with DAPI. The graphs represent the percentage of MHC-positive cells in each treatment obtained from three independent experiments. C, 4.1R silencing reduces myotube formation. sh4.1R or control stable C2C12 cells were grown for 4 days in differentiation medium with or without doxycycline. Cells were stained for MHC and DAPI. Fusion indices were calculated as the percentage of cells containing three or more nuclei within the myosin-positive myocytes. Each experiment was repeated three times, and S.D. values were determined. D, 4.1R silencing decreases myogenic related protein expression. Stable sh4.1R C2C12 lines were grown in growth medium (GM) and switched to differentiation medium (DM) in the presence or absence of doxycycline. Cell lysates were collected at the indicated time course and analyzed for the presence of 4.1R as well as the myogenic differentiation marker MHC, MLC, myofusion marker Cav3, and MRFs (Myf5, MyoD, and myogenin). Each was detected with its respective Ab. Tubulin served as a loading control. Note that to detect several proteins from the same set of experiments, an equal amount of lysate was fractionated in more than one gel and transferred to its respective membrane. When the same membrane was used for more than one antibody, the membrane was stripped between each probing with a limitation of 3–4 strippings per membrane. Each membrane was probed with tubulin as a loading control. Error bars, S.D.
The effects of 4.1R silencing on C2C12 myoblast differentiation were first examined using the differentiation marker MHC in an immunofluorescent staining assay. 4.1R shRNA and control shRNA stable lines, grown in differentiation medium for 3 days in the presence or absence of doxycycline, were immunostained for the presence of MHC. Approximately 30% of the control cells exhibited positive MHC staining regardless of whether they were grown in the absence or presence of doxycycline (Fig. 2B). Similarly, ∼30% of 4.1R shRNA cells grown in the absence of doxycycline were MHC-positive (Fig. 2B). In contrast, only ∼5% of 4.1R shRNA cells stained positive for MHC when grown in the presence of doxycycline with reduced 4.1R levels (Fig. 2B). These results suggest that decreased 4.1R expression significantly affects myoblast differentiation. We further examined the fusion index, expressed as MHC-positive cells with three or more nuclei, in C2C12 stable lines grown for 4 days in differentiation medium in the presence or absence of doxycycline. Nearly 14% of MHC-positive cells displayed three or more nuclei in control lines treated with or without doxycycline. 4.1R shRNA lines grown in the absence of doxycycline showed a similar fusion index compared with the control cells (Fig. 2C). However, a considerable reduction in MHC-positive cells with three or more nuclei (∼4%) was observed in 4.1R shRNA cells treated with doxycycline (Fig. 2C). These results suggest a role for 4.1R in C2C12 myoblast differentiation and myotube nuclear fusion.
We next analyzed the effects of 4.1R silencing on myogenic differentiation markers, including MHC and myosin light chain (MLC), and myofusion marker caveolin-3 in 4.1R shRNA stable lines grown in growth medium and subsequently switched to differentiation medium for 1–5 days in the absence or presence of doxycycline. 4.1R was effectively knocked down by 80–90% when grown in doxycycline-containing medium during differentiation (Fig. 2D). Both differentiation and myofusion markers were expressed in d2–d5 cells grown in differentiation medium lacking doxycycline when 4.1R was expressed (Fig. 2D). However, MHC and MLC proteins were not detected in d2 4.1R shRNA cells grown in the presence of doxycycline when 4.1R was silenced. This reduction persisted through d5 with only ∼10–20% of MHC and MLC expressed in 4.1R silenced cells compared with that of non-silenced cells. Similarly, caveolin-3 levels were reduced by ∼80% in 4.1R-depleted cells on d2 and remained depressed throughout differentiation (Fig. 2D).
The formation of skeletal muscle is orchestrated by a family of MRFs, including MyoD, Myf5, myogenin, and MRF4 (12, 13). MRFs regulate myosin and caveolin-3 expression. The observation that 4.1R depletion decreases MHC, MLC, and caveolin-3 levels in C2C12 myotubes prompted further examination of whether expression of MRFs is also compromised in the absence of 4.1R. The same samples collected from the above analyses were examined for the presence of Myf5, MyoD, and myogenin. Both Myf5 and MyoD were detected in cells grown in both growth and differentiation medium throughout the differentiation process. Their expressions were not affected by the depletion of 4.1R, as evidenced by the nearly equal amounts of Myf5 and MyoD detected in samples with or without 4.1R (Fig. 2D). Conversely, myogenin protein levels were affected by 4.1R expression levels. Myogenin was not detected in cells grown in growth medium. After switching to differentiation medium lacking doxycycline, myogenin was detected when 4.1R was expressed. Myogenin levels were drastically reduced to ∼10–20% in 4.1R silenced cells from d1 through d5 of differentiation when compared with that of 4.1R non-silenced cells (Fig. 2D). These results suggest that 4.1R is a critical factor necessary for myoblast differentiation and acts by affecting myogenin expression.
4.1R Is Important for MyoD-induced Conversion of Fibroblasts into Myoblasts
Ectopic expression of MyoD and myogenin can convert non-muscle cells into myoblasts (34, 35). However, MyoD is more efficient than myogenin at initiating expression of the skeletal muscle gene and gives rise to myosin-positive cells at a higher frequency in many non-myogenic cells (36). In MEF cells, there were ∼4 times more myosin-positive cells for MyoD (30%) when compared with that of myogenin (8%). MyoD activates myogenin (37). Expression of MyoD in MEF cells resulted in reproducible production of myogenin when switched to differentiation medium (data not shown). To accurately analyze the effect of 4.1R on myogenesis, we chose to express MyoD to provide a more adequate differentiation frequency.
To further investigate the role of 4.1R in myogenesis, we tested whether 4.1R could affect MyoD-mediated myogenic conversion in MEF cells isolated from 4.1R+/+ (WT) or 4.1R−/− (KO) mice. RNA analysis of 4.1R−/− MEF showed a complete deletion of exons 2–4, which contain both translation initiation sites encoding the 135- or 80-kDa 4.1R forms (Fig. 3A). These MEF cells showed no muscle-specific protein expression either when grown in growth medium or after switching to differentiation medium (data not shown). We transduced MEF cells with pTRIPO-3FLAG-MyoD (MEF-MyoD) in which the expression of MyoD can be induced with doxycycline (Fig. 3B). MEF-MyoD cells grown in growth medium were transitioned to doxycycline-containing growth medium for 24 h. Cells were subsequently switched to doxycycline-containing differentiation medium for an additional period of time.
FIGURE 3.

Deletion of 4.1R reduces MyoD-induced differentiation and nuclear fusion in MEFs. A, validation of 4.1R deletion in 4.1R−/− MEF cells. Total RNA isolated from the 4.1R+/+ (WT) and 4.1R−/− (KO) cells were analyzed for the presence of exons 2–4, which contains the translation initiation sites for the 135- and 80-kDa isoforms of 4.1R. GAPDH served as an RT-PCR control. B, WT or KO MEF cells transduced with pTRIPO-3FLAG-MyoD were stably selected with puromycin. Expression of MyoD was analyzed after cells were incubated in differentiation medium in the presence or absence of doxycycline for 24 h. Actin served as a loading control. C, 4.1R deletion reduces myogenic differentiation. 4.1R WT-MyoD and KO-MyoD MEF stable lines were grown in differentiation medium in the presence of doxycycline for 48 h. Differentiation was analyzed by immunofluorescence staining for MHC with an anti-MF20 Ab and Texas Red-conjugated secondary Ab and assessed with Zeiss microscopy (×40). Nuclei were counterstained with DAPI. D, graphical presentation of the effect of 4.1R deletion on differentiation and fusion indices. Left graph, the differentiation indices were calculated as the percentage of MHC-positive cells in each treatment obtained from three independent experiments. Right graph, the fusion indices were calculated as the percentage of cells containing three or more nuclei within the MHC-positive cells. Each experiment was repeated three times, and S.D. values (error bars) were determined. E, 4.1R depletion decreases MHC, MLC, Cav3, and Myog protein expression. WT-MyoD and KO-MyoD MEF cells were grown in growth medium (GM) in the presence or absence of doxycycline for 24 h. Doxycycline-treated MEFs in growth medium were then transited to differentiation medium (DM) in the presence of doxycycline for the indicated periods of time. Cell lysates were collected and immunoblotted for the presence of the indicated protein with its respective Abs. Tubulin served as a loading control.
The effects of 4.1R on MyoD-mediated myogenic differentiation in MEF-MyoD cells were scored by positive MHC staining and nuclear fusion indices after 48 h in doxycycline-containing differentiation medium. Myogenic differentiation was significantly impaired in 4.1R−/− MEF-MyoD cells, as reflected by a reduced number of MHC positive cells (Fig. 3C). Approximately 30% of 4.1R+/+ and ∼5% of 4.1R−/− cells stained positive for MHC (Fig. 3D, left). Moreover, only ∼5% of MHC-positive cells possessed three or more than three nuclei in 4.1R−/− cells compared with ∼18% in 4.1R+/+ cells (Fig. 3D, right).
The levels of differentiation and myofusion markers were further examined for 24–60 h during MyoD-mediated myogenic differentiation. MyoD was not detected when doxycycline was omitted (Fig. 3E, lanes 1 and 2) but was detected in the presence of doxycycline in both growth and differentiation medium (Fig. 3E, lanes 3–12). MHC, MLC, and caveolin-3 proteins were expressed after ∼30 h in doxycycline-containing differentiation medium. Levels of these proteins were significantly reduced in 4.1R−/− cells when compared with that of 4.1R+/+ cells. 4.1R−/− cells had about 5–10-fold suppression of these protein levels when compared with those in 4.1R+/+ cells (Fig. 3E).
We next examined the expression of upstream MRFs. Whereas Myf5 levels in 4.1R+/+ or 4.1R−/− MEF-MyoD cells did not change during the course of differentiation, myogenin protein levels in 4.1R−/− were reduced to ∼10–20% compared with that of 4.1R+/+ cells (Fig. 3E). Consistent with the results from depletion of 4.1R by shRNA in the C2C12 system, MHC, MLC, caveolin-3, and myogenin levels were repressed, but there was no effect on the protein levels of Myf5. Together, these results suggest that the effect of 4.1R on MyoD-stimulated conversion of fibroblasts into myoblasts is exerted via its regulation on myogenin expression.
4.1R Isoforms Expressed in C2C12
Protein 4.1R is composed of many alternatively spliced exons, the expression of which has been shown to be regulated in a cell type- or differentiation stage-specific manner. To understand the function of 4.1R in modulating myogenic differentiation and myogenic protein expression, we first characterized 4.1R isoforms expressed in day 0 and day 8 differentiated C2C12.
4.1R cDNA libraries were constructed from day 0 and day 8 C2C12 RNA and cloned into TA vectors as described previously (7). A total of 192 clones from each library was initially screened with probes specific for AUG-1 containing exon 2′ that distinguishes the 135- and 80-kDa isoforms. Subsequently, primer pairs flanking the alternatively spliced exons were used to detect specific exon compositions. Notably, ∼95% of the 192 4.1R cDNA clones from either day 0 or day 8 cells included exon 2′, indicating that 4.1R isoforms predominantly originate from AUG-1. Furthermore, no major differences in exon composition were found in day 0 and day 8 cells. These results suggest that no relevant differentiation stage-specific alternative splicing occurs during C2C12 myogenic development. Similar to the forms expressed in skeletal muscle, the most predominant form, which is composed of all of the known alternatively spliced exons with the exception of exons 14, 15, and 17b (Fig. 4B), constituted 75% of the 4.1R cDNAs. Conversely, minor forms of protein 4.1R have varying inclusions of exons 14, 15, 16, and 17a (Fig. 4B).
FIGURE 4.

4.1R isoforms expressed in day 0 undifferentiated and day 8 differentiated C2C12. A, schematic diagram of protein 4.1R and its domains. HP, headpiece; MBD, membrane binding domain; SAB, spectrin-actin binding domain; CTD, C-terminal domain. Previously characterized constitutive sequences are indicated as solid gray boxes, and alternatively spliced cassettes are depicted as shaded boxes. Exon numbers are indicated. B, percentile prevalence of the major 4.1R isoforms identified in day 0 and day 8 differentiated C2C12. 4.1R cDNA libraries constructed from RNA isolated from day 0 and day 8 C2C12 cells were screened with an exon 2′-specific oligonucleotide probe that distinguishes between 135 and 80 kDa. Over 95% of 192 cDNA clones from day 0 and day 8 cells include exon 2′ and encode for 135-kDa forms. The exon compositions of the 135-kDa forms were further PCR-screened using specific oligonucleotides flanking the alternatively spliced exons.
4.1R Expression in 4.1R Knockdown C2C12 Cells Rescues Myogenic Differentiation and Myogenic Protein Expression
To investigate whether the phenotypes associated with 4.1R depletion in C2C12 could be reversed, we reconstituted 4.1R expression in pTRIPZ-4.1R shRNA C2C12 stable lines with an shRNA-resistant 4.1R rescue construct. The rescue construct is composed of all alternatively spliced exons except for exons 14, 15, and 17b (7).
Transfection of either a vector control plasmid or the 4.1R rescue construct did not alter the levels of the endogenous 4.1R (Fig. 5A) when compared with that of parental pTRIPZ-4.1R shRNA C2C12 (Fig. 2A). In the presence of doxycycline, the endogenous 4.1R levels were reduced to ∼20% compared with levels of 4.1R seen in the absence of doxycycline (Fig. 5A). The 4.1R rescue construct expressed approximately the same levels of 4.1R as that of endogenous 4.1R in the absence of doxycycline (Fig. 5A).
FIGURE 5.
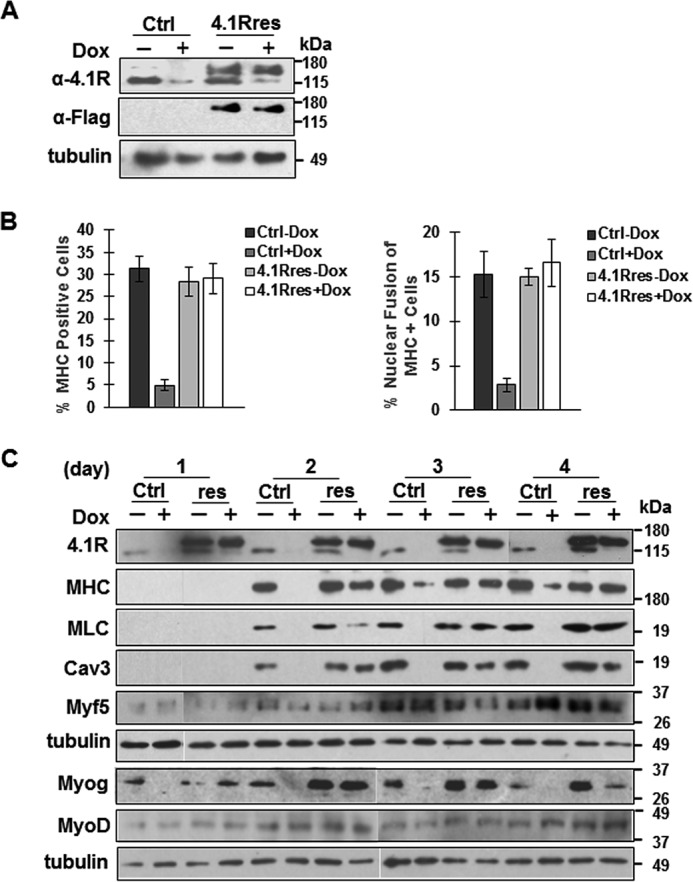
Expression of a 4.1R shRNA-resistant construct rescues the phenotypes associated with 4.1R depletion in C2C12 cells. A control vector (Ctrl) or 4.1R shRNA-resistant construct (4.1Rres) introduced into pTRIPZ-4.1R shRNA C2C12 stable lines was grown in the absence or presence of doxycycline in differentiation medium for the indicated time and analyzed. A, expression of exogenous 4.1R proteins was analyzed after cells were incubated in differentiation medium in the presence or absence of doxycycline for 24 h. Anti-4.1R Ab detects both endogenous and exogenous 4.1R, whereas anti-FLAG Ab detects exogenous 4.1R. Tubulin served as a loading control. B, re-expression of 4.1R reconstitutes myogenic differentiation and myotube formation. Control vector (Ctrl)- or 4.1R shRNA-resistant construct (4.1Rres)-transfected cells were grown for 3 days in differentiation medium in the presence or absence of doxycycline. Differentiation and fusion indices were obtained from three independent experiments. Each experiment was repeated three times, and S.D. values (error bars) were determined. C, re-expression of 4.1R reconstitutes myogenic related protein expression. Control vector- or 4.1R shRNA-resistant construct (res)-transfected cells were grown in differentiation medium in the presence or absence of doxycycline. Cell lysates were collected at the indicated time course and analyzed for the presence of 4.1R as well as MHC, MLC, Cav3, and MRFs (Myf5, MyoD, and myogenin). Each was detected with its respective Ab. Tubulin served as a loading control.
The control vector- and 4.1R rescue construct-transfected cells switched to differentiation medium for 3 days were analyzed for differentiation and nuclear fusion indices in the presence or absence of doxycycline. In a manner consistent with our previous observations, control vector-transfected cells in the absence of doxycycline had differentiation and fusion indices of 30 and 15%, respectively. 4.1R depletion in the presence of doxycycline reduced the differentiation index to 5% and the fusion index to 4% (Fig. 5B). The rescue construct cells were then examined. Regardless of whether they were grown in the absence or presence of doxycycline, 4.1R rescued cells exhibited differentiation (∼28%) and nuclear fusion (16%) indices almost equal to those of the control non-doxycycline-treated cells (Fig. 5B). These results suggest that 4.1R re-expression in the 4.1R shRNA-depleted cells could reverse the phenotypes and that 4.1R shRNA knockdown is not due to an off-target effect.
Because the differentiation and nuclear fusion phenotypes could be reconstituted with 4.1R re-expression, we further examined whether 4.1R re-expression could also restore the levels of myogenin and its downstream targets MHC, MLC, and caveolin-3. In control vector-transfected cells, 4.1R levels were reduced by >90% in the presence of doxycycline throughout d1–d4 differentiation (Fig. 5C). Similar to the parental pTRIPZ-4.1R shRNA C2C12, the reduction of 4.1R also drastically reduced myogenin, MHC, MLC, and caveolin-3 in control vector-transfected cells (Fig. 5C). MHC, MLC, and caveolin-3 were not detected in day 1 differentiated cells. Levels of these proteins were significantly reduced in doxycycline-treated cells when compared with that of non-treated cells. The reduced levels of these proteins persisted through d4 differentiation (Fig. 5C).
In 4.1R rescued lines, endogenous 4.1R was almost absent, whereas exogenous 4.1R was expressed in the presence of doxycycline (Fig. 5C). The re-expression of 4.1R correlated with the reappearance of MHC, MLC, and caveolin-3 to levels equivalent to that of non-doxycycline-treated cells (Fig. 5C). These results suggest that the presence of 4.1R is critical for maintaining the protein levels of myogenin, MHC, MLC, and caveolin-3.
The presence of 4.1R is critical for C2C12 myogenic differentiation and myogenic protein levels, prompting us to further investigate whether increased levels of 4.1R or myogenin in normal C2C12 cells could accelerate the differentiation processes. When exogenous 4.1R or myogenin was expressed at the same physiologic endogenous level (Fig. 6, A and B) and grown in the differentiation medium for 3 days, no significant differences in differentiation or fusion index between the vector and 4.1R or myogenin overexpression cells were observed (Fig. 6C). These results suggest that 4.1R or myogenin expressed at 2 times endogenous levels was not sufficient to accelerate the differentiation of C2C12.
FIGURE 6.

Overexpression 4.1R or myogenin has no effect on normal C2C12 differentiation. FLAG-4.1R or T7-myogenin introduced into C2C12 was grown in differentiation medium for the indicated number of days and analyzed. A, expression of FLAG-4.1R. Anti-4.1R Ab detects both endogenous and exogenous 4.1R. Tubulin served as a loading control. B, expression of T7-myogenin. Anti-myogenin Ab detects both endogenous and exogenous myogenin. Anti-T7 Ab detects exogenously expressed myogenin. Tubulin served as a loading control. C, overexpression of 4.1R or myogenin had no effect on normal C2C12 differentiation. 4.1R- or myogenin-transfected C2C12 cells were grown for 3 days in differentiation medium. Differentiation and fusion indices were obtained from three independent experiments. Each experiment was repeated three times, and S.D. values (error bars) were determined.
4.1R Stabilizes Myogenin Protein
To determine the functional significance of 4.1R in myogenin expression, we first examined whether 4.1R affected myogenin mRNA transcription. Semiquantitative RT-PCR analyses of RNA samples collected from Figs. 2D and 3E indicated that depletion of 4.1R in C2C12 or MyoD-transduced MEF cells had no effect on the levels of myogenin transcripts (Fig. 7, A and D). We then evaluated the impact of 4.1R on the steady-state level of myogenin protein. To examine myogenin stability, 4.1R shRNA stable C2C12 cells grown in the presence or absence of doxycycline-containing differentiation medium for 2 days were incubated with cycloheximide (CHX) for various times, and myogenin levels were measured. The presence of doxycycline reduced 4.1R expression by 80–90% (Fig. 2A). 4.1R knockdown in C2C12 led to greatly reduced stability of myogenin, as evidenced by a decrease in half-life from ∼65 min in 4.1R-positive samples to ∼32 min in 4.1R-depleted samples (Fig. 7, B and C). Similarly, myogenin levels were measured in MyoD-transduced 4.1R+/+ or 4.1R−/− MEFs grown in doxycycline-containing differentiation medium for 36 h and incubated with CHX for various times. In 4.1R+/+ cells, the half-life of myogenin is ∼35 min; 4.1R depletion shortened the half-life to ∼18 min (Fig. 7, E and F). These results suggest a role of 4.1R in myogenin protein stability.
FIGURE 7.

Reduced myogenin protein stability in C2C12 and MEF cells lacking 4.1R. A–C, analyses of myogenin in C2C12. A, C2C12 sh4.1R stable lines were cultured in growth medium (GM) or differentiation medium (DM) for the indicated number of days in the presence or absence of doxycycline. Total RNA was isolated and amplified by RT-PCR with limited cycles and examined for the presence of Myog. GAPDH served as a control. B, C2C12 sh4.1R stable cells were cultured in differentiation medium in the absence (−Dox) or presence (+Dox) of doxycycline for 3 days. Cells were exposed to 50 μg/ml CHX for the indicated time, and lysates were probed via Western blotting with an anti-myogenin Ab. Actin served as a loading control. C, Western blots from B were quantified, and the intensity of the myogenin bands was normalized to β-actin and expressed as a percentage of the density measured at time 0. Each experiment was repeated three times, and S.D. values (error bars) were determined. D–F, analyses of myogenin in MEF cells. D, stable 4.1R WT-MyoD and KO-MyoD MEF cells were cultured in the presence or absence of doxycycline-containing growth medium for 24 h. The doxycycline-treated cells were then switched to doxycycline-containing differentiation medium for the indicated times and analyzed for the presence of Myog and GAPDH mRNA by RT-PCR analyses. E, Western blotting analyses for the stability of myogenin. WT-MyoD and KO-MyoD MEF cells cultured in doxycycline-containing differentiation medium for 36 h were treated with CHX over a 60-min time course. The expression of myogenin at the indicated times was analyzed by Western blotting using an anti-myogenin Ab. Actin was used as a loading control. F, the half-life of myogenin was quantified as stated in C. Each experiment was repeated three times, and S.D. values were determined.
4.1R Associates and Co-localizes with VHL
VHL E3 ligase has been shown to destabilize the myogenin protein via the ubiquitin-proteasome pathway (26). We explored the possibility that 4.1R associates with the VHL-myogenin complex and plays a role in stabilizing myogenin by interfering with VHL-mediated polyubiquitination and degradation of myogenin.
We first determined if 4.1R associated with VHL and/or myogenin. To probe these interactions, FLAG-4.1R and HA-VHL or FLAG-4.1R and T7-myogenin were co-transfected into C2C12, and immunoprecipitates were analyzed for the presence of the associated proteins. As shown in Fig. 8A, FLAG-4.1R and HA-VHL were abundantly detected in anti-FLAG immunoprecipitates. In a reverse co-immunoprecipitation assay, HA-VHL and FLAG-4.1R were also present in anti-HA precipitates (Fig. 8A). These results suggest an association between HA-VHL and FLAG-4.1R. However, we were unable to detect the association between 4.1R and myogenin in the co-immunoprecipitation assay. To further evaluate the association of endogenous 4.1R and VHL, we performed immunoprecipitation assays using C2C12 lysates isolated from cells grown in differentiation medium for 2 days, when myogenin is expressed with anti-4.1R and anti-VHL antibodies. Consistent with the finding in Fig. 8A, endogenous 4.1R and VHL associated together (Fig. 8B). Once again, no association between endogenous 4.1R and myogenin was detected.
FIGURE 8.

4.1R associated and co-localized with VHL. A, C2C12 cells were co-transfected with HA-VHL and FLAG-4.1R and analyzed for the association of VHL and 4.1R in a co-immunoprecipitation assay. Left, cell extracts were precipitated (IP) with a control mouse IgG (mIg) or an anti-FLAG (Flag) Ab. The input extracts and immunoprecipitates were examined by immunoblotting (IB) with anti-HA and anti-FLAG Ab for the presence of HA-VHL and FLAG-4.1R, respectively. Right, extracts were precipitated with a control rabbit IgG (rIg) or an anti-HA Ab and examined with anti-HA and anti-FLAG Abs for the presence of HA-VHL and FLAG-4.1R, respectively. B–D, the association of endogenous VHL and 4.1R was analyzed using day 2 differentiated C2C12 (B), undifferentiated C2C12 (C), and mouse adult skeletal muscle (D) cell lysates in an immunoprecipitation assay. Left, cell extracts were immunoprecipitated with a control mouse IgG or an anti-VHL Ab and examined with anti-4.1R and anti-VHL Abs for the presence of 4.1R and VHL, respectively. Right, extracts were precipitated with rabbit Ig or an anti-4.1R Ab and examined with anti-4.1R and anti-VHL Abs for the presence of 4.1R and VHL, respectively. E, C2C12 cells were immunostained with anti-VHL (green) and anti-4.1R (red) and revealed with Zeiss microscopy (×100). DAPI stains DNA (blue). F, blot overlay assays of 4.1R and VHL. Top, 4.1R, GST, BSA, actin, and BL21 lysates are shown by Coomassie Blue staining. The GST-4.1R fusion protein is highly insoluble in the bacteria expression system. Attempts to extract the fusion protein also co-extracted several host proteins. The arrow indicates the 4.1R cleaved from GST-4.1R with PreScission protease. The smaller bands are the contaminated host proteins. Last lane, 50 μg of BL21 lysates was loaded and served as a negative control for co-purified host proteins from 4.1R preparations. Bottom, a duplicate gel with the same amount of proteins as in the Coomassie Blue-stained gel was separated by SDS-PAGE, overlaid with purified His-VHL, and followed by Western blotting with an anti-His antibody. Arrow, 4.1R cleaved from GST-4.1R with PreScission protease.
To evaluate whether the association of 4.1R and VHL is C2C12 differentiation-specific, immunoprecipitation was analyzed using undifferentiated C2C12. Similar to the results obtained from day 2 differentiated cells, 4.1R was abundantly detected in the precipitates of VHL and vice versa (Fig. 8C). They also associated together in physiologic conditions when muscle tissue lysates were analyzed (Fig. 8D).
To examine the subcellular distributions of 4.1R and VHL, we analyzed the endogenous VHL and 4.1R proteins in C2C12 cells by double-labeled immunofluorescence microscopy. VHL localized in the cytoplasm. In addition to the cytoplasm localization, where 4.1R also partially co-localized with VHL, only 4.1R was also found to be concentrated in the nucleus (Fig. 8E). These results suggest that VHL and 4.1R primarily associate in the cytoplasm.
To investigate whether 4.1R interacts directly with VHL, a blot overlay assay was used. 4.1R cleaved from GST-4.1R fusion proteins, as well as isolated GST proteins, was analyzed by SDS-PAGE and overlaid with purified His-VHL. BSA and actin were included as negative controls. GST-4.1R was insoluble, and attempts to extract the fusion protein co-purified several additional lower molecular weight proteins, as shown in Fig. 8F. These proteins are most likely bacteria host proteins, because only the high molecular weight band, and not the smaller molecular weight bands, reacted with an anti-4.1R antibody (data not shown). Due to contamination in the GST-4.1R preparation, cell lysates from host BL21 were also included in the assay to rule out the possibility of nonspecific binding. The binding of VHL to protein on the membrane was analyzed by Western blotting with an anti-His antibody (Fig. 8F, bottom). VHL interacted directly with 4.1R. Nonspecific interactions between VHL and control proteins were not observed. A replica of the gel was stained with Coomassie Blue for protein loading (Fig. 8F, top).
Inhibition of the Association of Myogenin and VHL by 4.1R
The association of 4.1R with VHL, combined with the reduced half-life of myogenin in the absence of 4.1R, suggests a possibility that 4.1R interferes with the VHL-mediated myogenin degradation pathway.
VHL interacts with myogenin (26) and 4.1R (Fig. 8F); however, these associations are most likely mutually exclusive because myogenin was not detected in 4.1R precipitates. Conversely, 4.1R was not present in myogenin precipitates. We examined whether binding of 4.1R to VHL impedes the association of VHL and myogenin. HEK-293 cells were co-transfected with a fixed amount of HA-VHL and T7-myogenin and increasing amounts of FLAG-4.1R. Cell lysates were precipitated with an anti-HA antibody, and the co-precipitated T7-myogenin and FLAG-4.1R were analyzed with anti-T7 and anti-FLAG antibody, respectively. When cell lysates were analyzed, comparable amounts of HA-VHL and T7-myogenin were both expressed in each transfection. Increasing amounts of FLAG-4.1R plasmids resulted in increased expressions of the exogenously expressed FLAG-4.1R (Fig. 9, lysate). In the absence of 4.1R, myogenin co-precipitated with VHL (Fig. 9, IP:HA). With increasing FLAG-4.1R, increased association of 4.1R with HA-VHL was noted. Conversely, the association of T7-myogenin with HA-VHL decreased in a FLAG-4.1R concentration-dependent manner (Fig. 9, IP:HA). These results suggest that a possible competition exists between 4.1R and myogenin for binding to VHL.
FIGURE 9.
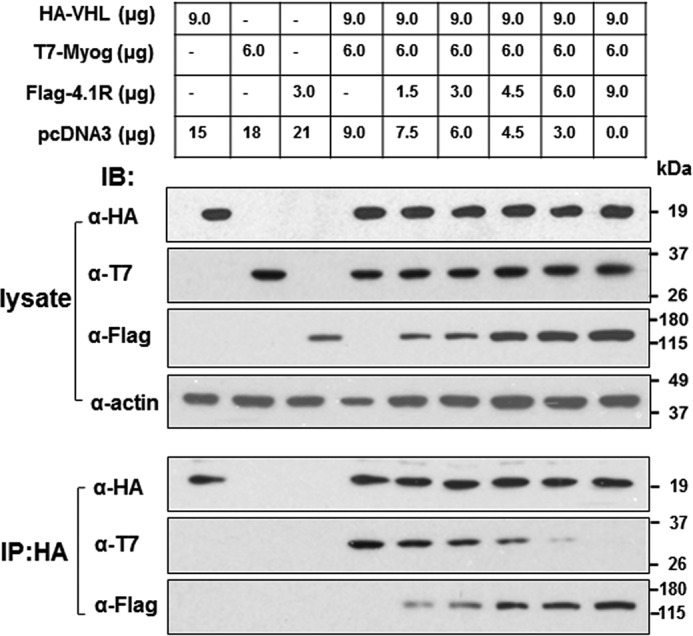
4.1R competitively interrupts the association of VHL and myogenin. C2C12 cells grown in 10-cm dishes were co-transfected with various combinations of HA-VHL, T7-myogenin, FLAG-4.1R, and empty vector pcDNA3, with a total of 24 μg of DNA, as indicated. Cells were lysed in IP buffer 48 h post-transfection and immunoprecipitated (IP) with an anti-HA antibody. lysate, equal amounts of lysates were analyzed via Western blotting (IB) for the exogenously expressed HA-VHL, T7-Myog, and FLAG-4.1R. IP:HA, anti-HA immunoprecipitates were equally divided and analyzed for HA-VHL and its associated T7-Myog and FLAG-4.1R with their respective antibodies.
4.1R Counteracts the Effect of VHL on Ubiquitination of Myogenin
VHL has been shown to catalyze the assembly of the polyubiquitin chain on the myogenin protein in a ubiquitination assay (26). We analyzed whether 4.1R+/+ MEF-MyoD and 4.1R−/− MEF-MyoD cells grown in doxycycline-containing differentiation medium have different degrees of ubiquitination on myogenin. Lysates from MEF-MyoD were precipitated with an anti-myogenin antibody and detected via Western blotting for ubiquitinated myogenin and myogenin (Fig. 10A). The intensities of the ubiquitinated myogenin and myogenin were compared from the 4.1R+/+ and 4.1R−/− samples. Although the amount of the precipitated myogenin in the 4.1R+/+ cells had greater intensity than that of 4.1−/− cells, a much more intense and broader band characteristic of ubiquitinated myogenin was observed in 4.1R−/− cells when compared with 4.1R+/+ cells (Fig. 10A). This result suggests that a more robust ubiquitination of myogenin occurs in cells lacking 4.1R.
FIGURE 10.

4.1R inhibits VHL-mediated ubiquitination and myogenin degradation via the ubiquitin-proteasome pathway. A, 4.1R knockdown increases polyubiquitinated myogenin. WT-MyoD and KO-MyoD MEF cells were cultured in differentiation medium in the presence of doxycycline for 48 h and analyzed for myogenin levels and its status of ubiquitination. Top, ubiquitination of myogenin was analyzed by immunoprecipitation (IP) of cell lysates with an anti-myogenin Ab and immunoblotting (IB) with an anti-Ub antibody. (Ub)n-MyoG, ubiquitinated myogenin. Bottom, the amount of immunoprecipitated myogenin was detected using an anti-myogenin Ab. B, decrease in polyubiquitinated myogenin by 4.1R overexpression. HEK-293 cells were transfected with combinations of His-Ub, T7-Myog, HA-VHL, and FLAG-4.1R as indicated. Left panel, lysates fractionated on an SDS-polyacrylamide gel were blotted with an anti-His antibody. The presence of the exogenously expressed proteins in cell lysates was confirmed by immunoblotting with an anti-T7 Ab for myogenin, anti-HA for VHL, and anti-FLAG for 4.1R. Actin served as a loading control. Right, extracts were immunoprecipitated with an anti-T7 antibody and immunoblotted with an anti-His Ab. The presence of T7-myogenin was confirmed by immunoblotting using an anti-T7 Ab. C, VHL increases the ubiquitination of myogenin. HEK-293 cells were transfected with His-Ub and T7-Myog and in combination with VHL or VHL C162F. Left, extracts were blotted with an anti-His Ab for the presence of ubiquitinated proteins, anti-T7 Ab for T7-Myog, and anti-HA Ab for VHL or VHL C162F. Right, anti-T7 immunoprecipitates were blotted with an anti-His Ab for the ubiquitinated T7-myogenin and anti-T7 Ab for the precipitated T7-Myog. D, 4.1R reverses the ubiquitination and quantity of myogenin in a VHL-specific manner. HEK-293 cells were transfected with His-Ub, T7-Myog, and either HA-VHL or HA-VHL C162F in the presence or absence of FLAG-4.1R, as indicated. Lysates were detected for the presence of transfected proteins with their respective antibodies. Anti-T7 immunoprecipitates were blotted with an anti-T7 Ab for the presence of T7-Myog. E, effect of proteasome inhibitor MG132 on the ubiquitination status and quantity of myogenin. HEK-293 cells were transfected with His-Ub, T7-Myog, and HA-VHL and either in the presence of FLAG-4.1R or with treatment of the proteasome inhibitor MG132, as indicated. Left, lysates were blotted with an anti-His Ab. The presence of the exogenously expressed proteins was confirmed by immunoblotting with their respective antibodies. Actin served as a loading control. Right, extracts were immunoprecipitated with an anti-T7 Ab and immunoblotted with an anti-His Ab. The presence of T7-Myog was confirmed with an anti-T7 Ab. F, effect of MG132 on myogenin in 4.1R knockdown cells. pTRIPZ4.1R shRNA C2C12 stable lines were cultured in differentiation medium in the presence of doxycycline for 2 days and treated with or without MG132. The amounts of 4.1R and myogenin were determined by immunoblotting with its respective antibodies. β-Actin served as a loading control.
4.1R competed with myogenin for binding to VHL (Fig. 9), and lack of 4.1R increased ubiquitinated myogenin (Fig. 10A), suggesting that the effect of 4.1R on myogenin stability acts through a VHL-mediated degradation pathway. We further tested the possibility that 4.1R overexpression can reverse the effect of VHL on the ubiquitination of myogenin in an in vivo ubiquitination assay. HEK-293 cells were transfected with His-Ub and with various combinations of T7-myogenin, HA-VHL, and FLAG-4.1R. Intensities of myogenin and ubiquitinated myogenin in T7 immunoprecipitates were then analyzed. When lysates were assessed, extensive ubiquitinated proteins were detected in each combination of transfection (Fig. 10B, top left panel). In addition, the exogenously expressed proteins were detected in each transfection (Fig. 10B, bottom left panels). We then examined the effect of 4.1R overexpression on the ubiquitination status and the quantity of myogenin in each combination. When anti-T7 precipitates were probed with an anti-His antibody, smears of high molecular weight myogenin protein characteristic of ubiquitinated species were observed (Fig. 10B, top right panel). However, anti-T7 precipitates from a varied combination of transfections were noted to have a different degree of ubiquitination on T7-myogenin. In the absence of transfected HA-VHL, co-transfection of FLAG-4.1R reduced ubiquitination of T7-myogenin when compared with the sample without FLAG-4.1R (Fig. 10B, top right panel, lanes 2 and 3). When T7-myogenin was examined, a slightly more intense T7-myogenin band was noted in the presence of 4.1R compared with the sample lacking 4.1R (Fig. 10B, bottom right panel, lanes 2 and 3).
In the presence of transfected HA-VHL, a stronger contrast in T7-myogenin ubiquitination patterns was observed in the presence or absence of FLAG-4.1R. A significant increase in ubiquitinated T7-myogenin was present in T7-precipitates in the absence of FLAG-4.1R (Fig. 10B, top right panel, lanes 4 and 5). Conversely, a more intense T7-myogenin was detected in the presence of 4.1R (Fig. 10B, bottom right panel, lanes 4 and 5). The effects of 4.1R were reproduced in three separate experiments. Thus, a decrease in polyubiquitinated myogenin was clearly noted by 4.1R overexpression. These results suggest that VHL-mediated myogenin ubiquitination and degradation can be partially inhibited by the presence of 4.1R.
Whether the effect of 4.1R on reversing the ubiquitination of myogenin is specific for VHL was further examined. VHL C162F, a VHL mutant that is unable to interact with elongin C/Cullin2 and thus lacks activity to ubiquitinate the target protein (38), was used as a control. We first transfected HEK-293 cells with His-Ub and T7-myogenin (T7-Myog) in combination with either the WT-VHL or VHL C162F and analyzed for the extent of myogenin ubiquitination and quantity. When lysates were assessed, VHL C162F had an amount of ubiquitinated protein equal to that of the control. Approximately 5–10% more ubiquitinated proteins were detected in the presence of VHL (Fig. 10C, top left panel). Exogenously expressed proteins were detected in each transfection (Fig. 10C, bottom left panels). When T7 immunoprecipitates were examined, extensive ubiquitinated myogenin was detected in the presence of VHL, whereas lesser amounts of ubiquitinated myogenin were detected in the control and VHL C162F samples (Fig. 10C, top right panel). In addition, whereas comparable amounts of T7-myogenin were presented in the precipitates of control and VHL C162F, a significantly decreased amount (∼50%) of T7-myogenin was detected in the VHL sample compared with the control (Fig. 10C, bottom right panel). The mutant VHL exhibited almost no effect on myogenin ubiquitination and quantity (Fig. 10C), suggesting that polyubiquitination of myogenin by VHL is necessary for its degradation.
Furthermore, the reversing effect of 4.1R on myogenin ubiquitination and quantity is VHL-dependent. Co-transfection of 4.1R with VHL could recover the amount of myogenin because the intensity of myogenin was twice that of cells lacking 4.1R (Fig. 10D, lanes 1 and 2). However, co-transfection of 4.1R with VHL C162F did not affect myogenin quantity because equal amounts of myogenin were detected (Fig. 10D, lanes 3 and 4).
Inhibition of Ubiquitin-Proteasome Pathway-dependent Degradation of Myogenin by 4.1R
Proteasome inhibitor MG132 reduces the degradation of ubiquitin-conjugated proteins in mammalian cells (39). To further determine whether the effect of 4.1R on myogenin is mediated by the proteasome pathway, we first examined the effect of MG132 on ubiquitination and quantity of myogenin. HEK-293 cells were transfected with His-Ub, T7-myogenin, and HA-VHL in the presence of FLAG-4.1R or MG132 before harvesting. MG132 treatment resulted in a much more robust ubiquitination of proteins (Fig. 10E, top left panel) and increased T7-myogenin in lysate (Fig. 10E, bottom left panel) by ∼8-fold when compared with that of the control or 4.1R-treated samples. When the levels of ubiquitinated T7-myogenin were examined in T7 precipitates, an ∼8-fold increase in ubiquitinated myogenin was observed in MG132-treated samples when compared with that of the control (Fig. 10E, top right panel). As observed earlier (Fig. 10, B and D), 4.1R treatment reversed ubiquitination of myogenin by VHL and increased the quantity of myogenin by 2-fold (Fig. 10E, bottom right panel). Furthermore, MG132 treatment also increased precipitated T7-myogenin to ∼10-fold that of the control (Fig. 10E, bottom right panel). These results suggest an effect of MG132 in inhibiting the degradation of ubiquitinated myogenin.
Because 4.1R depletion reduced the half-life of myogenin (Fig. 7), we further examined whether MG132 treatment could restore the stability of endogenous myogenin in 4.1R-depleted C2C12. 4.1R shRNA stable C2C12 cells were grown in the presence of doxycycline-containing differentiation medium for 2 days and maintained for 4 h in the presence or absence of MG132, and myogenin levels were analyzed. In the presence of doxycycline, 4.1R expression was reduced to ∼20% of the levels noted in cells grown in the absence of doxycycline (Fig. 10F). The additional treatment with MG132 slightly increased 4.1R levels to ∼30–40% of the control non-doxycycline cell levels. Similar to the results obtained in Fig. 2D, myogenin expression was significantly reduced in doxycycline-treated samples where 4.1R levels were reduced. Conversely, the addition of the inhibitor abolished the effect of 4.1R depletion and restored the levels of myogenin to levels comparable with those obtained in the presence of 4.1R (Fig. 10D). These results suggest that the effect of 4.1R on myogenin stability is most likely through the proteasome ubiquitination and degradation pathway.
Discussion
This study reveals that, in addition to its functional role in myocytes, 4.1R influences skeletal muscle differentiation by stabilizing myogenin protein, preventing its breakdown from the ubiquitin degradation pathway. 4.1R associates with VHL. This appears to impede the association of myogenin and VHL in a dose-dependent manner. 4.1R depletion in both C2C12 and MyoD-induced MEF muscle differentiation systems delayed myoblast differentiation and myofusion due to reduced MHC and caveolin-3 expression, which are both downstream targets of myogenin. These findings indicate a novel role for 4.1R in the ubiquitination pathway and in skeletal muscle differentiation.
Protein 4.1R was detected in the cytoplasm of C2C12 throughout myogenesis. In addition, 4.1R also localizes to the nucleus of the proliferating myoblast and in the sarcomere of the differentiated myotubes. The nuclear localization of 4.1R has been detected in many cell types and implicated in mRNA splicing (1) and nuclear assembly (2). Consistent with our earlier observation in mouse skeletal muscle (7), 4.1R localizes to the A-band of the sarcomere in mature myotubes. However, cytoplasmic localization of 4.1R persists in differentiated C2C12 cells. 4.1R might associate with A-bands at a very late stage of differentiation that is not completely achieved in the C2C12 tissue culture model. Similar to the expression patterns of MHC and α-actinin, 4.1R expression considerably increases during myogenesis and probably, at least in part, contributes to the sarcomere structural organization.
We investigated the involvement of 4.1R in muscle differentiation using both C2C12 and MyoD-induced MEF systems. Gene knockdown by transfection of siRNA or plasmids coding for shRNA is often unsatisfactory due to the susceptibility of siRNA to nuclease degradation or prolonged depletion of the target genes, respectively. By using both cellular models, our experiments avoided complications associated with depletion or overexpression of the target genes that could be detrimental to the differentiation program. Using the inducible tet-on shRNA knockdown system, the effect of 4.1R on myotube differentiation was analyzed in cells with at least 75% 4.1R depletion at the desired time of differentiation in the C2C12 system. Similarly, the inducible tet-on MyoD expression system allowed for timed MyoD expression at the appropriate stage of MyoD-induced MEF differentiation. We observed very comparable results in both models.
This study indicated that 4.1R is positively involved in myogenic differentiation. MHC and caveolin-3 were greatly reduced in 4.1R-depleted cells. MHC is the motor protein of muscle thick filaments (40), and caveolin-3 is required for myoblast fusion and myotube formation (41). Reduction of these proteins impaired muscle formation.
Terminal differentiation of muscle, during which muscle-specific proteins such as MHC and caveolin-3 are induced, is stimulated by temporally specific expression of MRFs (42). We identified myogenin as the upstream target of 4.1R depletion; myogenin expression was compromised, whereas MyoD and Myf5 were not affected. 4.1R had no effect on the mRNA level of myogenin but was responsible for myogenin protein stability, as evidenced by the fact that the half-life of myogenin was drastically reduced in the absence of 4.1R.
MRFs are short lived transcription factors. Consistent with previous reports, we found that the myogenin protein has a short half-life of ∼65 min in C2C12 (27, 43). Steady-state levels of myogenin during differentiation are regulated by the ubiquitin-proteasome pathway. Anti-ubiquitination proteins participate in stabilizing myogenin (26, 27). TIP120B/CAND2 positively regulates myogenesis of C2C12 through binding to cullin and suppression of SCF-dependent ubiquitination and degradation of myogenin (27). EGLN3 prolyl hydroxylase interacts with and stabilizes myogenin by antagonizing VHL-mediated ubiquitination and degradation of myogenin (26). 4.1R also appears to protect myogenin from ubiquitin-proteasome degradation.
We detected a direct interaction between 4.1R and VHL, but no association between 4.1R and myogenin was noted. The interaction of VHL with myogenin or 4.1R thus appears to be mutually exclusive. It is conceivable that 4.1R competes with myogenin for the same binding site on VHL. The precise binding domain for VHL and myogenin is not known. We note that several conserved amino acid sequences exist within the membrane binding domain to the C-terminal domain of 4.1R and myogenin. We have yet to determine the exact sequences on 4.1R and myogenin that are responsible for interacting with VHL. An interaction between 4.1R and transcription elongation factor B polypeptide 1 (TCEB1) has been identified (44). TCEB1 is known to interact with VHL and is required for the function of VHL (45). This supports a possible indirect interaction between 4.1R and VHL through TCEB1.
It is puzzling that 4.1R homozygous knock-out mice are viable (46) despite the evidence from current studies that 4.1R is involved in the stabilization of a key transcription factor in the muscle differentiation pathway. It is worth noting that knock-out of other genes essential for muscle function and development is also compatible with viability. The mdx mouse is the most widely used model to study the pathogenesis of DMD. The mdx mouse displays some features of muscle degeneration, but the pathogenesis of disease is comparatively mild (47). The lack of dystrophin expression renders myofibers abnormally fragile. However, boys who harbor DMD mutations appear relatively normal until the age of 4 or 5 despite the absence of dystrophin at birth (48, 49).
It is worth noting that among the 4.1R paralogues, 4.1G and 4.1B are also expressed in muscle (6, 9). The membrane binding domain, spectrin-actin binding domain, and C-terminal domain are conserved among 4.1 family members. We speculate that the function of 4.1R in null mice might be replaced by redundant systems involving other 4.1 family members. Whether each of these family members also participates in myogenesis requires further investigation. It is possible that 4.1 members may also take part in other cellular processes involving ubiquitination, depending on the cell type and extracellular cues.
Protein 4.1R is a member of the FERM (4.1/ezrin/radixin/moesin) family of proteins. The FERM domain is a common protein module involved in localizing proteins to the plasma membrane, where they function as membrane-cytoskeletal linkers and regulators of multiple signaling pathways (50, 51). FERM family proteins have been shown to affect numerous processes, including cell polarization, migration, and proliferation (51). Some members of this family have also been implicated in tumor progression (52). Protein 4.1B expression is frequently lost in a variety of human tumors, including meningiomas (53), non-small-cell lung cancers (54), breast carcinomas (55), and prostate cancer (56). 4.1R was found to be involved in meningioma pathogenesis (57) and ependymal tumors (58). In addition, the ERM-like protein, merlin (the NF2 gene product) is a critical suppressor of meningiomas and schwannomas (59). Ezrin mutations enhance metastasis of bone and soft tissue sarcomas (60, 61).
Despite extensive research on the role of 4.1R as a tumor suppressor, the mechanism by which 4.1R suppresses tumorigenesis has remained elusive. Interestingly, tumor suppression by merlin has been shown to be linked to inhibition of the E3 ubiquitin ligase CRL4DCAF1. Merlin translocates from the cytoplasm and accumulates in the nucleus. Nuclear merlin binds to the E3 ubiquitin ligase CRL4DCAF1 and suppresses its activity to ubiquitinate target proteins (62). The subdomain A of the FERM domain of merlin consists of a ubiquitin-like fold (63) and may function as an inhibitory pseudosubstrate for CRLDCAF1 (62). Our preliminary results, however, suggest that 4.1R was not ubiquitinated and degraded in a VHL-dependent manner.3 It is tempting to speculate that 4.1R acts as an anti-ubiquitination molecule in a similar manner.
VHL E3 ligase complex has been shown to bind and catalyze ubiquitination of an array of proteins involved in many cellular processes. Among these are the seventh subunit of RNA polymerase II (64), atypical protein kinase, and cyclin D1 (65). It would be of interest to know whether 4.1R is involved in regulating the stability of these proteins.
In summary, this study demonstrates that 4.1R stabilizes the myogenic regulatory factor myogenin, partially by antagonizing VHL-mediated ubiquitination and degradation of myogenin. This in turn impacts the process of myogenesis. Further elucidation of the underlying mechanisms should provide important insights into the regulation of skeletal muscle differentiation and the biological role of 4.1R.
Experimental Procedures
Cell Culture and Transfection
C2C12 cells (ATCC catalog no. CRL-1772, ATCC, Manassas, VA) were grown in growth medium containing DMEM, 10% heat-inactivated fetal bovine serum (Sigma), penicillin (50 μg/ml), and streptomycin (50 μg/ml). To induce myogenic differentiation, C2C12 myoblasts at 70–90% confluence were shifted from growth medium to differentiation medium (DMEM containing 2% horse serum). The number of days of differentiation is denoted as d1, d2, etc., for the number of days following incubation in differentiation medium. C2C12 cells transduced with inducible lentiviral pTRIPZ-4.1R shRNA or control scramble shRNA were grown in growth medium. Inducible knockdown stable cell lines were selected with puromycin. shRNA expression was induced in the presence of 200 ng/ml doxycycline for 24 h. Subsequent differentiation was achieved by growing the cells in doxycycline-containing differentiation medium for additional days as indicated.
Immortalized MEF cells isolated from wild-type (4.1R+/+) and 4.1R knock-out (4.1R−/−) mice were provided by Drs. An and Mohandas (New York Blood Bank). MEF cells transduced with inducible lentiviral pTRIPO-MyoD or an empty vector were grown in growth medium. Stable lines were selected with puromycin. MyoD expression was induced in the presence of doxycycline in growth medium for 24 h. Subsequent differentiation was achieved by replacing the growth medium with doxycycline containing differentiation medium for an additional 24, 36, 48, and 60 h. HEK-293 (ATCC catalog no. CRL-1573) cells were maintained in growth medium and transiently transfected with a combination of plasmids using Lipofectamine transfection reagent (Invitrogen) according to the manufacturer's protocol.
Plasmids
Expression plasmids HA-VHL-pRc/CMV (NM_000551; Addgene plasmid 19999) and HA-VHL C162F-pRc/CMV (Addgene plasmid 22042) were gifts from William Kaelin (38, 66). pCI-His-hUbi (NM_021009.6; Addgene plasmid 31815) was a gift from Astar Winoto (67). pcDNA3.1-T7-Myog was constructed by cloning the mouse myogenin (NM_031189.2) in frame with T7 tag into pcDNA3.1(+) (Invitrogen). pcDNA3.1-3FLAG-4.1R was constructed by cloning group I of 135-kDa 4.1R, which is composed of all alternatively spliced exons except for exons 14, 15, and 17b (7) in frame with three copies of FLAG (3× FLAG) into pcDNA3.1(+). The 135-kDa 4.1R was cloned in frame with GST into pGEX6p1 (GE Healthcare) to form pGEX-4.1R. The tet-on inducible expression vector pTRIPO was generated by replacing the AgeI-MluI fragment of pTRIPZ lentiviral shRNAmir tet-on-inducible vector (Thermo Fisher Scientific) containing the TurboRFP tag and the 5′-mir30/3′-mir30 sequences by an AgeI-HpaI-XhoI-EcoR1-MluI polylinker sequence. Establishment of the inducible pTRIPO-3FLAG-MyoD was constructed by two-step cloning: mouse MyoD (NM_010866.2) was cloned in frame with 3× FLAG tag into pcDNA3.1(+), and the 3× FLAG and MyoD were subsequently removed and cloned into pTRIPO. pTRIPZ-4.1R shRNA was constructed by cloning the target sequences (CCAGACATGTCAGTGACCA located in exon 21) into the pTRIPZ lentiviral shRNAmir vector for the ability to regulate gene silencing according to the manufacturer's protocol. For 4.1R rescue construct pcDNA3.1–3FLAG-4.1Rres, the underlined mutations at the shRNA target sequences (CCAGACATGTCGGTCACCA) were incorporated into pcDNA3.1–3FLAG-4.1R.
Animal and Skeletal Muscle Tissue Preparation
Adult BALB/c mice obtained from Charles River Laboratory (Wilmington, MA) were euthanized in a CO2 chamber. For protein lysates, quadriceps muscle tissues were frozen in liquid nitrogen, ground to powder in the presence of liquid N2, and subsequently dissolved in triple lysis buffer (50 mm Tris-HCl, pH 7.5, 125 mm NaCl, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate). For immunolabeling experiments, longitudinally dissected quadriceps muscle tissues were fixed in 4% paraformaldehyde and embedded in OCT according to standard histology techniques. Semithin sections (6 μm) were prepared using a Leica CM3050 cryostat (Wetzlar, Germany) and used in immunofluorescence assays.
Indirect Immunofluorescence and Imaging
Cells grown on coverslips were fixed in PBS containing 4% paraformaldehyde for 15 min and then permeabilized in 0.25% Triton X-100 in PBS for 5 min at room temperature. Cells were then blocked in 10% goat serum for 30 min and exposed to primary and secondary antibodies at room temperature for 1 h each. All samples were counterstained with DAPI. Quadriceps muscle tissue was processed for immunolabeling experiments as described (7). The samples were viewed with a Zeiss Axiovert 200M inverted microscope (Zeiss). The images were collected using SlideBookTMversion 4.0 software and processed using Photoshop software (Adobe Systems, Inc.).
Differentiation and Fusion Indices
Differentiation was analyzed by immunofluorescence staining for MHC with an anti-MF20 Ab and FITC-conjugated secondary Ab and revealed with Zeiss microscopy. Nuclei were counterstained with DAPI. Differentiation indices were calculated as the percentage of MHC-positive cells. Fusion indices were calculated as the percentage of cells containing three or more nuclei within the myosin-positive myocytes. Each experiment was repeated three times, and S.D. values were determined.
Western Blotting Analysis
Cell lysates or immunoprecipitates were analyzed by 10 or 15% SDS-PAGE and electrotransferred onto a PVDF (Millipore) or nitrocellulose membrane (Maine Manufacturing, LLC). The detection of 4.1R, α-actinin, MHC, MLC, MyoD, Myf5, Myog, caveolin-3 (Cav3), VHL, Ub, anti-β-actin, and α-β-tubulin was carried out by immunoblotting with anti-4.1R-HP (4), anti-α-actinin (A7811, Sigma-Aldrich), anti-myosin (MF-20, Developmental Studies Hybridoma Bank), anti-myosin light chain (T14, Developmental Studies Hybridoma Bank), anti-MyoD (C-20, Santa Cruz Biotechnology, Inc.), anti-Myf5 (C-20, Santa Cruz Biotechnology), anti-Myog (M-225, Santa Cruz Biotechnology; D5F, Developmental Studies Hybridoma Bank), anti-Cav3 (610421, BD Transduction Laboratories), anti-VHL (2738, Cell Signaling; FL-181, Santa Cruz Biotechnology), anti-Ub (P4D1, Santa Cruz Biotechnology), anti-β-actin (A2228, clone AC-74, Sigma-Aldrich), and α-β-tubulin (T8328, clone AA2, Sigma-Aldrich) antibody diluted in either 4% milk in TBST (20 mm Tris-HCl, pH 7.6, 140 mm NaCl, 0.1% Tween 20) or in antibody enhancer diluent (Amresco, Inc., Solon, OH) and developed using an ECL detection kit (Amersham Biosciences).
The presence of exogenously expressed FLAG-4.1R, HA-VHL, His-Ub, and T7-Myog proteins was detected with anti-FLAG (F7425, F3165, Sigma-Aldrich), anti-HA (H6908, Sigma; clone 3F10, Roche Applied Science), anti-His (37-2900, clone 4A12E4, Thermo Fisher Scientific), and anti-T7 (69522-3, Novagen; T8823, Sigma) antibodies. VeriBlot secondary antibodies (ab131366 or ab131368, Abcam) were used for co-immunoprecipitation Western blotting analyses.
To detect several proteins from the same set of experiments, an equal amount of lysate was fractionated into more than one gel and transferred to its respective membrane. When the same membrane was used for more than one antibody, the membrane was stripped between the two probings. To avoid stripping away proteins bound to the membrane, a membrane was limited to 3–4 strippings. Each membrane was also probed with tubulin, which served as a loading control to ensure that no pipetting error was involved.
Gel Overlay Assays
GST and 4.1R/GST fusion proteins were affinity-purified via coupling to glutathione-Sepharose beads (GE Healthcare) as described previously (4). 4.1R was cleaved from 4.1R/GST fusions using PreScission Protease (GE Healthcare). Gel overlay assays were performed as described previously (7). 4.1R, GST, BSA, actin, and 50 μg of BL21 lysates were fractionated on 10% SDS-polyacrylamide gel and either stained with Coomassie Blue dye or electrotransferred onto a PVDF membrane. The membrane was incubated in blocking buffer (50 mm Tris-HCl, pH 7.5, 140 mm NaCl, 0.1% Tween 20, 0.5% Nonidet P-40, 3% BSA, 0.5% gelatin, and 2 mm DTT) for 12 h at 4 ºC and was overlaid with 3 μg/ml His-VHL (OriGene Technologies, Inc., Rockville, MD) in binding buffer (50 mm Tris-HCl, pH 7.5, 140 mm NaCl, 0.5% Nonidet P-40, 1% BSA, 0.25% gelatin, 2 mm ATP, and 2 mm DTT) for 3 h at 4 ºC. Then the membrane was washed with washing buffer (50 mm Tris-HCl, pH 7.5, 140 mm NaCl, 1.0% Nonidet P-40, and 2 mm DTT), followed by Western blotting using an anti-His antibody.
RT-PCR Analyses
Analysis of myogenin mRNA was performed using a limiting PCR cycle amplification protocol that obtains the PCR product within its linear range (68). RNAs were reverse-transcribed using oligo(dT). PCRs were performed with Ex2-S (GTCCCAACCCAGGAGATCAT) and Ex3-As (AGTTGGGCATGGTTTGGTCT) for myogenin and with GAPDH-S (AGGTCGGTGTGAACGGATTTG) and GAPDH-As (TGTAGACCATGTAGTTGAGGTCA) for GAPDH. PCR products were fractionated on 2% agarose gels and quantified using analytic software from the ChemiImagerTM 5500 system (Alpha Innotech Co.).
cDNA Cloning and Isoform Analyses
Total RNA isolated from day 0 and day 8 differentiated C2C12 were subjected to RT-PCR. The primers used in the PCR amplification were chosen to anneal to sequences present in exons 1 (gacaacgactacttctgg) and 21 (accttggtcactgacatgtctgg). The PCR products were subcloned into the TOPO TA vector (Invitrogen). One hundred ninety-two colonies were randomly selected and grown in 96-well microtiter plates; they were subsequently analyzed in a DNA dot blotting assay using primer exon 2′ (gaacatcatgacaacag) as described previously (7). The inclusion or exclusion of exon 2′ distinguishes the 135-kDa (exon 2′-positive) and 80-kDa (exon 2′-negative) isoforms. Identification of the alternatively spliced exons was performed using PCR with primer sets flanking the exon of interest: primers exon 2-S (tgcagctcctgaacctgagctca) and exon 6-As (tgcagctcctgaacctgagctca) for alternatively spliced exons 4 and 5; primers exon 7-S (tccacccgacccagcacaatt) and exon 9-As (aagtcagcctgagctggagt) for exon 8; primers exon 13-S (tgaagagaagcggggagaaga) and exon 17-As (actgatgctggcatggtgct) for exons 14, 15, and 16; primers exon 17-S (atcaacgggcaagtccctact) and 21-As (accttggtcactgacatgtctgg) for exons 17a, 17b, 18, 19, and 20.
Ubiquitination Assays
Cells were transiently transfected with a combination of plasmids encoding His ubiquitin (pCI-His-hUbi), T7-myogenin (pcDNA3.1-T7-MyoG), HA-VHL (HA-VHL-pRc/CMV), HA-VHL162 (HA-VHL C162F-pRc/CMV), and FLAG-4.1R (pcDNA3.1–3FLAG-4.1R). Cells were harvested and lysed in IP buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.5% Nonidet P-40, 0.2% sodium deoxycholate, 0.1% SDS) and subjected to immunoprecipitation using an anti-T7 antibody, followed by Western blotting analysis with an anti-ubiquitin or its respective primary antibody and VeriBlot secondary antibody.
Myogenin Decay Assays
pTRIPZ-4.1R shRNA- or control shRNA-transduced C2C12 stable lines were grown in doxycycline-containing growth medium for 24 h and switched to doxycycline-containing differentiation medium for 3 days. Cells were then exposed to 50 μg/ml CHX for various times as indicated. Similarly, MyoD-transduced 4.1R+/+ and 4.1R−/− MEF stable lines were cultured in doxycycline-containing growth medium and switched to differentiation medium containing doxycycline for 48 h. The half-life of myogenin was determined by treating cells with CHX over a 60-min time course. Cells were harvested at the indicated time points. Total cell lysates were subjected to Western blotting analysis with an anti-myogenin antibody. Actin served as a loading control. Western blots were quantified using analytic software from the ChemiImagerTM 5500 system (Alpha Innotech Co.). Each experiment was repeated three times, and S.D. values were determined. The intensities of the myogenin bands were normalized to β-actin and expressed as a percentage of the density measured at time 0.
Author Contributions
S.-C. H. designed the study, analyzed and interpreted the data, and wrote the paper. A. Z. designed the study, constructed vectors and established cell lines, performed initial studies, and contributed to the immunofluorescent studies (Figs. 1 (C–E), 2 (A and B), 3 (A and C), and 8E) and immunoprecipitation assays (Figs. 8 (A and B) and 10A). D. T. N. and H. S. Z. performed and analyzed the experiments (Figs. 1 (A and B), 2 (C and D), 3 (D and E), 4–7, 8 (C, D, and F), 9, and 10 (B–F)). E. J. B. designed the study and edited the paper.
Acknowledgments
We thank Drs. An and Mohandas (New York Blood Bank) for mouse embryonic fibroblast cells isolated from wild-type (4.1R+/+) and 4.1R knock-out (4.1R−/−) mice. We thank Penny Wang and Zachary McKenzie for technical assistance.
This work was supported by National Institutes of Health Grants HL24385 and HL44985 (to E. J. B.) and National Institutes of Health Grant HL61295 and the Claudia Barr Award (to S. C. H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
S.-C. Huang, A. Zhou, D. T. Nguyen, H. S. Zhang, and E. J. Benz, unpublished data.
- 4.1R
- protein 4.1R
- MRF
- myogenic regulatory factor
- Ub
- ubiquitin
- VHL
- von Hippel-Lindau
- MEF
- mouse embryonic fibroblast
- MHC
- myosin heavy chain
- MLC
- myosin light chain
- CHX
- cycloheximide
- DMD
- Duchenne muscular dystrophy
- d
- day
- Ab
- antibody
- Myog
- myogenin
- Cav3
- caveolin-3.
References
- 1. Lallena M. J., Martínez C., Valcárcel J., and Correas I. (1998) Functional association of nuclear protein 4.1 with pre-mRNA splicing factors. J. Cell Sci. 111, 1963–1971 [DOI] [PubMed] [Google Scholar]
- 2. Krauss S. W., Chen C., Penman S., and Heald R. (2003) Nuclear actin and protein 4.1: essential interactions during nuclear assembly in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 10752–10757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mattagajasingh S. N., Huang S. C., Hartenstein J. S., and Benz E. J. Jr. (2000) Characterization of the interaction between protein 4.1R and ZO-2: a possible link between the tight junction and the actin cytoskeleton. J. Biol. Chem. 275, 30573–30585 [DOI] [PubMed] [Google Scholar]
- 4. Mattagajasingh S. N., Huang S. C., Hartenstein J. S., Snyder M., Marchesi V. T., and Benz E. J. Jr. (1999) A nonerythroid isoform of protein 4.1R interacts with the nuclear mitotic apparatus (NuMA) protein. J. Cell Biol. 145, 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang S., Guo X., Debnath G., Mohandas N., and An X. (2009) Protein 4.1R links E-cadherin/β-catenin complex to the cytoskeleton through its direct interaction with β-catenin and modulates adherens junction integrity. Biochim. Biophys. Acta 1788, 1458–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taylor-Harris P. M., Keating L. A., Maggs A. M., Phillips G. W., Birks E. J., Franklin R. C., Yacoub M. H., Baines A. J., and Pinder J. C. (2005) Cardiac muscle cell cytoskeletal protein 4.1: analysis of transcripts and subcellular location: relevance to membrane integrity, microstructure, and possible role in heart failure. Mamm. Genome 16, 137–151 [DOI] [PubMed] [Google Scholar]
- 7. Kontrogianni-Konstantopoulos A., Huang S. C., and Benz E. J. Jr. (2000) A nonerythroid isoform of protein 4.1R interacts with components of the contractile apparatus in skeletal myofibers. Mol. Biol. Cell 11, 3805–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delhommeau F., Dalla Venezia N., Morinière M., Collin H., Maillet P., Guerfali I., Leclerc P., Fardeau M., Delaunay J., and Baklouti F. (2005) Protein 4.1R expression in normal and dystrophic skeletal muscle. C. R. Biol. 328, 43–56 [DOI] [PubMed] [Google Scholar]
- 9. Pinder J. C., Taylor-Harris P. M., Bennett P. M., Carter E., Hayes N. V., King M. D., Holt M. R., Maggs A. M., Gascard P., and Baines A. J. (2012) Isoforms of protein 4.1 are differentially distributed in heart muscle cells: relation of 4.1R and 4.1G to components of the Ca2+ homeostasis system. Exp. Cell Res. 318, 1467–1479 [DOI] [PubMed] [Google Scholar]
- 10. Stagg M. A., Carter E., Sohrabi N., Siedlecka U., Soppa G. K., Mead F., Mohandas N., Taylor-Harris P., Baines A., Bennett P., Yacoub M. H., Pinder J. C., and Terracciano C. M. (2008) Cytoskeletal protein 4.1R affects repolarization and regulates calcium handling in the heart. Circ. Res. 103, 855–863 [DOI] [PubMed] [Google Scholar]
- 11. Braun T., and Arnold H. H. (1996) Myf-5 and myoD genes are activated in distinct mesenchymal stem cells and determine different skeletal muscle cell lineages. EMBO J. 15, 310–318 [PMC free article] [PubMed] [Google Scholar]
- 12. Molkentin J. D., and Olson E. N. (1996) Defining the regulatory networks for muscle development. Curr. Opin. Genet. Dev. 6, 445–453 [DOI] [PubMed] [Google Scholar]
- 13. Arnold H. H., and Winter B. (1998) Muscle differentiation: more complexity to the network of myogenic regulators. Curr. Opin. Genet. Dev. 8, 539–544 [DOI] [PubMed] [Google Scholar]
- 14. Yamaguchi A. (1995) Regulation of differentiation pathway of skeletal mesenchymal cells in cell lines by transforming growth factor-β superfamily. Semin. Cell Biol. 6, 165–173 [DOI] [PubMed] [Google Scholar]
- 15. Auradé F., Pinset C., Chafey P., Gros F., and Montarras D. (1994) Myf5, MyoD, myogenin and MRF4 myogenic derivatives of the embryonic mesenchymal cell line C3H10T1/2 exhibit the same adult muscle phenotype. Differentiation 55, 185–192 [DOI] [PubMed] [Google Scholar]
- 16. Davis R. L., Weintraub H., and Lassar A. B. (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 [DOI] [PubMed] [Google Scholar]
- 17. Bisbal C., Silhol M., Laubenthal H., Kaluza T., Carnac G., Milligan L., Le Roy F., and Salehzada T. (2000) The 2′-5′ oligoadenylate/RNase L/RNase L inhibitor pathway regulates both MyoD mRNA stability and muscle cell differentiation. Mol. Cell. Biol. 20, 4959–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guttridge D. C., Mayo M. W., Madrid L. V., Wang C. Y., and Baldwin A. S. Jr. (2000) NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 [DOI] [PubMed] [Google Scholar]
- 19. Figueroa A., Cuadrado A., Fan J., Atasoy U., Muscat G. E., Muñoz-Canoves P., Gorospe M., and Muñoz A. (2003) Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol. Cell. Biol. 23, 4991–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin D., Hidaka K., Shirai M., and Morisaki T. (2010) RNA-binding motif protein 24 regulates myogenin expression and promotes myogenic differentiation. Genes Cells 15, 1158–1167 [DOI] [PubMed] [Google Scholar]
- 21. Olguín H. C. (2011) Regulation of Pax7 protein levels by caspase-3 and proteasome activity in differentiating myoblasts. Biol. Res. 44, 323–327 [PubMed] [Google Scholar]
- 22. Boutet S. C., Disatnik M. H., Chan L. S., Iori K., and Rando T. A. (2007) Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell 130, 349–362 [DOI] [PubMed] [Google Scholar]
- 23. Abu Hatoum O., Gross-Mesilaty S., Breitschopf K., Hoffman A., Gonen H., Ciechanover A., and Bengal E. (1998) Degradation of myogenic transcription factor MyoD by the ubiquitin pathway in vivo and in vitro: regulation by specific DNA binding. Mol. Cell. Biol. 18, 5670–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doucet C., Gutierrez G. J., Lindon C., Lorca T., Lledo G., Pinset C., and Coux O. (2005) Multiple phosphorylation events control mitotic degradation of the muscle transcription factor Myf5. BMC Biochem. 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindon C., Albagli O., Domeyne P., Montarras D., and Pinset C. (2000) Constitutive instability of muscle regulatory factor Myf5 is distinct from its mitosis-specific disappearance, which requires a D-box-like motif overlapping the basic domain. Mol. Cell. Biol. 20, 8923–8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu J., Menzies K., Freeman R. S., and Taubman M. B. (2007) EGLN3 prolyl hrdroxylase regulates skeletal muscle differentiation and myogenin protein stability. J. Biol. Chem. 282, 12410–12418 [DOI] [PubMed] [Google Scholar]
- 27. Shiraishi S., Zhou C., Aoki T., Sato N., Chiba T., Tanaka K., Yoshida S., Nabeshima Y., Nabeshima Y., and Tamura T. A. (2007) TBP-interacting protein 120B (TIP120B)/cullin-associated and neddylation-dissociated 2 (CAND2) inhibits SCF-dependent ubiquitination of myogenin and accelerates myogenic differentiation. J. Biol. Chem. 282, 9017–9128 [DOI] [PubMed] [Google Scholar]
- 28. Varshavsky A. (2012) The ubiquitin system and immense realm. Annu. Rev. Biochem. 81, 167–176 [DOI] [PubMed] [Google Scholar]
- 29. Reinstein E., and Ciechanover A. (2006) Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann. Intern. Med. 145, 676–684 [DOI] [PubMed] [Google Scholar]
- 30. Tintignac L. A., Lagirand J., Batonnet S., Sirri V., Leibovitch M. P., and Leibovitch S. A. (2005) Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 280, 2847–2856 [DOI] [PubMed] [Google Scholar]
- 31. Noy T., Suad O., Taglicht D., and Ciechanover A. (2012) HUWE1 ubiquitinates MyoD and targets it for proteasomal degradation. Biochem. Biophys. Res. Commun. 418, 408–413 [DOI] [PubMed] [Google Scholar]
- 32. Iwai K., Yamanaka K., Kamura T., Minato N., Conaway R. C., Conaway J. W., Klausner R. D., and Pause A. (1999) Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. U.S.A. 96, 12436–12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lisztwan J., Imbert G., Wirbelauer C., Gstaiger M., and Krek W. (1999) The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 13, 1822–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tapscott S. J., Davis R. L., Thayer M. J., Cheng P. F., Weintraub H., and Lassar A. B. (1988) MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 242, 405–411 [DOI] [PubMed] [Google Scholar]
- 35. Edmondson D. G., and Olson E. N. (1989) A gene with homology to the myc similarity region of MyoD1 is expressed during myogenesis and is sufficient to activate the muscle differentiation program. Genes Dev. 3, 628–640 [DOI] [PubMed] [Google Scholar]
- 36. Bergstrom D. A., and Tapscott S. J. (2001) Molecular distinction between specification and differentiation in the myogenic basic helix-loop-helix transcription factor family. Mol. Cell. Biol. 21, 2404–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thayer M. J., Tapscott S. J., Davis R. L., Wright W. E., Lassar A. B., and Weintraub H. (1989) Positive autoregulation of the myogenic determination gene MyoD1. Cell 58, 241–248 [DOI] [PubMed] [Google Scholar]
- 38. Lonergan K. M., Iliopoulos O., Ohh M., Kamura T., Conaway R. C., Conaway J. W., and Kaelin W. G. (1998) Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell. Biol. 18, 732–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee D. H., and Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 [DOI] [PubMed] [Google Scholar]
- 40. Wells L., Edwards K. A., and Bernstein S. I. (1996) Myosin heavy chain isoforms regulate muscle function but not myofibril assembly. EMBO J. 15, 4454–4459 [PMC free article] [PubMed] [Google Scholar]
- 41. Galbiati F., Volonte D., Engelman J. A., Scherer P. E., and Lisanti M. P. (1999) Targeted down-regulation of caveolin-3 is sufficient to inhibit myotube formation in differentiating C2C12 myoblasts: transient activation of p38 mitogen-activated protein kinase is required for induction of caveolin-3 expression and subsequent myotube formation. J. Biol. Chem. 274, 30315–30321 [DOI] [PubMed] [Google Scholar]
- 42. Biederer C. H., Ries S. J., Moser M., Florio M., Israel M. A., McCormick F., and Buettner R. (2000) The basic helix-loop-helix transcription factors myogenin and Id2 mediate specific induction of caveolin-3 gene expression during embryonic development. J. Biol. Chem. 275, 26245–26251 [DOI] [PubMed] [Google Scholar]
- 43. Edmondson D. G., Brennan T. J., and Olson E. N. (1991) Mitogenic repression of myogenin autoregulation. J. Biol. Chem. 266, 21343–21346 [PubMed] [Google Scholar]
- 44. Ewing R. M., Chu P., Elisma F., Li H., Taylor P., Climie S., McBroom-Cerajewski L., Robinson M. D., O'Connor L., Li M., Taylor R., Dharsee M., Ho Y., Heilbut A., Moore L., Zhang S., et al. (2007) Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brugarolas J. (2014) Molecular genetics of clear-cell renal cell carcinoma. J. Clin. Oncol. 32, 1968–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi Z. T., Afzal V., Coller B., Patel D., Chasis J. A., Parra M., Lee G., Paszty C., Stevens M., Walensky L., Peters L. L., Mohandas N., Rubin E., and Conboy J. G. (1999) Protein 4.1R-deficient mice are viable but have erythroid membrane skeleton abnormalities. J. Clin. Invest. 103, 331–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Banks G. B., and Chamberlain J. S. (2008) The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Curr. Top. Dev. Biol. 84, 431–453 [DOI] [PubMed] [Google Scholar]
- 48. Sussman M. (2002) Duchenne muscular dystrophy. J. Am. Acad. Orthop. Surg. 10, 138–151 [DOI] [PubMed] [Google Scholar]
- 49. Infante J. P., and Huszagh V. A. (1999) Mechanisms of resistance to pathogenesis in muscular dystrophies. Mol. Cell Biochem. 195, 155–167 [DOI] [PubMed] [Google Scholar]
- 50. Shaw R. J., Paez J. G., Curto M., Yaktine A., Pruitt W. M., Saotome I., O'Bryan J. P., Gupta V., Ratner N., Der C. J., Jacks T., and McClatchey A. I. (2001) The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev. Cell. 1, 63–72 [DOI] [PubMed] [Google Scholar]
- 51. Bretscher A., Edwards K., and Fehon R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586–599 [DOI] [PubMed] [Google Scholar]
- 52. Perry A., Gutmann D. H., and Reifenberger G. (2004) Molecular pathogenesis of meningiomas. J. Neurooncol. 70, 183–202 [DOI] [PubMed] [Google Scholar]
- 53. Gutmann D. H., Donahoe J., Perry A., Lemke N., Gorse K., Kittiniyom K., Rempel S. A., Gutierrez J. A., and Newsham I. F. (2000) Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Hum. Mol. Genet. 9, 1495–1500 [DOI] [PubMed] [Google Scholar]
- 54. Yageta M., Kuramochi M., Masuda M., Fukami T., Fukuhara H., Maruyama T., Shibuya M., and Murakami Y. (2002) Direct association of TSLC1 and DAL-1, two distinct tumor suppressor proteins in lung cancer. Cancer Res. 62, 5129–5133 [PubMed] [Google Scholar]
- 55. Kuns R., Kissil J. L., Newsham I. F., Jacks T., Gutmann D. H., and Sherman L. S. (2005) Protein 4.1B expression is induced in mammary epithelial cells during pregnancy and regulates their proliferation. Oncogene 24, 6502–6515 [DOI] [PubMed] [Google Scholar]
- 56. Wong S. Y., Haack H., Kissil J. L., Barry M., Bronson R. T., Shen S. S., Whittaker C. A., Crowley D., and Hynes R. O. (2007) Protein 4.1B suppresses prostate cancer progression and metastasis. Proc. Natl. Acad. Sci. U.S.A. 104, 12784–12789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robb V. A., Li W., Gascard P., Perry A., Mohandas N., and Gutmann D. H. (2003) Identification of a third Protein 4.1 tumor suppressor, Protein 4.1R, in meningioma pathogenesis. Neurobiol. Dis. 13, 191–202 [DOI] [PubMed] [Google Scholar]
- 58. Rajaram V., Gutmann D. H., Prasad S. K., Mansur D. B., and Perry A. (2005) Alterations of protein 4.1 family members in ependymomas: a study of 84 cases. Mod. Pathol. 18, 991–997 [DOI] [PubMed] [Google Scholar]
- 59. Pećina-Šlaus N. (2013) Merlin, the NF2 gene product. Pathol. Oncol. Res. 19, 365–373 [DOI] [PubMed] [Google Scholar]
- 60. Khanna C., Wan X., Bose S., Cassaday R., Olomu O., Mendoza A., Yeung C., Gorlick R., Hewitt S. M., and Helman L. J. (2004) The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat. Med. 10, 182–186 [DOI] [PubMed] [Google Scholar]
- 61. Yu Y., Khan J., Khanna C., Helman L., Meltzer P. S., and Merlino G. (2004) Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat. Med. 10, 175–181 [DOI] [PubMed] [Google Scholar]
- 62. Li W., You L., Cooper J., Schiavon G., Pepe-Caprio A., Zhou L., Ishii R., Giovannini M., Hanemann C. O., Long S. B., Erdjument-Bromage H., Zhou P., Tempst P., and Giancotti F. G. (2010) Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4 (DCAF1) in the nucleus. Cell 140, 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shimizu T., Seto A., Maita N., Hamada K., Tsukita S., Tsukita S., and Hakoshima T. (2002) Structural basis for neurofibromatosis type 2: crystal structure of the merlin FERM domain. J. Biol. Chem. 277, 10332–10336 [DOI] [PubMed] [Google Scholar]
- 64. Na X., Duan H. O., Messing E. M., Schoen S. R., Ryan C. K., di Sant'Agnese P., Golemis E. A., and Wu G. (2003) Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel-Lindau protein. EMBO J. 22, 4249–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zatyka M., da Silva N. F., Clifford S. C., Morris M. R., Wiesener M. S., Eckardt K. U., Houlston R. S., Richards F. M., Latif F., and Maher E. R. (2002) Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel-Lindau disease. Cancer Res. 62, 3803–3811 [PubMed] [Google Scholar]
- 66. Iliopoulos O., Kibel A., Gray S., and Kaelin W. G. (1995) Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1, 822–826 [DOI] [PubMed] [Google Scholar]
- 67. Young J. A., Sermwittayawong D., Kim H. J., Nandu S., An N., Erdjument-Bromage H., Tempst P., Coscoy L., and Winoto A. (2011) Fas-associated death domain (FADD) and the E3 ubiquitin-protein ligase TRIM21 interact to negatively regulate virus-induced interferon production. J. Biol. Chem. 286, 6521–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Deguillien M., Huang S. C., Morinière M., Dreumont N., Benz E. J. Jr., and Baklouti F. (2001) Multiple cis elements regulate an alternative splicing event at 4.1R pre-mRNA during erythroid differentiation. Blood 98, 3809–3816 [DOI] [PubMed] [Google Scholar]