

Abstract
In contrast to G protein-coupled receptors, for which chemical and peptidic inhibitors have been extensively explored, few compounds are available that directly modulate heterotrimeric G proteins. Active Gαq binds its two major classes of effectors, the phospholipase C (PLC)-β isozymes and Rho guanine nucleotide exchange factors (RhoGEFs) related to Trio, in a strikingly similar fashion: a continuous helix-turn-helix of the effectors engages Gαq within its canonical binding site consisting of a groove formed between switch II and helix α3. This information was exploited to synthesize peptides that bound active Gαq in vitro with affinities similar to full-length effectors and directly competed with effectors for engagement of Gαq. A representative peptide was specific for active Gαq because it did not bind inactive Gαq or other classes of active Gα subunits and did not inhibit the activation of PLC-β3 by Gβ1γ2. In contrast, the peptide robustly prevented activation of PLC-β3 or p63RhoGEF by Gαq; it also prevented G protein-coupled receptor-promoted neuronal depolarization downstream of Gαq in the mouse prefrontal cortex. Moreover, a genetically encoded form of this peptide flanked by fluorescent proteins inhibited Gαq-dependent activation of PLC-β3 at least as effectively as a dominant-negative form of full-length PLC-β3. These attributes suggest that related, cell-penetrating peptides should effectively inhibit active Gαq in cells and that these and genetically encoded sequences may find application as molecular probes, drug leads, and biosensors to monitor the spatiotemporal activation of Gαq in cells.
Keywords: G protein, inhibitor, p63, peptide interaction, phospholipase C
Introduction
G protein-coupled receptors (GPCRs)3 are the largest family of cell surface receptors in eukaryotes and regulate essentially all physiological processes (1–4). Scores of neurotransmitters, hormones, and other extracellular signaling molecules promote conserved conformational changes in GPCRs that favor interaction with heterotrimeric G proteins (Gαβγ) on the inner leaflet of the plasma membrane. The subsequent exchange of GTP for GDP alters three switch regions within Gα subunits to promote dissociation from Gβγ (5, 6). These activated subunits then directly stimulate downstream effectors, leading to propagation and amplification of signaling pathways (7, 8).
Signaling cascades downstream of several hundred different GPCR are activated by the cognate agonist of these receptors. Many of these GPCRs, particularly those activated by neurotransmitters, also have been targeted by receptor subtype-selective agonists and antagonists. Indeed, up to a quarter of current top-prescribed, Food and Drug Administration-approved drugs take advantage of GPCR-selective targeting. In contrast, few molecular probes are available for dissection of the downstream signaling cascades emanating from these GPCRs. Most notably, bacterially derived cholera and pertussis toxins contributed enormously to the initial identification of Gαs and Gαi and were subsequently applied to differentiate Gαs- and Gαi-mediated signaling pathways (9). However, only a few small molecules have been identified that target Gα subunits, and reliable pharmacological agents that selectively inhibit these signaling proteins are not generally available (10). For example, BIM-46174 and BIM-46187 apparently interact with most Gα subunits (11, 12), and some selectivity for Gαq has been reported for BIM-46187 (13). In contrast, YM-254890 (14), and related natural products such as FR900359 (15, 16), specifically inhibit Gα subunits of the Gq family (17, 18), and a crystal structure of a G protein heterotrimer bound to YM-254890 illustrates that this molecule prevents GDP release and heterotrimer dissociation (19). Unfortunately, synthesis of cyclic depsipeptides is difficult, and YM-254890 remains unavailable from commercial sources. The mechanism of this molecule also precludes inhibition against Gα subunits in the GTP-bound state; for example, on mutant proteins that are constitutively active because of loss of GTPase function.
Linear peptides also have been identified that selectively target Gα subunits (20). For example, the G protein regulatory (GPR or GoLoco) motif of proteins preferentially binds GDP-bound Gαi subunits with relatively high affinity to decrease spontaneous nucleotide exchange (21). Small peptides based on this motif were applied to inhibit D2 dopamine receptor-stimulated potassium channel currents (22). Phage display in combination with Gαi1 was also used to generate peptides that specifically bind Gα subunits dependent on the state of bound nucleotide (23–25). One of these peptides preferentially binds active Gαi and Gαt and was shown to inhibit the capacity of Gαi1 to be inactivated by RGS12 and to reduce activation of cGMP phosphodiesterase by Gαt in membrane preparations.
A molecular understanding of the binding of Gα subunits with their downstream effectors potentially informs the development of peptides to pharmacologically antagonize functional interaction of these signaling cohorts. For example, rearrangement of the switch regions of Gα subunits creates a hydrophobic cleft between switch 2 and α helix 3 that is a major site of interaction with effectors (26) and potentially exists as a site for drug targeting. The best understood Gα/effector interaction is between Gαq and its effectors, the phospholipase C-β (PLC-β) isozymes and the related Dbl family proteins p63RhoGEF, Trio, and kalirin (27–29). These effectors engage the hydrophobic cleft of Gαq using almost identical sets of interactions derived from a helix-turn-helix (HTH) substructure (Fig. 1) that is induced in the effectors upon engagement with Gαq. The HTH of PLC-β3 and p63RhoGEF forms the major interface with Gαq in crystal structures of these complexes, whereas secondary interactions orient the complexes at membranes for efficient effector activation (30, 31).
FIGURE 1.

Gαq uses a conserved mechanism to engage effectors. A, a helix-turn-helix (Hα1/Hα2, cylinders) in either PLC-β3 (blue) or p63RhoGEF (yellow) engages a shallow groove on activated Gαq (green with switch regions in red) between switch II and α3. The structures of Gαq bound to either PLC-β3 (PDB code 3OHM) (30) or p63RhoGEF (PDB code 2RGN) (31) were superimposed using Gαq. For clarity, only a single Gαq is shown. The expanded region highlights residues in PLC-β3 that directly contact Gαq; contacts boxed in the corresponding sequence alignment. C-term, C terminus; N-term, N terminus. B and C, peptides derived from the helix-turn-helix of PLC-β3 selectively bind active Gαq. B, fluorescence polarization (ΔmP, change in millipolarization) was used to measure the binding of the indicated Gα subunits to either a TAMRA-labeled peptide (27-mer(I860A)) derived from the helix-turn-helix of PLC-β3 or the equivalent peptide also containing L859E (27-mer(L859E+I860A)). All Gα subunits were activated with aluminum fluoride except where indicated (no AlF4). C, the above assay was used to measure affinities of active Gαq for the TAMRA-labeled peptides listed. Substitutions include I860A (red) and M869Nle (underlined in red).
Here we show that linear peptides based on the HTH of PLC-β3 specifically bind activated Gαq to prevent engagement and activation of downstream effectors by Gαq. These peptides have no appreciable affinity for either GDP-bound Gαq or for activated forms of other Gα subunits and should be useful reagents for interdicting signaling cascades controlled by Gαq. Indeed, we show that microinjection of HTH-based peptides into mouse neurons of the prefrontal cortex prevents depolarization downstream of muscarinic cholinergic receptor-dependent activation of Gαq.
Results
Canonical Effector Interactions with Gαq Drive Complex Formation
Recent structures of activated Gαq bound to either PLC-β3 (30) or p63RhoGEF (31) highlight an essentially identical mechanism of effector engagement. In both cases, an HTH of the effector occupies the canonical effector-binding site of Gαq composed of a shallow groove between the α3 helix and switch II (Fig. 1A). The HTH is presumed to provide the majority of favorable interactions promoting complex formation and buries ∼810 Å2 of Gαq (5% of the accessible surface) to produce the largest contiguous interface between proteins in these structures. Deletion of the HTH of PLC-β3 (30) or p63RhoGEF (29) abrogates interaction with Gαq; conversely, grafting the HTH of PLC-β3 onto PLC-δ1 engenders complex formation with Gαq (30).
To more accurately quantify interactions between activated Gαq and HTH peptides, changes in fluorescence polarization were measured for a set of TAMRA-labeled peptides spanning the HTH of PLC-β3 and titrated with various Gα subunits (Fig. 1, B and C). A 27-residue peptide (27mer(I860A)), derived from the complete HTH of PLC-β3, bound to activated Gαq with high affinity (Kd ∼400 nm), and complex formation was dependent upon activation because little change in fluorescence occurred in the absence of aluminum fluoride (Fig. 1B). This interaction was also specific for Gαq because activation of other Gα subunits (Gαi1, Gαt, and Gαs) did not result in binding to the HTH-based peptide. A residue on the HTH that is a major contributor to this interaction is leucine at position 859 of PLC-β3, and mutating this residue to glutamic acid abolished binding of the TAMRA-labeled 27-mer(I860A) (Fig. 1B).
The 27-mer(I860A) peptide contains two substitutions relative to the wild-type sequence: the oxidation-prone methionine was replaced with isosteric l-norleucine, and isoleucine at position 860 of full-length PLC-β3 was replaced with alanine (Fig. 1C). This second substitution (I860A) was shown previously to increase the phospholipase activity of PLC-β3 in response to Gαq (30) and was assumed to increase the affinity of the complex. Indeed, the substitutions contributed to an approximate 10-fold increase in affinity for activated Gαq compared with the equivalent peptide (27-mer) without substitutions (Fig. 1C). Truncation of the 27-mer by removal of two alanines at its C terminus (25-mer) led to a minor reduction (< 2-fold) in affinity for Gαq. In contrast, shorter peptides lacking most (13-mer) or all (10-mer) of the C-terminal helix of the HTH did not bind activated Gαq with measurable affinity.
The robust increase in fluorescence polarization produced by the complex of TAMRA-27-mer(I860A) with active Gαq was used in a series of competition binding experiments to assess the relative affinities of unlabeled peptides for active Gαq (Fig. 2). The capacity of unlabeled 27-mer(I860A) to interact with Gαq was quantified by titrating this peptide into a solution containing 0.4 μm TAMRA-labeled peptide and active Gαq. The resulting decrease in fluorescence polarization was quantified, and a Ki (∼0.3 μm) for unlabeled 27-mer(I860A) was calculated as described under “Experimental Procedures” (Fig. 2A). This Ki was essentially identical to the Kd (∼0.4 μm) determined for direct binding of the TAMRA-labeled 27-mer(I860A) to active Gαq. Thus, the presence of the TAMRA moiety does not appreciably affect Gαq-HTH complex formation.
FIGURE 2.

Competition assay to measure affinities of peptides and effectors for Gαq. A and B, individual unlabeled peptides were titrated into a solution of Gαq bound to TAMRA-27-mer(I860A), and fluorescence polarization was measured. Ki values were calculated using an equation to convert IC50 to Ki values in fluorescence-based competition assays (38). The sequence alignment is annotated as in Fig. 1. Inset, circular dichroism spectra for select peptides. Substitutions include I860A (red) and M869Nle (underlined in red). C, competition assay used to measure affinities of the indicated effectors for Gαq activated with aluminum fluoride.
A shorter, unlabeled peptide (21-mer(I860A)) that retained all PLC-β3 residues that contact Gαq in the complex (30) also effectively inhibited (Ki ∼1 μm) the binding of TAMRA-27-mer(I860A). Binding was also more favorable with this peptide than with the equivalent 21-mer without substitution (Fig. 2A), which again supports the idea that the I860A substitution enhances complex formation. The ∼3-fold decrease in inhibitory activity of 21-mer(I860A) relative to 27-mer(I860A) is consistent with direct binding measurements that showed a slight decrease in the affinity of the TAMRA-labeled 25-mer relative to the equivalent 27-mer. This trend also held in comparisons of the activity of unlabeled, unsubstituted 21-mer of PLC-β3 (Ki ∼3 μm) with that of the related 36-mer (Ki ∼1 μm) (Fig. 2B).
The reason(s) why the shorter peptides exhibit a small but consistent decrease in affinity for Gαq relative to the longer forms is unclear. One possibility is that loss of residues either proximal to or contained within the helices of the HTH motif destabilizes the secondary structure needed for complex formation with Gαq. This destabilization is likely to manifest only after formation of the complex because, irrespective of their length, the isolated peptides exhibit minimal helical content based on circular dichroism spectroscopy (Fig. 2A, inset). If true, this destabilization would result in higher off rates for the shorter peptides in complex with Gαq.
Peptides (21-mers) corresponding to the HTH portions of the four PLC-β isozymes exhibited a 25-fold range of inhibitory activities (Fig. 2B). Interestingly, these relative activities correlate with the general capacity of Gαq to activate these isozymes (PLC-β1 ∼ β3 > β2 > β4) (32, 33), suggesting that Gαq-mediated activation of PLC-β isozymes is principally a function of complex formation driven by the affinity of Gαq for the HTH region. Equivalent 21-mers from p63RhoGEF and the related GEFs Trio and kalirin exhibited Ki values in the range of ∼25–50 μm, which is similar to the inhibitory activity observed with the HTH of PLC-β4. Relative to the equivalent portion in PLC-β3, the first helix of the HTH is substantially longer in the structure of p63RhoGEF bound to Gαq. Consistent with previous results, extension of a peptide of p63RhoGEF to include the entire first helix increased inhibitory activity ∼15-fold relative to the cognate 21-mer (Fig. 2B).
The competition assay was also useful for quantifying relative affinities of active Gαq and full-length effectors. For example, full-length PLC-β3 prevented engagement of active Gαq by TAMRA-27-mer(I860A) with an IC50 of ∼250 nm (Fig. 2C). This inhibitory activity was ∼3-fold greater than that of the 36-mer of PLC-β3, indicating that most of the energetically favorable interactions with Gαq reside within the HTH of PLC-β3.
The C-terminal domain of PLC-β isozymes is required for phospholipase activation by Gαq at membranes; however, measurements of the contribution of the C-terminal domain to binding to Gαq have ranged from essentially no effect (30, 34) to promoting complex formation by at least 2 orders of magnitude (35). Related to this discrepancy, a truncated form (residues 1–886) of PLC-β3, which retains the HTH but lacks the entire C-terminal domain, competed for complex formation (Fig. 2C) with an inhibitory activity within 2-fold of that observed with full-length PLC-β3. This result is consistent with previous affinity measurements using surface plasmon resonance assays that indicated that the C-terminal domain of PLC-β3 is responsible for less than a 2-fold increase in affinity for Gαq relative to PLC-β3(1–886) (30). Conversely, a single substitution (L859E) within the HTH of full-length PLC-β3 that was shown previously to prevent activation of PLC-β3 by Gαq (30) completely abrogated the capacity of full-length PLC-β3 to compete with the TAMRA-labeled peptide for engagement of Gαq (Fig. 2C). In addition, PLC-δ1 was not responsive to Gαq and was incapable of competing with TAMRA-27-mer(I860A) (Fig. 2C).
Finally, a large fragment (residues 155–493) of p63RhoGEF that includes its HTH effectively competed (IC50 ∼500 nm) with TAMRA-27-mer(I860A) for engagement of Gαq (Fig. 2C). A relative Ki of 1.9 μm was observed for binding of the isolated HTH of p63RhoGEF to Gαq, and, therefore, as was observed with PLC-β3, the majority of favorable interactions for complex formation with Gαq reside within the HTH of p63RhoGEF.
Inhibition of Gαq-mediated Signaling in Vitro
Because peptides derived from the HTH of PLC-β3 effectively compete with effectors for engagement of Gαq, these peptides should also prevent the Gαq-dependent activation of these effectors. As illustrated in Fig. 3A, TAMRA-27-mer(I860A) indeed efficiently blocked Gαq-mediated activation of purified PLC-β3 in reconstituted lipid vesicles. Inhibition was dependent on the concentration of peptide as well as Gαq. A 5-fold increase in the concentration of Gαq required a corresponding increase in peptide concentration to achieve equivalent inhibition of phospholipase activity with no change in the shape of the concentration-effect curve. These results are strongly indicative of direct competitive inhibition by the peptide to prevent functional engagement of PLC-β3 by Gαq, which is expected if both peptide and effector share a common interface on Gαq.
FIGURE 3.

27-mer(I860A) inhibits active Gαq in lipid vesicles. A and B, lipid vesicles containing [3H]PIP2 were reconstituted with PLC-β3 and Gαq prior to measuring hydrolysis of PIP2 in the presence of increasing concentrations of the indicated peptides. 120 nm Gαq was used in B. C, TAMRA-27-mer(I860A) did not inhibit the capacity of Gβ1γ2 to activate PLC-β3 in lipid vesicles but prevented synergy with Gαq.
Although the presence of TAMRA had no apparent effect on HTH-Gαq binding in solution, removal of the TAMRA moiety from the peptide reduced inhibition of phospholipase activity by ∼5-fold (IC50 ∼200 nm) (Fig. 3B), suggesting that the fluorescent group has nonspecific affinity for membranes. This idea is also supported by the observation that TAMRA-27-mer(L859E+I860A) did not inhibit Gαq-mediated activation of PLC-β3 under identical conditions and that, therefore, TAMRA per se does not intrinsically inhibit activation. TAMRA-27-mer(I860A)-mediated inhibition of phospholipase activity was specific for Gαq because the peptide did not inhibit the equivalent activation of PLC-β3 by Gβ1γ2 (Fig. 3C). Indeed, under conditions of synergistic activation of PLC-β3 by both Gβ1γ2 and Gαq, only the Gβγ-mediated component of activation appeared to be preserved in the presence of TAMRA-27-mer(I860A).
Because the canonical effector-binding surface of Gαq is required for activation of both PLC isozymes and RhoGEFs, the capacity of 27-mer(I860A) to prevent activation of p63RhoGEF by Gαq was examined. Fluorescent GDP was preloaded onto RhoA, and the capacity of Gαq to stimulate p63RhoGEF-promoted guanine nucleotide exchange by Rho was quantified in the presence of increasing concentrations of 27-mer(I860A) (Fig. 4). Activity was sharply reduced by 5 μm 27-mer(I860A) and completely inhibited by 12.5 μm 27-mer(I860A).
FIGURE 4.
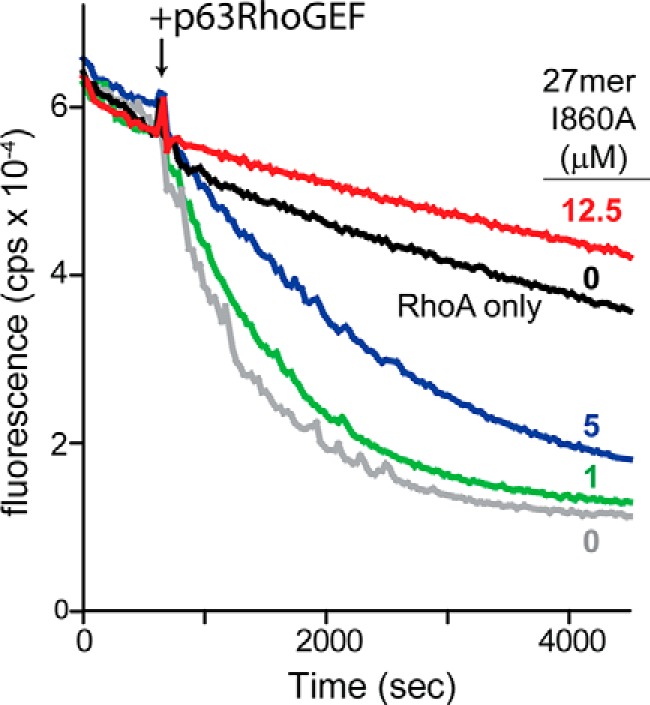
Peptides derived from the helix-turn-helix of PLC-β3 inhibit the capacity of Gαq to activate p63RhoGEF. RhoA (100 nm) preloaded with BODIPY-GDP was incubated with unlabeled GDP (2 μm), Gαq (100 nm), aluminum fluoride, and the indicated amounts of 27-mer(I860A) prior to measuring nucleotide exchange using fluorescence (cps, counts per second). p63RhoGEF (100 nm) was added as indicated. The trace marked RhoA only contains no Gαq or p63RhoGEF and indicates spontaneous nucleotide exchange.
Inhibition of Gαq-mediated Signaling in Cells
Given the robust capacity of synthetic versions of the HTH of PLC-β3 to inhibit effector activation by Gαq using purified components, versions of the 27-mer were expressed in HEK293 cells and assessed for their capacity to inhibit Gαq-promoted signaling (Fig. 5).
FIGURE 5.

Gαq-dependent PLC-β3 activity in HEK293 cells is specifically inhibited by genetically encoded versions of the helix-turn-helix of PLC-β3. A–C, HEK293 cells were co-transfected with expression vectors encoding the 5HT2A receptor (100 ng) and increasing amounts of PLC-β3 (A), YFP-27mer-CFP (B), or YFP-27mer-CFP-CAAX (C) variants prior to metabolic labeling of inositide pools, receptor activation with DOI (2 μm), and quantification of inositol phosphates. Protein expression and cellular totals (actin) were verified with Western blotting as shown. Cells without addition of agonist (−DOI) were also tested.
Activity initially was assessed after expression of full-length versions of PLC-β3 rendered catalytically inactive by substitution (H332A) of the active site of the lipase (Fig. 5A). This approach presumed that heterologous expression of PLC-β3(H332A) would act in a dominant-negative fashion to prevent activation of endogenous PLC-β in response to agonist (DOI)-promoted stimulation of the co-expressed, Gαq-coupled 5HT2A receptor. Indeed, expression of increasing amounts of PLC-β3(H332A) resulted in essentially complete inhibition of DOI-stimulated inositol phosphate accumulation. Importantly, this inhibitory activity was not observed (Fig. 5A) with PLC-β3(H332A+L859E), which introduces an additional substitution, L859E, shown previously to prevent complex formation of PLC-β3 with Gαq.
Given the results with PLC-β3(H332A), we examined the inhibitory activity of a construct consisting of the sequence equivalent to 27-mer(I860A) flanked by two fluorescent proteins to foster intracellular stability. This construct also potently blocked 5HT2A receptor-dependent activation of phospholipase C in HEK293 cells (Fig. 5B), and introduction of L859E into this construct abrogated this inhibitory activity. Modification of the expression construct to direct posttranslation prenylation of the chimeric HTH peptide resulted in ∼10-fold more efficient inhibitory activity of the 27-mer(I860A) sequence, and activity again was lost by introduction of the L859E mutation into the 27-mer(I860A) sequence (Fig. 5C).
An ultimate goal of this work is to develop peptidomimetic inhibitors of Gαq-promoted signaling in vivo. Toward this goal, the capacity of these peptides to block neuronal depolarization was assessed (Fig. 6). Peptides were introduced into neuronal cells from slices of mouse prefrontal cortex via patch clamp prior to monitoring depolarization in response to muscarinic cholinergic receptor-promoted activation of a Gαq by the muscarinic agonist carbachol. Carbachol-stimulated current was similar to previously recorded activity (36) and was not affected by injection of 10 μm 27-mer(L859E+I860A). In contrast, 10 μm 27-mer(I860A) completely blocked carbachol-stimulated depolarization, and this inhibition was similar to that observed after the simultaneous application of the muscarinic antagonist pirenzepine and 27-mer(L859E+I860A). Thus, a 27-mer based on the HTH of PLC-β3 blocks Gαq-dependent signaling in a native neuronal system, and this inhibitory activity is lost after introduction of a mutation into the 27-mer known to result in loss of interaction of PLC-β3 with this G protein.
FIGURE 6.

27-mer(I860A) inhibits Gαq-mediated neuronal depolarization in the prefrontal cortex of mice. A, pretreatment of a representative prefrontal cortex neuron with 27-mer(L859E+I860A) had no effect on action potential firing in response to CCh (10 μm) (top panel). In contrast, depolarization was lost when 27-mer(I860A) was used (center panel) or the M1 receptor antagonist pirenzepine (2 μm) was applied with 27-mer(L859E+I860A) (bottom panel). B, the inward current induced by carbachol after indicated pretreatments. Sample sizes included six neurons for the active and inactive peptides and four neurons for the active peptide with agonist. *, p < 0.05 from baseline. **, p < 0.05 between the different experiments n.s., not significant.
Discussion
The Gq-regulated effectors provide a striking illustration of converging evolution in disparate signaling proteins. We previously concluded that the HTH imparts most, if not all of, the binding energy for the complex of Gαq and PLC-β3, and this idea is confirmed here. Indeed, the equivalent region of p63RhoGEF and related GEFs appears also to embody most of the binding energy for complexation of these effectors with Gαq.
Although the HTH of PLC-β3 is sufficient to recapitulate high-affinity interaction with active Gαq, complex formation mediated by the HTH is insufficient to elevate phospholipase activity (30). This follows from the additional requirement of the ∼250-residue carboxyl-terminal domain of the PLC, which, in combination with a membrane surface, is needed for activation. It remains unclear how these additional components impart activation, especially in light of the fact that the carboxyl-terminal domain likely provides no more than a 2-fold enhancement in affinity for Gαq. However, one distinct possibility is that the carboxyl-terminal domain productively orients PLC-β3 at membranes for efficient interaction with Gαq and hydrolysis of membrane-resident phosphatidylinositol 4,5-bisphosphate. Other PLC-β isozymes would be activated similarly. Indeed, a similar membrane-induced steering of p63RhoGEF and related GEFs with Gαq appears to be required for the efficient capacity of these GEFs to active RhoA (37).
Elucidation of the role of the HTH in Gαq binding to PLC-β isozymes together with the observation of dominant negative-like activity of a lipase-dead form of full-length PLC-β3 led us to investigate the cellular activity of a genetically encoded form of the HTH. Thus, a peptide optimized for binding to Gαq in vitro proved to markedly inhibit the stimulation of inositol lipid hydrolysis occurring downstream of the Gq-linked 5HT2A receptor. The increase in cellular potency of the peptide observed after introduction of a CAAX motif almost certainly occurs as a consequence of increasing the concentration of the peptide at the membrane, and we conclude that this form of the 27-mer(I860A) provides an outstanding genetically encoded reagent to interdict Gαq-stimulated signaling at the cellular level. It is likely that, although the HTH evolved for optimal physiological activation of PLC-β by Gαq, it did not evolve to provide the highest binding affinity. Indeed, a simple alanine substitution of Ile-860 in PLC-β3 was shown previously empirically to increase Gαq-mediated activation of the lipase, and we show here that such a substitution in HTH also significantly increases HTH binding affinity for Gαq. Thus, both rational and iterative screens of other substitutions in HTH should lead to additional increases in binding affinity in HTH-related peptides, and a long-term goal of this work is to generate Gαq-directed drugs consisting of these peptides packaged in cell-permeable forms. The remarkable capacity of the 27-mer(I860A) in a fluorescent protein-flanked form to block Gαq-stimulated inositol lipid signaling also suggests a starting point to develop, for example, biosensors that image the signaling dynamics of activated Gαq.
Experimental Procedures
Peptides
Peptides were either synthesized in-house using a PS3 solid-state peptide synthesizer (Protein Technologies, Inc.) or by Anaspec. Peptides in-house were purified with a C18 reversed phased HPLC system with a UV detector, and purity was determined to be >95% using analytical reversed phase HPLC. Correct molecular weights of peptides were determined using MALDI-TOF mass spectrometry.
A subset of TAMRA-labeled PLC-β3 peptides following the C2 domain include 9, 12, 25, and 27 residues (9-, 12-, 25-, and 27-mer). An additional TAMRA-27-mer(I860A) contains a point mutation, I860A, as well as a non-natural norleucine (Nle) substituted for methionine at residue 869. All peptides were amidated on the C terminus and acetylated or labeled with TAMRA on the N terminus. Synthesized peptides included different isoforms of PLC-β isozymes and Dbl family GEF proteins shown in Fig. 2B. All peptides were dissolved in 10 mm K2HPO4 at concentrations of 1–5 mm.
Fluorescence Polarization Assay
Fluorescence polarization assays were performed in small-volume cuvettes (Starna Cells, Inc. Atascadero, CA) in a Fluorolog-3 spectrophotometer (Horiba Scientific, Edison, NJ) that contained a final volume of 60 μl. Fluorescence polarization experiments used 400 nm TAMRA-labeled peptides and monitored the change in polarization at an excitation wavelength of 554 nm and an emission wavelength of 569 nm. Increasing amounts of Gα proteins were added to the TAMRA-labeled peptide solution to determine the affinity of Gαq for the peptides. Increasing amounts of unlabeled peptides or proteins were added with 400 nm TAMRA-27-mer(I860A) and 800 nm Gαq to determine the IC50 values of the unlabeled peptides. All experiments contained 10 mm HEPES (pH 7.0), 150 mm NaCl, and 50 mm MgCl2 with or without 150 μm AlCl3 plus 50 mm NaF. All Kd and IC50 values were calculated using Prism5 using a one-site binding hyperbola (Y = Bmax × X/Kd + X). For competition experiments, Ki values were calculated from IC50 values using an equation derived by Nikolovska-Coleska et al. (38).
Protein Expression and Purification
GαqΔ34, GαqΔ7, PLC-β3, and PLC-β3(1–886) were purified from High-Five insect cells. GαqΔ34 was engineered for high level of expression using a chimeric form of Gαq/i that contained an N-terminal hexahistidine tag followed by residues 1–28 of rat Gai1, a tobacco etch virus protease site, and residues 35–359 of mouse Gαq (GαqΔ34). GαqΔ7 was constructed similarly. Purification of GαqΔ34 and GαqΔ7 followed protocols published previously (30). PLC-β3 and PLC-β3 (1–886) were purified following protocols published previously (30). The truncated p63RhoGEF pleckstrin homology-Dbl homology (PH-DH) extension (155–493) was purified from Escherichia coli and followed protocols published previously (29).
Quantification of Phospholipase Activity of Purified PLC-β3 Isozymes
Basal phospholipase activity was determined by combining 330 μm l-α-phosphatidyl ethanolamine and 30 μm l-α-phosphatidylinositol-4,5-bisphosphate and drying the lipids under N2 in chloroform solution followed by resuspension by sonication in 20 mm HEPES (pH 7.2). Reactions were incubated for 10 min at 30 °C in a final buffer consisting of 30 mm HEPES (pH 7.2), 70 mm KCl, 2 mm DTT, 16.7 mm NaCl, 3 mm EGTA, 200 nm free Ca2+, 0.17 mg/ml fatty acid-free BSA, 10 mm NaF, 30 μm AlCl3, 2.7 mm MgCl2, and 20,000 dpm [3H]PIP2 with 200–300 pm of PLC-β3 to maintain the linearity of the enzyme assay. Increasing amount of peptide were added to the solution containing 60–200 nm Gαq or 20–60 nm Gβ1γ2.
Transfection of HEK293 Cells and Quantification of [3H]Inositol Phosphate Accumulation
HEK293 cell-based assays measured accumulation of [3H]inositol phosphates. HEK293 cells were maintained in high-glucose DMEM containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in an atmosphere of 95% air/5% CO2. HEK293 cells were transfected with 100 ng of DNA that encoded the 5HT2A receptor in the pcDNA vector and other indicted DNA vectors using FuGENE 6 (Promega, Madison, WI) according to the protocol of the manufacturer. The total amount of transfected DNA was 400 ng and included empty pcDNA vector to maintain equal amounts of DNA per well. Twenty-four hours post-transfection, media were exchanged, and cells were metabolically labeled with 1 μCi of myo-[2-3H(N)]inositol (American Radiolabeled Chemicals, St. Louis, MO) at 37 °C for 24 h. Metabolic labeling proceeded at 37 °C for 1 h in a water bath with 10 mm LiCl to inhibit phosphatases and 2 μm DOI to stimulate the 5HT2A receptor. Incubations were terminated by aspiration of the culture medium and subsequent addition of 50 mm formic acid for 20 min, followed by neutralization with 150 mm NH4OH. 3H-labeled inositol phosphates were isolated and quantified using Dowex chromatography.
Guanine Nucleotide Exchange Assays
The guanine nucleotide exchange activity of purified p63RhoGEF pleckstrin homology-Dbl homology extension (155–493) was determined using a kinetic fluorescence-based assay with RhoA that was preloaded with BODIPY FL-conjugated GDP (BODIPY-GDP, Molecular Probes) as described previously (39). Exchange assays were performed using a Fluorolog-3 (Horiba Scientific) with wavelengths at λexcitation = 500 nm (slits = 1 nm), λemission =511 nm (slits = 1 nm) and 1.5-ml quartz cuvettes thermostated at 20 °C while stirring constantly. Reactions were carried out in exchange buffer consisting of 20 mm Tris (pH 7.5), 150 mm NaCl, 10 mm MgCl2, 10 mm NaF, 30 μm AlCl3, and 10 μm GDP. Exchange assays contained BODIPY-GDP-preloaded RhoA (100 nm) equilibrated in exchange buffer at 20 °C, followed by addition of p63RhoGEF (100 nm), Gαq (100 nm), and the indicated amounts of peptides. Nucleotide exchange was monitored in real time for 2 h at 20 °C.
Electrophysiology
C57BL/6J mice were sacrificed via deep isoflurane anesthesia and rapid decapitation, and brain slices containing the prefrontal cortex (PFC) were prepared as described previously (40, 41). Briefly, brains were rapidly removed, and 300-μm slices were cut on a vibratome (Leica Biosystems, Buffalo Grove, IL) in a cold (1–4 °C) sucrose-based external solution. Slices were then immediately placed in normal artificial cerebrospinal fluid (194 mm sucrose, 20 mm NaCl, 4.4 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 1.2 mm NaH2PO4, 10 mm glucose, and 26 mm NaHCO3) saturated with 95% O2/5% CO2 and maintained at 30 °C and allowed to recover for at least 1 h. Individual slices were then placed in a holding chamber and continuously perfused with normal oxygenated artificial cerebrospinal fluid maintained at 30 °C at a rate of 2 ml/min. Neurons were visualized using infrared video microscopy (Olympus, Center Valley, PA). Recording electrodes (3–6 megohms) were pulled with a Flaming-Brown micropipette puller (Sutter Instruments, Novato, CA) using thin-walled borosilicate glass capillaries. Signals were acquired by a Multiclamp 700 B amplifier (Molecular Devices, Sunnyvale, CA), digitized at 10 kHz, and analyzed using Clampfit 10.2 software (Molecular Devices). Input resistance and access resistance were continuously monitored throughout all experiments, and those in which changes in access resistance exceeded 20% were excluded from all data analyses.
A previous report showed that activation of Gαq-coupled muscarinic acetylcholine 1 receptors (M1) in the PFC elicited a robust inward current in the PFC (36). Whole-cell slice electrophysiology was conducted to assess the ability of the Gαq inhibitory peptide to block Gαq-mediated M1 receptor signaling in the PFC in a voltage clamp using a potassium gluconate-based intracellular solution (135 mm K+ gluconate, 5 mm NaCl, 2 mm MgCl2, 10 mm HEPES, 0.6 mm EGTA, 4 mm Na2ATP, 0.4 mm Na2GTP (pH 7.3), 285–290 mosmol) containing spermine (0.1 mm) and the inactive or active peptide (10 μm). Cells were held at −70 mV. After achieving a stable 2-min baseline, we assessed the effects of the M1 receptor agonist carbachol (CCh, 10 μm, bath applied for 10 min) on the holding current in the presence of the active or inactive Gαq inhibitory peptide in the recording pipette. To confirm the effect observed with the inactive Gαq inhibitory peptide in the recording pipette was mediated by the M1 receptor, we assessed the ability of CCh to alter the holding current in the presence of the M1 receptor antagonist pirenzepine (2 μm, bath applied for 10 min).
The effects of CCh in the presence of the inactive or active peptide were analyzed by comparing the change in holding current during the final minute of drug application from the final minute of baseline. To eliminate the possibility that observed drug effects were due to cell rundown, the holding current during the final minute of washout was assessed, and any cells that did not recover baseline holding potential were removed from analyses. A maximum of two cells from each animal and one cell per slice were analyzed. t tests were used to analyze differences in the magnitude of the CCh response in the presence of the active versus inactive peptide as well as to analyze the difference in holding current in the presence of CCh relative to baseline for each peptide. Similar analyses were used to assess the effect of pirenzepine to block this effect.
Author Contributions
T. H. C. designed and conducted the majority of experiments. G. L. W. conducted the vesicle assay experiments (Fig. 3). E. G. L. G. conducted the neuronal depolarization experiments (Fig. 6). K. K. synthesized all peptides for this study and helped with the selection of specific mutations. Manuscript writing and editing were done by T. H. C., T. K. H., B. D. S., T. L. K., and J. S.
Acknowledgments
We acknowledge S. Hicks for help with experiments monitoring HTH peptides. We are thankful to M. Barrett for his assistance with cell and lipid vesicle experiments.
This work was supported by National Institutes of Health Grants R01-GM057391 (to J. S. and T. K. H), R01-AA019454 and U01-AA020911 (to T. L. K.), and AA022549 (to E. G. L. G.) and by a Melanoma Research Foundation grant (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein-coupled receptor
- PLC
- phospholipase C
- HTH
- helix-turn-helix
- Nle
- norleucine
- PFC
- prefrontal cortex
- CCh
- carbachol
- GEF
- guanine nucleotide exchange factor
- TAMRA
- carboxytetramethylrhodamine
- DOI
- 2,5-dimethoxy-4-iodoamphetamine
- PIP2
- phosphatidylinositol 4,5-bisphosphate.
References
- 1. Wettschureck N., and Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 [DOI] [PubMed] [Google Scholar]
- 2. Rozengurt E. (2007) Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 213, 589–602 [DOI] [PubMed] [Google Scholar]
- 3. Gainetdinov R. R., Premont R. T., Bohn L. M., Lefkowitz R. J., and Caron M. G. (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–144 [DOI] [PubMed] [Google Scholar]
- 4. Hubbard K. B., and Hepler J. R. (2006) Cell signalling diversity of the Gαq family of heterotrimeric G proteins. Cell Signal. 18, 135–150 [DOI] [PubMed] [Google Scholar]
- 5. Wall M. A., Coleman D. E., Lee E., Iñiguez-Lluhi J. A., Posner B. A., Gilman A. G., and Sprang S. R. (1995) The structure of the G protein heterotrimer Gi(α1β1γ2). Cell 83, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 6. Lambright D. G., Sondek J., Bohm A., Skiba N. P., Hamm H. E., and Sigler P. B. (1996) The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 379, 311–319 [DOI] [PubMed] [Google Scholar]
- 7. Adjobo-Hermans M. J., Goedhart J., van Weeren L., Nijmeijer S., Manders E. M., Offermanns S., and Gadella T. W. Jr. (2011) Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol. 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu J. Z., and Rasenick M. M. (2002) Real-time visualization of a fluorescent Gαs: dissociation of the activated G protein from plasma membrane. Mol. Pharmacol. 61, 352–359 [DOI] [PubMed] [Google Scholar]
- 9. Milligan G., and Kostenis E. (2006) Heterotrimeric G-proteins: a short history. Br. J. Pharmacol. 147, S46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smrcka A. V. (2013) Molecular targeting of Gα and Gβγ subunits: a potential approach for cancer therapeutics. Trends Pharmacol. Sci. 34, 290–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prévost G. P., Lonchampt M. O., Holbeck S., Attoub S., Zaharevitz D., Alley M., Wright J., Brezak M. C., Coulomb H., Savola A., Huchet M., Chaumeron S., Nguyen Q. D., Forgez P., Bruyneel E., et al. (2006) Anticancer activity of BIM-46174, a new inhibitor of the heterotrimeric Gα/Gβγ protein complex. Cancer Res. 66, 9227–9234 [DOI] [PubMed] [Google Scholar]
- 12. Ayoub M. A., Damian M., Gespach C., Ferrandis E., Lavergne O., De Wever O., Banères J. L., Pin J. P., and Prévost G. P. (2009) Inhibition of heterotrimeric G protein signaling by a small molecule acting on Gα subunit. J. Biol. Chem. 284, 29136–29145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmitz A. L., Schrage R., Gaffal E., Charpentier T. H., Wiest J., Hiltensperger G., Morschel J., Hennen S., Häußler D., Horn V., Wenzel D., Grundmann M., Büllesbach K. M., Schröder R., Brewitz H. H., et al. (2014) A cell-permeable inhibitor to trap Gα proteins in the empty pocket conformation. Chem. Biol. 21, 890–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taniguchi M., Nagai K., Arao N., Kawasaki T., Saito T., Moritani Y., Takasaki J., Hayashi K., Fujita S., Suzuki K., and Tsukamoto S. (2003) YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J. Antibiot. 56, 358–363 [DOI] [PubMed] [Google Scholar]
- 15. Zaima K., Deguchi J., Matsuno Y., Kaneda T., Hirasawa Y., and Morita H. (2013) Vasorelaxant effect of FR900359 from Ardisia crenata on rat aortic artery. J. Nat. Med. 67, 196–201 [DOI] [PubMed] [Google Scholar]
- 16. Miyamae A., Fujioka M., Koda S., and Morimoto Y. (1989) Structural studies of FR900359, a novel cyclic desipeptide from Ardisia crenata Sims (Myrsinaceae). J. Chem. Soc. Perkin Trans. 873–878 [Google Scholar]
- 17. Taniguchi M., Suzumura K., Nagai K., Kawasaki T., Takasaki J., Sekiguchi M., Moritani Y., Saito T., Hayashi K., Fujita S., Tsukamoto S., and Suzuki K. (2004) YM-254890 analogues, novel cyclic depsipeptides with Gαq/11 inhibitory activity from Chromobacterium sp. QS3666. Bioorg. Med. Chem. 12, 3125–3133 [DOI] [PubMed] [Google Scholar]
- 18. Takasaki J., Saito T., Taniguchi M., Kawasaki T., Moritani Y., Hayashi K., and Kobori M. (2004) A novel Gαq/11-selective inhibitor. J. Biol. Chem. 279, 47438–47445 [DOI] [PubMed] [Google Scholar]
- 19. Nishimura A., Kitano K., Takasaki J., Taniguchi M., Mizuno N., Tago K., Hakoshima T., and Itoh H. (2010) Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc. Natl. Acad. Sci. U.S.A. 107, 13666–13671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnston C. A., Willard F. S., Ramer J. K., Blaesius R., Roques C. N., and Siderovski D. P. (2008) State-selective binding peptides for heterotrimeric G-protein subunits: novel tools for investigating G-protein signaling dynamics. Comb. Chem. High Throughput Screen. 11, 370–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kimple R. J., De Vries L., Tronchère H., Behe C. I., Morris R. A., Gist Farquhar M., and Siderovski D. P. (2001) RGS12 and RGS14 GoLoco motifs are Gαi interaction sites with guanine nucleotide dissociation inhibitor activity. J. Biol. Chem. 276, 29275–29281 [DOI] [PubMed] [Google Scholar]
- 22. Webb C. K., McCudden C. R., Willard F. S., Kimple R. J., Siderovski D. P., and Oxford G. S. (2005) D2 dopamine receptor activation of potassium channels is selectively decoupled by Gα-specific GoLoco motif peptides. J. Neurochem. 92, 1408–1418 [DOI] [PubMed] [Google Scholar]
- 23. Johnston C. A., Ramer J. K., Blaesius R., Fredericks Z., Watts V. J., and Siderovski D. P. (2005) A bifunctional Gαi/Gαs modulatory peptide that attenuates adenylyl cyclase activity. FEBS Lett. 579, 5746–5750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnston C. A., Lobanova E. S., Shavkunov A. S., Low J., Ramer J. K., Blaesius R., Fredericks Z., Willard F. S., Kuhlman B., Arshavsky V. Y., and Siderovski D. P. (2006) Minimal determinants for binding activated Gα from the structure of a Gαi1-peptide dimer. Biochemistry 45, 11390–11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnston C. A., Willard F. S., Jezyk M. R., Fredericks Z., Bodor E. T., Jones M. B., Blaesius R., Watts V. J., Harden T. K., Sondek J., Ramer J. K., and Siderovski D. P. (2005) Structure of Gαi1 bound to a GDP-selective peptide provides insight into guanine nucleotide exchange. Structure 13, 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sprang S. R. (1997) G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66, 639–678 [DOI] [PubMed] [Google Scholar]
- 27. Harden T. K., Waldo G. L., Hicks S. N., and Sondek J. (2011) Mechanism of activation and inactivation of Gq/phospholipase C-β signaling nodes. Chem. Rev. 111, 6120–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aittaleb M., Boguth C. A., and Tesmer J. J. (2010) Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rojas R. J., Yohe M. E., Gershburg S., Kawano T., Kozasa T., and Sondek J. (2007) Gαq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J. Biol. Chem. 282, 29201–29210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Waldo G. L., Ricks T. K., Hicks S. N., Cheever M. L., Kawano T., Tsuboi K., Wang X., Montell C., Kozasa T., Sondek J., and Harden T. K. (2010) Kinetic scaffolding mediated by a phospholipase C-β and Gq signaling complex. Science 330, 974–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lutz S., Shankaranarayanan A., Coco C., Ridilla M., Nance M. R., Vettel C., Baltus D., Evelyn C. R., Neubig R. R., Wieland T., and Tesmer J. J. (2007) Structure of Gαq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923–1927 [DOI] [PubMed] [Google Scholar]
- 32. Jhon D. Y., Lee H. H., Park D., Lee C. W., Lee K. H., Yoo O. J., and Rhee S. G. (1993) Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-β3. J. Biol. Chem. 268, 6654–6661 [PubMed] [Google Scholar]
- 33. Smrcka A. V., and Sternweis P. C. (1993) Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C-β by G protein α and βγ subunits. J. Biol. Chem. 268, 9667–9674 [PubMed] [Google Scholar]
- 34. Lyon A. M., Dutta S., Boguth C. A., Skiniotis G., and Tesmer J. J. (2013) Full-length Gαq-phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat. Struct. Mol. Biol. 20, 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lyon A. M., Tesmer V. M., Dhamsania V. D., Thal D. M., Gutierrez J., Chowdhury S., Suddala K. C., Northup J. K., and Tesmer J. J. (2011) An autoinhibitory helix in the C-terminal region of phospholipase C-β mediates Gαq activation. Nat. Struct. Mol. Biol. 18, 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carr D. B., and Surmeier D. J. (2007) M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J. Neurophysiol. 97, 3432–3438 [DOI] [PubMed] [Google Scholar]
- 37. Shankaranarayanan A., Boguth C. A., Lutz S., Vettel C., Uhlemann F., Aittaleb M., Wieland T., and Tesmer J. J. (2010) Gαq allosterically activates and relieves autoinhibition of p63RhoGEF. Cell Signal. 22, 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nikolovska-Coleska Z., Wang R., Fang X., Pan H., Tomita Y., Li P., Roller P. P., Krajewski K., Saito N. G., Stuckey J. A., and Wang S. (2004) Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 332, 261–273 [DOI] [PubMed] [Google Scholar]
- 39. Rojas R. J., Kimple R. J., Rossman K. L., Siderovski D. P., and Sondek J. (2003) Established and emerging fluorescence-based assays for G-protein function: Ras-superfamily GTPases. Comb. Chem. High Throughput Screen. 6, 409–418 [DOI] [PubMed] [Google Scholar]
- 40. Mozhui K., Karlsson R. M., Kash T. L., Ihne J., Norcross M., Patel S., Farrell M. R., Hill E. E., Graybeal C., Martin K. P., Camp M., Fitzgerald P. J., Ciobanu D. C., Sprengel R., Mishina M., et al. (2010) Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. J. Neurosci. 30, 5357–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li C., Pleil K. E., Stamatakis A. M., Busan S., Vong L., Lowell B. B., Stuber G. D., and Kash T. L. (2012) Presynaptic inhibition of γ-aminobutyric acid release in the bed nucleus of the stria terminalis by κ opioid receptor signaling. Biol. Psychiatry 71, 725–732 [DOI] [PMC free article] [PubMed] [Google Scholar]