

Abstract
FOXO3a, a member of the forkhead homeobox type O (FOXO) family of transcriptional factors, regulates cell survival in response to DNA damage, caloric restriction, and oxidative stress. The von Hippel-Lindau (VHL) tumor suppressor gene encodes a component of the E3 ubiquitin ligase complex that mediates hypoxia-inducible factor α degradation under aerobic conditions, thus acting as one of the key regulators of hypoxia signaling. However, whether FOXO3a impacts cellular hypoxia stress remains unknown. Here we show that FOXO3a directly binds to the VHL promoter and up-regulates VHL expression. Using a zebrafish model, we confirmed the up-regulation of vhl by foxo3b, an ortholog of mammalian FOXO3a. Furthermore, by employing the clustered regularly interspaced short palindromic repeats (CRISPR)-associated RNA-guided endonuclease Cas9 (CRISPR/Cas9) technology, we deleted foxo3b in zebrafish and determined that expression of hypoxia-inducible genes was affected under hypoxia. Moreover, foxo3b-null zebrafish exhibited impaired acute hypoxic tolerance, resulting in death. In conclusion, our findings suggest that, by modulating hypoxia-inducible factor activity via up-regulation of VHL, FOXO3a (foxo3b) plays an important role in survival in response to hypoxic stress.
Keywords: FOXO, hypoxia, hypoxia-inducible factor (HIF), stress, transcription regulation, zebrafish, pVHL
Introduction
FOXO3a (also known as FKHRL1), together with FOXO1 (also known as FKHR), FOXO4 (also known as AFX1), and FOXO6 comprise the conserved forkhead box O (FOXO)4 gene family, which is a subclass of the forkhead family of transcription factors. The FOXO family is an evolutionarily conserved group of proteins and of vital importance in the control of cell and organism growth, development, metabolism, and longevity (1). FOXO proteins affect various cellular processes, including cell cycle regulation, cell survival, proliferation, and apoptosis (1). In their functional capacity as transcription factors, FOXO proteins bind to the consensus sequence (5′-TTGTTTAC-3′) and induce expression of their downstream targets. Several upstream signaling pathways modulate FOXO activity via nuclear-cytoplasmic shuttling and posttranslational modifications of FOXO proteins (2–4).
Multiple lines of evidence have shown that FOXO transcription factors are key players in stress signaling (1, 5, 6). The critical role of FOXO transcription factors in oxidative stress has been well defined (7, 8). For example, FOXO3a protects quiescent cells from oxidative stress by inducing expression of manganese superoxide dismutase (Mn-SOD) (9). Under oxidative stress, the function of FOXO3a is regulated by SIRT1 and SIRT2 deacetylation (10, 11), FOXO3a can regulate reactive oxygen metabolism by inhibiting mitochondrial gene expression (12), and association of β-catenin with FOXO4 or FOXO3a enhances FOXO transcriptional activity (13). In addition, redox-dependent formation of cysteine-thiol disulfide modulates FOXO activity (14, 15). It is also evident that the role of FOXO family members in oxidative stress signaling might account for their function in aging and cancer (16, 17).
The von Hippel-Lindau (VHL) gene is a classic tumor suppressor, and its inactivation is linked to the development of clear-cell renal cell carcinomas, hemangioblastomas, pheochromocytomas, and tumors in other organs (18). The best characterized function of pVHL is its ability to function as a substrate recognition subunit of a multiprotein E3 ubiquitin ligase complex that targets proline hydroxylated hypoxia-induced factor (HIF) 1/2α for proteasomal degradation under normoxic conditions (18, 19). Therefore, pVHL acts as a key regulator in hypoxia signaling.
Despite cellular stress induced by reactive oxygen, cellular oxygen is a fundamental factor that controls developmental processes as well as normal tissue homeostasis. Oxygen is required for metabolism, ATP production, and cell survival (20, 21). To maintain oxygen homeostasis, organisms use the hypoxia signaling pathway for adapting to low-oxygen conditions (22). The hypoxia-inducible factors HIF-1α and HIF-2α coordinate the cellular response to hypoxic conditions by inducing gene expression (23). The prolyl hydroxylase (PHD) gene family and the pVHL E3 ligase complex control HIF activity through oxygen-dependent hydroxylation and subsequent proteasomal degradation (24).
Research has shown that mitochondrially derived reactive oxygen species (ROS) can activate hypoxia signaling (25). Because FOXO3a, as well as FOXO4, can up-regulate Mn-SOD and other antioxidant enzymes (9, 11), FOXO transcription factors can inhibit HIF-1 activation by reducing the levels of mitochondrially derived ROS, suggesting a connection between FOXO transcription factors and hypoxia signaling. Moreover, FOXO3a has been shown to up-regulate CITED2 for inhibiting HIF1-induced apoptosis in response to hypoxic stress (2) and mediates the suppressor role of the tumour-suppressor phosphatase with tensin homology (PTEN) on p300-dependent HIF-1 activity (10). In addition, FOXO3a has been shown to promote metabolic adaptation to hypoxia by antagonizing Myc function (18). Although these observations preliminarily outline the role of FOXO3a in hypoxia signaling, the subtle process and the underlying mechanisms are still largely unknown.
In zebrafish, there are two orthologs of the mammalian FOXO3 gene, foxo3a and foxo3b. Zebrafish foxo3b was initially identified and named as zFKHR/foxo5, whose protein shared 55% identity with that of human FOXO3a (26). The phylogenetic analysis of vertebrate FOXO protein sequences showed that, compared with foxo3a, foxo3b was much closer to mammalian FOXO3a (26).
To elucidate the function of FOXO3a in hypoxia signaling, we examined the regulation of VHL by FOXO3a. In this study, we found that VHL was a direct downstream target of FOXO3a. Using a zebrafish model, we not only corroborate that vhl is regulated by foxo3b but also reveal that foxo3b could modulate hypoxia signaling through regulating vhl.
Results
VHL Is a Direct Downstream Target of FOXO3a
To investigate whether VHL was transcriptionally regulated by FOXO3a, promoter assays were undertaken. As shown in Fig. 1A, luciferase reporters containing the full-length or truncated portions of the human VHL promoter (−2101 to +66, −989 to +66, −599 to +66, −499 to +66, and −371 to +66; the transcription start site is designated +1) were transiently transfected into HEK293T cells together with a Myc empty vector or Myc tagged-FOXO3a expression vector. Overexpression of FOXO3a induced activity of promoter reporters including −2101 to +66, −989 to +66, and −599 to +66 but not of promoter reporters including −499 to +66 and −371 to +66 (Fig. 1B). Notably, among three FOXO3a-inducible reporters (−2101 to +66, −989 to +66, and −599 to +66), the reporter −599 to +66 had the highest activity, suggesting that the FOXO DNA binding element (DBE) might locate in the region between −599 to +66. After carefully searching, we identified one potential FOXO DBE at position −567 to −561 (TTGTTAC) in the VHL promoter (9) (Fig. 1C). A single G-to-C substitution in the putative FOXO DBE completely abolished FOXO3a- and FOXO3a-3A-mediated up-regulation of VHL promoter reporter activity (Fig. 1D) (FOXO3a-3A is an active form of FOXO3a with a T32A/S253A/S315A triple mutation (2)).
FIGURE 1.

VHL is a direct downstream target of FOXO3a. A, schematic of VHL promoter constructs. B, promoter reporter activities were measured after transient transfection with a Myc empty vector or Myc-FOXO3a expression vector in HEK293T cells. C, one suboptimal FOXO DBE, TTGTTAC (bold and underlined), was localized to the −567 to −561 region of the VHL promoter. D, compared with the wild-type VHL promoter reporter (−599/+66), the mutant VHL promoter reporter (single G-to-C substitution in the putative FOXO DBE) was not up-regulated by overexpression of FOXO3a and FOXO3a-3A in HEK 293T cells. E, expression of FOXO3a-3A-ER was confirmed by Western blotting assays. F, the mutant VHL promoter reporter (single G-to-C substitution in the putative FOXO DBE) was not up-regulated by FOXO3a activation. HEK 293T cells that constitutively express FOXO3a-A3-ER fusion protein were transfected with the wild-type or mutant VHL promoter reporters in the absence or presence of 4-HT (500 nm).
To further confirm the above observations, we established an inducible FOXO3a-expressing human embryonic kidney cell line (HEK293T) using a lentivirus expressing a conditionally active FOXO3a-A3-ER fusion protein that consisted of a T32A/S253A/S315A triple mutant fused with the ligand-binding domain of the estrogen receptor (ER) (2, 9). Expression of FOXO3a-A3-ER fusion protein in HEK 293T cells was confirmed by Western blotting assays (Fig. 1E). Treatment of the cells with 4-hydroxy-tamoxifen (4-HT), a modified ligand for the ER, for 16–20 h (27) significantly increased luciferase activity of the wild-type VHL promoter reporter (p < 0.01) but not of the mutated VHL promoter reporter (Fig. 1F), further validating the induction and binding region of FOXO3a on the VHL promoter.
To determine whether FOXO3a can directly bind to the VHL promoter, we designed a pair of primers that could amplify a fragment encompassing the potential FOXO DBE and performed ChIP using anti-FOXO3a antibody. As shown in Fig. 2, A and B, in vivo binding of FOXO3a to the VHL promoter region encompassing the FOXO DBE was observed. Further semiquantitative RT-PCR assays validated this binding ability of FOXO3a (Fig. 2C).
FIGURE 2.

FOXO3a directly regulates VHL expression. A–C, chromatin immunoprecipitation analysis confirmed that FOXO3a directly interacted with the VHL promoter region harboring the FOXO DBE (A and B), which was confirmed by semiquantitative real-time RT-PCR assays (C). D, semiquantitative real-time RT-PCR assays revealed that activation of FOXO3a with 500 nm 4-HT caused VHL mRNA to be increased. E, induction of pVHL by FOXO3a was confirmed by Western blotting assays.
Subsequently, we determined the induction of VHL by FOXO3a in the inducible HEK293T cell line. After treatment of cells with 4-HT for 24 h, a 5-fold increase in VHL mRNA was observed (Fig. 2D). The induction of pVHL protein by activating FOXO3a was further verified (Fig. 2E).
To determine the effect of endogenous FOXO3a on VHL expression, we used two sets of FOXO3a shRNA to knock down endogenous FOXO3a via lentivirus infection. First, we confirmed the efficiency of shRNA-mediated FOXO3a knockdown via semiquantitative RT-PCR assays in HEK293T and H1299 cells (Fig. 3, A and C). By contrast, knockdown of FOXO3a reduced the VHL mRNA level in HEK293T and H1299 cells (Fig. 3, B and D). Moreover, reduction of the pVHL protein level and enhancement of the HIF-1α protein level when FOXO3a was knocked down was validated (Fig. 3, E and F). Taken together, these results suggest that VHL might be a direct downstream target of FOXO3a.
FIGURE 3.

Knockdown of FOXO3a causes VHL expression to be reduced. A, knockdown of FOXO3a by two sets of FOXO3a shRNA in HEK293T cells was confirmed by semiquantitative real-time RT-PCR assays. Luciferase shRNA was used as a control. B, knockdown of FOXO3a in HEK293T caused VHL expression to be reduced. C, knockdown of FOXO3a by two sets of FOXO3a shRNA in H1299 cells was confirmed by semiquantitative real-time RT-PCR assays. Luciferase shRNA was used as a control. D, knockdown of FOXO3a in H1299 cells caused VHL expression to be reduced. E, knockdown of FOXO3a resulted in reduction of endogenous pVHL and induction of endogenous HIF-1α in HEK293T cells, as confirmed by Western blotting assays. F, knockdown of FOXO3a resulted in reduction of endogenous pVHL and induction of endogenous HIF-1α in H1299 cells, as confirmed by Western blotting assays.
FOXO3a Suppresses Hypoxia-inducible Gene Expression via VHL
To understand the biological consequence of FOXO3a up-regulating VHL, we examined whether FOXO3a has impacts on the function of VHL. To date, the well defined function of VHL is to modulate hypoxia-inducible gene expression via mediating oxygen-dependent HIF-α proteasomal degradation (19). Therefore, we examined the effect of FOXO3a on hypoxia-inducible gene expression. Overexpression of FOXO3acould inhibit expression of typical hypoxia-inducible genes, SLC2A1 and LDHA (28), in HEK293T cells under normoxia (Fig. 4, A and B, left columns).
FIGURE 4.

FOXO3a suppresses hypoxia-inducible gene expression via VHL. A, overexpression of FOXO3a caused SLC2A1 expression to be reduced in HEK293T cells under normoxia and hypoxia (2% O2). B, overexpression of FOXO3a caused LDHA expression to be reduced in HEK293T cells under normoxia and hypoxia (2% O2). C, knockdown of FOXO3a caused SLC2A1 expression to be increased in H1299 cells under normoxia and hypoxia (2% O2). D, knockdown of FOXO3a caused LDHA expression to be increased in H1299 cells under normoxia and hypoxia (2% O2). E, knockdown of endogenous VHL by transient transfection with two sets of VHL shRNA was confirmed by Western blotting assays. F, when VHL was knocked down, the suppressive effect of FOXO3a on SLC2A1 expression was abrogated under normoxia. G, knockdown of VHL abolished the suppressive effect of FOXO3a on SLC2A1 expression under hypoxia.
However, we noticed that, under normoxia, the base level of hypoxia-inducible genes, including SLC2A1 and LDHA, was very low because of lack of HIF-α. Therefore, it was difficult to evaluate the effect of FOXO3a on hypoxia-inducible gene expression because of the low values detected. Of note, in some cases, it appeared that PHD still had a partial effect on HIF-α function under certain hypoxic conditions (29, 30); thus, VHL could affect HIF-α function as well under these kinds of hypoxic conditions. Because HIF-α protein was enhanced, resulting in a higher expression level of hypoxia-inducible genes, it became much easier to evaluate the effect of these gene expressions (29, 31), particularly for evaluating the suppression role. To get a clear picture of the role of FOXO3a in hypoxia-inducible gene expression, we further examined the effect of FOXO3a under hypoxia. Similar to the tendency detected under normoxia, overexpression of FOXO3a also inhibited expression of SLC2A1 and LDHA under hypoxia (Fig. 4, A and B, right columns). In addition, the inhibitory effect of FOXO3a on hypoxia-inducible gene expression was more obvious under hypoxia compared with normoxia, which might result from a higher base level of hypoxia-inducible genes under this kind of hypoxic condition.
By contrast, knockdown of FOXO3a in H1299 cells resulted in up-regulation of SLC2A1 and LDHA under normoxia and hypoxia (Fig. 4, C and D). To validate whether the suppression of FOXO3a on expression of hypoxia-inducible genes is mediated by VHL, we knocked down VHL by shRNA (32) and then conducted further assays (Fig. 4E). Fig. 4F shows that, when VHL was knocked down, the inhibitory role of FOXO3a on SLC2A1 expression was abrogated under normoxia. Moreover, knockdown of VHL also abolished the inhibitory role of FOXO3a on SLC2A1 expression under hypoxia (Fig. 4G), further indicating that, under the hypoxic conditions of our experiments in this study, VHL still had some effects on HIF-α function. These data suggest that FOXO3a might modulate hypoxia signaling via regulation of VHL.
Zebrafish vhl Is a Downstream Target of foxo3b
To further determine the regulation of VHL by FOXO3a and the underling biological consequences in vivo, we took advantage of the zebrafish model. Initially, we examined the expression patterns of both foxo3b mRNA and vhl mRNA in different organs or tissues of adult zebrafish (3 months old) via semiquantitative RT-PCR assays. Foxo3b was highly expressed in muscle and then in the brain, heart, and liver (Fig. 5A). Interestingly, vhl was also highly expressed in muscle and then in the brain, heart, and liver (Fig. 5B). The similar expression patterns in foxo3b and vhl suggested that these two genes might be functionally correlated.
FIGURE 5.

The expression pattern of vhl in zebrafish and the effect of foxo3b on vhl promoter activity. A and B, semiquantitative real-time RT-PCR assays demonstrated the expression pattern of foxo3b (A) and vhl (B) in different tissues of adult zebrafish. C, zebrafish vhl promoter activity was induced after injection of the foxo3b expression vector in embryos. D, injection of a foxo3b-3A mutant induced zebrafish vhl promoter activity more dramatically (third column versus second column). The vhl promoter was not activated after injection of a foxo3b dominant-negative mutant (dn-foxo3b, p = 0.089).
Subsequently, we conducted promoter assays in embryos to determine whether ectopic expression of foxo3b could activate the vhl promoter. Injection of an HA-foxo3b expression vector in zebrafish embryos induced zebrafish vhl promoter reporter activity (Fig. 5C). Moreover, the T30A/S223A/S289A foxo3b-3A mutant stimulated vhl promoter reporter activity to a greater extent than that observed for wild-type foxo3b, which was similar to a mammalian FOXO3a-3A mutant (Fig. 5D) (2). By contrast, vhl promoter reporter activity was not induced by the dominant-negative foxo3b (1–277 amino acids) (Fig. 5D).
Next, we employed whole-mount in situ hybridization (WISH) assays for determining the regulation of vhl by foxo3b. The expression pattern of foxo3b by WISH has been reported previously (26). The expression patterns of vhl by WISH during zebrafish embryogenesis are shown in Fig. 6. During early embryogenesis, expression of vhl was ubiquitous. However, in the later stages of embryogenesis, vhl was expressed specifically in the otic vesicle, pectoral fin, inner ear, swimming bladder, kidney, and liver (Fig. 6). Ectopic expression of the zebrafish foxo3b by mRNA injection increased vhl expression in a dose-dependent manner (Fig. 7A). The induction of vhl by ectopic expression of foxo3b was further validated by semiquantitative RT-PCR assays (Fig. 7B). By contrast, knockdown of foxo3b using morpholino oligonucleotides, validated previously for specificity and efficiency (26), reduced vhl expression (Fig. 7C). Similarly, injection of dominant-negative foxo3b mRNA reduced vhl expression (Fig. 7D).
FIGURE 6.

The expression pattern of vhl during zebrafish embryogenesis as detected by WISH. A–F, vhl was ubiquitously expressed before the 10-somite stage. G–I, 25 hpf), vhl was highly expressed in the head and anterior mesoderm. J–M, 48 hpf, vhl was highly expressed in the otic vesicle, pectoral fin, and posterior mesoderm. N–Q, 3.5 dpf, vhl was highly expressed in the swimming bladder, inner ear, and kidney. R–T, 4.5 dpf, vhl was highly expressed in the swimming bladder, liver, inner ear, and kidney.
FIGURE 7.

Zebrafish foxo3b enhances vhl expression. A, ectopic expression of foxo3b by mRNA injection induced vhl expression in embryos, as detected by WISH. GFP mRNA was used as a control. B, ectopic expression of foxo3b by mRNA injection enhanced vhl expression in embryos, as revealed by semiquantitative real-time RT-PCR assays. GFP mRNA was used as a control. C, knockdown of foxo3b in embryos by morpholino oligonucleotide injection reduced vhl expression. a, standard morpholino oligonucleotide control (STD-MO, 8 ng/individual embryo). b, foxo3b translation-blocking (ATG code) morpholino oligonucleotide (foxo3b-ATG-MO, 8 ng/individual embryo). c, foxo3b splicing-blocking morpholino oligonucleotide (foxo3b-sp-MO, 8 ng/individual embryo). D, injection of dominant-negative foxo3b mRNA (dn-foxo3b) in zebrafish embryos reduced vhl expression (b) compared with those injected with GFP mRNA (a).
Targeted Disruption of foxo3b in Zebrafish Enhances Hypoxia-inducible Gene Expression and Impairs Zebrafish Hypoxia Tolerance
To further determine the regulation of vhl by foxo3b and the function of foxo3b in response to hypoxia, we deleted foxo3b in zebrafish via CRISPR/Cas9 technology. To disrupt the foxo3b gene in zebrafish, we designed an sgRNA targeting a region in exon 2 of foxo3b (Fig. 8A). After microinjecting the synthesized sgRNA and Cas9 mRNA into one-cell-stage embryos, we initially employed a heteroduplex mobility assay (HMA) to examine the efficiency of sgRNA/Cas9-mediated foxo3b disruption in the F1 generation (Fig. 8B) and then conducted a sequencing confirmation in the F1 generation. After screening, we obtained one mutant in the foxo3b gene (hereafter designated MT) (Fig. 8C). We crossed foxo3b+/−×foxo3b+/− and obtained offspring with the foxo3b+/+,foxo3b+/−, and foxo3b−/− genetic background at Mendel's ratio (1:2:1). From the embryonic stages to adulthood, foxo3b−/− homozygosity was indistinguishable from wild-type siblings. Overall, no obvious phenotypes were observed in foxo3b−/− zebrafish under normal conditions. Semiquantitative RT-PCR analysis indicated that foxo3b mRNA was largely reduced in foxo3b-null zebrafish under either normoxia or hypoxia (Fig. 8D), which suggested that foxo3b was effectively disrupted. Similarly, vhl mRNA was also reduced in foxo3b-null zebrafish under either normoxia or hypoxia, but the reduction rate was not as dramatic as that of foxo3b (Fig. 8E). These data suggested that vhl was transactivated by foxo3b in vivo and that other FOXO family members in zebrafish might have a partially redundant function for inducing vhl expression.
FIGURE 8.

Generation of foxo3b−/− zebrafish via CRISPR/Cas9 technology. A, targeting strategy for generating mutations in exon 2 of foxo3b. The sequence difference in foxo3b between the mutants and their wild-type siblings is indicated. B, verification of zebrafish foxo3b disruption by HMA. Genomic DNA was prepared from the mutants and their wild-type siblings. C, the predicted protein products of foxo3b in the mutants and their wild-type siblings. TAD, transactivation domain. D and E, expression of foxo3b (D) or vhl (E) in wild-type (foxo3b+/+) and foxo3b-null(foxo3b−/−) zebrafish embryos under normoxia and hypoxia.
Subsequently, we examined whether some common hypoxia-inducible genes were affected following disruption of foxo3b. As shown in Fig. 9, A–D, expression of vegf, pou5f1, pai1, and il11a was enhanced in foxo3b-null zebrafish under hypoxia (28, 33). These data suggested that foxo3b affected the expression of hypoxia-inducible genes. Consistently, when the embryos were treated with DMOG, a hydroxylase inhibitor (34), expression of vegf and pou5f1 was also enhanced in foxo3b-null zebrafish (Fig. 9, E and F). Taken together, these data indicated that disruption of foxo3b in zebrafish enhanced the expression of hypoxia-inducible genes and that foxo3b could modulate hypoxia signaling.
FIGURE 9.

Zebrafish foxo3b suppresses hypoxia-induced gene expression. A, foxo3b-null embryos (foxo3b−/−) exhibit higher vegf expression compared with their wild-type siblings (foxo3b +/+) under normoxia and hypoxia. B, foxo3b-null embryos (foxo3b−/−) exhibit higher pou5f1 expression compared with their wild-type siblings (foxo3b +/+) under normoxia and hypoxia. C, foxo3b-null embryos (foxo3b−/−) exhibit higher pai1 expression compared with their wild-type siblings (foxo3b +/+) under normoxia and hypoxia. D, foxo3b-null embryos (foxo3b−/−) exhibit higher il11a expression compared with their wild-type siblings (foxo3b +/+) under normoxia and hypoxia. E, foxo3b-null embryos (foxo3b−/−) exhibit higher vegf expression compared with their wild-type siblings (foxo3b +/+) with or without DMOG (100 μm) treatment. F, foxo3b-null embryos (foxo3b−/−) exhibit higher pai1 expression compared with their wild-type siblings (foxo3b +/+) with or without DMOG (100 μm) treatment.
To determine whether the effect of foxo3b on hypoxia-inducible gene expression was indeed mediated by the regulation of vhl, we took advantage of vhl-null zebrafish embryos (35). In wild-type sibling embryos, injection with foxo3b mRNA inhibited the expression of four hypoxia-inducible genes (vegf, ldha, cited2, and il11a) significantly compared with those injected with GFP mRNA (Fig. 10, A–D). By contrast, in vhl-null embryos, injection with foxo3b mRNA had no obvious effect on expression of the four hypoxia-inducible genes (vegf, ldha, cited2, and il11a) compared with those injected with GFP mRNA (Fig. 10, A–D). Expression of injected foxo3b and GFP mRNA was further confirmed by Western blotting assays (Fig. 10E) and fluorescence stereomicroscope, respectively (Fig. 10F). These data suggested that the suppressive role of foxo3b in expression of hypoxia-inducible genes was mediated by up-regulation of vhl.
FIGURE 10.

Zebrafish foxo3b suppresses hypoxia-induced gene expression through vhl. A, ectopic expression of foxo3b by mRNA injection suppressed vegf expression in wild-type sibling embryos (vhl+/+) but not in vhl-null embryos (vhl−/−), as revealed by semiquantitative real-time RT-PCR assays. GFP mRNA injections were used as controls. B, ectopic expression of foxo3b by mRNA injection suppressed ldha expression in the wild-type sibling embryos (vhl+/+) but not in vhl-null embryos (vhl−/−), as revealed by semiquantitative real-time RT-PCR assays. GFP mRNA injections were used as controls. C, ectopic expression of foxo3b by mRNA injection suppressed cited2 expression in wild-type sibling embryos (vhl+/+) but not in vhl-null embryos (vhl−/−), as revealed by semiquantitative real-time RT-PCR assays. GFP mRNA injections were used as controls. D, ectopic expression of foxo3b by mRNA injection suppressed il11a expression in wild-type sibling embryos (vhl+/+) but not in vhl-null embryos (vhl−/−), as revealed by semiquantitative real-time RT-PCR assays. GFP mRNA injections were used as controls. E, expression of myc-tagged foxo3b mRNA injected into embryos was confirmed by Western blotting assays. F, expression of control GFP mRNA injected into embryos was examined under a fluorescence stereomicroscope (Leica M205).
To determine the biological consequences of foxo3b modulating hypoxia signaling, we compared the acute hypoxia tolerance in foxo3b-null zebrafish and their wild-type siblings. Under 2% oxygen environmental conditions, foxo3b-null fry (15 dpf) died earlier than their wild-type siblings (Fig. 11). Similarly, with adult zebrafish, under 10% oxygen environmental conditions, foxo3b-null adults (3 months old) also died earlier than their wild-type siblings (supplemental Movies 1 and 2). These data suggested that foxo3b potentially affected zebrafish survival via modulation of hypoxia signaling.
FIGURE 11.
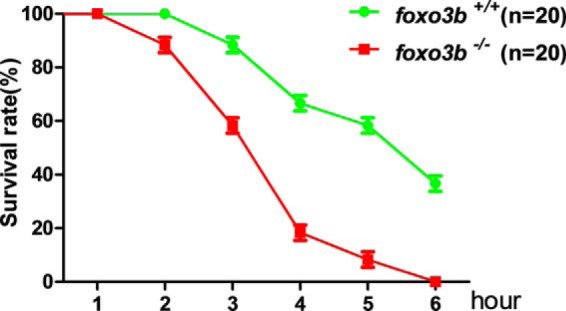
Foxo3b is required for hypoxia tolerance in zebrafish. Three groups of foxo3b-null zebrafish fry (15 dpf, 20/group) and their wild-type siblings (15 dpf, 20/group) were placed in the hypoxia chamber (Ruskinn INVIVO2400). The oxygen concentration was adjusted to 2% O2 ahead of time. The dead fry counted once an hour until the death of the last fry with one genotype (foxo3b−/−).
Discussion
Low oxygen tensions activate hypoxia signaling by promoting the protein stability of HIF-1α and HIF-2α (19, 21). As the key transcriptional regulators of hypoxia signaling, the regulation of HIF-1α and HIF-2α is well documented. Under normoxia, PHD1–3 use oxygen as a substrate to hydroxylate key proline residues of HIF-1α and HIF-2α, resulting in subsequent proteasomal degradation by the pVHL E3 ubiquitin ligase complex (36–38). Under hypoxia, PHD activity is inhibited, resulting in HIF-1α and HIF-2α stabilization and translocation to the nucleus for activating downstream genes involved in systematic and cellular adaptation to hypoxia (39). Therefore, PHDs (PHD1–3) and pVHL have been recognized as the major regulators for modulating hypoxia signaling through regulating the protein stability of HIF-1α and HIF-2α. In fact, it is evident that some factors could affect hypoxia signaling via regulation of PHDs and pVHL (29, 31, 40, 41, 43). Although FOXO3a has been reported to affect hypoxia signaling, it has not been shown to directly modulate the major proteins involved in hypoxia signaling, including PHDs and pVHL. Here we show that FOXO3a (foxo3b in zebrafish) transactivates pVHL (vhl in zebrafish) expression, resulting in suppression of hypoxia signaling, which provides additional evidence for supporting the role of FOXO3a in hypoxia signaling. Intriguingly, FOXO3a, together with other FOXO family members, exhibits tumor-suppressive functions (17, 44). It is noteworthy that pVHL is a well known classic tumor suppressor (45). Thus, FOXO3a, and other related FOXO proteins, might function as tumor suppressors by up-regulating pVHL expression. Confirmation of whether pVHL is transactivated by other FOXO family genes will further expand our knowledge about the function of the FOXO family in tumor suppression.
As a component of an E3 ubiquitin ligase complex, the targets of pVHL other than HIF-α have been identified (35, 46–48). Notably, the regulation of pVHL at the protein level through protein-protein interactions or posttranslational modifications has been revealed, which can affect VHL function in mediating its targets for proteasomal degradation (15, 31, 49). However, the regulation of VHL at the transcriptional level has largely remained underreported (50). Here we provide data to show the transcriptional regulation of VHL by FOXO3a. Future studies to identify additional transcription factors that can transactivate VHL will further our understanding of the regulation of VHL and the underlying mechanism thoroughly.
Even though many genes have been reported to affect hypoxia signaling directly or indirectly, it is still unclear whether these genes have any impact on animal hypoxic tolerance (acute hypoxia) or hypoxic adaptation (chronic hypoxia) (43). In addition, a key question regarding the functionality of these genes under animal hypoxic tolerance or hypoxic adaptation remains unanswered. Based on studies of human high-altitude adaptation, especially Tibetan adaptation studies, positive directional selection is identified in the HIF pathway and hypoxia-related genes (51). Among these genes, HIF-2α and PHD2 are particularly noteworthy because of the consistency with which they have been observed by different investigators and the biochemical evidence that shows the adaptive changes of these two genes and their effect on HIF signaling (51–54). These observations further support the vital role of the HIF pathway in hypoxic adaptation (51). It is likely that the genes affecting HIF signaling contribute to hypoxic tolerance or hypoxic adaptation (51). In this study, we showed that disruption of foxo3b in zebrafish led to impaired hypoxic tolerance, which might also be due to the influence of foxo3b on HIF activity via regulation of vhl. However, it should be acknowledged that the correlation between HIF activity and hypoxia tolerance or adaptation is undoubtedly complicated (51). The change of the PHD2 allele resulting in loss of function has been suggested to account for either hypoxic adaptation or hypoxic sensitivity (55, 56). On the other hand, change of the allele resulting in gain of function has also been suggested to respond to either hypoxic adaptation or hypoxic sensitivity (56, 57). In this regard, predicting the consequence of disruption of the genes involved in hypoxia signaling may not be simple. Given that HIF-1α and HIF-2α are key factors that orchestrate the hypoxic response, further delineation of the hypoxic response phenotypes of genes directly or indirectly involved in the HIF pathway in knockout and overexpression status will uncover animal hypoxic tolerance or hypoxic adaptation mechanistically.
Of note, mitochondrial ROS are required for hypoxic activation of HIFs, indicating a connection between two vital cellular stress signals (25). FOXO3a activates Mn-SOD expression to protect cells from oxidative stress (8, 9). In this study, we found that FOXO3a (foxo3b) could activate VHL (vhl) expression to enhance zebrafish hypoxic tolerance. Therefore, FOXO3a may serve as a vital factor that orchestrates cells in response to reactive oxygen stress and hypoxic stress conditions.
Experimental Procedures
Plasmid Construction
The human VHL promoters were amplified by PCR and subcloned into the pGL3-Basic vector (Promega). The deletion and DBE mutants of the VHL promoter were obtained by PCR and subcloned into the pGL3-Basic vector. FOXO3a-A3-ER was cloned into the lentivirus vector pHAGE-CMV-MCS-IZsGreen. FOXO3a shRNA was cloned into the lentivirus vector LentiLox3.7. The FOXO3a shRNA targeting sequences were 5′-GAGCTCTTGGTGGATCATC-3′ and 5′-GCACAGAGTTGGATGAAGT-3′. Two sets of VHL shRNA targeting human VHL and GFP shRNA targeting GFP have been described previously (32).
The zebrafish vhl promoter was amplified by PCR from zebrafish genomic DNA using the following primers: forward, 5′-ATATCGGTACCGACTTGGAAGTCTCGGAAGG-3′; reverse, 5′-ATATCCTCGAGCGTCAAAGACAGGACAGTTCC-3′. The resultant product was subcloned into the pGL3-Basic vector. All plasmids were verified by sequencing.
Cell Line and Culture Conditions
HEK293T and H1299 cell lines were originally obtained from the ATCC. All cell lines were cultured in DMEM supplemented with 10% FBS. HEK293T cells that constitutively expressed the FOXO3a-A3-ER fusion protein were obtained by infecting HEK293T cells with a lentivirus expressing FOXO3a-A3-ER.
Luciferase Reporter Assay
HEK293T cells were grown in 24-well plates and transfected with the indicated luciferase reporter, including pTK-Renilla as an internal control, using VigoFect reagent (Vigorous Biotech, Beijing, China). Luciferase activity was assayed 16–28 h after transfection using the Dual-Luciferase reporter assay system (Promega). Data were normalized to Renilla luciferase. Data are reported as mean + S.E. of three independent experiments performed in triplicate. The statistical analysis was performed using GraphPad Prism 5 (unpaired t test) (GraphPad Software Inc.).
For luciferase reporter assays in zebrafish, embryos were injected with the indicated plasmids and homogenized after 8–10 h. Luciferase activity was also determined using the Dual-Luciferase reporter assay system.
Semiquantitative Real-time RT-PCR
Total RNA was extracted by TRIzol reagent (Invitrogen), and cDNA synthesis was carried out using a first strand cDNA synthesis kit (Fermentas). The following primers were used to assess human VHL mRNA expression: forward, 5′-TCAGAGATGCAGGGACACACGATG-3′; reverse, 5′-ACCTGACGATGTCCAGTCTCCTGTA-3′. The following primers were used to assess human FOXO3a mRNA expression: forward, 5′-TGCTAAGCAGGCCTCATCTC-3′; reverse, 5′-CTTGTGTCAGTTTGAGGGTCTG-3′. The following primers were used to evaluate the internal control, 18s RNA forward, 5′-TCAACTTCGATGGTAGTCGCCGT-3′; reverse, 5′-TCCTTGGATGTGGTAGCCGTTCT-3′. The following primers were used to assess zebrafish genes: foxo3b-RT, TCATCTTCAAGGAGGAATGC (forward) and GGATGGAGTTCTTCCAACCA (reverse); β-actin-RT, TACAATGAGCTCCGTGTTGC (forward) and ACATACAATGGCAGGGGTGTT (reverse); vhl-RT, GTGGGACATCCATGGATGTT (forward) and CGTCAGCACAGGCAATGTGA (reverse); pou5f1-RT, TGAGGAAGAGGAGACTCTGA (forward) and GACTGAACATTTTGCCATAC (reverse); pai1-RT, ATTCCAAGGTTCTCCATGGA (forward) and GGTTCCTCAGTAGTAATGCG (reverse); ldha-RT, CCTTCTCAAGGATCTGACCG (forward) and ACACTGTAATCTTTATCCGC (reverse); il11a-RT, CCGGGTGTTTAGTACAGAGATT (forward) and CATGGAGCTGAGAAAGAGTAGG (reverse); cited2-RT, GTTCCGAGACAGTATCGCTAAG (forward) and CATCAAGACCTCCTCGTCAATAA (reverse); and vegf-RT, TGCTCCTGCAAATTCACACAA (forward) and ATCTTGGCTTTTCACATCTGCAA (reverse).
Data are reported as mean + S.E. of three independent experiments performed in triplicate. The statistical analysis was performed using GraphPad Prism 5 (unpaired t test) (GraphPad Software Inc.).
Chromatin Immunoprecipitation
An anti-FOXO3a antibody was purchased from Epitomics. The following primers were used for amplifying the VHL promoter region: forward, 5′-ATAAGCGTGATGATTGGGTGTTC-3′; reverse, 5′-CCCGAGTAGTTGGTACTGTAGGC-3′. The primers for amplifying β-actin and the procedure for ChIP were described previously (58).
Western Blotting Assays
The following antibodies were used for Western blotting: Myc (9E10, Santa Cruz Biotechnology) and α-tubulin (Upstate). The human pVHL polyclonal antibody was generated against synthesized peptides of pVHL (Abmart, Shanghai, China). The pVHL (A0377), FOXO3a (A0102), and HIF-1α (A6265) antibodies were purchased from ABclonal Co. The procedures for Western blotting assays were described previously (59). A Fuji Film LAS4000 mini luminescent image analyzer was used to image the blots.
Zebrafish Embryo Manipulation and Whole-mount in Situ Hybridization
Zebrafish (Danio rerio) strain AB was raised, maintained, reproduced, and staged according to standard protocols. The probes for foxo3b and vhl were amplified by PCR from cDNA pools using the appropriate sets of primers. The primers for foxo3b probe were described previously (26). The following primers were used for the vhl probe: vhl forward, 5′-CTTTAGTCTAACTCGGTGGT-3′; vhl reverse, AGGCAATGTGATCTTGG-3′. The foxo3b antisense morpholino oligonucleotides (foxo3b-ATG-MO and foxo3b-sp-MO) and their validation were also described previously (26). The procedure for whole-mount in situ hybridization was described previously (60).
Generation of foxo3b-null Zebrafish
Disruption of foxo3b in zebrafish was accomplished via CRISPR/Cas9 technology. Zebrafish foxo3b sgRNA was designed using the tools provided in the CRISP Design web site (http://crispr.mit.edu). The zebrafish-Codon-Optimized Cas9 plasmid (61) was digested with XbaI, purified, and transcribed using the T7 mMessage Machine Kit (Ambion). The pUC19-gRNA vector was used for amplifying the sgRNA template (62). The primers for amplifying gRNA template were 5′-GTAATACGACTCACTATAGGACAACGGCAGCCCAAGCCGTTTTAGAGCTAGAAATAGC-3′ and 5′-AAAAGCACCGACTCGGTGCC-3′. sgRNA was synthesized using the Transcript Aid T7 High Yield Transcription Kit (Fermentas). Cas9 RNA and sgRNA were mixed and injected into embryos at the one-cell stage. Cas9 RNA and sgRNA were injected at 0.75–1.25 ng/embryo and 0.075 ng/embryo, respectively.
After the injected embryos were incubated at 28.5 °C for 24 h, the genomic DNA was extracted from 20–30 embryos by heating the embryos at 94 °C for 40 min in lysis solution (50 mm NaOH) (the reaction was terminated by adding 1 m Tris-HCl (pH 8.0)). Mutant detection was followed by HMA as described previously (42). If the results were positive, the remaining embryos were raised to adulthood and treated as F0, which were backcrossed with the wild-type zebrafish for generating F1, which were genotyped by HMA initially and confirmed by sequencing of target sites. The F1 zebrafish harboring the mutations were backcrossed with the wild-type zebrafish to obtain F2. The F2 adult zebrafish with the same genotype (+/−) were intercrossed to generate F3 offspring, which should contain wild-type (+/+), heterozygous (+/−), and homozygous (−/−) offspring. The primers for detecting mutants were 5′-TGGACATTGCCATTGATCCAG-3′ (forward) and 5′-CCATGCATTCCTCCTTGAAGA-3′ (reverse).
Hypoxia Treatments
In pilot experiments, we noticed that zebrafish fry were more resistant to hypoxic treatment than adult zebrafish, as judged by survival time in the hypoxia chamber (Ruskinn INVIVO2400). Thus, we used 2% oxygen for zebrafish fry treatment and 10% oxygen for adult zebrafish treatment.
For zebrafish fry, two flasks were filled with 250 ml of water. Foxo3b-null fry (15 dpf, n = 20) were put into one flask, and their wild-type siblings (15 dpf, n = 20) were put in the second flask. Then, two flasks were put into the hypoxia chamber simultaneously. The oxygen concentration of the chamber was adjusted to 2% O2 ahead of time. The dead fry were counted once an hour until the death of the last zebrafish with one genotype (foxo3b−/−). This experiment was repeated three times.
For the adult zebrafish experiment, the zebrafish were initially weighed. During pilot experiments, we noticed that the body weight of the zebrafish could dramatically affect their acute hypoxic tolerance. To avoid interference by body weight in acute hypoxia tolerance determination, zebrafish of similar weight were chosen for further experiments. Subsequently, two flasks were filled with 250 ml of water. Three foxo3b-null zebrafish (0.36, 0.37, and 0.39 g) were put into one flask, and three wild-type siblings (0.36, 0.37, and 0.38 g) were put into the second flask. Before immersion in the hypoxia chamber (Ruskinn INVIVO2400), the oxygen concentration in the flask water was measured by an HQd portable meter (HACH Co.). The oxygen concentration in the foxo3b-null zebrafish flask was 7.10 mg/liter, and the oxygen concentration in the wild-type sibling flask was 7.04 mg/liter. The oxygen concentration in the hypoxia chamber was adjusted to 10% ahead of time. After putting the flasks containing zebrafish into the hypoxia chamber, the behavior of the zebrafish was closely monitored. When one genotypic zebrafish was expired, we took out the flasks from the chamber immediately and measured the oxygen concentration in the flask water. At that moment, the oxygen concentration in the foxo3b-null zebrafish flask was 1.48 mg/liter, and the oxygen concentration in the wild-type sibling flask was 1.54 mg/liter.
Author Contributions
W. X. designed the study and wrote the manuscript. X. L., X. C., Z. M., and B. H. designed the study, conducted the experiments, and analyzed the data. D. Z. generated the foxo3b-null zebrafish. G. O., J. W., and W. Z. contributed the reagents and analyzed the data. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Peter Ratcliffe, William Kaelin, Frank Lee, Navdeep Chandel, William Tansey, Katja Knauth, Boudewijn Burgering, and Linda Penn for the generous gift of reagents.
The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movies 1 and 2.
- FOXO
- forkhead box O
- Mn-SOD
- manganese superoxide dismutase
- VHL
- von Hippel-Lindau
- HIF
- hypoxia-inducible factor
- PHD
- prolyl hydroxylase
- ROS
- reactive oxygen species
- DBE
- DNA binding element
- ER
- estrogen receptor
- 4-HT
- 4-hydroxy-tamoxifen
- WISH
- whole-mount in situ hybridization
- HMA
- heteroduplex mobility assay
- MT
- mutant
- dpf
- days post-fertilization
- hpf
- hours post-fertilization
- CRISPR/Cas9
- clustered regularly interspaced short palindromic repeats (CRISPR)-associated RNA-guided endonuclease Cas9
- FKHR
- forkhead in rhabdomyosarcoma
- sgRNA
- single guide RNA
- DMOG
- dimethyl-oxalylglycine.
References
- 1. Eijkelenboom A., and Burgering B. M. (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 [DOI] [PubMed] [Google Scholar]
- 2. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., and Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 3. Essers M. A., Weijzen S., de Vries-Smits A. M., Saarloos I., de Ruiter N. D., Bos J. L., and Burgering B. M. (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 23, 4802–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brenkman A. B., de Keizer P. L., van den Broek N. J., Jochemsen A. G., and Burgering B. M. (2008) Mdm2 induces mono-ubiquitination of FOXO4. PLoS ONE 3, e2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hergovich A., Lisztwan J., Barry R., Ballschmieter P., and Krek W. (2003) Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat. Cell Biol. 5, 64–70 [DOI] [PubMed] [Google Scholar]
- 6. Tran H., Brunet A., Grenier J. M., Datta S. R., Fornace A. J. Jr, DiStefano P. S., Chiang L. W., and Greenberg M. E. (2002) DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296, 530–534 [DOI] [PubMed] [Google Scholar]
- 7. Storz P. (2011) Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signal. 14, 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Essers M. A., de Vries-Smits L. M., Barker N., Polderman P. E., Burgering B. M., and Korswagen H. C. (2005) Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 9. Kops G. J., Dansen T. B., Polderman P. E., Saarloos I., Wirtz K. W., Coffer P. J., Huang T. T., Bos J. L., Medema R. H., and Burgering B. M. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 10. Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., et al. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 11. Wang F., Nguyen M., Qin F. X., and Tong Q. (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514 [DOI] [PubMed] [Google Scholar]
- 12. Ferber E. C., Peck B., Delpuech O., Bell G. P., East P., and Schulze A. (2012) FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 19, 968–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gordan J. D., Lal P., Dondeti V. R., Letrero R., Parekh K. N., Oquendo C. E., Greenberg R. A., Flaherty K. T., Rathmell W. K., Keith B., Simon M. C., and Nathanson K. L. (2008) HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 14, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dansen T. B., Smits L. M., van Triest M. H., de Keizer P. L., van Leenen D., Koerkamp M. G., Szypowska A., Meppelink A., Brenkman A. B., Yodoi J., Holstege F. C., and Burgering B. M. (2009) Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 5, 664–672 [DOI] [PubMed] [Google Scholar]
- 15. Putker M., Madl T., Vos H. R., de Ruiter H., Visscher M., van den Berg M. C., Kaplan M., Korswagen H. C., Boelens R., Vermeulen M., Burgering B. M., and Dansen T. B. (2013) Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol. Cell 49, 730–742 [DOI] [PubMed] [Google Scholar]
- 16. Salih D. A., and Brunet A. (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 20, 126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dansen T. B., and Burgering B. M. (2008) Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 18, 421–429 [DOI] [PubMed] [Google Scholar]
- 18. Kaelin W. G. (2007) Von Hippel-Lindau disease. Annu. Rev. Pathol. 2, 145–173 [DOI] [PubMed] [Google Scholar]
- 19. Kaelin W. G., Jr. (2008) The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 8, 865–873 [DOI] [PubMed] [Google Scholar]
- 20. Semenza G. L. (2004) Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology 19, 176–182 [DOI] [PubMed] [Google Scholar]
- 21. Semenza G. L. (2013) HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 123, 3664–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Semenza G. L. (2010) HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaelin W. G. Jr., and Ratcliffe P. J. (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 [DOI] [PubMed] [Google Scholar]
- 24. Shen C., and Kaelin W. G. Jr. (2013) The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 23, 18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamanaka R. B., and Chandel N. S. (2009) Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 21, 894–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie X. W., Liu J. X., Hu B., and Xiao W. (2011) Zebrafish foxo3b negatively regulates canonical Wnt signaling to affect early embryogenesis. PLoS ONE 6, e24469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Littlewood T. D., Hancock D. C., Danielian P. S., Parker M. G., and Evan G. I. (1995) A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 23, 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Z., Liu X., Mei Z., Wang Z., and Xiao W. (2014) EAF2 suppresses hypoxia-induced factor 1α transcriptional activity by disrupting its interaction with coactivator CBP/p300. Mol. Cell Biol. 34, 1085–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Núñez-O'Mara A., Gerpe-Pita A., Pozo S., Carlevaris O., Urzelai B., Lopitz-Otsoa F., Rodríguez M. S., and Berra E. (2015) PHD3-SUMO conjugation represses HIF1 transcriptional activity independently of PHD3 catalytic activity. J. Cell Sci. 128, 40–49 [DOI] [PubMed] [Google Scholar]
- 30. Nakayama K., Frew I. J., Hagensen M., Skals M., Habelhah H., Bhoumik A., Kadoya T., Erdjument-Bromage H., Tempst P., Frappell P. B., Bowtell D. D., and Ronai Z. (2004) Siah2 regulates stability of prolyl-hydroxylases, controls HIF1 α abundance, and modulates physiological responses to hypoxia. Cell 117, 941–952 [DOI] [PubMed] [Google Scholar]
- 31. Kim J. J., Lee S. B., Jang J., Yi S. Y., Kim S. H., Han S. A., Lee J. M., Tong S. Y., Vincelette N. D., Gao B., Yin P., Evans D., Choi D. W., Qin B., Liu T., et al. (2015) WSB1 promotes tumor metastasis by inducing pVHL degradation. Genes Dev. 29, 2244–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J., Zhang W., Ji W., Liu X., Ouyang G., and Xiao W. (2014) The von Hippel-Lindau protein suppresses androgen receptor activity. Mol. Endocrinol. 28, 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Onnis B., Fer N., Rapisarda A., Perez V. S., and Melillo G. (2013) Autocrine production of IL-11 mediates tumorigenicity in hypoxic cancer cells. J. Clin. Invest. 123, 1615–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Asikainen T. M., Schneider B. K., Waleh N. S., Clyman R. I., Ho W. B., Flippin L. A., Günzler V., and White C. W. (2005) Activation of hypoxia-inducible factors in hyperoxia through prolyl 4-hydroxylase blockade in cells and explants of primate lung. Proc. Natl. Acad. Sci. U.S.A. 102, 10212–10217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Du J., Zhang D., Zhang W., Ouyang G., Wang J., Liu X., Li S., Ji W., Liu W., and Xiao W. (2015) pVHL negatively regulates antiviral signaling by targeting MAVS for proteasomal degradation. J. Immunol. 195, 1782–1790 [DOI] [PubMed] [Google Scholar]
- 36. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., and Ratcliffe P. J. (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 37. Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., and Kaelin W. G. Jr. (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 [DOI] [PubMed] [Google Scholar]
- 38. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., and Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 39. Schödel J., Oikonomopoulos S., Ragoussis J., Pugh C. W., Ratcliffe P. J., and Mole D. R. (2011) High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117, e207–e217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gerez J., Tedesco L., Bonfiglio J. J., Fuertes M., Barontini M., Silberstein S., Wu Y., Renner U., Páez-Pereda M., Holsboer F., Stalla G. K., and Arzt E. (2015) RSUME inhibits VHL and regulates its tumor suppressor function. Oncogene 34, 4855–4866 [DOI] [PubMed] [Google Scholar]
- 41. Yang F., Zhou L., Wang D., Wang Z., and Huang Q. Y. (2015) Minocycline ameliorates hypoxia-induced blood-brain barrier damage by inhibition of HIF-1α through SIRT-3/PHD-2 degradation pathway. Neuroscience 304, 250–259 [DOI] [PubMed] [Google Scholar]
- 42. Ota S., Hisano Y., Muraki M., Hoshijima K., Dahlem T. J., Grunwald D. J., Okada Y., and Kawahara A. (2013) Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells 18, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ratcliffe P. J. (2013) Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J. Physiol. 591, 2027–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paik J. H., Kollipara R., Chu G., Ji H., Xiao Y., Ding Z., Miao L., Tothova Z., Horner J. W., Carrasco D. R., Jiang S., Gilliland D. G., Chin L., Wong W. H., Castrillon D. H., and DePinho R. A. (2007) FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gossage L., Eisen T., and Maher E. R. (2015) VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 15, 55–64 [DOI] [PubMed] [Google Scholar]
- 46. Okumura F., Uematsu K., Byrne S. D., Hirano M., Joo-Okumura A., Nishikimi A., Shuin T., Fukui Y., Nakatsukasa K., and Kamura T. (2016) Parallel regulation of von Hippel-Lindau disease by pVHL-mediated degradation of B-Myb and hypoxia-inducible factor α. Mol. Cell Biol. 36, 1803–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen J., Liu F., Li H., Archacki S., Gao M., Liu Y., Liao S., Huang M., Wang J., Yu S., Li C., Tang Z., and Liu M. (2015) pVHL interacts with ceramide kinase like (CERKL) protein and ubiquitinates it for oxygen-dependent proteasomal degradation. Cell Signal. 27, 2314–2323 [DOI] [PubMed] [Google Scholar]
- 48. Gamper A. M., Qiao X., Kim J., Zhang L., DeSimone M. C., Rathmell W. K., and Wan Y. (2012) Regulation of KLF4 turnover reveals an unexpected tissue-specific role of pVHL in tumorigenesis. Mol. Cell 45, 233–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chitalia V. C., Foy R. L., Bachschmid M. M., Zeng L., Panchenko M. V., Zhou M. I., Bharti A., Seldin D. C., Lecker S. H., Dominguez I., and Cohen H. T. (2008) Jade-1 inhibits Wnt signalling by ubiquitylating β-catenin and mediates Wnt pathway inhibition by pVHL. Nat. Cell Biol. 10, 1208–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ji W., Wang J., Zhang W., Liu X., Ouyang G., and Xiao W. (2014) pVHL acts as a downstream target of E2F1 to suppress E2F1 activity. Biochem. J. 457, 185–195 [DOI] [PubMed] [Google Scholar]
- 51. Bigham A. W., and Lee F. S. (2014) Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 28, 2189–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song D., Li L. S., Arsenault P. R., Tan Q., Bigham A. W., Heaton-Johnson K. J., Master S. R., and Lee F. S. (2014) Defective Tibetan PHD2 binding to p23 links high altitude adaption to altered oxygen sensing. J. Biol. Chem. 289, 14656–14665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simonson T. S., Yang Y., Huff C. D., Yun H., Qin G., Witherspoon D. J., Bai Z., Lorenzo F. R., Xing J., Jorde L. B., Prchal J. T., and Ge R. (2010) Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75 [DOI] [PubMed] [Google Scholar]
- 54. Petousi N., and Robbins P. A. (2014) Human adaptation to the hypoxia of high altitude: the Tibetan paradigm from the pregenomic to the postgenomic era. J. Appl. Physiol. 116, 875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beall C. M. (2007) Two routes to functional adaptation: Tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. U.S.A. 104, 8655–8660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beall C. M., Cavalleri G. L., Deng L., Elston R. C., Gao Y., Knight J., Li C., Li J. C., Liang Y., McCormack M., Montgomery H. E., Pan H., Robbins P. A., Shianna K. V., Tam S. C., et al. (2010) Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. U.S.A. 107, 11459–11464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yi X., Liang Y., Huerta-Sanchez E., Jin X., Cuo Z. X., Pool J. E., Xu X., Jiang H., Vinckenbosch N., Korneliussen T. S., Zheng H., Liu T., He W., Li K., Luo R., et al. (2010) Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Feng X., Liu X., Zhang W., and Xiao W. (2011) p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. EMBO J. 30, 3397–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou J., Feng X., Ban B., Liu J., Wang Z., and Xiao W. (2009) Elongation factor ELL (eleven-nineteen lysine-rich leukemia) acts as a transcription factor for direct thrombospondin-1 regulation. J. Biol. Chem. 284, 19142–19152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu J. X., Hu B., Wang Y., Gui J. F., and Xiao W. (2009) Zebrafish eaf1 and eaf2/u19 mediate effective convergence and extension movements through the maintenance of wnt11 and wnt5 expression. J. Biol. Chem. 284, 16679–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu D., Wang Z., Xiao A., Zhang Y., Li W., Zu Y., Yao S., Lin S., and Zhang B. (2014) Efficient gene targeting in zebrafish mediated by a zebrafish-codon-optimized cas9 and evaluation of off-targeting effect. Yi Chuan Xue Bao 41, 43–46 [DOI] [PubMed] [Google Scholar]
- 62. Chang N., Sun C., Gao L., Zhu D., Xu X., Zhu X., Xiong J. W., and Xi J. J. (2013) Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 23, 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.