

Abstract
Background
Asthma is characterized by type 2 T-helper cell (Th2) inflammation, goblet cell hyperplasia, airway hyperreactivity, and airway fibrosis. Monocyte chemoattractant protein-1 (MCP-1 or CCL2) and its receptor, CCR2, have been shown to play important roles in the development of Th2 inflammation. CCR2-deficient mice have been found to have altered inflammatory and physiologic responses in some models of experimental allergic asthma, but the role of CCR2 in contributing to inflammation and airway hyperreactivity appears to vary considerably between models. Furthermore, MCP-1-deficient mice have not previously been studied in models of experimental allergic asthma.
Methods
To test whether MCP-1 and CCR2 are each required for the development of experimental allergic asthma, we applied an Aspergillus antigen-induced model of Th2 cytokine-driven allergic asthma associated with airway fibrosis to mice deficient in either MCP-1 or CCR2. Previous studies with live Aspergillus conidia instilled into the lung revealed that MCP-1 and CCR2 play a role in anti-fungal responses; in contrast, we used a non-viable Aspergillus antigen preparation known to induce a robust eosinophilic inflammatory response.
Results
We found that wild-type C57BL/6 mice developed eosinophilic airway inflammation, goblet cell hyperplasia, airway hyperreactivity, elevations in serum IgE, and airway fibrosis in response to airway challenge with Aspergillus antigen. Surprisingly, mice deficient in either MCP-1 or CCR2 had responses to Aspergillus antigen similar to those seen in wild-type mice, including production of Th2 cytokines.
Conclusion
We conclude that robust Th2-mediated lung pathology can occur even in the complete absence of MCP-1 or CCR2.
Background
Monocyte chemoattractant protein-1 (MCP-1, also known as CCL2) and its receptor, CCR2, have been the focus of intense interest due to increasing awareness of their association with debilitating human diseases, including asthma [1-3] and pulmonary fibrosis [4-7]. Since MCP-1 attracts and activates a variety of cells, including monocytes, immature dendritic cells, basophils, natural killer cells, and a subset of T lymphocytes [8-17], MCP-1 may have multiple roles in the immune response. Models of Th1 or Th2 inflammation applied to mice deficient in either MCP-1 or CCR2 have clearly shown important roles for this chemokine and its receptor in the development of inflammation [18-24]. However, results obtained using allergen-induced models of asthma (ovalbumin and cockroach antigen) in CCR2-deficient mice are varied, showing either increased, decreased or unchanged Th2 inflammation and airway hyperreactivity (AHR) [25-27], possibly due to differences in the allergen models or strains of mice used. These experiments with CCR2-deficient mice do not directly address the role of MCP-1, which is just one of several MCP chemokines that can bind to CCR2. Although MCP-1-deficient mice have been reported to have defects in Th2 responses [18,19], the effects of MCP-1 deletion in allergen-induced allergic experimental asthma have not been previously reported.
In addition to Th2 inflammation, airway fibrosis is another important feature of human asthma. Blease and colleagues [28,29] examined the contributions of MCP-1 and CCR2 to the development of fibrosis following intratracheal administration of Aspergillus fumigatus conidia to A. fumigatus sensitized mice. Airway fibrosis was significantly increased in mice treated with MCP-1 neutralizing antibody and in CCR2-deficient mice. However, these increases in fibrosis were seen in the setting of impaired clearance of conidia and a markedly increased neutrophilic inflammatory response, suggesting that the increased fibrosis might be attributable simply to an impaired antifungal response. Previous studies involving other models of allergic asthma applied to CCR2-deficient mice did not examine whether airway fibrosis occurred in these models or whether development of fibrosis was dependent on CCR2 expression [25-27]. Consequently, the role of MCP-1 and CCR2 in the development of allergen-induced lung fibrosis is not well established.
In this study, we hypothesized that the effects of MCP-1 are mediated through CCR2 and that MCP-1 and CCR2 are independently required for the development of experimental allergic asthma. To test this hypothesis, we subjected mice deficient in either MCP-1 or CCR2 to an Aspergillus antigen model of Th2-cytokine-driven allergic asthma associated with significant airway fibrosis and measured pulmonary inflammation, cytokine production, AHR and fibrosis.
Methods
Mice
Breeding pairs of Mcp-1+/+ and Mcp-1-/- mice [19] and Ccr2+/+ and Ccr2-/- mice [21] were generated as previously described. Mice were bred and maintained under specific pathogen-free conditions in the Laboratory Animal Resource Center at San Francisco General Hospital. All mice were backcrossed nine times with C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). Deletion of Mcp-1 or Ccr2 genes was confirmed by PCR. Similar numbers of male and female six-week-old mice were used for the study. The UCSF Institutional Animal Care and Use Committee approved all experimental protocols.
Aspergillus Antigen Sensitization Protocol
The Aspergillus fumigatus antigen preparation consisted of a mixture of culture filtrate (300 μg protein/mouse) and mycelial extract (80 μg protein/mouse) in PBS (Cellgro by Mediatech, Inc, Herndon, VA). Culture filtrates and mycelial extract were prepared as described previously [30]. For sensitization, anesthetized six-week old mice were given 50 μl of Aspergillus antigen intranasally five times at four-day intervals. Control mice were given 50 μl of PBS according to the same schedule as Aspergillus antigen-treated mice. All measurements and samples were obtained from mice four days after the final Aspergillus antigen administration, which was 20 days after the first challenge. Our group has previously found that airway reactivity measured four days after the final Aspergillus antigen challenge was similar to reactivity measured at earlier time points (on the same day as the final challenge or one day after the final challenge) [30].
Determination of Airway Reactivity
Mice were anesthetized and paralyzed by intraperitoneal injection of etomidate (28 mg/kg) (Bedford Laboratories, Bedford, OH) and pancuronium bromide (0.1 mg/kg) (Baxter Healthcare Corporation, Irvine, CA). A tracheal cannula was inserted via a midcervical incision and the mice were ventilated using a Harvard model 683 rodent ventilator (9 μl/g tidal volume, 150 breaths per minute) (Harvard Apparatus, Holliston, MA). Using a whole body plethysmograph, airflow resistance was calculated during baseline breathing and in response to serially increasing doses of intravenous acetylcholine chloride (0.032, 0.100, 0.316, 1.00, and 3.16 μg/gm body weight) (Sigma, St. Louis, MO). The log of the concentration of acetylcholine (μg/gm) required for a 200% increase in total lung resistance, designated log PC200, was reported.
Bronchoalveolar Lavage (BAL)
After completion of the airway physiology measurements, the lungs were lavaged five times with 0.8-ml aliquots of sterile PBS. The lavage fluid was pooled and centrifuged, and the cell pellet was treated with red-blood-cell lysing buffer (Sigma, Saint Louis, MO). After being washed, the samples were resuspended in PBS. Total leukocytes were counted using a hemacytometer. Differential cell counts were determined by cytocentrifugation and Diff-Quik staining (Dade Behring Inc., Newark, DE) followed by microscopic examination of at least 300 cells.
Thoracic Lymph Node Isolation and Lung Histology
Thoracic lymph nodes were harvested from mice exposed to Aspergillus antigen. Lungs were then removed en bloc and the left mainstem bronchus was firmly sutured closed. The left lung was removed by cutting the left mainstem distal to the suture. It was then frozen in liquid nitrogen and stored at -70°C until processed for hydroxyproline content. The right lung was inflated to 20 cm water pressure with 10% neutral buffered formalin (VWR Scientific Products, West Chester, PA) and fixed in 10% formalin for more than 48 h. Fixed lungs were embedded in paraffin, sectioned at 5 μm thickness, and stained with either hematoxylin and eosin (H&E), periodic acid Schiff (PAS), or trichrome by the Pathology Department of San Francisco General Hospital using standard protocols. The proportion of peribronchial inflammatory cells that were eosinophils was determined by counting inflammatory cells surrounding airways with lumens of 100–200 μm (measured on the short axis) on H&E stained sections. We analyzed 500 total cells (100 cells from each of five airways) for each animal studied.
Analysis of Cytokine Production by Cells
To prepare single-cell suspensions for cytokine analyses, isolated lymph nodes were gently minced using a syringe plunger and cells were passed through 70-μm cell strainers. Red blood cells were removed by hypotonic lysis at room temperature. Lymph node cells were counted, centrifuged, and resuspended in RPMI medium 1640 (Cellgro by Mediatech, Inc, Herndon, VA) supplemented with FCS (10% vol/vol) (Hyclone, Logan, UT), penicillin (100 U/ml) (Cellgro by Mediatech, Inc, Herndon, VA), streptomycin (100 μg/ml) (Cellgro by Mediatech, Inc, Herndon, VA), phorbol 12 myristate 13-acetate (PMA) (25 ng/m) (Sigma, Saint Louis, MO), and ionomycin (1 μg/ml) (Sigma, Saint Louis, MO) to a final concentration of 5 million cells per ml. Cells were then aliquoted into 96-well plates and incubated at 37°C. After 40 h, cell supernatants were harvested and stored at -70°C until they were analyzed. ELISA for IL-4, -5, -13 and IFN-γ was performed on stimulated lymph node cell supernatant per the manufacturer's protocols (R&D Systems, Minneapolis, MN).
Determination of MCP-1
For quantitation of MCP-1 levels in BAL fluid, C57BL/6 wild-type mice were treated with the previously described Aspergillus antigen protocol. Four days after the final Aspergillus antigen administration, lungs from Aspergillus antigen- and PBS-treated mice were lavaged two times with 0.6-ml aliquots of sterile PBS. The samples were centrifuged and the supernatants were collected and stored at -70°C until analysis. ELISA for MCP-1 was performed on cell-free BAL fluid per the manufacturer's protocol (R&D Systems, Minneapolis, MN).
Measurement of Serum Total IgE Concentration
Sera were obtained from blood collected by cardiac puncture from Aspergillus antigen- or PBS-treated mice after airway responsiveness measurement. Serum total IgE concentration was determined by a sandwich ELISA using complementary antibody pairs for mouse IgE (clone R35-72 and R35-118) obtained from Pharmingen (Pharmingen, San Diego, CA) according to the manufacturer's instructions. Color development was achieved using streptavidin-conjugated horseradish peroxidase (Pharmingen, San Diego, CA) followed by addition of HRP substrate (ABTS, Sigma, Saint Louis, MO).
Determination of Lung Hydroxyproline Content
Lungs were analyzed for hydroxyproline content as previously described [31] with slight modification. Lungs were homogenized in distilled water and incubated with 50% trichloroacetic acid on ice for 20 min. Samples were centrifuged and the pellet was mixed with 12 N hydrochloric acid and baked at 110°C for 14–18 h until samples were charred and dry. The samples were resuspended in 2 ml deionized water by incubating for 72 h at room temperature applying intermittent vortexing. Serial dilutions of trans-4-hydroxy-L-proline standard (Sigma, Saint Louis, MO) were prepared. 200 μl of vortexed sample (or standard) was added to 500 μl 1.4% chloramine T/0.5 M sodium acetate/10% isopropanol (Fisher Scientific, Pittsburgh, PA) and incubated for 20 min at room temperature. Next, 500 μl of Ehrlich's solution (1.0 M p-dimethylaminobenzaldehyde, 70% isopropanol/30% perchloric acid) (Fisher Scientific, Pittsburgh, PA) was added, mixed, and incubated at 65°C for 15 min. After samples returned to room temperature, the optical density of each sample and standard was measured at 550 nm and the concentration of lung hydroxyproline was calculated from the hydroxyproline standard curve.
Statistical Analysis
Statistical significance for treatment effect was determined by analysis of variance with post-ANOVA t tests corrected for multiple comparisons using Bonferroni adjustment. These statistical analyses were performed using statistical software STATA 5.0 (Stata Corporation, College Station, TX) and R [32] (The R Foundation for Statistical Computing, Vienna, Austria). All tests were two-tailed with a p-value of 0.05 for statistical significance.
Results
Aspergillus antigen airway challenge induces MCP-1 production
We used a model system that involved repeated intranasal challenges with Aspergillus antigen over a 20-day period. To determine whether antigen challenge induces MCP-1 production in the airway, we measured MCP-1 protein levels in BAL fluid from wild-type C57BL/6 mice on day 20. MCP-1 levels were markedly higher in Aspergillus antigen-treated mice (46.3 ± 12.7 pg/ml, mean ± SE) than in PBS-treated mice (5.8 ± 1.3 pg/ml), (P = 0.01).
MCP-1- and CCR2-deficient mice develop airway inflammation in response to Aspergillus antigen
The Aspergillus antigen induction of MCP-1 was accompanied by a significant degree of lung inflammation as assessed by BAL fluid cell counts and lung histology. In wild-type mice, Aspergillus antigen induced a >20-fold increase in BAL fluid cell numbers (Fig. 1) and the development of prominent infiltrates in peribronchovascular spaces and scattered infiltrates in the lung parenchyma (Fig. 2A and 2B). The inflammatory infiltrates consisted of numerous eosinophils as well as other cell types.
Figure 1.
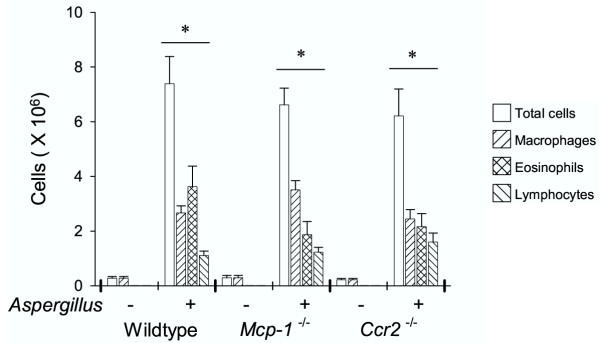
Aspergillus antigen induced similar increases in BAL fluid cell counts in wild-type, Mcp-1-/- and Ccr2-/- mice. Total cells, macrophages, eosinophils, and lymphocytes are expressed as mean BAL fluid total cell counts ± SE from wild-type, Mcp-1-/- and Ccr2-/- mice (PBS-treated, N = 5 mice/group; Aspergillus antigen-treated, N = 8 mice/group; Aspergillus antigen exposure and sample collection are described in methods). Neutrophils represented <0.5% of total cells for all groups. The data shown are from one experiment and representative of three separate experiments. Asterisks (*) indicate values that are statistically significantly different (p < 0.001) compared to PBS controls.
Figure 2.
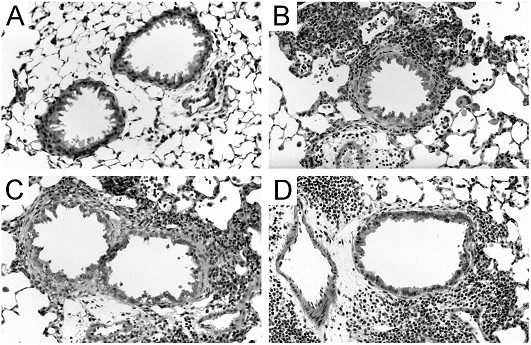
Aspergillus antigen-induced lung inflammation appears similar in wild-type, Mcp-1-/- and Ccr2-/- mice. H&E stained lung sections from PBS- or Aspergillus antigen-treated wild-type, Mcp-1-/- and Ccr2-/- mice. Representative normal airway from wild-type control mice (A) (similar findings from Mcp-1-/- and Ccr2-/- control mice are not shown). Representative lung sections from Aspergillus antigen-treated wild-type (B), Mcp-1-/- (C) and Ccr2-/- mice (D) demonstrate intense peribronchiolar and perivascular inflammation. Aspergillus antigen exposure and sample collection are described in methods. Magnification: 20× objective.
To determine the airway inflammatory response to Aspergillus antigen in the absence of MCP-1 or its receptor, CCR2, we used mice with targeted disruptions of the genes that encode MCP-1 and CCR2. Since mouse strain differences are associated with major differences in antigen reactivity in many model systems, the mice used here were produced by extensive backcrossing into a C57BL/6 genetic background. Both MCP-1- and CCR2-deficient mice developed marked airway inflammation in response to Aspergillus antigen (Figs. 2C and 2D). The BAL fluid cell counts from Aspergillus antigen-treated MCP-1- and CCR2-deficient mice revealed significantly greater numbers of all cell types than in PBS-treated controls (p < 0.001). The numbers of macrophages, lymphocytes and neutrophils were not significantly different from those in Aspergillus antigen-treated wild-type mice (Fig. 1). The BAL fluid eosinophil response in MCP-1- and CCR2-deficient mice was slightly (~30–40%) smaller than in wild-type mice, but this difference did not reach statistical significance (Fig. 1). The fraction of peribronchial inflammatory cells that were eosinophils was not significantly different among wild-type mice (51 ± 13%, mean ± standard deviation), CCR2-deficient mice (52 ± 6%), and MCP-1-deficient mice (37 ± 13%) (N = 5 mice/group). These findings indicate that there was a robust inflammatory response to Aspergillus antigen even in the absence of MCP-1 or CCR2.
MCP-1- and CCR2-deficient mice develop AHR and produce mucus in response to Aspergillus antigen
To determine airway reactivity to acetylcholine in mice exposed to Aspergillus antigen or to vehicle (PBS) alone, we compared airway reactivity of PBS- and Aspergillus-antigen-treated mice 4 days after the final challenge as described in the methods section. Measurements from this time point were previously found to be comparable to those from earlier time points [30]. In the experiment shown in Fig. 3, the PBS-treated group included a mixture of wild-type, Mcp-1-/-, and Ccr2-/- mice since preliminary experiments showed similar airway reactivity between PBS-treated wild-type, Mcp-1-/-, and Ccr2-/- mice (not shown). Aspergillus-antigen-treated wild-type, Mcp-1-/-, and Ccr2-/- mice each had significantly lower PC200 values than did PBS-treated controls (P < 0.001), indicating the development of AHR (Fig. 3). Although there appeared to be a trend toward less airway reactivity in Aspergillus-antigen-treated Mcp-1-/- and Ccr2-/- mice than in Aspergillus-antigen-treated wild-type mice, this trend was not statistically significant and was not observed in two additional Aspergillus-antigen-challenge experiments comparing wild-type mice to either Mcp-1-/- or Ccr2-/- mice separately (data not shown).
Figure 3.
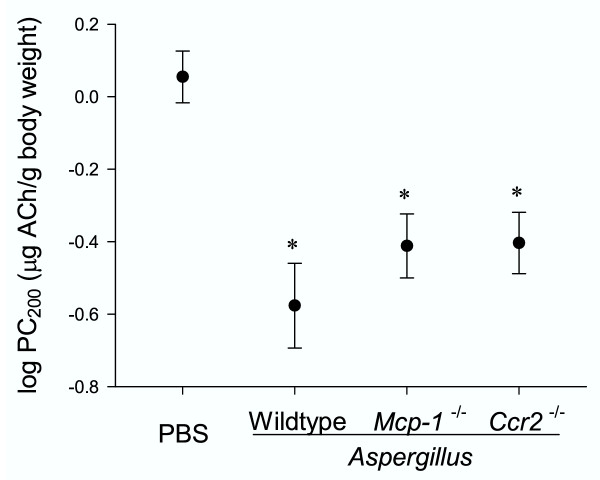
Aspergillus antigen induced AHR in wild-type, Mcp-1-/- and Ccr2-/- mice. Airway reactivity in response to intravenous acetylcholine was measured invasively. Data are expressed as log PC200 and lower values indicate higher airway response. Aspergillus antigen exposure and the airway measurement protocol are described in methods (PBS-treated, N = 12 mice; Aspergillus antigen-treated, N = 8–10 mice/group;). The data shown are from one experiment and representative of three separate experiments. Asterisks (*) indicate values that are statistically significantly different (p < 0.001) compared to PBS controls.
To determine if Aspergillus-antigen challenge results in increased mucus production, we analyzed lung histology by PAS-staining. As shown in Fig. 4A, there was minimal PAS staining in the airway epithelium of control mice. In contrast, Aspergillus-antigen-treated mice from all three groups showed accumulation of PAS-stained material in epithelial cells (Fig. 4B,4C,4D), indicating that Aspergillus antigen airway challenge resulted in mucus production by goblet cells. These findings indicate that Aspergillus antigen induces AHR and mucus production even in the absence of MCP-1 or CCR2.
Figure 4.
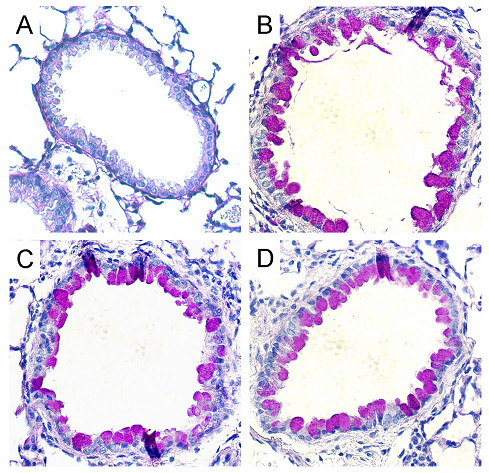
Aspergillus antigen induced goblet cell hyperplasia in wild-type, Mcp-1-/- and Ccr2-/- mice. Representative PAS-stained lung sections from PBS-treated wild-type mice (A) showed minimal PAS-positive staining (similar findings from Mcp-1-/- and Ccr2-/- control mice are not shown). Aspergillus antigen-treated wild-type (B), Mcp-1-/- (C) and Ccr2-/- mice (D) showed magenta staining in epithelial cells, which represents mucus. Aspergillus antigen exposure and sample collection are described in methods. Magnification, 40× objective.
Th2 cytokine and IgE production is similar in Aspergillus antigen-treated wild-type, Mcp-1-/- and Ccr2-/- mice
To determine if deletion of MCP-1 or CCR2 alters the cytokine response to Aspergillus antigen, we assayed Th1 and Th2 cytokines in stimulated cell supernatants prepared from thoracic lymph nodes isolated from Aspergillus antigen-treated mice. (PBS-treated mice had much smaller thoracic lymph nodes and it was not possible to reliably obtain sufficient numbers of cells from these mice for comparison.) MCP-1- and CCR2-deficient mice had concentrations of the cytokines IL-4, IL-5, IL-13 and IFN-γ generally similar to those in wild-type mice (Fig. 5A,5B,5C,5D). There was a trend toward lower IL-4 production in cells from Ccr2-/- mice, but this difference was not statistically significant. In addition, sera from Aspergillus-antigen-treated mice and control mice were assayed for serum total IgE levels. As shown in Fig. 5E, Aspergillus antigen induced increases in serum IgE in wild-type, Mcp-1-/-, and Ccr2-/- mice similar to those in control mice.
Figure 5.
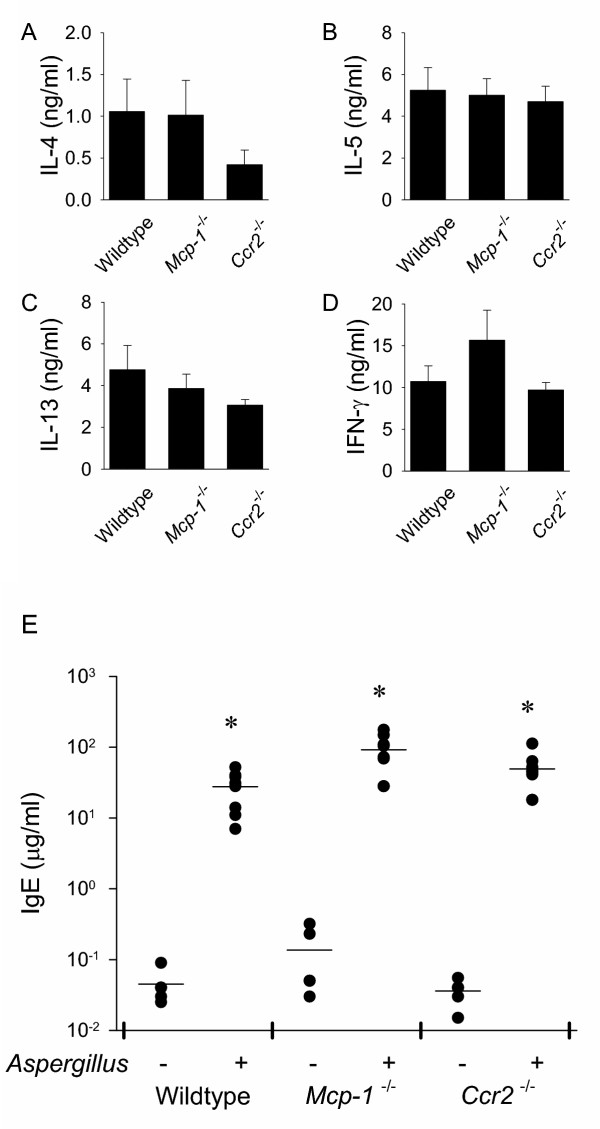
Aspergillus antigen-treated wild-type, Mcp-1-/- and Ccr2-/- mice demonstrated intact Th2 cytokine production and induction of IgE. For cytokine determination, draining lymph node cells from Aspergillus antigen-treated wild-type, Mcp-1-/- and Ccr2-/- mice were isolated and stimulated with PMA/ionomycin for 40 hr and cytokine levels for IL-4 (A), IL-5 (B), IL-13 (C), and IFN-γ (D) were quantitated by ELISA. Serum IgE (E) from Aspergillus antigen-treated wild-type, Mcp-1-/- and Ccr2-/- mice and control mice were measured by ELISA. In (A-D), bars represent mean ± SE; in (E), results are expressed as the common log of IgE concentration where each circle represents a single PBS- or Aspergillus antigen-treated mouse and horizontal lines represent the mean of each group (PBS-treated, N = 5 mice/group; Aspergillus antigen-treated, N = 8–9 mice/group). Aspergillus antigen exposure and sample collection are described in methods. Asterisks (*) indicate values that are statistically significantly different (p < 0.001) compared to PBS controls.
Aspergillus antigen-induced lung fibrosis develops in the absence of MCP-1 or CCR2
To determine whether Aspergillus antigen-induced airway fibrosis develops in the absence of MCP-1 or CCR2, we measured lung hydroxyproline content in PBS- and Aspergillus-antigen-challenged mice (Fig. 6). Aspergillus antigen treatment resulted in a two-fold increase in lung hydroxyproline, a measure of collagen content. This effect was very similar in wild-type, Mcp-1-/-, and Ccr2-/- mice. Histopathologically, lung sections from PBS-treated mice had normal lung architecture and minimal evidence of trichrome staining (Fig. 7A). Lung sections from mice treated with Aspergillus antigen had clear increases in trichrome staining in a peribronchiolar distribution (Fig. 7B,7C,7D). There were no apparent differences in trichrome staining in wild-type mice as compared to either MCP-1- or CCR2-deficient mice after allergen challenge.
Figure 6.
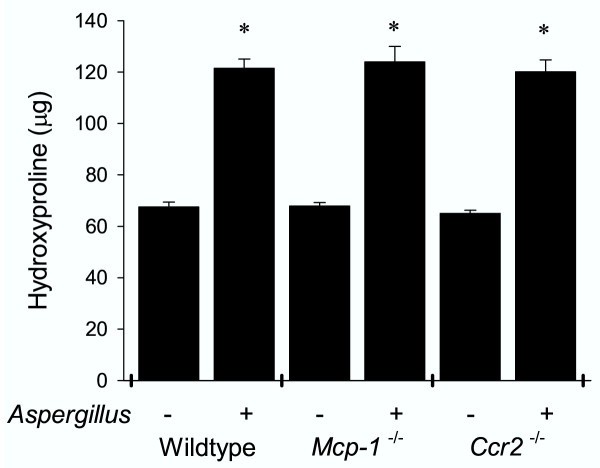
Aspergillus antigen induced similar lung fibrosis in wild-type, Mcp-1-/- and Ccr2-/- mice. Left lungs from Aspergillus antigen- or PBS-treated wild-type, Mcp-1-/- and Ccr2-/- mice were analyzed for total hydroxyproline content as described in methods. Results are expressed as mean ± SE (N = 10 mice/group). Aspergillus antigen exposure and sample collection are described in methods; data are representative of two separate experiments. Asterisks (*) indicate values that are statistically significantly different (p < 0.001) compared to PBS controls.
Figure 7.
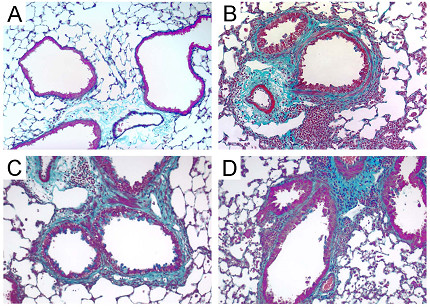
Increased airway subepithelial collagen deposition after treatment with Aspergillus antigen. Representative lung sections from PBS-treated mice show minimal trichrome staining around small airways (A) (similar findings from Mcp-1-/- and Ccr2-/- control mice are not shown). Increased trichrome staining is noted around small airways in Aspergillus antigen-treated wild-type (B), Mcp-1-/- (C) and Ccr2-/- (D) mice. Blue staining around airways represents collagen. Aspergillus antigen exposure and sample collection are described in methods. Magnification, 20× objective.
Discussion
We hypothesized that MCP-1 and its receptor, CCR2, are independently required for the development of Aspergillus-antigen-induced allergic asthma. We found that wild-type C57BL/6 mice challenged with Aspergillus antigen developed robust Th2 responses associated with pulmonary inflammation, AHR, mucus production and fibrosis. Surprisingly, neither MCP-1 nor CCR2 was critical for the development of these lung pathologies, since robust responses were also seen in mice with deletions of genes encoding either protein. These results demonstrate that neither MCP-1 nor CCR2 are required for the development of experimental allergic asthma induced by exposure to Aspergillus antigen.
Our results stand in contrast to some previous reports showing important roles for MCP-1 or CCR2 in other models of allergic asthma [25,27,33]. Although the precise explanation of these differences is not clear, there are several experimental factors that may contribute. For example, the choice of antigen and the route of sensitization differ between models. We used antigens prepared from Aspergillus, an important allergen in some people with asthma, and administered it exclusively to the respiratory tract, presumably a relevant route for sensitization in asthma. Previous studies have used ovalbumin [25,26,33] or cockroach antigen [27] and have used intraperitoneal antigen injections to sensitize prior to antigen challenge. CCR2-deficient mice have been shown to have defects in recruitment of antigen-presenting cells to the peritoneum [21,34,35], suggesting that CCR2 could be important for sensitization when antigen is administered to the peritoneum. Another factor that differs between studies is timing. We studied mice at 4 days after the final allergen challenge, when all aspects of the Aspergillus antigen-induced experimental asthma phenotype are present. Campbell et al. found that the administration of MCP-1 antibody could inhibit AHR in cockroach antigen sensitized and challenged mice at very early time points (1 and 8 h post challenge) but not later (24 h after challenge) [27]. The effect on AHR at 1 and 8 h was ascribed to MCP-1's ability to activate mast cells, which are important in some asthma models but not in others [36]. Genetic background may also be an important factor, since mouse strains vary widely in their response to airway antigen challenge [37]. Previous experimental asthma studies involving CCR2-deficient mice have used mice of mixed genetic backgrounds [25-27], whereas we used mice that had been backcrossed nine times to C57BL/6 and therefore have a more homogenous genetic background. Some of the specifics of our experimental system may therefore account for the lack of a requirement for MCP-1 and CCR2. However, MacLean et al. [26] used an allergic asthma model involving ovalbumin, intraperitoneal sensitization, and mice of mixed genetic backgrounds and found that CCR2-deficient mice had intact responses to allergen challenge. This indicates that the lack of a requirement for CCR2 is not unique to a single asthma model. It also highlights the difficulty in pinpointing the experimental factors that account for the diverse results reported by various investigators.
Of note, neither MCP-1 nor CCR2 was critical for inflammatory cell migration to the lungs after Aspergillus antigen challenge. We found that Aspergillus antigen-induced monocyte recruitment (as measured by counting BAL fluid macrophages) was intact in both MCP-1- and CCR2-deficient mice. While intact alveolar macrophage recruitment in response to airway instillation of Saccharopolyspora rectivirgula has been reported in CCR2-deficient mice [38], other in vivo models have demonstrated requirements for MCP-1 and CCR2 in monocyte/macrophage recruitment [19,39-42]. Our finding indicates that other chemoattractants are sufficient for maximal monocyte/macrophage recruitment in this Aspergillus antigen model. In support of this observation, a recent microarray-based analysis of gene expression changes in a similar asthma model found that 14 different chemokines (including MCP-1/JE) were induced by Aspergillus antigen challenge [43]. However, we did find that MCP-1 and CCR2 may have indirect effects on eosinophil recruitment in response to Aspergillus antigen. While there was marked eosinophil recruitment to the lungs in MCP-1- and CCR2-deficient mice, there was a trend toward fewer eosinophils than in wild-type mice. Since MCP-1 is not a chemoattractant for eosinophils (which lack CCR2), this trend suggests that MCP-1 may have indirect effects on eosinophil recruitment in this model. A more dramatic decrease of eosinophil recruitment has been seen following neutralization of MCP-1 in another model, but that effect was associated with other signs of impaired Th2 immunity [33]. Although there may be some role for MCP-1 and CCR2 in eosinophil recruitment, robust inflammatory responses to Aspergillus antigen occurred even in the complete absence of either of these molecules.
In contrast to our results indicating a robust Th2 response in MCP-1- and CCR2-deficient mice after Aspergillus antigen challenge, diminished Th2 cytokine production has been reported in studies of MCP-1 neutralization or deletion in different models [19,20,33,44,45]. In studies involving CCR2-deficient mice, the results have been more heterogenous, suggesting that CCR2 deletion may increase [25,28], decrease [24], or have no effect on Th2 responses [26]. As mentioned previously, the explanation for these different Th2 responses in CCR2-deficient mice is not clear, and may suggest that complex pathways involving other CCR2 ligands or MCP-1 receptors [46] are operational in different models of inflammation. However, if these pathways exist and were important in the model we used, we would have expected to find that deletion of MCP-1 and CCR2 had different effects. Instead, we observed that MCP-1- and CCR2-deficient mice were similar in all respects, including cytokine production, IgE production, and AHR. Our results support the idea that the role of MCP-1 and CCR2 in the development of allergic responses may be dependent upon the experimental model used.
The role of MCP-1 and CCR2 in the development of allergen-induced airway fibrosis has not been extensively explored. Previous findings of increased pulmonary fibrosis in CCR2-deficient mice compared to wild-type mice after treatment with Aspergillus conidia were accompanied by neutrophilic inflammation and the inability of CCR2-deficient mice to clear the organism normally [28,29]. Consequently, the persistence of Aspergillus organisms in the airway may have altered the fibrotic response. Other studies involving different experimental systems have suggested that MCP-1 and CCR2 may directly or indirectly contribute to the development of fibrosis. Gharaee-Kermani et al. [47] found that MCP-1 directly induced increased production of collagen by cultured fibroblasts, although the role of CCR2 was not explored in that report. MCP-1 and CCR2 may also indirectly influence fibrosis via their effects on inflammatory cells. Previous studies showed that CCR2-deficient mice developed less pulmonary fibrosis in response to three different stimuli, including intratracheal bleomycin instillation, than did wild-type mice [48,49]; however, those studies did not test the requirement for MCP-1 in the development of fibrosis. In C57BL/6 mice, bleomycin induces a robust inflammatory response that consists of neutrophils and lymphocytes, with a smaller component of eosinophils [50], in contrast to our allergen model. Thus, it is possible that the relative abundance or types of recruited cells in response to a particular airway challenge greatly influence the character or extent of lung fibrosis mediated by MCP-1 or CCR2.. Therefore, based on these previously published results we might have expected MCP-1 and CCR2 to be critical to the development of allergen-mediated fibrosis. However, we found that MCP-1-deficient and CCR2-deficient mice each developed marked fibrosis following Aspergillus antigen challenge, similar to wild-type mice. Our result, in contrast to the reported requirement for CCR2 in the development of bleomycin-induced pulmonary fibrosis, suggests that different cell types and mediators may be operational in allergen-induced airway fibrosis than those observed in bleomycin-induced lung fibrosis.
Conclusions
In conclusion, this study demonstrates that pulmonary inflammation, Th2 immune responses, Th2-mediated airway pathology, and lung fibrosis are remarkably intact despite the complete absence of MCP-1 or CCR2 in an Aspergillus antigen-driven model of allergic airway disease. Previous studies have demonstrated roles for MCP-1 and CCR2 in other models of inflammation and fibrosis, including different allergic airway disease models [25,27,33]. Those findings indicate that the role of MCP-1 and CCR2 in allergic responses and in fibrosis depends on the models used, although it is difficult to identify which experimental factors determine whether MCP-1 and CCR2 are required. Both MCP-1 and CCR2 may be good therapeutic targets for some diseases. However, the variable involvement of these potential targets in animal models indicates that it may be extremely challenging to predict which human diseases are most likely to benefit from this approach.
Abbreviations
AHR, airways hyperreactivity; BALF, bronchoalveolar lavage fluid.
Authors' contributions
LLK conceived of the experiment, carried out all experiments and prepared the manuscript. MWR assisted in collection and analysis of mouse samples. XLB performed all mouse airway measurements. SC and XH performed antigen challenge and assisted in collection and analysis of mouse samples. IFC and BJR provided the targeted knock-out mice, provided expert advice and interpretation of the study's results. DJE participated in the study's design, coordination and final revisions of the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We thank Yee Hwa Yang for statistical assistance and Dean Sheppard for helpful comments.
Contributor Information
Laura L Koth, Email: lkoth@itsa.ucsf.edu.
Madeleine W Rodriguez, Email: mwrod@itsa.ucsf.edu.
Xin Liu Bernstein, Email: xinliub@itsa.ucsf.edu.
Salina Chan, Email: salinac@itsa.ucsf.edu.
Xiaozhu Huang, Email: benny@itsa.ucsf.edu.
Israel F Charo, Email: icharo@gladstone.ucsf.edu.
Barrett J Rollins, Email: Barrett_Rollins@dfci.harvard.edu.
David J Erle, Email: erle@itsa.ucsf.edu.
References
- Sousa AR, Lane SJ, Nakhosteen JA, Yoshimura T, Lee TH, Poston RN. Increased expression of the monocyte chemoattractant protein-1 in bronchial tissue from asthmatic subjects. Am J Respir Cell Mol Biol. 1994;10:142–147. doi: 10.1165/ajrcmb.10.2.8110469. [DOI] [PubMed] [Google Scholar]
- Alam R, York J, Boyars M, Stafford S, Grant JA, Lee J, Forsythe P, Sim T, Ida N. Increased MCP-1, RANTES, and MIP-1alpha in bronchoalveolar lavage fluid of allergic asthmatic patients. Am J Respir Crit Care Med. 1996;153:1398–1404. doi: 10.1164/ajrccm.153.4.8616572. [DOI] [PubMed] [Google Scholar]
- Tonnel AB, Gosset P, Tillie-Leblond I. Characteristics of the Inflammatory response in bronchial lavage fluids from patients with status asthmaticus. Int Arch Allergy Immunol. 2001;124:267–271. doi: 10.1159/000053729. [DOI] [PubMed] [Google Scholar]
- Antoniades HN, Neville-Golden J, Galanopoulos T, Kradin RL, Valente AJ, Graves DT. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 1992;89:5371–5375. doi: 10.1073/pnas.89.12.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A. Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Crit Care Med. 1994;149:655–659. doi: 10.1164/ajrccm.149.3.8118632. [DOI] [PubMed] [Google Scholar]
- Iyonaga K, Takeya M, Saita N, Sakamoto O, Yoshimura T, Ando M, Takahashi K. Monocyte chemoattractant protein-1 in idiopathic pulmonary fibrosis and other interstitial lung diseases. Hum Pathol. 1994;25:455–463. doi: 10.1016/0046-8177(94)90117-1. [DOI] [PubMed] [Google Scholar]
- Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J. 1999;14:376–382. doi: 10.1034/j.1399-3003.1999.14b23.x. [DOI] [PubMed] [Google Scholar]
- Valente AJ, Graves DT, Vialle-Valentin CE, Delgado R, Schwartz CJ. Purification of a monocyte chemotactic factor secreted by nonhuman primate vascular cells in culture. Biochemistry. 1988;27:4162–4168. doi: 10.1021/bi00411a039. [DOI] [PubMed] [Google Scholar]
- Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med. 1989;169:1485–1490. doi: 10.1084/jem.169.4.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Robinson EA, Tanaka S, Appella E, Kuratsu J, Leonard EJ. Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J Exp Med. 1989;169:1449–1459. doi: 10.1084/jem.169.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994;91:3652–3656. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P, Bianchi G, Zhou D, van Damme J, Jilek P, Sozzani S, Mantovani A. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur J Immunol. 1994;24:3233–3236. doi: 10.1002/eji.1830241249. [DOI] [PubMed] [Google Scholar]
- Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Activation of NK cells by CC chemokines. Chemotaxis, Ca2+ mobilization, and enzyme release. J Immunol. 1996;156:322–327. [PubMed] [Google Scholar]
- Bischoff SC, Krieger M, Brunner T, Dahinden CA. Monocyte chemotactic protein 1 is a potent activator of human basophils. J Exp Med. 1992;175:1271–1275. doi: 10.1084/jem.175.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuna P, Reddigari SR, Rucinski D, Oppenheim JJ, Kaplan AP. Monocyte chemotactic and activating factor is a potent histamine-releasing factor for human basophils. J Exp Med. 1992;175:489–493. doi: 10.1084/jem.175.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam R, Lett-Brown MA, Forsythe PA, Anderson-Walters DJ, Kenamore C, Kormos C, Grant JA. Monocyte chemotactic and activating factor is a potent histamine-releasing factor for basophils. J Clin Invest. 1992;89:723–728. doi: 10.1172/JCI115648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol. 1997;158:4129–4136. [PubMed] [Google Scholar]
- Lloyd C. Chemokines in allergic lung inflammation. Immunology. 2002;105:144–154. doi: 10.1046/j.1365-2567.2002.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. 2000;404:407–411. doi: 10.1038/35006097. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- Traynor TR, Herring AC, Dorf ME, Kuziel WA, Toews GB, Huffnagle GB. Differential roles of CC chemokine ligand 2/monocyte chemotactic protein-1 and CCR2 in the development of T1 immunity. J Immunol. 2002;168:4659–4666. doi: 10.4049/jimmunol.168.9.4659. [DOI] [PubMed] [Google Scholar]
- Warmington KS, Boring L, Ruth JH, Sonstein J, Hogaboam CM, Curtis JL, Kunkel SL, Charo IR, Chensue SW. Effect of C-C chemokine receptor 2 (CCR2) knockout on type-2 (schistosomal antigen-elicited) pulmonary granuloma formation: analysis of cellular recruitment and cytokine responses. Am J Pathol. 1999;154:1407–1416. doi: 10.1016/S0002-9440(10)65394-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Sung S, Kuziel WA, Feldman S, Fu SM, Rose CE., Jr Enhanced airway Th2 response after allergen challenge in mice deficient in CC chemokine receptor-2 (CCR2) J Immunol. 2001;166:5183–5192. doi: 10.4049/jimmunol.166.8.5183. [DOI] [PubMed] [Google Scholar]
- MacLean JA, De Sanctis GT, Ackerman KG, Drazen JM, Sauty A, DeHaan E, Green FH, Charo IF, Luster AD. CC chemokine receptor-2 is not essential for the development of antigen-induced pulmonary eosinophilia and airway hyperresponsiveness. J Immunol. 2000;165:6568–6575. doi: 10.4049/jimmunol.165.11.6568. [DOI] [PubMed] [Google Scholar]
- Campbell EM, Charo IF, Kunkel SL, Strieter RM, Boring L, Gosling J, Lukacs NW. Monocyte chemoattractant protein-1 mediates cockroach allergen-induced bronchial hyperreactivity in normal but not CCR2-/- mice: the role of mast cells. J Immunol. 1999;163:2160–2167. [PubMed] [Google Scholar]
- Blease K, Mehrad B, Standiford TJ, Lukacs NW, Gosling J, Boring L, Charo IF, Kunkel SL, Hogaboam CM. Enhanced pulmonary allergic responses to Aspergillus in CCR2-/- mice. J Immunol. 2000;165:2603–2611. doi: 10.4049/jimmunol.165.5.2603. [DOI] [PubMed] [Google Scholar]
- Blease K, Mehrad B, Lukacs NW, Kunkel SL, Standiford TJ, Hogaboam CM. Antifungal and airway remodeling roles for murine monocyte chemoattractant protein-1/CCL2 during pulmonary exposure to Asperigillus fumigatus conidia. J Immunol. 2001;166:1832–1842. doi: 10.4049/jimmunol.166.3.1832. [DOI] [PubMed] [Google Scholar]
- Corry DB, Grunig G, Hadeiba H, Kurup VP, Warnock ML, Sheppard D, Rennick DM, Locksley RM. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol Med. 1998;4:344–355. doi: 10.1007/s008940050092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–889. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. R: A language for data analysis and graphics. Journal of Computational and Graphical Statistics. 1996;5:299–314. [Google Scholar]
- Gonzalo JA, Lloyd CM, Wen D, Albar JP, Wells TN, Proudfoot A, Martinez AC, Dorf M, Bjerke T, Coyle AJ, Gutierrez-Ramos JC. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J Exp Med. 1998;188:157–167. doi: 10.1084/jem.188.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, Gelfand EW. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med. 1997;186:449–454. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lamm WJ, Albert RK, Chi EY, Henderson WR, Jr, Lewis DB. Influence of the route of allergen administration and genetic background on the murine allergic pulmonary response. Am J Respir Crit Care Med. 1997;155:661–669. doi: 10.1164/ajrccm.155.2.9032210. [DOI] [PubMed] [Google Scholar]
- Schuyler M, Gott K, Cherne A. Experimental hypersensitivity pneumonitis:role of MCP-1. J Lab Clin Med. 2003;142:187–195. doi: 10.1016/S0022-2143(03)00107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus UA, Waelsch K, Kuziel WA, Delbeck T, Mack M, Blackwell TS, Christman JW, Schlondorff D, Seeger W, Lohmeyer J. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol. 2003;170:3273–3278. doi: 10.4049/jimmunol.170.6.3273. [DOI] [PubMed] [Google Scholar]
- Rutledge BJ, Rayburn H, Rosenberg R, North RJ, Gladue RP, Corless CL, Rollins BJ. High level monocyte chemoattractant protein-1 expression in transgenic mice increases their susceptibility to intracellular pathogens. J Immunol. 1995;155:4838–4843. [PubMed] [Google Scholar]
- Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol. 1995;155:5769–5776. [PubMed] [Google Scholar]
- Gunn MD, Nelken NA, Liao X, Williams LT. Monocyte chemoattractant protein-1 is sufficient for the chemotaxis of monocytes and lymphocytes in transgenic mice but requires an additional stimulus for inflammatory activation. J Immunol. 1997;158:376–383. [PubMed] [Google Scholar]
- Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, Hamid Q, Rothenberg ME. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI200317912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chensue SW, Warmington KS, Ruth JH, Sanghi PS, Lincoln P, Kunkel SL. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formation: relationship to local inflammation, Th cell expression, and IL-12 production. J Immunol. 1996;157:4602–4608. [PubMed] [Google Scholar]
- Nakajima H, Kobayashi M, Pollard RB, Suzuki F. Monocyte chemoattractant protein-1 enhances HSV-induced encephalomyelitis by stimulating Th2 responses. J Leukoc Biol. 2001;70:374–380. [PubMed] [Google Scholar]
- Schecter AD, Berman AB, Yi L, Ma H, Daly CM, Soejima K, Rollins BJ, Charo IF, Taubman MB. MCP-1-dependent signaling in CCR2(-/-)aortic smooth muscle cells. J Leukoc Biol. 2004;75:1079–1085. doi: 10.1189/jlb.0903421. [DOI] [PubMed] [Google Scholar]
- Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- Moore BB, Paine R, 3rd, Christensen PJ, Moore TA, Sitterding S, Ngan R, Wilke CA, Kuziel WA, Toews GB. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol. 2001;167:4368–4377. doi: 10.4049/jimmunol.167.8.4368. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Ma B, Zheng T, Homer RJ, Lee CG, Charo IF, Noble P, Elias JA. IL-13-induced chemokine responses in the lung: role of CCR2 in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2002;168:2953–2962. doi: 10.4049/jimmunol.168.6.2953. [DOI] [PubMed] [Google Scholar]
- Hao H, Cohen DA, Jennings CD, Bryson JS, Kaplan AM. Bleomycin-induced pulmonary fibrosis is independent of eosinophils. J Leukoc Biol. 2000;68:515–521. [PubMed] [Google Scholar]