

Abstract
Myeloid-derived suppressor cells (MDSCs) increase late sepsis immunosuppression and mortality in mice. We reported that microRNA (miR) 21 and miR-181b expression in Gr1+CD11b+ myeloid progenitors increase septic MDSCs in mice by arresting macrophage and dendritic cell differentiation. Here, we report how sepsis regulates miR-21 and miR-181b transcription. In vivo and in vitro binding studies have shown that C/EBPα transcription factor, which promotes normal myeloid cell differentiation, binds both miRNA promoters in Gr1+CD11b+ cells from sham mice. In contrast, in sepsis Gr1+CD11b+ MDSCs miR-21 and miR-181b promoters bind both transcription factors Stat3 and C/EBPβ, which co-imunoprecipitate as a single complex. Mechanistically, transcription factor Rb phosphorylation supports Stat3 and C/EBPβ accumulation at both miRNA promoters, and C/EBPβ or Stat3 depletion by siRNA in sepsis Gr1+CD11b+ MDSCs inhibits miR-21 and miR-181b expression. To further support this molecular path for MDSC accumulation, we found that Stat3 and C/EBP binding at miR-21 or miR-181b promoter was induced by IL-6, using a luciferase reporter gene transfection into naive Gr1+CD11b+ cells. Identifying how sepsis MDSCs are generated may inform new treatments to reverse sepsis immunosuppression.
Keywords: sepsis, MDSCs, microRNA, Stat3, C/EBPβ, myeloid cell differentiation
Introduction
Myeloid-derived suppressor cells (MDSCs) are a heterogenous population of immature myeloid cells, mainly progenitors and precursors of monocytes/macrophages, granulocytes, and dendritic cells.1,2 In mice, MDSCs are broadly identified by expression of the myeloid markers Gr1 and CD11b.1 Since the Gr1 antigen is comprised of two epitopes, Ly6G and Ly6C, murine MDSCs can be divided into two categories based on morphology and expression of Ly6G and Ly6C.2,3 Granulocytic MDSCs resemble granulocytes and are CD11b+Ly6G+Ly6Clow and monocytic MDSCs resemble monocytes and are CD11b+Ly6G−Ly6Chigh.4 Because the Gr1 marker is not expressed on human myeloid cells, human MDSCs are broadly defined as CD11b+CD33+HLA-DRlow/−, which can be further classified as granulocytic MDSCs expressing CD15 and monocytic MDSCs expressing CD14.5–7 MDSCs expand and accumulate under most acute and chronic inflammatory conditions, including sepsis and cancer2,8,9 and are potently immunosuppressive, as they suppress both innate and adaptive immune responses.1,8,10 MDSCs suppress the immune resposes via production of nitric oxide (NO), reactive oxygen species, and arginase, as well as pro- and anti-inflammatory/immunosuppressive cytokines such as IL-10 and TGFβ.1,4,11 Different MDSC subsets may use different mechanisms to suppress the host immunity. For example, monocytic MDSCs produce NO and arginase to inactivate the immune response and suppress T cells,4,12 whereas granulocytic MDSCs use ROS to suppress immune cells.4 MDSCs may accumulate when stress-induced mediators disrupt normal myeloid cell differentiation, leading to MDSC expansion/accumulation. In support of this, immature myeloid cells from bone marrow of naive mice differentiate normally into mature innate cells.1,13 Although cytokine and growth factors, including IL-6, M-CSF, G-CSF and VEGF, support MDSC expansion,14 the responsible molecular mechanism remains unclear.
MicroRNAs (miRNAs) as regulators of post-transcriptional gene expression 15,16 contribute to cell development, differentiation and homeostasis.17,18 MiRNAs, which are small (~22-nucleotides), single-stranded noncoding RNAs encoded by genomic DNA, are most commonly transcribed by RNA polymerase II.19,20 Once transcribed, the primary miRNA transcript, which is capped with polyadenylated RNA, is cleaved by the RNase III enzyme Drosha in combination with DGCR8 enzyme to produce a hairpin precursor miRNA of ~65-nucleotides.19,21 The precursor miRNA is then exported to the cytoplasm by exportin 5 and is further cleaved by the RNase III enzyme Dicer to produce a miRNA duplex. The Dicer loaded miRNA duplex is then recruited to the RNA-binding protein argonaute 2 by TRBP to form the miRNA-induced silencing complex (miRISC), which also includes regulatory, target-specific RNA-binding proteins.15,19 Within miRISC, the miRNA duplex is unwound where one strand (the passenger strand) is cleaved leaving a single-stranded mature miRNA (the guide strand) to guide the miRISC to the 3′ untranslated region (UTR) of its target mRNA, resulting in inhibition of its translation and/or a decrease in its stability.19,22 The specific effect of a miRNA is thought to be influenced by the “seed” region, which includes nucleotide numbers 2–8 located at the 5′ end of the miRNA sequence and nucleates its interaction with the target 3′ UTR.23 Perfect base-pairing between the “seed” region and the target 3′ UTR induces the target mRNA instability and degradation, whereas imperfect base-pairing can inhibit the mRNA translation.19,24 Recent studies indicate that miRNA expression can be regulated at the transcriptional level by cell-specific transcription factors and also at the transcript processing levels by other protein cofactors, which can be induced during inflammation or cellular stress.15,25
We reported that miR-21 and miR-181b expression is required for MDSC accumulation in late septic mice.26 Here, we investigated how miR-21 and miR-181b induction is regulated during sepsis. We find that Stat3 and C/EBPβ together activate miR-21 and miR-181b promoters in MDSCs, and that promoter binding requires Rb protein phosphorylation.
RESULTS
Stat3 phosphorylation and C/EBPβ protein levels increase during sepsis and induce miR-21 and miR-181b expression in MDSCs
Stat3 and the CCAAT/enhancer-binding protein (C/EBP) β regulate myeloid cell development and differentiation.27,28 We reported that accumulation of Gr1+CD11b+ MDSCs in septic mice is due to arrested differentiation of the Gr1+CD11b+ myeloid progenitors. We also discovered that Stat3 phosphorylation at tyrosine705 and C/EBPβ protein levels are increased during sepsis, when measured in whole bone marrow cells.26 We hypothesized that the increases in Stat3 phosphorylation and C/EBPβ protein expression may lead to the dysregulation of myeloid cell differentiation and maturation via increasing miR-21 and miR-181b, resulting in Gr1+CD11b+ MDSC accumulation.
First, we determined Stat3 phosphorylation (p-Stat3) and C/EBPβ levels in purified Gr1+CD11b+ cells isolated from the bone marrow of sham (normal) and septic mice. We also measured levels of C/EBPα, which like C/EBPβ binds the same C/EBP consensus site on target gene promoters,29 and has been implicated in myeloid cell development under normal physiological conditions.27 Western blot analysis showed that Stat3 phosphorylation was induced in septic Gr1+CD11b+ cells while Stat3 protein levels remained unchanged (Fig. 1A). In addition, C/EBPβ protein was induced markedly, while C/EBPα expression was not changed (Fig. 1B). We observed that p-Stat3 and C/EBPβ levels were higher during late sepsis. This may reflect changes in the number of MDSCs during the course of sepsis inflammation. We previously showed that late sepsis Gr1+CD11b+ cells contain more immature, immunosuppressive Gr1+CD11b+ cells (i.e., MDSCs) than Gr1+CD11b+ cells from early septic mice, which unlike late sepsis MDSCs, can differentiate readily ex vivo.8 Thus, these results suggest that the increases in Stat3 phosphorylation and C/EBPβ protein levels occur only in Gr1+CD11b+ MDSCs.
Figure 1. Stat3 and C/EBPβ activate miR-21 and miR-181b expression in sepsis Gr1+CD11b+ MDSCs.

Sepsis was induced by cecal ligation and puncture (CLP). (A and B) Bone marrow cells were harvested from septic mice that were moribund and sacrificed at days 1–5 (representing early sepsis) and at days 6–28 (representing late sepsis) as well as sham mice. Gr1+CD11b+ cells were then purified using magnetic beads and pooled from 6 mice per group. Cell lysates were prepared and levels of total and phosphorylated (p-Stat3; Tyr705) Stat3 (A), and C/EBPβ proteins (B) were determined by immunoblot. The results are representative of two experiments. Lower panel in A shows densitometry of the p-Stat3 bands. Values were normalized to β-actin and are presented relative to sham, which is set at 1-fold. (C) Knockdown of Stat3 or C/EBPβ in sepsis MDSCs inhibits miR-21 and miR-181b expression. Gr1+ CD11b+ cells were isolated from the bone marrow of late septic mice, pooled (n = 6 mice per group), and transfected with Stat3-specific, C/EBPβ-specific, or control siRNA. After 36 hr in culture, cells were harvested and levels of miR-21 and miR-181 were determined by northern blot. Levels of the U6 RNA were also measured as an internal control. The knockdown was confirmed by western blotting (not shown). (D) Phosphorylation of Stat3 in the bone marrow during sepsis is restricted to the Gr1+CD11b+ MDSCs. CD3+ T cells were isolated from whole bone marrow cells by positive selection using anti-CD3 magnetic beads. To isolate CD19+ cells, whole bone marrow cells from late septic mice were first depleted of Gr1−CD11b+ cells (which mostly consists of CD19+ and CD11c+ cells). CD19+ cells were then positively selected from the depleted cell population using anti-CD19 magnetic beads. The results are representative of two immunoblots using two different cell preparations. (E) A schematic diagram depicting the Stat3 and C/EBPβ binding sites in the miR-21 and miR-181b promoters. (F) The miR-21 and miR-181b promoter fragments used in the luciferase gene constructs (see Fig. 6). The Stat3 and C/EBP binding sites are underlined. The core nucleotides of the consensus binding sites are bolded. Lower case letters indicate the mutation introduced in the binding site.
To determine whether the increases in Stat3 phosphorylation and C/EBPβ protein levels play a role in the induction of miR-21 and miR-181b expression, we performed northern blot analysis using late sepsis MDSCs with Stat3 and C/EBPβ knockdown. As shown in Fig. 1C, knockdown of Stat3 or C/EBPβ reduced levels of miR-21 and miR-181b markedly. Knockdown of C/EBPα did not affect the miRNA expression (data not shown). These results suggest that p-Stat3 and C/EBPβ may induce miR-21 and miR-181b expression during sepsis. In addition, western blot analysis revealed that the induction of Stat3 phosphorylation was specific to the Gr1+CD11b+ MDSCs, because we did not detect p-Stat3 in CD3+ T cells or CD19+ B cells from septic mice (Fig. 1D).
p-Stat3 and C/EBPβ bind the miR-21 and miR-181 promoters in MDSCs
Examination of miRBase database (http:mirbase.org/search) revealed that mouse miR-181b is encoded in two independent transcripts, each has two paralogs: miR-181a-1/b-1, which contains miR-181a-1 and miR-181b-1 and is located on chromosome 1 and miR-181a-2/b-2, which contains miR-181a-2 and miR-181b-2 and is located on chromosome 2. Whereas miR-181b-1 and miR-181b-2 primary transcripts have slightly different sequence homology, they produce the same mature miR-181b. Our previous microarray study showed that miR-181b-1 (also known as miR-181b) was induced during sepsis. 26 On the other hand, miR-21 is located on chrmosome 11. Using Ensembl database (www.ensembl.org), we identified 10 kb DNA fragments upstream of the miR-21 and miR-181b-1 transcripts (assigned +1) as putative regulatory elements of miR-21 and miR-181b-1.
To study the transcription regulation of miR-21 and miR-181 expression, we performed bioinformatic analysis using MatInspector Software (Genomatix Software GmbH, Munich, Germany) to identify transcription factor consensus binding sites within the 10 kb upstream of the primary transcripts (+1) and also 5 kb downstream sequences of the miR-21 and miR-181b transcripts. The analysis revealed several binding sites for the Stat3 and C/EBP transcription factors within the sequences upstream of miR-21 and miR-181b transcripts. We were interested in Stat3 and C/EBP binding sites because the knockdown of Stat3 or C/EBPβ reduced miR-21 and miR-181b expression (Fig. 1C). Further analysis of these regulatory elements revealed two sets of Stat3 and C/EBP binding sites on both promoters. One set is close to the miRNA transcription start site and the second set is located further upstream. The upstream binding sites in both promoters (Fig. 1E), unlike the proximal sites, are in close proximity of one another and have the highest binding affinity score, and therefore were analyzed in detail. These consensus binding sites and their flanking sequences are shown in Fig. 1F. Importantly, the core sequences of the Stat3 (TTCC) and C/EBP (GGAA) binding sites are conseved in miR-21 and miR-181b promoters.
To determine whether the Stat3 or C/EBP binding sites are directly involved in the transcription activation of miR-21 and miR-181b during sepsis, we performed chromatin immunoprecipitation (ChIP) assays to examine protein-DNA interactions at the miRNA promoters in vivo. Real-time PCR analysis of the immunoprecipitated DNA showed that the miR-21 promoter sequence containing the Stat3 and C/EBP binding sites at -2133 and -2197, respectively, were enriched in the p-Stat3 and C/EBPβ protein complexes in Gr1+CD11b+ MDSCs (i.e, during sepsis only). The levels of p-Stat3 and C/EBPβ recruitment to the miR-21 and miR-181b promoters were significantly higher in late sepsis compared with early sepsis, as numbers of the immature CD31+ cells are increased during late sepsis.8 On the other hand, the promoter DNA was enriched in the C/EBPα immunoprecipitate isolated only from the Gr1+CD11b+ cells of sham mice (Fig. 2A; left panel). Closely similar DNA enrichment was observed for the miR-181 promoter, except that the levels were slightly higher than those from the miR-21 promoter. In addition, we did not detect p-Stat3 or C/EBPβ protein binding to the proximal Stat3 and C/EBP sites on either promoter (data not shown).
Figure 2. Stat3 and C/EBPβ, but not C/EBPα, bind to miR-21 and miR-181 promoter in sepsis Gr1+CD11b+ MDSCs.

(A) Protein binding in total Gr1+CD11b+ cell population. Gr1+CD11b+ cells were isolated and pooled (n = 6 mice per group) from the bone marrow of sham and septic mice. Cells were fixed in 1% formaldehyde to cross-link protein-DNA complexes. Cells were lyzed and the pelleted nuclei were digested with chromatin shearing enzymatic cocktail. The chromatin solution was then immunoprecipitated with antibodies specific to p-Stat3 (Tyr705), C/EBPβ, C/EBPα, p-Rb (Ser780), or IgG isotype control antibody. Next, chromatin cross-links were reversed to recover the protein-bound DNA. The purified DNA was amplified by qPCR to measure the level of enrichment of miR-21 and miR-181b sequences in the immunoprecipitated complexes using promoter-specific primer/probe sets. PCR reactions were performed in triplicate. Samples were normalized to the “input” DNA (i.e., DNA isolated before immunoprecipitation) and are presented as fold enrichment relative to the IgG-immunoprecipitated samples (set at 1-fold). Data are expressed as mean ± s.d. (*p ≤ 0.05) of three experiments. *, compare with early sepsis. (B) Stat3 and C/EBPβ binding to the miR-21 and miR-181 promoters in Gr1+CD11b+ MDSCs is restriced to the CD31+ subset. The CD31+ cell were then purified from the total Gr1+CD11b+ cell population by positive selection. Cells were treated, and chromatin was immunoprecipitated as described above. PCR was performed to measure the levels of the miR-21 and miR-181b promoter DNA sequence enrichment in chromatin immunoprecipitated from the CD31+-enriched Gr1+CD11b+ cells from the bone marrow and spleens. Data are expressed as mean ± s.d. of three experiments.
p-Stat3 and C/EBPβ binding to the miR-21 and miR-181b promoters is restricted to the CD31+ subset of the Gr1+CD11b+ MDSCs
MDSCs include myeloid progenitors and precursors at differenent maturation stages.1 We previously showed that the CD31+ cell subset, a more immature stage of Gr1+CD11b+ cells, increases from 34% in early sepsis to 79% in late sepsis.8 We hypothesized that the CD31+ subset are mostly Gr1+CD11b+ MDSCs in septic mice, because of dysregulated differentiation and maturation capacity,8 due to increased miR-21 and miR-181b expression. We performed ChIP to determine p-Stat3 and C/EBP protein bindings to the miRNA promoters using CD31+-enriched MDSCs. The CD31+ cells were selected from the total Gr1+CD11b+ cells isolated from the bone marrow. As shown in Fig. 2B, both p-Stat3 and C/EBPβ were bound to their putative sites on the miR-21 and miR-181b promoters only in cells from septic mice, whereas C/EBPα binding was detected only in cells derived from sham mice. The levels of p-Stat3 and C/EBPβ bindings were nearly similar in early and late sepsis, because we used equal numbers of purified CD31+ MDSCs, whereas total MDSCs from early and late sepsis (Fig. 2A) contain varying numbers of CD31+ cells. MDSCs also accumulate in the spleen under inflammatory conditions, with some studies suggesting that they may be dervied from the bone marrow due to accelerated myelopoiesis or by extramedullary hematopoiesis in the spleen.11 We next examined the p-Stat3 and C/EBPβ binding to the miRNA promoters in CD31+-enriched MDSCs isolated from spleens of late septic mice. We observed p-Stat3 and C/EBP proteins binding at the miR-21 and miR-181b promoters, a pattern similar to that in bone marrow CD31+ cells (data not shown). In addition, p-Stat3 and C/EBPβ did not bind the miR-21 or miR-181b promoters in sepsis Gr1+CD11b+ cells that lack the CD31+ subset (i.e., CD31+-depleted MDSCs) (data not shown). On the other hand, we detected C/EBPα binding in sham and sepsis cells, which was significantly higher in sham cells. These results suggest that the miR-21 and miR-181b promoter binding by p-Stat3 and C/EBPβ occurs only in the CD31+ subset of sepsis Gr1+ CD11b+ cells.
We next performed electrophoretic mobility shift assay (EMSA) to confirm the specificity of p-Stat3 and C/EBPβ binding in vitro. DNA oligonucleotides specific to the Stat3 and C/EBP binding sites in the miR-21 and miR-181b promoters were incubated with nuclear extracts from CD31+-enriched MDSCs isolated from the bone marrow of late septic mice. As shown in Fig. 3A, we detected binding to the DNA probe containing the Stat3 and C/EBP sites in the miR-21 promoter, as indicated by the formation of a DNA-protein complex (lane 2). Supershift analysis by the addition of the p-Stat3 or C/EBPβ specific antibody revealed specific binding by p-Stat3 and C/EBPβ proteins (lane 3). Specific mutations in the Stat3 or C/EBP site prevented the DNA-protein interactions (lane 4). In addition, binding to the Stat3 or C/EBP site was competed out with an excess of unlabeled DNA native probe (lane 5) but was not competed out with an excess of mutated probe (lane 6). Of note, the C/EBP probe formed two bands. The upper (minor) band appears to be non-specific becaused it was not shifted with the C/EBPβ antibody. Similar DNA-protein interactions were detected with probes covering the Stat3 and C/EBP sites on the miR-181b promoter (Fig. 3B). In addition, the Stat3 probe from miR-21 or miR-181b promoters failed to form DNA-protein complexes with protein extracts from CD31+ cells isolated from sham mice (Supplementary Fig. 1). Using the same extracts, we observed a specific DNA-protein complex with the C/EBP probe, which was supershifted with C/EBPα antibody (Supplementary Fig. 2) but not C/EBPβ antibody (data not shown). Together, these results show that the transcription factors Stat3 and C/EBPβ bind specifically to the miR-21 and miR-181b promoters in vivo and in vitro in sepsis MDSCs, and suggest that their binding activity is limited to the CD31+ subset of MDSCs.
Figure 3. Electrophoretic mobility shift assay of Stat3 and C/EBPβ binding specificity to the miR-21 and miR-181b promoters.

EMSA was performed to determine the Stat3 and C/EBPβ binding specificity to the miR-21 and miR-181b promoters. Nuclear proteins were isolated from CD31+ cells purified from the bone marrow Gr1+CD11b+ cells from late septic mice. Protein extract was incubated with biotin-labeled double-stranded DNA oligos containing the Stat3 or C/EBP binding sites in the miR-21 and miR-181b promoters. The protein-DNA complexes were then electrophoresed on 6% native polyacrylamide gel, immunoblotted, and the DNA was visualized with streptavidin-conjugated antibody and chemiluminescent reagent. (A) Stat3 and C/EBPβ binding to miR-21 promoter. In lane 3, 3 μg of antibody specific to p-Stat3 or C/EBPβ was incubated with the nuclear protein for 5 min before the addition of the labeled DNA probe. In lanes 5 and 6, 200-fold excess of unlabeled wild-type or mutated probe, respectively, was used as a competitor to confirm the binding specificity. Results are representative of three experiments. (B) Stat3 and C/EBPβ binding to miR-181b promoter. The short arrow with an asterisk indicates a non-specific band, which migrated at the same rate as the supershifted complex. The results are representative of two experiments.
p-Stat3 and C/EBPβ may bind the miR-21 and miR-181b promoters as a single complex
We set out to investigate the kinetics of p-Stat3 and C/EBPβ protein interactions at the miR-21 and miR-181b promoters. We performed co-immunoprecipitation experiments using p-Stat3 and p-Rb antibodies. We were interested in the transcription factor Rb because our previous experiments suggested that it may be involved in the MDSC accumulation during sepsis. Interaction of Rb with other transcription factors is regulated by phosphorylation on the Rb protein by cyclin-dependent kinase (CDK) protein complexes with cyclins.30–32 We find that the cyclin D1 forms a complex with CDK4 in sepsis MDSCs and that this complex is disrupted upon NFI-A knockdown.33 NFI-A protein is induced downstream of miR-21 and miR-181b, and we have shown that NFI-A knockdown in sepsis Gr1+CD11b+ MDSCs restores their ability to differentiate into macrophages and dendritic cells.26 We reasoned that elevated levels of NFI-A protein may facilitate the cyclin D1-CDK4 complex formation, which then function in a feedback loop to phosphorylate Rb and thus promotes miR-21 and miR-181b expression. To test this possibility, we first measured Rb protein levels and phosphorylation (p-Rb; Ser780) in Gr1+CD11b+ cells from sham and septic mice. While Rb protein was expressed in sham and sepsis Gr1+CD11b+ cells, p-Rb was detected in cells from septic mice only (Fig. 4A).
Figure 4. Rb is phosphorylated by cyclin D1-CDK4 protein complex in sepsis Gr1+CD11b+ cells and binds to C/EBPα, whereas p-Stat3 binds to C/EBPβ.

(A) Gr1+CD11b+ cells were isolated from the bone marrow of sham and septic mice and levels of Rb and p-Rb proteins were determined by immunoblot. (B and C) Knockdown of cyclin D1 or CDK4 inhibits Rb phosphorylation. CD31+ cells were purified from total Gr1+CD11b+ MDSCs isolated from the bone marrow of late septic mice. Cells were transfected with cyclin D1-, CDK4-specific or control siRNA. After 36 hr, cells were harvested and cell lysates were prepared for immunoblot. (B) Levels of the cyclin D1 and CDK4 proteins after the knockdown. Lower panel shows densitometry of the cyclin D1 and CDK4 protein bands. Sample values were normalized to β-actin levels. (C) levels of p-Stat3, C/EPBβ, C/EBPα, and p-Rb proteins after cyclin D1 or CDK4 knockdown. The results are representative of three experiments. (D) Co-immunoprecipitation analysis of protein binding to Rb. Bone marrow Gr1+CD11b+ cell lysates were prepared and immunoprecipitated with Rb-specific or IgG isotype control antibody using protein G-agarose beads. The immunoprecipitated protein complexes were resolved on denaturing polyacrylamide gel and then immunoblotted with specific antibody against Rb, p-Rb (Ser780), p-Stat3 (Tyr705), C/EBPβ or C/EBPα. (E) Co-immunoprecipitation analysis of protein binding to Stat3. Cell lysates were immunoprecipitated with Stat3-specific antibody and immunoblotted as in D. The results are representative of three experiments. (F) Protein levels in the crude (input) extract before the immunoprecipitation. The results are representative of two experiments.
We next assessed p-Stat3, p-Rb and C/EBPβ protein levels in late sepsis CD31+-enriched Gr1+CD11b+ MDSCs after preventing the cyclin D1-CDK4 complex formation via siRNA-mediated knockdown of cyclin D1 or CDK4, which was confirmed by western blotting (Fig. 4B). As expected, the knockdown of cyclin D1 or CDK4 inhibited Rb phosphorylation. However, it did not affect Stat3 phosphorylation or C/EBPβ or C/EBPα protein levels. (Fig. 4C). Although we did not detect p-Rb binding at the miR-21 or miR-181b promoter in ChIP assay (Fig. 2), we hypothesized that it may affect the p-Stat3 and/or C/EBP protein bindings. Co-immunoprecipitation experiments revealed that C/EBPα, but not p-Stat3 or C/EBPβ protein, was detected with p-Rb in the immunoprecipitated protein complex in sepsis Gr1+CD11b+ MDSCs (Fig. 4D). In contrast, C/EBPβ, but not C/EBPα or p-Rb, was detected in the Stat3 protein complex (Fig. 4E). These results show differential protein-protein interactions of p-Stat3 and C/EBP proteins in sepsis MDSCs and suggest that p-Stat3 and C/EBPβ may bind the miR-21 and miR-181b promoters as a single complex, since they were detected simultaneously at the miRNA promoters in ChIP assay.
Lack of Rb phosphorylation prevents p-Stat3 and C/EBPβ from binding the miR-21 and miR-181b promoters
In the ChIP assay, C/EBPα, which promotes normal, steady-state myelopoiesis and myeloid differentiation,27 binds the miR-21 and miR181b promoters in sham but not sepsis Gr1+CD11b+ cells (Fig. 2). However, C/EBPα protein was detected in the immunoprecipitated p-Rb protein complex in sepsis Gr1+CD11b+ cells (Fig. 4D), at which time p-Stat3 and C/EBPβ bind the miRNA promoters (Fig. 2). We hypothesized that phosphorylation on Rb may cause these differential bindings. To test this, we examined C/EBPα, p-Stat3 and C/EBPβ binding to the miRNA promoters in sepsis Gr1+CD11b+ cells lacking phosphorylated Rb, using ChIP assay. The knockdown of cyclin D1 or CDK4 reduced p-Rb levels markedly (Fig. 5A). Semi-quantitative and real-time qPCR analysis of the ChIP DNA revealed that p-Stat3 and C/EBPβ binding at the miR-21 and miR-181b promoters was disrupted (Fig. 5B and C). Interestingly, this was paralleled by a simultaneous binding of C/EBPα.
Figure 5. Knockdown of cyclin D1, CDK4, or Rb disrupts Stat3 and C/EBPβ binding and simultaneously induces C/EBPα binding to the miR-21 and miR-181b promoters in sepsis Gr1+CD11b+ MDSCs.

Gr1+CD11b+ cells were isolated from the bone marrow of late septic mice and transfected with cyclin D1-, CDK4-, Rb-specific or control siRNA for 36 hr. (A) Knockdown of cyclin D1 or CDK4 inhibits Rb phosphorylation. Levels of Rb and p-Rb in cell lysate were determined by immunoblot. Cells were fixed, lyzed, and chromatin immunoprecipitation (ChIP) was performed with antibody specific to p-Stat3, C/EBPβ or C/EBPα, as described in Fig. 2. (B and D) the recovered, ChIP DNA was amplified by semi-quantitative PCR using primers that flank the Stat3 and C/EBP binding sites in the miR-21 and miR-181b promoters. An IgG-immunoprecipitated samples is shown as a negative control. The results are representative of three experiments. (C and E) the recovered DNA was amplified by real-time PCR. Samples values were normalized to the “input” DNA values and are presented as fold enrichment relative to the IgG-immunoprecipitated samples (set at 1-fold). Data are expressed as mean ± s.d. of three experiments. (F) Levels of c-Myc protein after the Rb knockdown in sepsis Gr1+CD11b+ cells isolated from the bone marrow. The results are representative of two experiments.
We confirmed the role of p-Rb in regulating the C/EBPα, p-Stat3 and C/EBPβ binding activity using sepsis Gr1+CD11b+ cells lacking Rb expression due to Rb knowdown. ChIP analysis revealed that lack of Rb prevented p-Stat3 and C/EBPβ binding, while facilitating the C/EBPα binding to the miR-21 and miR-181b promoters (Fig. 5D and E). These results support that phosphorylated Rb protein regulates the C/EBPα, Stat3, and C/EBPβ binding at the miR-21 and miR-181b promoters.
The Stat3 and C/EBP binding sites on the miR-21 and miR-181b promoters activate reporter gene expression in naive Gr1+CD11b+ cells
Having determined that p-Stat3 and C/EBPβ bind to their putative sites on the miR-21 and miR-181b promoters in sepsis Gr1+CD11b+ MDSCs, we next investigated the functional significance of their binding using reporter gene assays. A miR-21 promoter fragment (106 bp) or a miR-181b promoter fragment (58 bp) containing the Stat3 and C/EBP binding sites (see Fig. 1F) were cloned upstream of the luciferase reporter gene driven by the CMV mini-enhancer, which activates the luciferase expression to a certain extent. The reporter constructs were transfected into Gr1+CD11b+ cells isolated from the bone marrow of naive mice, which do not express miR-21 or miR-181.26 The transfected cells were stimulated with IL-6 to induce Stat3 phosphorylation and C/EBPβ expression 5. As shown in Fig. 6A, the construct containing native Stat3 and C/EBP binding sites from the miR-21 promoter exhibited a significantly higher luciferase gene expression over that induced by the CMV mini-enhancer alone. The level of luciferae expression was slightly lower than the control construct containing the CMV promoter plus mini-enhancer, which was designed to produce maximum luciferase gene expression. Site-directed mutagenesis of the Stat3 and C/EBP sites, individually or in combination, reduced the luciferase expression significantly compared with the construct containing the wild-type Stat3 and C/EBP sites, and to almost the same level induced by the mini-emhancer alone. In the absence of IL-6, however, the Stat3 and C/EBP combined binding sites failed to increase the luciferase expression. We also observed a similar pattern of the luciferase gene activation with the reporter constructs containing the Stat3 and C/EBP sites from the miR-181b promoter (Fig. 6A). In addition, similar kinetic of luciferase gene expression was observed in sham and sepsis Gr1+CD11b+ cells, with slightly higher luciferase levels (Supplementary Fig. 3). However, luciferase gene was expressed to some extent even in sepsis cells, which already possess high levels of p-Stat3, C/EBPβ (Fig. 1), and p-Rb (Fig. 4A) that were required for the miRNA promoter activation. We also observed activation of luciferase gene expression after stimulation with IL-10 (data not shown), which also activates the Stat3 pathway.34,35 These results suggest that the Stat3 and C/EBP binding sites can activate the miR-21 and miR-181b promoters in the presence of IL-6 or IL-10 signal.
Figure 6. The Stat3 and C/EBP binding sites from miR-21 or miR-181b promoters enhance reporter gene expression.

(A) Gr1+CD11b+ cells were isolated from the bone marrow of naive mice. Cells were transfected with a renilla luciferase plasmid plus a luciferase plasmid containing native or mutated Stat3 and C/EBP binding sites. Cells were incubated for 24 hr without or with 10 ng/ml of recombinant mouse IL-6. Transfection with a luciferase plasmid that contains the CMV promoter only served as a control for maximum luciferase activity. Transfection with a luciferase plasmid that contains the miniCMV enhancer only (no inserts) served as a positive control. Luciferase values were normalized to renilla luciferase and are presented relative to the positive control (set at 100%). Data are expressed as mean ± s.d. (*p ≤ 0.05) of three experiments. *, compare with positive control; **, compare with Stat3 wt + Cebp wt site construct. (B) Rb knockdown in sepsis Gr1+CD11b+ MDSCs diminishes the reporter gene expression that is induced by the Stat3 and C/EBP binding sequences. Gr1+CD11b+ cells were isolated from the bone marrow of late septic mice. Cells were first transfected with pools of control or Rb-specific siRNA for 24 hr. Cells were then washed, transfected with luciferase plasmids containing the Stat3 and C/EBP binding sites from the miR promoters, and treated as described in A. Cells were incubated for 24 hr without or with 10 ng/ml of mouse rIL-6. Data are expressed as mean ± s.d. )*p ≤ 0.05) of three experiments and are presented relative to the positive control, which contains no Stat3 or C/EBP binding sites. *, compare with positive control; **, compare with Rb siRNA. (C) IL-6 induces phosphorylation of Stat3 and Rb proteins during sepsis. Gr1+CD11b+ cells were isolated from the bone marrow of late septic mice and stimulated with IL-6. Levels of p-Stat3 and p-Rb were determined by immunoblot. The results are representative of two experiments.
Rb knockdown in sepsis Gr1+CD11b+ cells abolishes the miRNA promoter-mediated activation of the reporter gene expression
The results presented in Fig. 5D reveal that Rb regulates the binding of p-Stat3 and C/EBPβ proteins to the miRNA promoters in Gr1+CD11b+ MDSCs during sepsis. Knockdown of Rb abolished binding of p-Stat3 and C/EBPβ to the miR-21 and miR-181b promoters, as they were replaced by C/EBPα, similar to what we observed in sham cells (Fig. 2). To examine the functional aspects of the Rb effect on the miR promoters, we measured the luciferase activity in sepsis Gr1+CD11b+ MDSCs after Rb knockdown, cells with Rb knockdown were transfected with the luciferase construct containing the Stat3 and C/EBP binding sites, which showed high levels of the luciferase gene activation in naive cells (Fig. 6A). The results showed that Rb knockdown significantly decreased the luciferase gene expression driven by the Stat3 and C/EBP sites (Fig. 6B). Absence of IL-6 stimulation did not inhibit the luciferase expression completely as we observed in naive cells (Fig. 6A), because these cells already possess high levels of p-Stat3. To delineate the role of IL-6 in the activation of the miR promoters, we performed immunoblotting using protein extracts from sham and sepsis Gr1+CD11b+ cells stimulated with IL-6. IL-6 stimulation induced Stat3 phosphorylation in sham cells (Fig. 6C), As expected, cells from sepsis mice expressed p-Stat3, and its level was further increased after stimulation. Also, Rb phosphorylation was further increased in sepsis cells after stimulation with IL-6. Together, these results suggest that p-Rb regulates the miR-21 and miR-181b promoter activity during sepsis and that this process requires IL-6-mediated signaling.
DISCUSSION
Induction of miR-21 and miR-181b expression during the immune response to sepsis increases the numbers of MDSCs by arresting the Gr1+CD11b+ myeloid progenitor differentiation and maturation 26. In this study, we identify Stat3 and C/EBPβ as critical transcription factors in the induction of miR-21 and miR-181b in Gr1+CD11b+ cells in septic mice. We demonstrate that Stat3 and C/EBPβ activate the miR-21 and miR-181b promoters in a similar fashion, and by a mechanism that is mediated by IL-6 signaling and requires phosphorylation on the Rb protein, which requires Stat3 and C/EBPβ binding at the miR-21 and miR-181b promoters. These findings uncover the mechanism of miR-21 and miR-181b transcription regulation and thus reveal the molecular path leading to MDSCs expansion during murine sepsis.
While our findings that miR-21 and miR-181b are involved in sepsis pathogenesis is novel, a recent study by Iliopoulos et. al.36 reported that miR-21 and miR-181b can function as part of a feedback loop that links inflammation to cancer. They showed that IL-6 mediated activation of Stat3 in human breast epithelial cell line increased miR-21 and miR-181b expression and maintained the transformed state by preventing their differentiation. In addition, Stat3 bound three putative Stat3 binding sites on the miR-21 and miR-181b promoters, and Stat3 knockdown reduced miR-21 and miR-181b expression.36 Our detailed bioinformatic analysis revealed several Stat3 binding sites on the miR-21 and miR-181b promoters. However, only a distal Stat3 site at -2133 and -1473, respectively, on miR-21 and miR-181b promoters bound p-Stat3 in vivo and in vitro in Gr1+CD11b+ cells from septic mice (Figs. 2 and 3, and data not shown). This site was also neccessary for the activation of the reporter gene expression. A recent study reported Stat3 binding to an enhancer region in miR-21 promoter ~800 bp upstream of the transcription start site in human multiple myeloma cells.37 However, we could not detect Stat3 binding at this site in Gr1+CD11b+ cells. Given that the miR-21 promoter is rich in Stat3 binding sites, it is possible that binding to some of these sites may be cell-specific. In Gr1+CD11b+ cells, we detected Stat3 binding only at the Stat3 site that is in very close proximity to the C/EBP consensus site, which binds the C/EBPβ and C/EBPα proteins. Our results suggested that Stat3 and C/EBPβ may bind the miR-21 promoter in one protein complex, and the same pattern was detected for the miR-181 promoter, which is also rich in Stat3 binding sites. Thus, it is possible that the close proximity of this distal Stat3 site to the C/EBP site determines the Stat3 binding to this site in the Gr1+CD11b+ cells, especially we observed that both sites are required for full miR-21 or miR-181 promoter-mediated reporter gene activation.
Our finding that C/EBPβ synergizes with Stat3 to activate the miR-21 and miR-181b promoters is particularly important. Both Stat3 and C/EBPβ contribute to MDSC expansion/accumulation.5 The C/EBPβ is upregulated in murine bone marrow cells after inflammatory cytokine treatments or fungal infection and increases the production of granulocyte progenitors, whereas C/EBPα increases the progenitor numbers in the absence of infection or inflammatory signal.27 This study suggested that C/EBPα can promote granulopoiesis under normal/ steady-state conditions, while C/EBPβ is required for emergency/enhanced generation of granulocyte progenitors.27 During emergency, such as infection/excessive inflammation, generation and mobilization of myeloid precursors is necessary to compensate for lost innate immune cells. In support of this theory, a recent study has reported that Stat3 was necessary for accelerating cell cycle progression of the hematopoietic progenitors and increased production of granulocyte progenitors.28 Stat3 mediated these effects by ehancing the expression of C/EBPβ. In line with this, mice deficient in Stat3 had reduced generation of granulocyte progenitors, and phenotypes of these progenitors resembled myeloid progenitors lacking C/EBPβ, as they did not produce granulocyte precursors.27,28 In addition, inhibition of Stat3 activation with the tyrosine kinase inhibitor sunitinib in tumor bearing mice reduced MDSC numbers.38 Morever, a recent study has shown that generation of MDSCs in vitro from mouse and human bone marrow cells depended on C/EBPβ and required IL-6 stimulation.39 While none of these studies investigated a miRNA involvement, this information clearly implicates Stat3 and CEBPβ in promoting myelopoiesis and thus suggests that they can expand MDSCs under pathophysiological conditions. In support of this notion, MDSC generation has been shown to be impaired in C/EBPβ knockout mice.27,28 Our results showed that Stat3 phoshorylation (activation) and C/EBPβ protein levels were increased and bound the miR-21 and miR-181b promoters in sepsis Gr1+CD11b+ cells, which cannot differentiate into innate immune cells and are immunosuppressive.8 In contrast, C/EBPα binding at the miR-21 and miR-181b promoters was observed only in normal Gr1+CD11b+ from sham mice, which can differentiate ex vivo and are not immunosuppressive. Activation of the reporter gene expression in the naive Gr1+CD11b+ cells with sequences containing the Stat3 and C/EBP binding site depended on IL-6 signal, and importantly both sites were required for full activation of the reporter gene expression. Morever, we find that the knockdown of Stat3 or C/EBPβ in sepsis Gr1+CD11b+ cells inhibits miR-21 and miR-181b expression. In addition, both Stat3 and C/EBP binding sites were required for full activation of the luciferase gene expression, indicating a combinatorial effect by Stat3 and C/EBPβ. While Stat338,40 and C/EBPβ41,42 are reported to regulate target gene expression independently, our study shows that they regulate the miR-21 and miR-181b promoters in a cooperative manner during sepsis. The close proximity of the Stat3 and C/EBP binding sites may facilitate the bindings of p-Stat3 and C/EBPβ in one protein complex, as observed in this study, and thus may explain the cooperative effect of p-Stat3 and C/EBPβ on the miR-21 and miR-181b promoter activation. Taken together, these experiments demonstrate that Stat3 and C/EBPβ synergize to induce miR-21 and miR-181b expression and thus promote MDSC accumulation during sepsis.
This study also reveals an additional layer in the transcriptional regulation of miR-21 and miR-181b during sepsis. We find that the Rb protein is phosphorylated in sepsis Gr1+CD11b+ cells. Rb activity is regulated by phosphorylation and has been implicated in transcription regulation of target genes via mediating protein-protein interaction at target gene promoters.43,44 We observed that Rb phosphorylation was induced by the cyclin D1-CDK4 protein complex. Knockdown of cyclin D1 or CDK4 abolished Rb phosphorylation. Importantly, lack of Rb phosphorylation, similar to the knockdown of Rb itself, led to displacement of Stat3 and C/EBPβ from the miR-21 and miR-181b promoters. This resulted in binding of C/EBPα, thus producing a pattern similar to what we observed in the normal Gr1+CD11b+ cells from sham mice.
The mechanism of Rb-mediated, differential binding of C/EBPβ and C/EBPα at the miRNA promoters in Gr1+CD11b+ cells provides new insight into sepsis regulation. Phosphorylated Rb promotes cell cycle progression and proliferation44,45 by activating transcription factor E2F1, which supports G1/S cell cycle progression.44,45 In contrast, dephosphorylated Rb promotes cell cycle arrest and differentiation by inactivating E2F proteins.46 In sepsis inflammation, Rb is phosphorylated at Serine780 and cannot bind C/EBPβ but, instead, forms a complex with C/EBPα to promote myeloid cell differentiation. Thus, the dual role of Rb in regulating cell proliferation and differentiation is mediated by its phosphorylation status.45 Both cyclin D1-CDK4 and cyclin E-CDK2 phosphorylate Rb during the G1/S cell cycle progression, and cyclin D1-CDK4 specifically phosphorylates Rb at Serine780 in vitro in human fibroblasts.31 We previously reported that sepsis inhibits cyclin-dependent kinase inhibitor p21 expression due to increases in the NFI-A protein.33 P21 inhibits the cyclin D1-CDK4 protein complex, which facilitates cell cycle arrest and promotes cell differentiation.47,48 In this study, knockdown of cyclin D1 or CDK4 in sepsis Gr1+CD11b+ cells inhibited Rb phosphorylation at Serine780 (Fig. 5A), Rb knockdown displaced C/EBPβ from the miR promoters and simultaneously induced C/EBPα binding (Fig. 5D) and abolished the miR promoter-driven reporter gene expression (Fig. 6B). Thus, sequestration of C/EBPα by p-Rb may override the inhibitory effect of C/EBPα on myeloid cell expansion to direct Gr1+CD11b+ cell accumulation during sepsis.
C-Myc protein regulates emergency granulopoiesis, which expands myeloid cell progenitor pools.27 A recent study showed that the c-Myc promoter is co-regulated by Stat3, C/EBPβ, and C/EBPα in murine myeloid progenitors.28 Although all three proteins were recruited to the c-Myc promoter, C/EBPα dissociated shortly after stimulation with the growth factor G-CSF, which induced Stat3 phosphorylation and C/EBPβ expression. Our results suggest the possibility that a switch between C/EBPβ and C/EBPα is sufficient to turn on and off a target promoter, since c-Myc protein levels did not change in sepsis Gr1+CD11b+ cells following Rb knockdown (Fig. 5F). Given that c-Myc promoter is a target of both C/EBPβ and C/EBPα, our results suggest that p-Rb regulates the miR-21 and miR-181b promoters during sepsis by binding and sequestering C/EBPα protein, which allows C/EBPβ and p-Stat3 binding to the miR promoters.
We propose a model (Fig. 7) of miR-21 and miR-181b transcription regulation during sepsis, wherein C/EBPα normally binds to the miR-21 and miR-181 promoters and keeps them under tight control. During sepsis, however, Rb protein is phosphorylated and forms a protein complex with C/EBPα, preventing promoter binding, and allowing C/EBPβ and p-Stat3 to activate miR-21 and miR-181b promoters. While the path leading to Rb phosphorylation requires further investigation, our results clearly indicate that Rb plays an important role in the regulation of the transcription factor bindings at and activation of the miR-21 and miR-181b promoters during sepsis.
Figure 7. A diagram depicting transcription regulation of miR-21 and miR-181b promoters in Gr1+CD11b+ cells.
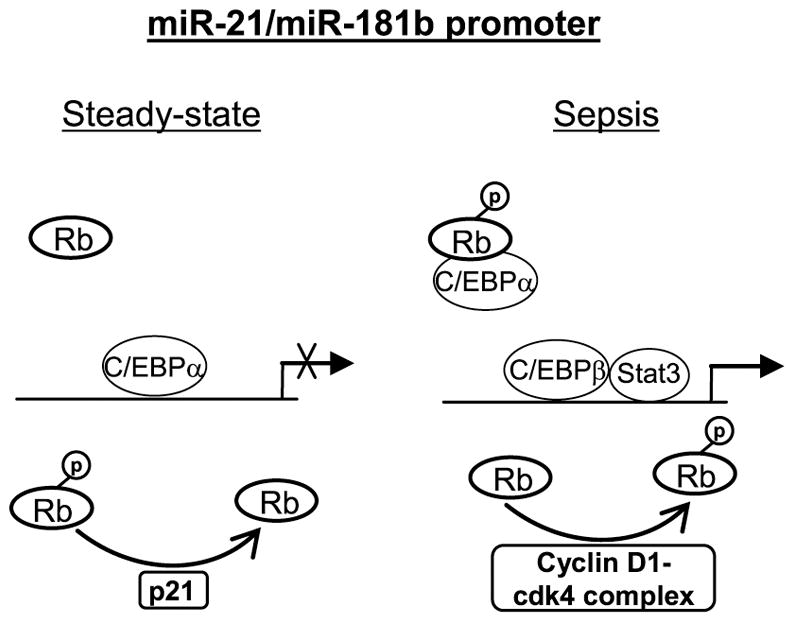
Under normal conditions (i.e., steady-state myelopoiesis), Rb protein is de-phosphorylated via cyclin-dependent kinase (cdk) inhibitor p21-dependent feedback mechanism and thus cannot interact with the C/EBPα protein, allowing it to bind and inactivate the miR-21 and miR-181b promoters. During sepsis, Rb is phosphorylated by a cyclin D1-cdk4 protein complex and interacts with and displaces the C/EBPα protein, thus allowing the Stat3 and C/EBPβ proteins to bind and activate the miR-21 and miR-181b promoters. This differential regulatory pathway requires an IL-6-mediated signal.
The early hyperinflammatory reaction of sepsis, if not treated early, can progress to a prolonged state of immunosuppression, which increases the mortality rate in animals and humans with sepsis.49,50 This sepsis-associated immunosuppression is characterized by increased apoptosis and attenuated functions of innate and adaptive immunity cells, expansion of regulatory T cells with immunosuppressive activities and elevated levels of immunosuppressive cytokines.51–53 These pathophysiological changes diminish immune function and promote seconday/opportunistic infections. Recent studies suggest that miR-21 can modulate the immune responses by promoting anti-inflammation and immunosuppression, as increased expression of miR-21 occurs during chronic bacterial and viral infections.54 MDSCs are repressors during sepsis,8,26 and blocking miR-21 and miR-181b in vivo reduces MDSC numbers and improves late sepsis survival.26 By uncovering the mechanism of miR-21 and miR-181b transcription regulation, the present study not only provides insights into the regulation of miR-21 and miR-181b expression during sepsis but also adds important information on how MDSCs are generated during sepsis and perhaps in the broader context of immune repressor phenotypes.
METHODS
Mice
Male BALB/c mice, 8 weeks old were purchased from Harlan Sprague Dawley (Indianapolis, IN). The mice were housed in a pathogen-free facility and were acclimated to the new environment for a week before surgery. All experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the East Tennessee State University Animal Care and Use Committee.
Induction of sepsis
Polymicrobial sepsis, which emulates general peritonitis occurring in humans,55,56 was induced by cecal ligation and puncture (CLP) as described previously 57, a model that creates a prolonged infection and develops into early and late sepsis phases with ~90% mortality over 4 weeks. Briefly, mice were anesthetized via inhalation with 2.5% isoflurane (Abbott Laboratories, Abbott Park, IL). A midline abdominal incision was made and the cecum exteriorized, ligated distal to the ileocecal valve, and then punctured twice with a 21-gauge needle. A small amount of feces was extruded into the abdominal cavity. The abdominal wall and skin were sutured in layers with 3-0 silk. Sham-operated mice were treated identically except that the cecum was not ligated or punctured. For fluid resuscitation, mice received (i.p.) 1 ml lactated Ringers plus 5% dextrose for fluid resuscitation. To generate late sepsis, mice were subcutaneously administered antibiotic (Imipenem; 25 mg/kg body weight) or an equivalent volume of 0.9% saline. To establish intra-abdominal infection and approximate the clinical situation of early human sepsis where there often is a delay between the onset of sepsis and the delivery of therapy,58 injections of Imipenem were given at 8 and 16 hr after CLP, which results in low mortality (25–30%) during the early phase and high mortality (~60–70%) during the late/chronic phase.57 The presence of early sepsis was confirmed by transient systemic bacteremia and elevated circulating proinflammatory cytokines in the first 5 days after CLP. Late/chronic sepsis (after day 5) was confirmed by enhanced peritoneal bacterial overgrowth and reduced circulating proinflammatory cytokines. Moribund mice, those suffering deep hypothermia (<34°C), lethargy, and weight loss (>30%), were sacrificed and analyzed. Mice that were found dead before sacrifice were not included in the analysis.
Isolation of Gr1+CD11b+ cells
Bone marrow and spleen Gr1+CD11b+ cells were harvested from sham and septic mice immediately after mice were sacrificed, using magnetically assisted cell sorting according to the manufacturer’s protocol (Miltenyi Biotech, Auburn, CA). The bone marrow cells were flushed out of the femurs with RBMI 1640 medium (without serum) under aseptic conditions.57 The spleens were minced in RBMI 1640 medium. A single cell suspension of the bone marrow or spleen was made by pipetting up and down and filtering through a 70-μm nylon strainer followed by incubation with erythrocyte lysis buffer. After washing, total Gr1+CD11b+ cells were purified by subjecting the single cell suspension to positive selection of the Gr1+CD11b+ cells by incubating with biotinylated mouse anti-Gr1 antibody (Clone RB6-8C5; eBioscience, San Diego, CA) for 15 min at 4°C. After washing, the cells were incubated with anti-biotin magnetic beads for 20 min at 4°C and subsequently applied to the MS column. The selection process was repeated using anti-CD11b antibody. Purified Gr1+CD11b+ cells were then washed and resuspended in sterile saline. Cell purity was determined by flow cytometry and was > 90%.
To enrich for CD31+ MDSCs (for differentiation experiments), purified Gr1+ cells were incubated with anti-CD31 antibody (eBioscience) and then selected with magnetic beads (Milteny), after washing away the beads used for Gr1+ selection with a special buffer. Cell purity was determined by flow cytometry and was higher than 94%. Cells were then washed and resuspended in sterile saline.
Cell culture
Gr1+CD11b+ cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine (all from Hyclone Laboratories, Logan, UT), and 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) at 37°C and 5% CO2.
MiRNA analysis
Levels of miR-21 and miR-181b in whole bone marrow cells were measured using a miRNA Northern Blot Assay kit per the manufacturer’s instructions (Signosis, Santa Clara, CA). Briefly, 2 μg of miRNA-enriched RNA were separated by 15% TBE Urea-gels, transferred to a nylon membrane, and then immobolized by UV cross-linking. Membranes were hybridized overnight at 42°C with a biotin-labeled miRNA probe complementary to miR-21 or miR-181b. Membranes were blocked for 30 min at room temperature and then incubated with Streptavidin-HRP for 45 min at room temperature. After washing, miRNAs were detected with a chemiluminescence substrate, the bands were visualized using the ChemiDoc XRS System (Bio-Rad, Hercules, CA), and the images were captured with the Image Lab Software V3.0 (Bio-Rad).
Protein extracts
To prepare total protein extract, cells were lysed in 1x RIPA buffer containing 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholic acid, and 1 mM EDTA (Millipore, Temecula, CA) plus 1x protease inhibitor cocktail. After 30 min on ice, cell lysate was cleared by centrifugation for 5 min at 4°C and 14,000 rpm. Protein concentations were determined by Bradford assay (Bio-Rad), and aliquots were kept at −20°C.
To isolate nuclear proteins, we used the NE-PER nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL) per the manufacturer’s instructions. Immediately after harvesting, cells were washed in PBS and resuspended in CER1 lysis buffer with protease inhibitor cocktail and incubated on ice for 1 min. CER2 buffer was added and the incubation continued for 5 min. Supernatants (cytoplasmic proteins) were removed by centrifugation for 5 min at 4°C and 14,000 rpm. The nuclear pellets were resuspended in NER lysis buffer with protease inhibitor cocktail and incubated for 40 min on ice with occasional vortexing. The nuclear proteins were recovered by centrifugation for 10 min at 4°C and 14,000 rpm.
Immunoprecipitation
Immunoprecipitation (IP) of Stat3 or Rb protein complexes was performed as described previously59 with some modifications. Briefly, total protein extracts were pre-cleared by incubation with pre-blocked protein G-agarose beads for 1 h at 4°C. Beads were pre-blocked by incubation for 1 h with 100 μg/ml of BSA. Pre-blocked beads were washed with buffer C (250 mM sucrose, 10 mM Tris-HCl [pH 7.5], 25 mM KCl, 5 mM MgCl2, 2 mM DTT, 30 U/ml RNase inhibitor, and 1x protease inhibitor cocktail). Extract was centifuged at 2,000 rpm for 5 min and supernatant (900 μl) was added to 100 μl of pre-blocked protein G-agarose beads that were coated with 10 μl antibody against p-Stat3 (phospho tyrosine705), p-Rb (phospho serine780), or IgG istype control (Santa Cruz Biotechnology, Santa Cruz, CA). After overnight incubation (with rotation) at 4°C, the beads were centrifuged and washed three times with buffer C. Aliquots of bound protein complexes were used for protein analysis by immunoblot as described below.
Immunoblots
Equal amounts of total protein extract or immunoprecipitated protein complexes (IP) were mixed with 5x Laemmeli sample buffer, separated by a SDS-10% polyacrylamide gel (Bio-Rad) and subsequently transferred to nitrocellulose membranes (Thermo Scientific, Waltham, MA ). After blocking with 5% milk in Tris-buffered saline/Tween-20 for 1 hr at room temperature, membranes were probed overnight at 4°C with the appropriate primary antibody. After washing, blots were incubated with the appropriate HRP-conjugated secondary antibody (Life Technologies, Grand Island, NY) for 2 hr at room temperature. Proteins were detected with the enhanced chemiluminescence detection system (Thermo Scientific). The developed bands were visualized using the ChemiDoc XRS Detection System (Bio-Rad) and the images were captured with the Image Lab Software V3.0. Membranes were stripped and re-probed with b-actin antibody (Sigma-Aldrich) as a loading control.
siRNA-mediated knockdown
Purified Gr1+CD11b+ cells or CD31+-enriched Gr1+CD11b+ cells were transfected with pools of cyclin D1-specific, CDK4-specific, Rb-specific or scrambled (control) siRNAs at a 0.5 μM final concentration (Santa Cruz Biotechnology), using HiPerFect transfection reagent per the manufacturer’s instructions (Qiagen, Valencia, CA) and the Gene Pulser MXCell system (Bio-Rad, Herclues, CA). After 36 hr, cells were harvested and analyzed by immunoblot or ChIP.
Chromatin immunoprecipitation (ChIP)
ChIP was performed to assess in vivo DNA-protein interactions at the miR-21 and miR-181b promoters using ChIP-IT Express Enzymatic Shearing kit according to the manufacturer’s instructions (Active Motif, Carlsbad, CA). Briefly, cells were harvested and protein-DNA complexes were cross-linked by fixation in 1% formaldehyde in minimal culture medium for 10 min at room temperature. After washing with cold PBS, cells were lysed in 1x lysis buffer containing protease inhibitor cocktail. Cell lysate was cleared by centrifugation at 5,000 rpm for 10 min at 4°C. The pelleted nuclei were then resuspended in digestion buffer and incubated with the enzymatic shearing cocktail at 37°C for 10 min. The sheared chromatin solution was recovered by centrifugation at 15,000rpm for 10 min at 4°C. Ten microliter of the chromatin solution was reserved as “input” DNA sample. The remaining chromatin solution was immunoprecipitated overnight at 4°C with protein G magnetic beads and 3 μg of antibody specific to p-Stat3, C/EBPα, C/EBPβ, p-Rb, or isotype control antibody (Santa Cruz Biotechnology). The chromatin/antibody complexes captured on the beads were washed three times in ChIP buffer and then eluted by incubation for 15 min in 50 μl elution buffer. Next, the DNA-protein cross-links were reveresed by incubating the eluted chromatin with 50 μl of reverese cross-linking buffer. The supernatant containing the DNA was then incubated, along with the “input” DNA samples at 95°C for 15 min. After treatment with proteinase K for 1 h at 37°C, the reaction was stopped and the resulting DNA was stored at −20°C until analyzed by PCR as described below.
Quantitative real-time PCR (RT-qPCR)
RT-qPCR was used to measure enrichment of miR-21 and miR-181b promoter sequences in the ChIPed DNA using primer and fluorescently labeled internal probe sequences specific to each promoter (Integrated DNA Technologies, Coralville, IA). The primers and their coordinates are shown in Supplementary Table 1. Samples were analysed in duplicates. The PCR reaction (25 μl) contained 5 μl ChIP DNA, 12.5 μl of 2x TaqMan real-time PCR Master Mix containing DNA polymerase and dNTPs (Applied Biosystem, Foster City, CA) and 100 nM of primer/probe mix (Integrated DNA Technologies). The PCR conditions were: 2 min at 50°C, 10 min at 95°C, followed by 40 cycles with 15s at 95°C and 1 min at 60°C (combined annealing and extension), using the Bio-Rad CFX96 Real-Time System. Relative enrichment of DNA sequences was calculated by normalizing averaged cycle threshold (Ct) values to the input DNA values. These values are presented as fold change relative to DNA from the IgG-immunoprecipitated samples (set at 1-fold).
Semiquantitative PCR
In some experiments, standard PCR was performed to measure enrichment of miR-21 and miR-181b promoter sequences in the ChIPed DNA using the same primers described for the real-time PCR, which generate a 140-bp fragment of miR-21 promoter and a 110-bp frangment of miR-181b promoter. PCR reaction was performed in a 50-μl volume containing 5 μl ChIP DNA, 1 μM of each primer, 2 mM MgCl2, 0.2 μM dNTPs and 0.04 U/μl AmpliTag Gold DNA polymerase (Applied Biosystems). The PCR conditions were as follows: 1 cycle at 94°C for 10 min, 30 cycles at 94°C, 58°C, and 72°C for 30 s each, and a final cycle at 72°C for 5 min. Equal amounts of PCR products were run on 1.2% ethidium bromide-stained agarose gel. The bands were visualized using the ChemiDoc XRS detection System (Bio-Rad) and the images were captured with the Image Lab Software V3.0 (Bio-Rad). The PCR primers were designed to amplify a 137-bp sequence in the miR-21 promoter and a 107-bp sequence in the miR-181b promoter.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed to determine transcription factor binding at the miR-21 and miR-181 promoters using the LightShift Chemiluinescent EMSA Kit according to the manufacturer’s instructions (Thermo Scientific). Binding reaction was carried out in 20 μl volume containing 10 μg of nuclear protein, 1 pmol biotin-labeled double-stranded, synthetic oligonucleotide probe (Integrated DNA Technologies), 1X binding buffer, 5% v/v glycerol, 5 mM MgCl2, 50 ng/μl Poly(dI.dC), and 0.05% NP-40. For competition experiments, 200-fold molar excess of the unlabeled probe was added to determine binding specficity. For supershift experiments, 3 μg of antibody that recognizes Stat3 or C/EBPβ was incubated with the nuclear proteins for 5 min before the addition of the probe. The binding reaction was incubated for 20 min at room temperature and then stopped by adding 5 μl of 5X loading buffer. DNA-protein complexes were electrophoresed on a 6% native polyacrylamide gel in 0.5X TBE buffer for 50 min at 4°C, transferred onto a positively charged nylon membrane (Thermo Scientific), and the DNA was fixed on the membrane by UV crosslinking. The biotin-labeled probe was detected with streptavidin-horseradish peroxidase and a chemiluminescent reagent. The DNA bands were visualized using the ChemiDoc XRS Detection System (Bio-Rad). The DNA probes included 30–31 nucleotide sequences containing the wild type or mutated Stat3 or C/EBP binding site in the miR-21 and miR-181b promoters and are shown in Supplementary Table 2.
Transient transfection and luciferase reporter assay
Sequences containing the Stat3 and C/EBP binding sites from miR-21 or miR-181b promoters (Fig. 1F) were cloned in the pEZX-M02 vector upstream of the luciferase gene, in which the luciferase gene expression is constitutively activated by the mini-CMV enhancer. Normal Gr1+CD11b+ cells (1 × 106 cells/well) in 24-well plates were transfected using using the HiPerFect transfection reagent per the manufacturer’s instructions (Qiagen, Valencia, CA) with 2 μg of luciferase plasmid plus 0.1 μg of Renilla luciferase expression plasmid as a control for transfection efficiency (GeneCopoeia, Rockville, MD). Cells were incubated for 24 hr without or with 10 ng/ml of recombinant mouse IL-6. Cells were harvested, and luciferase and renilla activities were determined with the dual luciferase reporter assay system (Promega, Madison, WI). A pEZX luciferase expression plasmid with no Stat3 or C/EBP binding site inserts served as a positive control. The pEZX-M02 luciferase plasmid, in which the luciferase gene is controlled by the CMV promoter and mini-enhancer served as a control for the maximum luciferase gene activity. Luciferase values were normalized to Renilla activity and are presented relative to the luciferase positive control (set at 100%).
Statistical analysis
Data were analyzed with Microsoft Excel V3.0. Differences between 2 groups were analyzed by use of unpaired student’s t test. One-way ANOVA with Turkey’s multiple comparison tests was used to analyze the data with more 2 two groups. The data are presented as mean ± s.d.. Statistical significance is reported for p-values ≤ 0.05.
Supplementary Material
The CD31+ cells were isolated from the total Gr1+CD11b+ cell population, and EMSA was performed as described in Fig. 4. The results are representative of two experiments.
The CD31+ cells were isolated from the total Gr1+CD11b+ cell population, and EMSA was performed as described in Fig. 4. The results are representative of two experiments. Note the antibody not only shifted most of the protein complex but also enhanced the C/EBPα protein binding to the probe.
Cells were isolated, transfected with luciferase plasmids, and treated as described in Fig. 9. Luciferase values are expressed as mean ± s.d. (*p ≤ 0.05) from two experiments and are presented relative to the positive control (set at 100%). *, compare with positive control; **, compare with Stat3 wt + Cebp wt site.
Acknowledgments
This work was supported by a National Institutes of Health Grants R01GM103887 (to M. E.) and C06RR0306551 (to College of Med.).
ABBREVIATIONS
- MDSC
myeloid-derived suppressor cell
- CLP
cecal ligation and puncture
- IL
interleukin
- miRNA
microRNA
- siRNA
small interfering RNA
- ChIP
chromatin immunopresciptation
- EMSA
electrophoretic Mobility Shift Assay (EMSA)
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bronte V. Myeloid-derived suppressor cells in inflammation: uncovering cell subsets with enhanced immunosuppressive functions. Eur J Immunol. 2009;39:2670–2672. doi: 10.1002/eji.200939892. [DOI] [PubMed] [Google Scholar]
- 4.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trikha P, Carson WE., III Signaling pathways involved in MDSC regulation. Biochim Biophys Acta. 2014;1846:55–65. doi: 10.1016/j.bbcan.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vuk-Pavlovic S, Bulur PA, Lin Y, Qin R, Szumlanski CL, Zhao, et al. Immunosuppressive CD14+HLA-DRlow/- monocytes in prostate cancer. Prostate. 2010;70:443–455. doi: 10.1002/pros.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun. 2012;80:2026–2034. doi: 10.1128/IAI.00239-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17:281–292. doi: 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong YY, Fuchsberger M, Xiang SD, Apostolopoulos V, Plebanski M. Myeloid derived suppressor cells and their role in diseases. Curr Med Chem. 2013;20:1437–1444. doi: 10.2174/0929867311320110006. [DOI] [PubMed] [Google Scholar]
- 11.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 13.Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol. 2003;74:186–196. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- 14.Saleem SJ, Conrad DH. Hematopoietic cytokine-induced transcriptional regulation and Notch signaling as modulators of MDSC expansion. Int Immunopharmacol. 2011;11:808–815. doi: 10.1016/j.intimp.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 16.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 17.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 18.Williams AE. Functional aspects of animal microRNAs. Cell Mol Life Sci. 2008;65:545–562. doi: 10.1007/s00018-007-7355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 20.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 21.Ballarino M, Pagano F, Girardi E, Morlando M, Cacchiarelli D, Marchioni M, et al. Coupled RNA processing and transcription of intergenic primary microRNAs. Mol Cell Biol. 2009;29:5632–5638. doi: 10.1128/MCB.00664-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009;21:452–460. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 25.Tsitsiou E, Lindsay MA. microRNAs and the immune response. Curr Opin Pharmacol. 2009;9:514–520. doi: 10.1016/j.coph.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, et al. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun. 2014;82:3816–3825. doi: 10.1128/IAI.01495-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landschulz WH, Johnson PF, McKnight SL. The DNA binding domain of the rat liver nuclear protein C/EBP is bipartite. Science. 1989;243:1681–1688. doi: 10.1126/science.2494700. [DOI] [PubMed] [Google Scholar]
- 30.Hu X, Zuckerman KS. Cell cycle and transcriptional control of human myeloid leukemic cells by transforming growth factor beta. Leuk Lymphoma. 2000;38:235–246. doi: 10.3109/10428190009087015. [DOI] [PubMed] [Google Scholar]
- 31.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Tamai K, Kato J, et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 32.Riley DJ, Lee EY, Lee WH. The retinoblastoma protein: more than a tumor suppressor. Annu Rev Cell Biol. 1994;10:1–29. doi: 10.1146/annurev.cb.10.110194.000245. [DOI] [PubMed] [Google Scholar]
- 33.McClure C, Ali E, Youssef D, Yao ZQ, McCall CE, El Gazzar M. NFI-A disrupts myeloid cell differentiation and maturation in septic mice. J Leukoc Biol. 2016;99:201–211. doi: 10.1189/jlb.4A0415-171RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braun DA, Fribourg M, Sealfon SC. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J Biol Chem. 2013;288:2986–2993. doi: 10.1074/jbc.M112.386573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: recent developments and future challenges. Brief Funct Genomics. 2013;12:489–498. doi: 10.1093/bfgp/elt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39:493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller AK, Burger R, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 38.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–2513. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Li P, Grgurevic S, Liu Z, Harris D, Rozorski U, Calin GA, et al. Signal transducer and activator of transcription-3 induces microRNA-155 expression in chronic lymphocytic leukemia. PLoS One. 2013;8:e64678. doi: 10.1371/journal.pone.0064678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta AK, Kone BC. CCAAT/enhancer binding protein-beta trans-activates murine nitric oxide synthase 2 gene in an MTAL cell line. Am J Physiol. 1999;276:F599–F605. doi: 10.1152/ajprenal.1999.276.4.F599. [DOI] [PubMed] [Google Scholar]
- 42.Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- 43.Chen PL, Riley DJ, Chen-Kiang S, Lee WH. Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proc Natl Acad Sci USA. 1996;93:465–469. doi: 10.1073/pnas.93.1.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 45.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–138. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 46.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–M56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 48.Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22:33–41. doi: 10.1016/j.tcb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun. 2010;78:1582–1592. doi: 10.1128/IAI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCall CE, El Gazzar M, Liu T, Vachharajani V, Yoza B. Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. J Leukoc Biol. 2011;90:439–446. doi: 10.1189/jlb.0211075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shubin NJ, Monaghan SF, Ayala A. Anti-inflammatory mechanisms of sepsis. Contrib Microbiol. 2011;17:108–124. doi: 10.1159/000324024. [DOI] [PubMed] [Google Scholar]
- 54.Sheedy FJ. Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response. Front Immunol. 2015;6:19–28. doi: 10.3389/fimmu.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belikoff B, Hatfield S, Sitkovsky M, Remick DG. Adenosine negative feedback on A2A adenosine receptors mediates hyporesponsiveness in chronically septic mice. Shock. 2011;35:382–387. doi: 10.1097/SHK.0b013e3182085f12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 57.Brudecki L, Ferguson DA, Yin D, Lesage GD, McCall CE, El Gazzar M. Hematopoietic stem-progenitor cells restore immunoreactivity and improve survival in late sepsis. Infect Immun. 2012;80:602–611. doi: 10.1128/IAI.05480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mazuski JE, Sawyer RG, Nathens AB, DiPiro JT, Schein M, Kudsk KA, et al. The Surgical Infection Society guidelines on antimicrobial therapy for intra-abdominal infections: an executive summary. Surg Infect (Larchmt ) 2002;3:161–173. doi: 10.1089/109629602761624171. [DOI] [PubMed] [Google Scholar]
- 59.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The CD31+ cells were isolated from the total Gr1+CD11b+ cell population, and EMSA was performed as described in Fig. 4. The results are representative of two experiments.
The CD31+ cells were isolated from the total Gr1+CD11b+ cell population, and EMSA was performed as described in Fig. 4. The results are representative of two experiments. Note the antibody not only shifted most of the protein complex but also enhanced the C/EBPα protein binding to the probe.
Cells were isolated, transfected with luciferase plasmids, and treated as described in Fig. 9. Luciferase values are expressed as mean ± s.d. (*p ≤ 0.05) from two experiments and are presented relative to the positive control (set at 100%). *, compare with positive control; **, compare with Stat3 wt + Cebp wt site.