

Abstract
The objective of this review is to evaluate the evidence that recreational methamphetamine exposure might damage dopamine neurones in human brain, as predicted by experimental animal findings. Brain dopamine marker data in methamphetamine users can now be compared with those in Parkinson's disease, for which the Oleh Hornykiewicz discovery in Vienna of a brain dopamine deficiency is established. Whereas all examined striatal (caudate and putamen) dopamine neuronal markers are decreased in Parkinson's disease, levels of only some (dopamine, dopamine transporter) but not others (dopamine metabolites, synthetic enzymes, vesicular monoamine transporter 2) are below normal in methamphetamine users. This suggests that loss of dopamine neurones might not be characteristic of methamphetamine exposure in at least some human drug users. In methamphetamine users dopamine loss was more marked in caudate than in putamen, whereas in Parkinson's disease the putamen is distinctly more affected. Substantia nigra loss of dopamine-containing cell bodies is characteristic of Parkinson's disease, but similar neuropathological studies have yet to be conducted in methamphetamine users. Similarly, it is uncertain whether brain gliosis, a common feature of brain damage, occurs after methamphetamine exposure in humans. Preliminary epidemiological findings suggest that methamphetamine use might increase risk of subsequent development of Parkinson's disease. We conclude that the available literature is insufficient to indicate that recreational methamphetamine exposure likely causes loss of dopamine neurones in humans but does suggest presence of a striatal dopamine deficiency that, in principle, could be corrected by dopamine substitution medication if safety and subject selection considerations can be resolved.
Keywords: Methamphetamine, dopamine, neurotoxicity, striatum, substantia nigra, gliosis, levodopa, Parkinson's disease
Graphical abstract
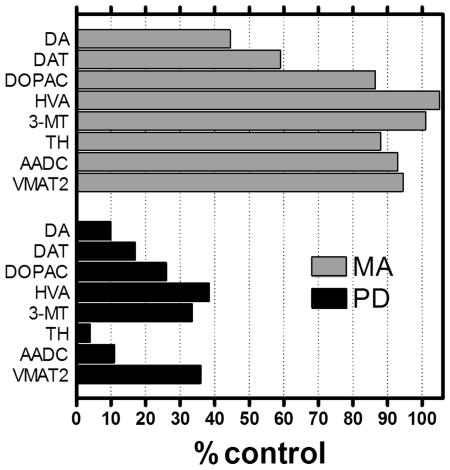
Striatal dopamine (DA) marker changes in Parkinson's disease (PD) vs. methamphetamine (MA) users. In autopsied brain of persons with PD striatal levels of all DA markers (DA and metabolites DOPAC, HVA and 3-MT, synthesizing enzymes TH and AADC, and transporters VMAT2 and DAT) are low whereas in MA users only two markers (DA and DAT) are below normal. This suggests that any loss of brain dopamine neurons in at least a subgroup of recreational MA users, if at all present, might not be substantial.
Introduction and Objective
Methamphetamine (MA) is a dopaminergic stimulant drug widely used recreationally (for review see (Kish, 2014)). MA can harm humans in part because some recreational users of the drug become addicted to the stimulant (Kish, 2008). Our own epidemiological findings from a large California cohort study also suggest, for example, that there is increased rate of all-cause mortality for MA users (standardized mortality rate, 4.67) in comparison to individuals of similar age, race, and sex in the general population (Callaghan et al., 2012b).
A still open question is whether chronic MA exposure can harm or “damage” neurones in the human brain that use dopamine as a neurotransmitter – i.e., whether MA is “neurotoxic” to those neurones. MA/amphetamine promotes release of dopamine from dopamine neurones in human brain ((Laruelle et al., 1995; Cardenas et al., 2004); see also (Pifl et al., 1995; Rothman & Baumann, 2003; Sulzer et al., 2005)) through an action on both the plasma membrane dopamine transporter and the vesicular monoamine transporter. Experimental animal reports of MA-induced structural damage in dopamine neurons assessed by histological procedures (swollen/distorted axons, Fink-Heimer silver staining) and loss of dopamine markers (see (Kish, 2014)) predict that the stimulant, at some unknown, probably high, dose, should also damage dopamine neurones in human brain. Historically, the general consensus in the animal literature has been that MA selectively damages dopamine nerve terminals (in the dopamine-rich striatum) but with a sparing of cell bodies in the substantia nigra (e.g., (Ryan et al., 1990)); however, some experimental data suggest that (structural) damage may also extend to dopaminergic cell bodies in the nigra (see (Ares-Santos et al., 2014)).
The objective of this review is to evaluate, in a brief update summary, the evidence for and against the possibility that MA use in humans might be associated with damage to dopamine neurones. This is a summary update to an extensive review on the “neurotoxicity” of MA up to approximately 2012/2013 (Kish, 2014). Whenever possible, MA findings will be compared with those in idiopathic Parkinson's disease, as Parkinson's disease remains the “gold standard” dopamine deficiency disorder in the human. This manuscript is submitted in honour of Professor Oleh Hornykiewicz, who made the extraordinary discovery 56 years ago in Vienna of a brain dopamine deficiency in Parkinson's disease, and who is celebrating this year his 90th birthday.
We believe that effort aimed at understanding whether MA, a widely used stimulant, damages human brain is justified, in part because this is a public health and forensic medicine issue and especially because the information obtained might inform new approaches to therapy. Further, demonstration of dopamine neurone damage in brain of recreational MA users would raise some concern that such damage might also occur in young people receiving the MA metabolite, amphetamine, for therapeutic purposes (e.g., attention deficit hyperactivity disorder, (Ricaurte et al., 2005)).
Dopamine
If dopamine neurone damage has occurred in human brain, it would be expected that levels of brain dopamine, as well as those of all dopamine markers, should be lower than those levels in persons who do not use MA.
Parkinson's disease: The dopamine deficiency discovery
The autopsied human brain investigation of Oleh Hornykiewicz in Vienna provided the proof that a brain dopamine deficiency is the defining biochemical feature of Parkinson's disease.
Hermann Blaschko, the British Council Scholarship supervisor of Hornykiewicz in Oxford England from 1956 to 1958, was the original “inspiration” for Hornykiewicz to be involved in studies of dopamine: Whereas dopamine was previously felt to be only of limited interest as a metabolic intermediate in the pathway to noradrenaline, Blaschko speculated that dopamine might have “some regulatory functions of its own which are not yet known” – and asked Hornykiewicz to continue to work with dopamine (Hargittai & Hargittai, 2005). Back in Vienna, Hornykiewicz, on his own, decided to focus on dopamine's possible role in the brain. Soon, armed with the experimental findings that brain dopamine was highly localized in the striatum (caudate and putamen) (Bertler & Rosengren, 1959; Sano et al., 1959), and aware of Arvid Carlsson's pioneering animal work demonstrating that the “tranquilizing” effect of reserpine, which can cause immobility in animals and a Parkinson's like syndrome (akinesia) in humans, could be abolished by the catecholamine precursor levodopa (Carlsson et al., 1957), and the further demonstration that reserpine actually depletes the animal brain of catecholamines whereas levodopa preferentially restored the dopamine levels to normal (Carlsson et al., 1958; Weil-Malherbe & Bone, 1958), Hornykiewicz abandoned his brain dopamine work in the animal (Holzer & Hornykiewicz, 1959) and moved quickly to the (autopsied) human brain itself (see (Hornykiewicz, 2010) for a review).
Logistical difficulties for the postmortem brain investigation were in part technical – a dopamine iodine assay set up in a “semi-dark” basement of the Vienna Pharmacological Institute, and Dowex ion exchange columns requiring multiple football bladders – as a constant pressure pump was lacking. More difficult was obtaining a source of autopsied brain of Parkinson's patients (from Vienna's Wien-Lainz hospital) given that, at that time, neurochemical investigation of postmortem human brain was not considered to be a reasonable approach.
Hornykiewicz describes the moment of discovery: “In April 1959, about eight weeks after reading the Bertler and Rosengrin report in Experientia (Bertler & Rosengren, 1959), we received the first brain of a patient who had died with Parkinson's disease. After carrying the sample of the caudate nucleus and putamen of this patient, together with control samples, through the extraction procedure, the thrilling moment of performing the von Euler and Hamberg color (iodine) reaction arrived. Instead of the pink color in the control samples, indicating presence of dopamine, the reaction vials containing the Parkinson material showed hardly a tinge of pink discoloration. For the first time ever, and before even placing the reaction vials into the colorimeter, I could see the brain dopamine deficiency in Parkinson's disease literally with my own naked eye!” (Hargittai & Hargittai, 2005).
One year later Hornykiewicz had collected additional material and confirmed a striatal dopamine deficiency in a representative number of persons with Parkinson's disease (Ehringer & Hornykiewicz, 1960).
Professor David Marsden (Marsden, 1990) said this of the discovery in Vienna: “The recent history of Parkinson's disease has been marked by momentous discoveries. Before the 1960s, the clinical features and neuropathology of the disorder had been established, anticholinergic drugs and stereotaxic surgery were popular, but the illness progressed relentlessly and was a cause of miserable disability….The discovery of selective striatal dopamine deficiency in the parkinsonian brain in the early 1960s changed everything.”
Methamphetamine users: One (Toronto) laboratory reports low brain dopamine
Given the extent of worldwide use of MA as a recreational drug, it is surprising that, to our knowledge, information on the status of dopamine in postmortem brain of chronic MA users is still limited to findings from our Toronto laboratory (Wilson et al., 1996a; Moszczynska et al., 2004). Part of the logistical difficulty is certainly explained by the need to obtain MA brain material from medical examiners/coroners – most have little or no “extra” time to help out in a research investigation and, as a practical matter, often are not aware at autopsy that the decedent ever used MA until weeks (or more) later at which time blood drug measurements confirm drug use and the brain material has already been processed. Also, as the world has become much more litigious, medical examiners willing to go beyond their normal governmental duties and help out in a research study are now being asked to “sign off” on provocative “indemnification” statements in case of liability.
Our 2004 report (Moszczynska et al., 2004) summarizes our findings (from 1990 to 2004; (Wilson et al., 1996a; Moszczynska et al., 2004)) of dopamine measurement (by HPLC-electrochemical detection) in postmortem striatum of 20 chronic MA users and 14 control subjects having similar age and postmortem time. All MA users tested positive for MA and/or its metabolite amphetamine in blood and brain obtained at autopsy, meaning that the subjects had last used the drug “recently” (within approximately five days preceding death). In addition, the MA data were compared with those of 12 subjects with neuropathologically confirmed Parkinson's disease (in collaboration with Professor Hornykiewicz), being the gold standard dopamine deficiency disorder (Wilson et al., 1996b).
We found that dopamine levels in most of the individual MA users were below the lower limit of the control range in both the caudate and putamen striatal subdivisions. On average, dopamine concentration in the MA users was decreased by 61% and 50% in caudate and putamen, respectively. Previously, Hornykiewicz showed that the inter- (and intra-) regional striatal dopamine loss in Parkinson's disease has a highly characteristic pattern (Bernheimer et al., 1973). As expected, in this new Parkinson's brain material, serving as a neurological control for the MA users, striatal dopamine levels were severely decreased in caudate (-82%) and to a greater extent in putamen (-97%) of the Parkinson's disease subjects with no overlap whatsoever between individual control and Parkinson's disease subjects (Moszczynska et al., 2004). In principle, low striatal dopamine levels in the MA users could be explained by a loss of dopamine in intact neurones (e.g., caused by excessive MA-induced dopamine release and depletion) and/or by actual loss of dopaminergic neurones containing the neurotransmitter.
Unfortunately, our finding (Wilson et al., 1996a; Moszczynska et al., 2004) of low dopamine in some MA users still awaits replication by an independent laboratory and must therefore be considered “preliminary”. The explanation for the lack of replication investigations is likely due in part to the practical difficulty in obtaining (frozen) autopsied brain of MA users from different medical examiner centres with the necessary short postmortem time. Further, recreational MA use in, for example, the United States is highly prevalent only in certain geographical areas (e.g., U.S. west coast). It should be noted however that “many” (11) years passed before the 1960 Hornykiewicz dopamine deficiency in Parkinson's disease discovery (Ehringer & Hornykiewicz, 1960) was replicated in an independent laboratory (Fahn et al., 1971), in which only two cases were analyzed. The explanation for the replication delay in the Parkinson's disease literature could well be due, at least in part, to the appreciation in the scientific community that his findings just “had to be correct” and did not require any replication.
Dopamine Metabolite Homovanillic Acid (HVA)
Parkinson's disease: Striatal HVA is low, but not as low as that of dopamine
Following the 1960 discovery, there was some concern in the scientific community that the finding of low dopamine in Parkinson's disease might have been a pre- or postmortem artifact (Hornykiewicz, 1994). Hornykiewicz hoped to refute this proposition through demonstration of a “control” neurochemical that was not decreased in brain of the subject: “We continued to look for neurotransmitter parameters that were not reduced in Parkinson brain.” (Hornykiewicz, 1993). Subsequent investigation however disclosed low brain levels in neurotransmitters noradrenaline and serotonin (Ehringer & Hornykiewicz, 1960; Bernheimer et al., 1961), prompting this comment from Hornykiewicz: “No help from noradrenaline and serotonin.” (Hornykiewicz, 1992a).
Given data suggesting that the dopamine metabolite homovanillic acid (HVA) is “stable” in autopsied human brain (Bernheimer, 1964), attention was turned to measurement of this dopamine marker. As expected, striatal HVA levels in Parkinson's disease were lower than those in the control subjects (Bernheimer & Hornykiewicz, 1965). Loss of this dopamine marker provided additional support for a dopamine deficiency in Parkinson's disease; however, as noted by Hornykiewicz, the magnitude of the striatal HVA reduction was less than that of dopamine (see below). This finding of increased (neurotransmitter turnover) ratio of dopamine metabolite vs. dopamine suggested a compensatory increase in activity of the remaining dopamine neurones in Parkinson's disease in which neurotransmitter synthesis, release, and metabolism might be accelerated (Hornykiewicz & Kish, 1987). [This notion of “neurochemical compensation” was later extended by Zigmond and colleagues in their elegant investigations employing an animal model of Parkinson's disease (Zigmond et al., 1984).] The Pifl-Hornykiewicz team (see (Pifl et al., 2014)) later obtained data suggesting that the increased HVA/dopamine ratio could also be explained in part by impaired dopamine sequestration in vesicles and consequent increased metabolism of dopamine.
Methamphetamine users: Striatal homovanillic acid concentration (and dopamine biosynthetic enzymes) are normal
Unlike the situation in Parkinson's disease, characterized by marked dopamine loss and reduced HVA, in postmortem brain of dopamine deficient MA users we found that striatal HVA levels were surprisingly and distinctly normal, as were concentrations of two other dopamine metabolites (3,4-dihydroxyphenylacetic acid [DOPAC], 3-methoxytyramine [3-MT]), and two biosynthetic enzymes (tyrosine hydroxylase, dopa decarboxylase) (Wilson et al., 1996a; Moszczynska et al., 2004), which are all markedly decreased in Parkinson's disease (Zhong et al., 1995; Wilson et al., 1996b).
It is possible that normal HVA in the presence of low dopamine could be explained by greatly accelerated release and metabolism of dopamine (to HVA) in the MA users – all having recently used the stimulant. However, the lack of any substantial reduction in striatal HVA (or those of the other dopamine markers mentioned above), even in those MA users having very severely decreased dopamine, provides little support for any “marked” loss of entire dopamine neurones in MA user brain, as is the case in Parkinson's disease.
Dopamine Transporter
Parkinson's disease: Striatal dopamine transporter is, as expected, low
As expected, in both postmortem brain studies (Wilson et al., 1996b) and imaging investigations of living brain (Frost et al., 1993; Lee et al., 2000), striatal levels of the dopamine transporter are decreased in Parkinson's disease as compared to those in matched controls. The loss of this dopamine marker provides further evidence supporting the loss of entire dopamine-containing neurones in Parkinson's disease.
Methamphetamine users: Striatal dopamine transporter is reduced: Why?
In 1996 we showed that levels of the striatal dopamine transporter, determined by both radioligand binding and Western blotting procedures, were decreased in postmortem brain of MA users (Wilson et al., 1996a). This is the most replicated brain dopamine marker observation in the human methamphetamine literature, with similar findings observed in a postmortem brain immunohistochemical study (Kitamura et al., 2007) and in seven independent PET/SPECT imaging studies of living brain (McCann et al., 1998; Sekine et al., 2001; Volkow et al., 2001a; Volkow et al., 2001b; Sekine et al., 2003; Johanson et al., 2006; Chou et al., 2007; McCann et al., 2008; Schouw et al., 2013; Yuan et al., 2014).
The explanation for the dopamine transporter reduction in MA users is still argued (a compensatory phenomenon in response to low dopamine? (see (Kish, 2014)), and could be explained by loss of the transporter protein (which can change in concentration independently of dopamine neurone changes; (Wilson & Kish, 1996)) or by loss of part or all of the neurone containing the transporter (see also below). The follow-up brain imaging investigation by Volkow and colleagues (Volkow et al., 2001a) suggests that the striatal dopamine transporter reduction can normalize with extended drug abstinence (see also (Chou et al., 2007; Volkow et al., 2015)).
Vesicular Monoamine Transporter (VMAT2)
Parkinson's disease: Striatal VMAT2 levels are, as expected, low
The vesicular monoamine transporter (VMAT2) is localized to the membrane of intraneuronal vesicles and is responsible for transporting monoamine neurotransmitters (dopamine, serotonin, noradrenaline) from the neuronal cytoplasm into the storage vesicles. Although VMAT2 is not specific to mammalian brain dopamine neurones, most VMAT2 in the dopamine-rich striatum is likely present in dopamine neurones (see (Wilson et al., 1996b; Frey et al., 2001)).
Like all dopamine markers mentioned above and examined to date, striatal VMAT2 levels, using binding of radiolabeled tetrabenazine analogues or immunoreactivity, is low in both postmortem (see (Wilson et al., 1996b; Tong et al., 2011) and references therein) and living brain (by positron emission tomography, PET; (Frey et al., 1996; Lee et al., 2000)) of persons with Parkinson's disease.
Methamphetamine users: Striatal VMAT2 binding/immunoreactivity is normal/near-normal or elevated
Striatal VMAT2 has been measured in human MA users in two postmortem brain investigations (Wilson et al., 1996a; Kitamura et al., 2007) and in a total of three brain imaging studies (Johanson et al., 2006; Boileau et al., 2008; Boileau et al., 2016a). Taken together, the findings provide evidence for no or at most, a slight loss of VMAT2 in chronic human MA users.
In our own 1996 postmortem brain study we found that striatal VMAT2 binding at a high saturating substrate concentration was normal in MA users (Wilson et al., 1996a). Similarly, Kitamura and colleagues (Kitamura et al., 2007), also in a postmortem brain investigation, found no statistically significant difference in VMAT2 protein (by immunohistochemistry) in humans exposed to MA, although a trend for a reduction was observed in nucleus accumbens and putamen.
In the first brain imaging study (PET) striatal VMAT2 binding (at a very low non-saturating radiotracer dose) was slightly (by 10%) reduced in MA users scanned when (reputedly) withdrawn for three years from the drug (Johanson et al., 2006), whereas in the two more recent investigations (both from our laboratory) binding was increased in early abstinence (less than one week) but was normal in those withdrawn for 7-15 days (Boileau et al., 2008; Boileau et al., 2016a). We interpret the increased VMAT2 binding in very early drug abstinence as explained by reduced competition between intravesicular dopamine and the VMAT2 radioligand at the VMAT2 site at which (we believe) dopamine is likely to be low in MA users who had recently taken the drug. This reduced competition between dopamine and the radiolabeled VMAT2 ligand (11C-dihydrotetrabenazine) for the VMAT2 binding site (Tong et al., 2008) likely explains increased VMAT2 binding in very early abstinence. [Similarly, striatal VMAT2 binding is slightly above normal in persons with dopa-responsive dystonia, characterized by low dopamine due to a metabolic defect in intact dopamine neurones; (De La Fuente-Fernandez et al., 2003)]. In later MA abstinence, levels of dopamine might normalize to some extent, explaining the normal VMAT2 binding one to two weeks following drug withdrawal. As expected, striatal VMAT2 binding was markedly decreased in a small group of patients with Parkinson's disease, employed as a neurological control in the MA study (Boileau et al., 2008).
The findings for the striatal VMAT2 dopamine marker in MA users (elevated to slightly decreased) differ from those for Parkinson's disease (markedly reduced) and raise further uncertainty whether loss of dopamine neurones is a feature of MA use in the human.
Substantia Nigra Cellularity
Parkinson's disease: Decreased number of “dopaminergic” substantia nigra cell bodies
It would be expected that loss of entire neurones utilizing dopamine as a neurotransmitter – those originating in the human substantia nigra and innervating the dopamine-rich striatum – should be reflected by decreased number of substantia nigra compacta dopamine-containing cell bodies. As expected, Parkinson's disease is fundamentally characterized by loss of nigral cell bodies that contain dopamine markers (the pigment neuromelanin formed as part of dopamine metabolism; the biosynthetic enzyme tyrosine hydroxylase) (Bernheimer et al., 1973; Hirsch et al., 1988; Fearnley & Lees, 1991; Pakkenberg et al., 1991; Rudow et al., 2008). In general, the loss of nigral cell number (and nigral dopamine concentration; (Hornykiewicz, 1963; Tong et al., 2006)) in Parkinson's disease is less than that of dopamine concentration in the nerve terminal regions of the caudate (about 80%) and putamen (>95%) (Hornykiewicz, 1992b). This prompted Hornykiewicz to suggest that the primary degenerative process in Parkinson's disease might begin at the nerve ending – a “retrograde degeneration” dying back phenomenon (Hornykiewicz, 1992b).
Methamphetamine users: No quantitative neuropathological data on nigral cellularity are available
As the formalin fixed tissue collected from multiple sources was not appropriate for quantitative cell counting, no quantitative assessment of the substantia nigra compacta cell bodies was conducted in autopsied brain of the MA users of our studies (Wilson et al., 1996a; Moszczynska et al., 2004). However, qualitative assessment of the substantia nigra in our studies by a neuropathologist experienced in histopathological examination of patients with degenerative movement disorders (including Parkinson's disease, progressive supranuclear palsy, and multiple system atrophy) showed no “obvious” loss of nigral cells in the MA users. Kitamura, in his neuropathological investigation of MA users in Japan, did not comment on brainstem changes in his case material (Kitamura et al., 2007; Kitamura et al., 2010).
Brain Gliosis as an Index of Neurotoxicity
As “brain damage” can be commonly associated with activation and/or proliferation of glial cells, there has been interest in establishing whether brain astrogliosis or microgliosis is a feature of human MA users (see (Tong et al., 2014)), as occurs in experimental animals who have received high doses of the stimulant (O'Callaghan & Miller, 1994; Thomas et al., 2004). The special interest in this question is related to the possibility that brain gliosis, depending on the stage of damage, might be both helpful and/or detrimental (Sofroniew & Vinters, 2010; Streit, 2010) – allowing for the use of glial-specific therapeutics that could protect the MA-exposed brain from harm.
Parkinson's disease: Substantia nigra microgliosis – but astrogliosis still uncertain
As reviewed by Fellner and colleagues (Fellner et al., 2011), microgliosis in the degenerating substantia nigra is a common feature of Parkinson's disease. With respect to astrogliosis the situation in Parkinson's disease is much less certain (see (Bruck et al., 2016)) – and in our own study levels of astroglial marker proteins in brain homogenates of persons with Parkinson's disease were generally normal (Tong et al., 2015). In this regard, Halliday and Stevens (Halliday & Stevens, 2011) have suggested that decreased astroglial response in Parkinson's disease might exacerbate the neurodegeneration.
Methamphetamine users: Brain gliosis status is uncertain
To our knowledge, studies of brain gliosis in human MA users are limited to a brain imaging study and two postmortem brain investigations.
In the first investigation Sekine and colleagues (Sekine et al., 2008) reported in a brain imaging (PET) study a massive increase (250-1500%) in binding of 11C-PK11195, purportedly to activated microglial and astroglial cells, throughout the brain of MA users who had been withdrawn from the drug for at least six months. The data however are uncertain because of the high non-specific binding and difficulty in specific binding quantitation (see (Tong et al., 2014)).
The second study included quantitative measurement of microglial and astroglial markers in postmortem brain of MA users (Kitamura et al., 2010) with the obtained data generally “mixed”. Thus, number of striatal microglial cells were normal (CR3/43 positive) or increased (glucose transporter-5 positive) and those of astroglial cells (glial fibrillary acid protein (GFAP) and S100B) were normal. Kitamura noted that there was no proliferation of activated microglia and that most astrocytes did not exhibit reactive changes.
Our own postmortem brain investigation employed a striatal brain homogenate approach in which concentrations of protein makers of microglial and astroglial cells were measured by Western blotting. Although protein levels of the glial markers were, as expected, elevated in brain of the neurological controls (multiple system atrophy), concentrations of intact glial proteins were normal in the MA group (Tong et al., 2014). The only “hint” of a glial disturbance was our finding (of uncertain significance) of increased brain levels of two partially degraded astrocytic protein markers (heat shock protein-27, vimentin) in the MA users.
The above three studies taken together suggest that the status of brain gliosis in human MA users is still uncertain.
Does Methamphetamine Cause Clinical Parkinsonism in the Human?
If MA severely damages dopamine neurones, Parkinsonism should be a characteristic feature of at least a subgroup of humans who use MA. Assuming that MA users have a striatal dopamine deficiency, it might also be expected that a subgroup might be more susceptible to drug-induced Parkinsonism should they receive dopamine receptor blocking drugs for control of psychosis. However, suggestive evidence for the latter possibility appears limited to a single case report (Matthew & Gedzior, 2015).
Recently, we employed an epidemiological approach to address the above questions by making use of United States California statewide inpatient hospital data in which individuals with a MA/amphetamine related condition were followed for up to 16 years to determine whether the MA group was more likely than a matched (appendicitis) group and a drug control (cocaine) group to show increased risk of developing Parkinson's disease. Consistent with our hypothesis, the MA, but not the cocaine group, had increased hazard ratio (HR, 1.76; 95% CI 1.12-2.75) as compared with the proxy control group (Callaghan et al., 2010; Callaghan et al., 2012a). More recently, Curtin and colleagues (Curtin et al., 2015) reported similar findings using a Utah Statewide population database of MA users who had a higher risk (HR, 2.8; CI 1.6-4.8) of developing Parkinson's disease. These longitudinal association studies do not provide “proof” but are consistent with the possibility that MA might damage dopamine neurones in a subgroup of users in which Parkinsonism develops in middle/late age after some age-related loss of dopamine neurones. However, the findings also would suggest that only a very small proportion of MA users are subsequently diagnosed with Parkinson's disease.
Comment
Marked dopamine loss in some methamphetamine users could be “harmful”
The objective of this review was to evaluate the available literature to establish whether MA might damage dopamine neurones in the human.
Employing the term “damage” (“injury”,“harm”) in a very general sense it is reasonable, we believe, to conclude that the dopamine loss in some MA users could cause some behavioural abnormality, perhaps limited to those individuals in which the reduction was near-total. Thus, we think it likely that subjects #448 (95% caudate dopamine loss) and #678 (97% caudate dopamine loss) of our study (Moszczynska et al., 2004), who suffered a near-total loss of dopamine (95%, 97%, respectively) (which falls into the “Parkinsonian range”) would have had some behavioural impairment caused by the very low dopamine. To our knowledge, no behavioural data are available for individuals affected exclusively by a loss of dopaminergic innervation to the caudate, or selectively to the putamen. However, Hornykiewicz has emphasized the different overlapping roles in the human in which dopamine innervation to caudate nucleus can be expected to control aspects of “cognition” whereas that to the putamen (more affected than that to caudate in Parkinson's disease) is more likely involved in control of motor function (see (Kish et al., 1988)). Given that the caudate nucleus is affected more by dopamine loss in MA users, we suspect that the major dopamine-related harm in these drug users might be cognitive difficulties that would persist as long as the dopamine deficiency is severe.
The possibility that any cognitive deficits in MA users are explained by low dopamine, however, remains to be established. In our brain imaging VMAT2 binding study of MA users in early abstinence we did find an association between higher striatal VMAT2 binding (suggesting low dopamine) and poorer performance on some neuropsychological tests, but as a whole, only slight deficits in cognitive function were observed in the MA users as compared to control subjects (Boileau et al., 2016a). Also, we recently reported that a brain imaging index of dopamine neurotransmission (amphetamine-stimulated changes in dopamine D3/D2 receptor binding) was either enhanced (substantia nigra and globus pallidus) or unchanged (striatum), i.e., not blunted, in MA users withdrawn from the drug for at least seven days (Boileau et al., 2016b) – which would not be predicted in subjects having substantial dopamine reduction. However, the possibility still remains that dopamine neurone function in the striatum might be diminished in, but restricted to, those in very early abstinence when dopamine concentration is presumably much lower than that in later abstinence (Boileau et al., 2016a) or after more severe chronic MA use (Boileau et al., 2016b) (see also (Wang et al., 2012)).
But does methamphetamine cause loss of all or part of dopamine neurones?
Taking Parkinson's disease as the gold standard of a degenerative dopamine deficiency disorder, we can compare the loss of dopamine markers in Parkinson's disease vs. human MA users in our autopsied brain studies. As shown in the Table, whereas levels of striatal dopamine nerve terminal markers, without any exception, are (moderately to severely) decreased in Parkinson's disease, concentrations of only two dopamine markers (dopamine, dopamine transporter) are below normal in MA users and with some (dopamine metabolites, biosynthetic enzymes, VMAT2) normal. Although levels of dopamine markers can change independently of changes in neuronal number, the findings that, unlike Parkinson's disease, not all striatal dopamine markers are reduced suggest that any dopamine neuronal loss in MA, if at all present, is unlikely to be substantial.
Table. Brain dopamine marker changes in autopsied brain of patients with PD versus MA users.
| Dopamine markers | PD | MA |
|---|---|---|
| Dopamine | Decreased | Decreased |
| Metabolites (HVA, DOPAC, 3-MT) | Decreased | Normal |
| Biosynthetic enzymes (TH, AADC) | Decreased | Normal |
| VMAT2 | Decreased | Normal to slightly decreased |
| DAT | Decreased | Decreased |
| SNpc cellularity | Decreased | Unknown but no “obvious” cell loss |
AADC, aromatic L-amino acid decarboxylase; DAT, dopamine transporter; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; MA, methamphetamine; 3-MT, 3-methoxytyramine; PD, Parkinson's disease; SNpc, substantia nigra pars compacta; TH, tyrosine hydroxylase; VMAT2, Vesicular monoamine transporter subtype 2.
Dopamine substitution therapeutics in Parkinson's disease – any role in methamphetamine users?
From a public health perspective, most would agree that it is far more important to develop a therapeutic that will help people suffering from medical illnesses (e.g., Parkinson's disease, MA addiction) than to focus exclusively on the more limited “academic” question whether the illness is or is not associated with a particular pattern of brain damage or pathology. Similarly, the new focus on the glial cell (an index of toxicity) in MA users is not primarily on its use as a “toxicity” index, but rather on the hope that a helpful therapeutic could be developed based on neuroinflammatory actions/factors released by the glial cell.
As Hornykiewicz (Hornykiewicz, 2004) comments: “The discovery of the profound lack of dopamine in the parkinsonian brain opened the door to the next and most consequential step: that from the ‘dead (post mortem) human material to the live patient’, i.e. to the use of levodopa as dopamine substitution treatment in Parkinson's disease. To put this idea into effect, in January 1961, I asked Walter Birkmayer, who was in charge of the neurological ward of the Municipal Home for the Aged in Lainz (Vienna) to try out levodopa in his permanently housed Parkinson patients”. In November 1961 Birkmayer and Hornykiewicz (Birkmayer & Hornykiewicz, 1961) published a short paper in “Wiener Klinische Wochenschrift” providing the conclusive evidence that amelioration of parkinsonism could be achieved in patients with Parkinson's disease by intravenous levodopa, and that same month presented a dramatic film of the treatment to the Medical Association of Vienna. “…The high expectations of those present were not disappointed by our presentation, which was followed by a spirited discussion. The lucidity of the rationale for the use of L-DOPA and its convincing, if not to say miraculous, effect in patients, produced the definite feeling in the audience filling the traditional lecture hall of the “Billroth Haus” that they had just witnessed something that had all the marks of the out of the ordinary.” (Hornykiewicz, 2001)
The utility of levodopa is unquestionably established in Parkinson's disease. Although levodopa is not a “cure” [Following on an early suggestion of Hornykiewicz that the cause of the degeneration in Parkinson's disease might be due to a “reserpine-line” abnormality in dopamine storage (Hornykiewicz, 1964), the Vienna Hornykiewicz team later obtained postmortem data suggesting that a focus on abnormal dopamine vesicular storage in Parkinson's disease might actually point the way to a neuroprotective therapeutic; (Pifl et al., 2014)], this symptomatic treatment has transformed neurology practice.
Hornykiewicz reminds us: “Today it is difficult for people, even for the young neurologists, to realize what the situation of the neurologists was before the L-DOPA era. The Parkinson patients were hopeless patients; they were crowding the chronic wards in the hospitals and they ended up completely stiff and bedridden, they could not get up, they could not feed themselves, they could not move, and had to be cared for until they died. The doctors were powerless. They could not do anything for those patients” (Hargittai & Hargittai, 2005).
Assuming that the Toronto postmortem brain (Wilson et al., 1996a; Moszczynska et al., 2004) and brain imaging findings – suggesting that some MA users have a potentially harmful and severe brain dopamine deficiency – can be replicated and extended by independent groups, it seems reasonable to suggest that dopamine substitution therapy in methamphetamine users might help improve aspects of cognition subserved by caudate dopamine function in some drug users. It could also be argued that the striatal dopamine transporter reduction, consistently observed in brain of methamphetamine users, might actually represent a compensatory phenomenon to “correct” the dopamine deficiency by increasing synaptic dopamine levels via decreased neurotransmitter uptake. It needs to be emphasized however that because of the possible interaction between levodopa and MA resulting in brain toxicity, levodopa should probably not be administered while residual MA is present (Boileau et al., 2016a), making the usefulness of levodopa as dopamine substitution therapy in MA users logistically difficult. Also, whereas a severe striatal dopamine deficiency is present in all patients with Parkinson's disease, information on brain dopamine status --- to identify those drug users having a severe dopamine depletion --- is not yet available for MA users by using a brain imaging biomarker. Further, little information is available on the length of time in abstinence that a dopamine deficiency might persist, with our preliminary brain imaging data suggesting that a dopamine deficiency could be short-lived in some (low dose?) drug users (Boileau et al., 2016a). Finally, perhaps MA users might only require low doses of levodopa to be of benefit. In this context, MA users might suffer dopamine loss but no loss of dopamine neurones, and therefore need only very low doses of levodopa to be of benefit, as is the case with dopa-responsive dystonia (Furukawa et al., 1999).
Conclusions
We believe that it is premature, based on the available literature, to conclude whether recreational doses of MA cause or do not cause loss of, or damage to dopamine neurones in humans.
However, the fact that in our autopsied brain investigations not all dopamine markers were decreased in the dopamine-rich striatum of MA users – unlike the situation in Parkinson's disease (see Table) – leads us to speculate that MA, at the doses used by the subjects of our studies, was unlikely to have caused significant damage to brain dopamine neurones in the human. We further speculate that our findings in MA users who had recently used the drug, of very low striatal dopamine, as demonstrated by direct measurement (postmortem brain) and as inferred from high VMAT2 binding (living brain), are likely explained by a massive and acute drug-induced release and depletion of the neurotransmitter – that is at least partly reversible. Similarly, we feel that it is reasonable to speculate that the remarkably constant findings, by many different groups in the literature, of low striatal dopamine transporter in MA users is due to a compensatory (and reversible) attempt to maximize synaptic dopamine – a protein “downregulation” in the absence of neurone loss. We emphasize the above are only speculations that may well need to be revised in future.
Assuming for the sake of argument that nigral dopamine cellularity is spared in MA users, we also acknowledge the practical difficulty in quantitative measurement of striatal dopamine nerve terminal number/density in human brain without depending on a dopamine “marker” (to identify the neurone as “dopaminergic”) that has the potential of up- or down-regulation independently of any changes in neuropil concentration (for discussion see (Kish, 2014)). In fact, we argue that this “biochemical marker confound” will continue to raise uncertainty when interpreting findings in all studies of dopamine nerve terminal density in the human that aim to be “quantitative”. (For example, what is the evidence that 60% loss of striatal dopamine, dopamine transporter, VMAT2, or dopamine biosynthetic enzymes equals 60%, or indeed any loss, of dopamine neuropil?). The lack of “stability” of dopamine neurone markers is much less of an issue for those few animal studies that have qualitatively (only) assessed dopamine nerve terminal structural damage after MA exposure using classical histological neurotoxicity techniques (including electron microscopy) and reported evidence, for example, of tyrosine hydroxylase-positive neuronal processes with “swollen and either empty of organelles or filled with membranous debris” (Ryan et al., 1990) and “vacuolated tyrosine hydroxylase-immunoreactive fibers with degenerating morphology” (Ares-Santos et al., 2014). However, to our knowledge, such technically difficult investigations demonstrating structural damage to striatal dopaminergic terminals have yet to be conducted in postmortem brain of MA users. With respect to structural neuronal abnormalities in Parkinson's disease striatum, the literature also appears to be limited to qualitative reports of morphologically abnormal, “short and kinked” (resembling Alzheimer's disease neuropil threads), or “swollen”, “thickened non-beaded” tyrosine hydroxylase-positive neurites, which are probably undergoing degeneration, in the dopamine and tyrosine hydroxylase depleted striatum (Benzing et al., 1993; Huot et al., 2007; Bedard et al., 2011; Kordower et al., 2013) and pallidum (Jan et al., 2000) of autopsied brains from patients with Parkinson's disease and a report of “dystrophic neurites” in (biopsied) caudate nucleus of two patients with advanced Parkinson's disease – but with the authors' acknowledgement that it could not be determined whether the changes were actually dopaminergic (no dopamine marker staining was performed in the electron microscopy study) (Lach et al., 1992).
This review also raises the general question of the applicability of experimental animal findings to the human condition.
As mentioned above, Oleh Hornykiewicz was encouraged by data from animal experiments (e.g., levodopa reversal of reserpine akinesia) but since the predictability of animal findings to the human is always uncertain, the definitive study had to await his investigation of dopamine in humans with Parkinson's disease. In the case of MA, there remains the question whether animal findings of dopamine neurone damage following very high dose MA exposure (see (Kish, 2014)) will ever translate to human users of the drug. Had humans received the “equivalent”, often lethal, high doses of MA as those used in the animal studies, would we have observed in our autopsied brain studies more convincing evidence of dopamine neurone disturbance in which all examined dopamine markers would be markedly below normal and with prominent astroglial and microglial activation?
We do feel that our finding of low dopamine in MA users very recently withdrawn from the drug might have some clinical significance should the low dopamine (which we suggest is a reversible phenomenon) impact negatively on cognitive functioning and drug relapse. In this respect, the investigational use of levodopa as a dopamine substitution therapy in MA users in early drug withdrawal receives some general justification and would be a clear test of this hypothesis. However, more information on the safety and time course of any dopamine deficiency in MA users are needed, as well as the need to identify those users who are more likely to need and benefit from dopamine substitution therapy. Today, Parkinson's disease, owing to the efforts of Oleh Hornykiewicz in Vienna, still remains the gold standard of a levodopa-treatable neurodegenerative dopamine deficiency disorder.
Acknowledgments
This work was supported by the National Institute of Drug Abuse, National Institute of Health grants DA07182 and DA040066 to SJK.
References
- Ares-Santos S, Granado N, Espadas I, Martinez-Murillo R, Moratalla R. Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology. 2014;39:1066–1080. doi: 10.1038/npp.2013.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard C, Wallman MJ, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson's disease and Huntington's chorea. Parkinsonism Relat Disord. 2011;17:593–598. doi: 10.1016/j.parkreldis.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Benzing WC, Mufson EJ, Armstrong DM. Alzheimer's disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid beta-protein deposition is present. Brain Res. 1993;606:10–18. doi: 10.1016/0006-8993(93)91563-8. [DOI] [PubMed] [Google Scholar]
- Bernheimer H. Distribution of Homovanillic Acid in the Human Brain. Nature. 1964;204:587–588. doi: 10.1038/204587b0. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O. [Distribution of 5-hydroxytryptamine (serotonin) in the human brain and its behavior in patients with Parkinson's syndrome] Klin Wochenschr. 1961;39:1056–1059. doi: 10.1007/BF01487648. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Hornykiewicz O. [Decreased homovanillic acid concentration in the brain in parkinsonian subjects as an expression of a disorder of central dopamine metabolism] Klin Wochenschr. 1965;43:711–715. doi: 10.1007/BF01707066. [DOI] [PubMed] [Google Scholar]
- Bertler A, Rosengren E. Occurrence and distribution of dopamine in brain and other tissues. Experientia. 1959;15:10–11. doi: 10.1007/BF02157069. [DOI] [PubMed] [Google Scholar]
- Birkmayer W, Hornykiewicz O. [The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia] Wiener klinische Wochenschrift. 1961;73:787–788. [PubMed] [Google Scholar]
- Boileau I, McCluskey T, Tong J, Furukawa Y, Houle S, Kish SJ. Rapid Recovery of Vesicular Dopamine Levels in Methamphetamine Users in Early Abstinence. Neuropsychopharmacology. 2016a;41:1179–1187. doi: 10.1038/npp.2015.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau I, Payer D, Rusjan PM, Houle S, Tong J, McCluskey T, Wilson AA, Kish SJ. Heightened Dopaminergic Response to Amphetamine at the D3 Dopamine Receptor in Methamphetamine Users. Neuropsychopharmacology. 2016b doi: 10.1038/npp.2016.108. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau I, Rusjan P, Houle S, Wilkins D, Tong J, Selby P, Guttman M, Saint-Cyr JA, Wilson AA, Kish SJ. Increased vesicular monoamine transporter binding during early abstinence in human methamphetamine users: Is VMAT2 a stable dopamine neuron biomarker? J Neurosci. 2008;28:9850–9856. doi: 10.1523/JNEUROSCI.3008-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck D, Wenning GK, Stefanova N, Fellner L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol Dis. 2016;85:262–274. doi: 10.1016/j.nbd.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan RC, Cunningham JK, Sajeev G, Kish SJ. Incidence of Parkinson's disease among hospital patients with methamphetamine-use disorders. Mov Disord. 2010;25:2333–2339. doi: 10.1002/mds.23263. [DOI] [PubMed] [Google Scholar]
- Callaghan RC, Cunningham JK, Sykes J, Kish SJ. Increased risk of Parkinson's disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 2012a;120:35–40. doi: 10.1016/j.drugalcdep.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Callaghan RC, Cunningham JK, Verdichevski M, Sykes J, Jaffer SR, Kish SJ. All-cause mortality among individuals with disorders related to the use of methamphetamine: a comparative cohort study. Drug Alcohol Depend. 2012b;125:290–294. doi: 10.1016/j.drugalcdep.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Cardenas L, Houle S, Kapur S, Busto UE. Oral D-amphetamine causes prolonged displacement of [11C]raclopride as measured by PET. Synapse. 2004;51:27–31. doi: 10.1002/syn.10282. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M, Magnusson T, Waldeck B. On the presence of 3-hydroxytyramine in brain. Science. 1958;127:471. doi: 10.1126/science.127.3296.471. [DOI] [PubMed] [Google Scholar]
- Chou YH, Huang WS, Su TP, Lu RB, Wan FJ, Fu YK. Dopamine transporters and cognitive function in methamphetamine abuser after a short abstinence: A SPECT study. Eur Neuropsychopharmacol. 2007;17:46–52. doi: 10.1016/j.euroneuro.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Curtin K, Fleckenstein AE, Robison RJ, Crookston MJ, Smith KR, Hanson GR. Methamphetamine/amphetamine abuse and risk of Parkinson's disease in Utah: a population-based assessment. Drug Alcohol Depend. 2015;146:30–38. doi: 10.1016/j.drugalcdep.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Fuente-Fernandez R, Furtado S, Guttman M, Furukawa Y, Lee CS, Calne DB, Ruth TJ, Stoessl AJ. VMAT2 binding is elevated in dopa-responsive dystonia: visualizing empty vesicles by PET. Synapse. 2003;49:20–28. doi: 10.1002/syn.10199. [DOI] [PubMed] [Google Scholar]
- Ehringer H, Hornykiewicz O. Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des menches und ihr Verhalten bei Erkrankungen des extrapyramiden Systems. (Distribution of noradrenaline and dopamine (3-hydroxytyramine) in human brain: their behaviour in extrapyramidal system diseases) Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. (Republished in English translation in Parkinsonism and Related Disorders 1998;1234:1253-1257) [DOI] [PubMed] [Google Scholar]
- Fahn S, Libsch LR, Cutler RW. Monoamines in the human neostriatum: topographic distribution in normals and in Parkinson's disease and their role in akinesia, rigidity, chorea, and tremor. J Neurol Sci. 1971;14:427–455. doi: 10.1016/0022-510x(71)90178-x. [DOI] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Fellner L, Jellinger KA, Wenning GK, Stefanova N. Glial dysfunction in the pathogenesis of alpha-synucleinopathies: emerging concepts. Acta Neuropathol. 2011;121:675–693. doi: 10.1007/s00401-011-0833-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KA, Koeppe RA, Kilbourn MR. Imaging the vesicular monoamine transporter. Advances in neurology. 2001;86:237–247. [PubMed] [Google Scholar]
- Frey KA, Koeppe RA, Kilbourn MR, Vander Borght TM, Albin RL, Gilman S, Kuhl DE. Presynaptic monoaminergic vesicles in Parkinson's disease and normal aging. Ann Neurol. 1996;40:873–884. doi: 10.1002/ana.410400609. [DOI] [PubMed] [Google Scholar]
- Frost JJ, Rosier AJ, Reich SG, Smith JS, Ehlers MD, Snyder SH, Ravert HT, Dannals RF. Positron emission tomographic imaging of the dopamine transporter with 11C-WIN 35,428 reveals marked declines in mild Parkinson's disease. Ann Neurol. 1993;34:423–431. doi: 10.1002/ana.410340331. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Nygaard TG, Gutlich M, Rajput AH, Pifl C, DiStefano L, Chang LJ, Price K, Shimadzu M, Hornykiewicz O, Haycock JW, Kish SJ. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology. 1999;53:1032–1041. doi: 10.1212/wnl.53.5.1032. [DOI] [PubMed] [Google Scholar]
- Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson's disease. Mov Disord. 2011;26:6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- Hargittai B, Hargittai I. Oleh Hornykiewicz Candid Science V: Conversations with Famous Scientists. Imperial College Press; London: 2005. pp. 619–647. [Google Scholar]
- Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- Holzer G, Hornykiewicz O. [On dopamine (hydroxytyramine) metabolism in the rat brain] Naunyn-Schmiedebergs Archiv fur experimentelle Pathologie und Pharmakologie. 1959;237:27–33. [PubMed] [Google Scholar]
- Hornykiewicz O. [The tropical localization and content of noradrenalin and dopamine (3-hydroxytyramine) in the substantia nigra of normal persons and patients with Parkinson's disease] Wiener klinische Wochenschrift. 1963;75:309–312. [PubMed] [Google Scholar]
- Hornykiewicz O. The role of brain dopamine (3-hydroxytyramine) in parkinsonism. In: Trabucchi E, Paoletti R, Canal N, editors. Biochemical and neurophysiological correlations of centrally acting drugs. Pergamon; Oxford: 1964. pp. 57–68. [Google Scholar]
- Hornykiewicz O. From dopamine to Parkinson's disease: a personal research record. In: Samson F, Adelman G, editors. The Neurosciences: Paths of Discovery, II. Birkhäuser; Boston: 1992a. pp. 125–147. [Google Scholar]
- Hornykiewicz O. The primary site of dopamine neuron damage in Parkinson's disease: substantia nigra or striatum? Mov Disord. 1992b;7:288. [Google Scholar]
- Hornykiewicz O. Parkinson's disease and the adaptive capacity of the nigrostriatal dopamine system: possible neurochemical mechanisms. Advances in neurology. 1993;60:140–147. [PubMed] [Google Scholar]
- Hornykiewicz O. Levodopa in the 1960s: starting point Vienna. In: Poewe W, Lees AJ, editors. 20 Years of Madopar - New Avenues. Editiones Roche; Basel, Switzerland: 1994. pp. 1–17. [Google Scholar]
- Hornykiewicz O. How L-DOPA was discovered as a drug for Parkinson's disease 40 years ago. Wiener klinische Wochenschrift. 2001;113:855–862. [PubMed] [Google Scholar]
- Hornykiewicz O. Oleh Hornykiewicz. In: Squire LR, editor. The History of Neuroscience in Autobiography. Elsevier; Amsterdam: 2004. pp. 240–281. [Google Scholar]
- Hornykiewicz O. A brief history of levodopa. Journal of neurology. 2010;257:S249–252. doi: 10.1007/s00415-010-5741-y. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. Advances in neurology. 1987;45:19–34. [PubMed] [Google Scholar]
- Huot P, Levesque M, Parent A. The fate of striatal dopaminergic neurons in Parkinson's disease and Huntington's chorea. Brain. 2007;130:222–232. doi: 10.1093/brain/awl332. [DOI] [PubMed] [Google Scholar]
- Jan C, Francois C, Tande D, Yelnik J, Tremblay L, Agid Y, Hirsch E. Dopaminergic innervation of the pallidum in the normal state, in MPTP-treated monkeys and in parkinsonian patients. Eur J Neurosci. 2000;12:4525–4535. [PubMed] [Google Scholar]
- Johanson CE, Frey KA, Lundahl LH, Keenan P, Lockhart N, Roll J, Galloway GP, Koeppe RA, Kilbourn MR, Robbins T, Schuster CR. Cognitive function and nigrostriatal markers in abstinent methamphetamine abusers. Psychopharmacology (Berl) 2006;185:327–338. doi: 10.1007/s00213-006-0330-6. [DOI] [PubMed] [Google Scholar]
- Kish SJ. Pharmacologic mechanisms of crystal meth. Cmaj. 2008;178:1679–1682. doi: 10.1503/cmaj.071675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ. Chapter 08: The Pathology of Methamphetamine Use in the Human Brain. In: Madras BK, Kuhar M, editors. The Effects of Drug Abuse on the Human Nervous System. Elsevier Inc; Amsterdam: 2014. pp. 203–297. [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Kitamura O, Takeichi T, Wang EL, Tokunaga I, Ishigami A, Kubo S. Microglial and astrocytic changes in the striatum of methamphetamine abusers. Leg Med (Tokyo) 2010;12:57–62. doi: 10.1016/j.legalmed.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Kitamura O, Tokunaga I, Gotohda T, Kubo S. Immunohistochemical investigation of dopaminergic terminal markers and caspase-3 activation in the striatum of human methamphetamine users. Int J Legal Med. 2007;121:163–168. doi: 10.1007/s00414-006-0087-9. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lach B, Grimes D, Benoit B, Minkiewicz-Janda A. Caudate nucleus pathology in Parkinson's disease: ultrastructural and biochemical findings in biopsy material. Acta Neuropathol. 1992;83:352–360. doi: 10.1007/BF00713525. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck CH, Rosenblatt W, Zea-Ponce Y, Zoghbi SS, Baldwin RM, Charney DS, Hoffer PB, Kung HF, et al. SPECT imaging of striatal dopamine release after amphetamine challenge. J Nucl Med. 1995;36:1182–1190. [PubMed] [Google Scholar]
- Lee CS, Samii A, Sossi V, Ruth TJ, Schulzer M, Holden JE, Wudel J, Pal PK, de la Fuente-Fernandez R, Calne DB, Stoessl AJ. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson's disease. Ann Neurol. 2000;47:493–503. [PubMed] [Google Scholar]
- Marsden CD. Parkinson's disease. Lancet. 1990;335:948–952. doi: 10.1016/0140-6736(90)91006-v. [DOI] [PubMed] [Google Scholar]
- Matthew BJ, Gedzior JS. Drug-induced parkinsonism following chronic methamphetamine use by a patient on haloperidol decanoate. International journal of psychiatry in medicine. 2015;50:405–411. doi: 10.1177/0091217415612736. [DOI] [PubMed] [Google Scholar]
- McCann UD, Kuwabara H, Kumar A, Palermo M, Abbey R, Brasic J, Ye W, Alexander M, Dannals RF, Wong DF, Ricaurte GA. Persistent cognitive and dopamine transporter deficits in abstinent methamphetamine users. Synapse. 2008;62:91–100. doi: 10.1002/syn.20471. [DOI] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszczynska A, Fitzmaurice P, Ang L, Kalasinsky KS, Schmunk GA, Peretti FJ, Aiken SS, Wickham DJ, Kish SJ. Why is parkinsonism not a feature of human methamphetamine users? Brain. 2004;127:363–370. doi: 10.1093/brain/awh046. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- Pakkenberg B, Moller A, Gundersen HJ, Mouritzen Dam A, Pakkenberg H. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson's disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. 1991;54:30–33. doi: 10.1136/jnnp.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol Pharmacol. 1995;47:368–373. [PubMed] [Google Scholar]
- Pifl C, Rajput A, Reither H, Blesa J, Cavada C, Obeso JA, Rajput AH, Hornykiewicz O. Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci. 2014;34:8210–8218. doi: 10.1523/JNEUROSCI.5456-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricaurte GA, Mechan AO, Yuan J, Hatzidimitriou G, Xie T, Mayne AH, McCann UD. Amphetamine treatment similar to that used in the treatment of adult attention-deficit/hyperactivity disorder damages dopaminergic nerve endings in the striatum of adult nonhuman primates. J Pharmacol Exp Ther. 2005;315:91–98. doi: 10.1124/jpet.105.087916. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH. Monoamine transporters and psychostimulant drugs. Eur J Pharmacol. 2003;479:23–40. doi: 10.1016/j.ejphar.2003.08.054. [DOI] [PubMed] [Google Scholar]
- Rudow G, O'Brien R, Savonenko AV, Resnick SM, Zonderman AB, Pletnikova O, Marsh L, Dawson TM, Crain BJ, West MJ, Troncoso JC. Morphometry of the human substantia nigra in ageing and Parkinson's disease. Acta Neuropathol. 2008;115:461–470. doi: 10.1007/s00401-008-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan LJ, Linder JC, Martone ME, Groves PM. Histological and ultrastructural evidence that D-amphetamine causes degeneration in neostriatum and frontal cortex of rats. Brain Res. 1990;518:67–77. doi: 10.1016/0006-8993(90)90955-b. [DOI] [PubMed] [Google Scholar]
- Sano L, Gamo T, Kakimoto Y, Taniguchi K, Takesada M, Nishinuma K. Distribution of catechol compounds in the human brain. Biochim Biophys Acta. 1959;32:586–587. doi: 10.1016/0006-3002(59)90652-3. [DOI] [PubMed] [Google Scholar]
- Schouw ML, Caan MW, Geurts HM, Schmand B, Booij J, Nederveen AJ, Reneman L. Monoaminergic dysfunction in recreational users of dexamphetamine. Eur Neuropsychopharmacol. 2013;23:1491–1502. doi: 10.1016/j.euroneuro.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Iyo M, Ouchi Y, Matsunaga T, Tsukada H, Okada H, Yoshikawa E, Futatsubashi M, Takei N, Mori N. Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am J Psychiatry. 2001;158:1206–1214. doi: 10.1176/appi.ajp.158.8.1206. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Minabe Y, Ouchi Y, Takei N, Iyo M, Nakamura K, Suzuki K, Tsukada H, Okada H, Yoshikawa E, Futatsubashi M, Mori N. Association of dopamine transporter loss in the orbitofrontal and dorsolateral prefrontal cortices with methamphetamine-related psychiatric symptoms. Am J Psychiatry. 2003;160:1699–1701. doi: 10.1176/appi.ajp.160.9.1699. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, Kawai M, Takebayashi K, Yamamoto S, Matsuzaki H, Ueki T, Mori N, Gold MS, Cadet JL. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci. 2008;28:5756–5761. doi: 10.1523/JNEUROSCI.1179-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglial activation and neuroinflammation in Alzheimer's disease: a critical examination of recent history. Front Aging Neurosci. 2010;2:22. doi: 10.3389/fnagi.2010.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Progress in neurobiology. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Tong J, Ang LC, Williams B, Furukawa Y, Fitzmaurice P, Guttman M, Boileau I, Hornykiewicz O, Kish SJ. Low levels of astroglial markers in Parkinson's disease: relationship to alpha-synuclein accumulation. Neurobiol Dis. 2015;82:243–253. doi: 10.1016/j.nbd.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Boileau I, Furukawa Y, Chang LJ, Wilson AA, Houle S, Kish SJ. Distribution of vesicular monoamine transporter 2 protein in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab. 2011;31:2065–2075. doi: 10.1038/jcbfm.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Fitzmaurice P, Furukawa Y, Schmunk GA, Wickham DJ, Ang LC, Sherwin A, McCluskey T, Boileau I, Kish SJ. Is brain gliosis a characteristic of chronic methamphetamine use in the human? Neurobiol Dis. 2014;67:107–118. doi: 10.1016/j.nbd.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Tong J, Hornykiewicz O, Kish SJ. Inverse relationship between brain noradrenaline level and dopamine loss in Parkinson disease: a possible neuroprotective role for noradrenaline. Arch Neurol. 2006;63:1724–1728. doi: 10.1001/archneur.63.12.1724. [DOI] [PubMed] [Google Scholar]
- Tong J, Wilson AA, Boileau I, Houle S, Kish SJ. Dopamine modulating drugs influence striatal (+)-[11C]DTBZ binding in rats: VMAT2 binding is sensitive to changes in vesicular dopamine concentration. Synapse. 2008;62:873–876. doi: 10.1002/syn.20573. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci. 2001a;21:9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding YS, Logan J, Wong C, Miller EN. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001b;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Smith L, Fowler JS, Telang F, Logan J, Tomasi D. Recovery of dopamine transporters with methamphetamine detoxification is not linked to changes in dopamine release. Neuroimage. 2015;121:20–28. doi: 10.1016/j.neuroimage.2015.07.035. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Smith L, Volkow ND, Telang F, Logan J, Tomasi D, Wong CT, Hoffman W, Jayne M, Alia-Klein N, Thanos P, Fowler JS. Decreased dopamine activity predicts relapse in methamphetamine abusers. Mol Psychiatry. 2012;17:918–925. doi: 10.1038/mp.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil-Malherbe H, Bone AD. Effect of reserpine on the intracellular distribution of catecholamines in the brain stem of the rabbit. Nature. 1958;181:1474–1475. doi: 10.1038/1811474a0. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996a;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kish SJ. The vesicular monoamine transporter, in contrast to the dopamine transporter, is not altered by chronic cocaine self-administration in the rat. J Neurosci. 1996;16:3507–3510. doi: 10.1523/JNEUROSCI.16-10-03507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Rajput A, Ang L, Guttman M, Shannak K, Niznik HB, Hornykiewicz O, Pifl C, Kish SJ. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson's disease. Neurology. 1996b;47:718–726. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
- Yuan J, Lv R, Robert Brasic J, Han M, Liu X, Wang Y, Zhang G, Liu C, Li Y, Deng Y. Dopamine transporter dysfunction in Han Chinese people with chronic methamphetamine dependence after a short-term abstinence. Psychiatry Res. 2014;221:92–96. doi: 10.1016/j.pscychresns.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Zhong XH, Haycock JW, Shannak K, Robitaille Y, Fratkin J, Koeppen AH, Hornykiewicz O, Kish SJ. Striatal dihydroxyphenylalanine decarboxylase and tyrosine hydroxylase protein in idiopathic Parkinson's disease and dominantly inherited olivopontocerebellar atrophy. Mov Disord. 1995;10:10–17. doi: 10.1002/mds.870100104. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Acheson AL, Stachowiak MK, Stricker EM. Neurochemical compensation after nigrostriatal bundle injury in an animal model of preclinical parkinsonism. Arch Neurol. 1984;41:856–861. doi: 10.1001/archneur.1984.04050190062015. [DOI] [PubMed] [Google Scholar]