

Abstract
Uncovering the molecular context of dysregulated metabolites is crucial to understand pathogenic pathways. However, their system-level analysis has been limited owing to challenges in global metabolite identification. Most metabolite features detected by untargeted metabolomics carried out by liquid-chromatography-mass spectrometry cannot be uniquely identified without additional, time-consuming experiments. We report a network-based approach, prize-collecting Steiner forest algorithm13 for integrative analysis of untargeted metabolomics (PIUMet), that infers molecular pathways and components via integrative analysis of metabolite features, without requiring their identification. We demonstrated PIUMet by analyzing changes in metabolism of sphingolipids, fatty acids and steroids in a Huntington’s disease model. Additionally, PIUMet enabled us to elucidate putative identities of altered metabolite features in diseased cells, and infer experimentally undetected, disease-associated metabolites and dysregulated proteins. Finally, we established PIUMet’s ability for integrative analysis of untargeted metabolomics data with proteomics data, demonstrating that this approach elicits disease-associated metabolites and proteins that cannot be inferred by individual analysis of these data.
The development of effective therapeutic approaches requires a system-level understanding of the molecules altered in a disease, as well as complex interactions among them. Metabolites are of particular interest1, as global metabolite measurements reflect disease-associated cellular biochemical activities that are not detected by transcriptional analysis or other ‘omic’ experiments2. Therefore, the integrative analysis of metabolomics with other omic data is a crucial step to identifying disease etiology.
Systems-level analysis of metabolomic data has been limited, however. Global measurements of metabolites (untargeted metabolomics) are performed using liquid chromatography-mass spectrometry (LC-MS)3, which detects thousands of metabolite ‘peaks’ or ‘features’, defined by a unique combination of a mass-to-charge ratio (m/z) and retention time. Despite the high mass accuracy of modern instruments, several metabolites can match a peak. Therefore, unambiguous identification of features is a bottleneck in metabolomic studies4,5. Typically, relatively few of the features are characterized through tandem mass spectrometry (MS/MS) experiments that require additional time and incur additional cost6, and the majority of the features remain unknown. The sparsity of identified metabolites is an additional major obstacle to interpretation of metabolomic data7–9.
Current approaches for analyzing metabolomic data including network and pathway tools7–9 rely on the metabolite features that had been further characterized via MS/MS. A first step toward analyzing uncharacterized metabolite features involved inferring potential identities of metabolite features using a Gaussian graphical model that leveraged genomics10, but that approach only applied for large sets of samples with genomic data. The Mummichog algorithm11 used metabolic networks to resolve some unknown metabolite features11 but did not link these data to other system-level molecular information. Because each type of molecular data is biased toward different molecular processes12, integrative analysis of untargeted metabolomics is essential for biological insight and consequently new therapeutic approaches.
To overcome challenges in integrative analysis of untargeted metabolomics, we developed a network-based algorithm named PIUMet (available at http://fraenkel-nsf.csbi.mit.edu/PIUMet/). PIUMet infers pathways and experimentally undetected components from untargeted metabolite LC-MS peaks. PIUMet leverages an integrated network of over one million protein and metabolite interactions. Using advanced network optimization methods, PIUMet infers putative metabolites corresponding to features and molecular mechanisms underlying their dysregulation. Moreover, our approach provides statistical methods that account for uncertainty in the experimental data and the network. Additionally, PIUMet can be used to perform integrative analysis of untargeted metabolomics data with other large-scale data sets such as proteomics data, permitting the identification of a more complete picture of disease-associated pathways. We demonstrated the application of the algorithm by analyzing untargeted lipidomics and phosphoproteomics from a cell-line model of Huntington’s disease (HD), a genetic neurodegenerative disorder.
RESULTS
PIUMet overview
We developed the PIUMet algorithm to address challenges in system-level analysis of metabolomics. PIUMet adapts the prize-collecting Steiner forest algorithm13 for integrative analysis of untargeted metabolomics. Untargeted metabolomic experiments generate thousands of metabolite features (or peaks) characterized by an m/z value and a retention time. Typically, each feature matches several metabolites with masses in the right range6. The raw features that significantly differ between samples and controls using a user-selected statistical test are the input to PIUMet. PIUMet uses a machine-learning approach that leverages a database of protein-protein and protein-metabolite interactions (PPMI) to infer a network of dysregulated metabolic pathways (Fig. 1 and Supplementary Fig. 1). We demonstrated our approach in the context of uncovering pathways associated with disease, and we refer to altered metabolite peaks as ‘disease features’. In this context, PIUMet output consists of disease-associated proteins and metabolites (Fig. 1). We refer to components of this network whose identity was unknown before running PIUMet as ‘hidden’ components. Hidden metabolites directly connected to disease features represent their putative identities, and the remaining hidden metabolites and proteins identified by PIUMet are disease-associated proteins and metabolites that had not been measured directly by experiments. PIUMet can perform multi-omic analysis to reveal links between metabolomic dysregulation and other molecules such as proteins (Fig. 1). Although here we focused on disease-related data, PIUMet is very general, and can be applied to many biological settings.
Figure 1.
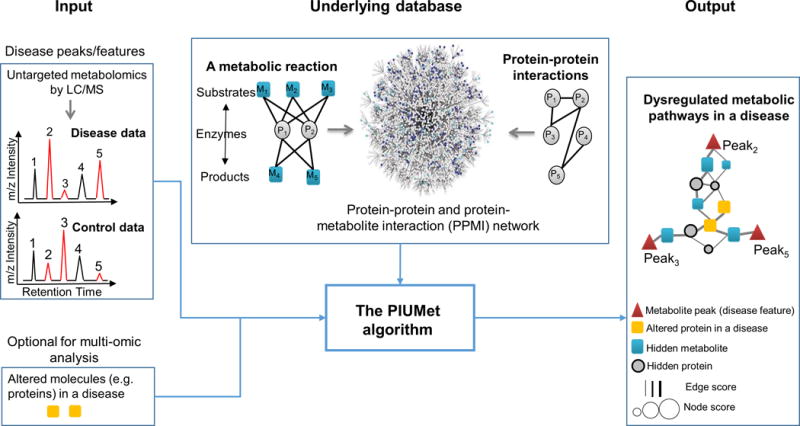
PIUMet identifies disease-associated pathways and hidden components from untargeted metabolomic data. The input to PIUMet are metabolomic peaks that differ between disease and control samples (peaks 2, 3 and 5 in the shown example). PIUMet then searches an underlying database. The PPMI nodes are proteins (circle nodes) or metabolites (square nodes). These nodes are connected via edges representing physical interactions among proteins, as well as substrate-enzyme and product-enzyme associations of metabolic reactions. PIUMet output is an optimum subnetwork of PPMI that connects disease features. This network represents dysregulated metabolic pathways in diseased cells, and its components display hidden proteins and metabolites that had not been detected in experiments. Hidden metabolites directly connected to disease features represent the putative identity of these features. Additionally, the resulting nodes and edges are scored based on their robustness to uncertainty in the underlying database. PIUMet also accept other omic data such as proteomics as an optional input.
The PIUMet database (the PPMI network) contains over 42,000 nodes connected by over one million edges (Supplementary Table 1). We built this network by integrating knowledge of biochemical reactions with curated interactions among proteins taken from three established databases. The result was a weighted graph, in which the nodes represent either metabolites or proteins, and the edges show the interactions between proteins as well as enzymatic and transporter reactions (Fig. 1). Each edge has a weight reflecting confidence in the reliability of the interaction (Online Methods).
To decipher the context of disease features, PIUMet does not require their prior identification; instead, it embraces the ambiguous identity of features. PIUMet represents each disease feature as a node, connected to all metabolites with masses similar to the disease features. The connecting edges have an arbitrary and equal weight (w; Fig. 2a). Using network optimization techniques described below, PIUMet identifies the subset of these metabolites most likely to correspond to disease features. It further calculates a score (R) for each one of the resulting metabolites indicating the degree to which its identification is robust to network parameters (w and the PPMI edge weights; Online Methods).
Figure 2.
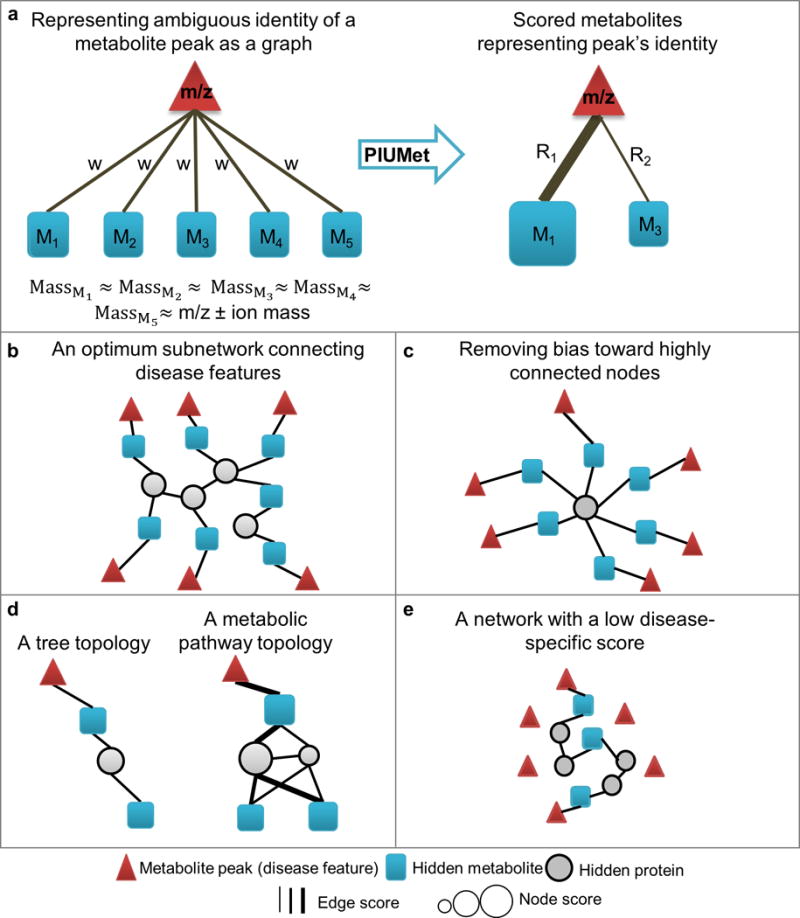
PIUMet. (a) PIUMet embraces the ambiguous identity of disease features. It first identifies putative metabolites matching a feature based on mass. It then represents each feature as a node (m/z), which is connected to the matched metabolites (M1–5). PIUMet reduces the ambiguity in the assignment and scores each of these metabolites. (b) In an optimum subnetwork of PPMI that links disease features, blue squares connected to triangles represent the inferred metabolites corresponding to the features. These metabolites are connected by high-confidence protein-protein and protein-metabolites interactions. (c) An example of an undesirable result that is biased toward highly connected nodes. (d) A comparison of a subnetwork in a tree structure with a subnetwork that captures the complex topology of metabolic reactions. (e) An example of a network generated from randomly chosen mock data sets, in which the majority of input nodes remain separated, and a few are connected via a long path of protein-protein and protein-metabolite interactions.
PIUMet searches the PPMI interactome for a subnetwork connecting disease features using high-probability protein-protein and protein-metabolite interactions (Fig. 2b). PIUMet optimizes the network using the prize-collecting Steiner forest algorithm, assigning prizes to disease features and costs to edge weights13,14. Node prizes reflect the significance of feature dysregulation (as determined by the user), and edge costs are anticorrelated with the edge confidence scores. The optimum solution balances the desire to include as many disease features as possible with a reluctance to use low-confidence edges. Specifically, we maximized the sum of the prizes from connected disease features, while minimizing the edge costs included in the final network.
PIUMet includes several features to improve its accuracy. First, it eliminates bias toward highly connected nodes in the PPMI interactome such as ATP or ubiquitin. As these high-degree nodes can connect almost any nodes in the PPMI interactome, they provide little insight into altered pathways (Fig. 2c). PIUMet penalizes the inclusion of high-degree nodes by assigning a penalty correlated with the node’s degree. Second, PIUMet generates a family of networks to infer the complex interconnections between substrates, enzymes and products that cannot be represented in tree structures (Fig. 2d). It merges solutions from many runs that differ by quantities of random noise that are added to the PPMI edge weights to find multiple, high-probability interactions that connect disease features. PIUMet then calculates a robustness score (R) for resulting nodes and edges to account for uncertainty in molecular interactions (Online Methods). Third, PIUMet calculates a disease-specific score for each resulting node and a score for each network. These scores are determined by generating a family of networks from randomly selected disease features that mimic the experimental data. A disease-specific score for each node is calculated by the frequency of the node in these ‘mock’ networks. We observed that in these mock networks, only a minority of the mock features were connected, and the connections were typically long paths (Fig. 2e). Thus, in contrast with real data, metabolites corresponding to randomly selected disease features were distributed apparently at random in the PPMI network. Based on this observation, we defined a disease-specific score for each network (Online Methods).
Identifying dysregulated pathways in HD
To test PIUMet, we performed integrative analysis of the untargeted lipidomic data from a cellular model of HD. HD is a genetic, neurodegenerative disorder caused by a CAG repeat expansion in the gene encoding the huntingtin protein. We used conditionally immortalized striatal cell lines (STHdh) derived from either wild-type embryos (STHdh Q7) or knock-in embryos, expressing a 111 CAG-expanded huntingtin gene (STHdh Q111)15. Measuring global levels of lipids using LC-MS, we found 115 metabolite features that differed significantly between the lines (P ≤ 0.01, determined by two-tailed student’s t-test). Of these, 37 had masses that matched to 296 potential metabolites in the PPMI network (Supplementary Table 2). PIUMet identified a network connecting more than 51% of these features via hidden metabolites and proteins (Figs. 3 and 4). The resulting networks had significantly higher disease-specific scores than control networks (P = 1.2 × 10‒37; Supplementary Fig. 2), and the nodes were specific to disease (disease-specific score ≥ 90%; Supplementary Fig. 3).
Figure 3.
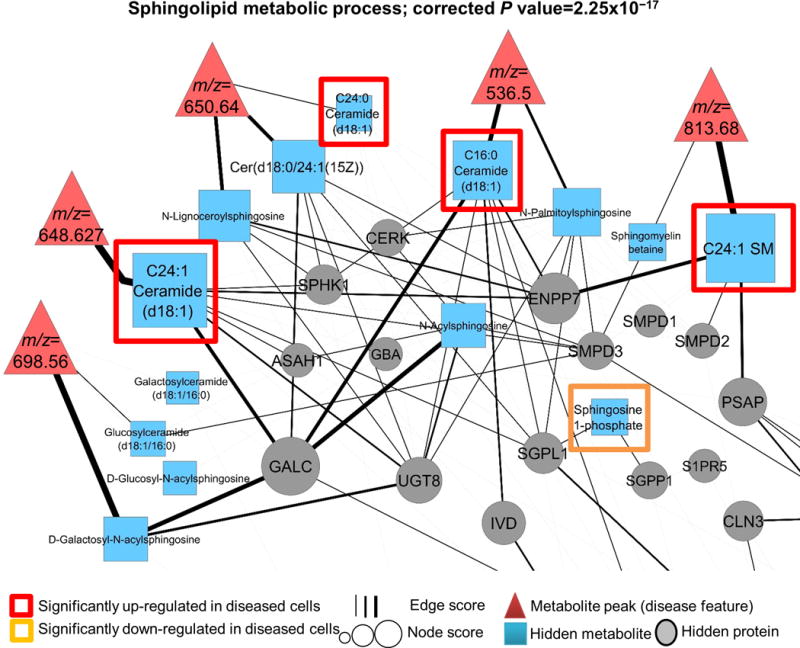
The PIUMet subnetwork showing altered sphingolipid metabolism in the STHdh cell line model of HD. PIUMet connects disease features via high-probability protein-protein and protein-metabolite interactions. Metabolites connected to disease features represent their putative identities. These metabolites along with the rest of the nodes are ranked based on the robustness scores, and are shown by different sizes. We experimentally verified the dysregulation of sphingolipids (upregulated and downregulated) using a targeted metabolomic platform. Also shown are hidden proteins that play a role in dysregulation of sphingolipids in diseased cells.
Figure 4.
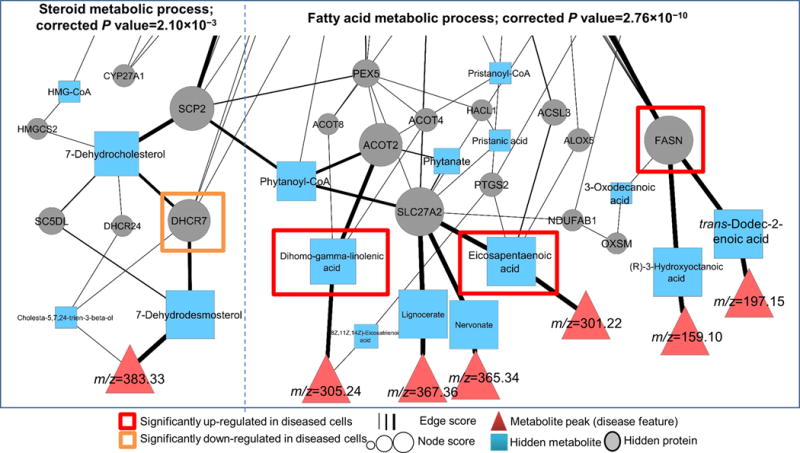
Altered fatty acid and steroid metabolic processes identified by PIUMet. Shown is a part of resulting network associated with these processes. Disease features are directly connected to metabolites representing their putative identities. The remaining nodes display hidden or experimentally undetected metabolites and hidden proteins of these pathways, with their sizes associated with robustness scores. We experimentally verified that the proteins and metabolites highlighted by red and orange boxes were altered in diseased cells.
Initially, the node for each disease feature is linked to all metabolites that potentially match based on mass. Metabolites remaining after PIUMet runs represent putative identities of the disease features, and their scores are associated with the probability of the predictions (Figs. 3 and 4). To verify the dysregulation of inferred metabolites corresponding to disease features, we used a targeted metabolomic platform that can identify several hundred predefined metabolites using reference standards. With this platform, we detected only eight of the 296 PPMI metabolites matching disease features (Supplementary Fig. 4), and all eight were identified by the targeted platform as dysregulated in diseased cells. PIUMet inferred six of these metabolites as putative identities of disease features (hypergeometric test P = 6.00 × 10‒4; Supplementary Fig. 4).
As PIUMet analyzes the disease features in a network that contains proteins and metabolites, it also elicits disease-associated proteins (hidden proteins) from untargeted metabolomic data. Gene ontology enrichment analysis with relevant background correction (Online Methods) showed that these proteins were significantly enriched in sphingolipid (corrected P = 2.25 × 10‒17; Fig. 3), fatty acid (corrected P = 2.76 × 10‒10; Fig. 4) and steroid (corrected P = 2.10 × 10‒3; Fig. 4) metabolic processes, which we further investigated.
In Figure 3 we show the dysregulated sphingolipid subnetwork, including disease-associated sphingolipids and proteins in this pathway. Targeted experiments demonstrated that four sphingolipids directly connected to disease features were significantly altered in diseased cells (Supplementary Fig. 5). Additionally, PIUMet identified sphingosine 1-phosphate (S1P) as a hidden metabolite in this pathway, to which there was no corresponding metabolite peak from LC-MS experiments. S1P is a key signaling molecule that activates antiapoptotic pathways by binding to cell-surface receptors16. We experimentally confirmed that S1P levels were significantly downregulated in diseased cells (P ≤ 0.001; Supplementary Fig. 6a). We also found that treating diseased cells with a S1P analog (FTY720-P) had protective effects, significantly decreasing apoptosis (P = 7.98 × 10‒5; Supplementary Fig. 6b,c). Although S1P has been previously examined in the context of HD17, those investigations had been motivated by the effect of S1P in other neurodegenerative diseases18,19, and did not identify the molecular mechanisms. In contrast, our approach inferred the underlying network of sphingolipid dysregulation and the role of S1P in diseased cells without any prior assumptions.
PIUMet also discovered an altered steroid metabolism network in our HD model (Fig. 4), consistent with previous reports20. Specifically, progressive alterations have been shown in sterol precursors of cholesterol in the R6/1 mouse model of HD21. However, the molecular mechanisms underlying these changes are unknown. We noted that one of the high-scoring nodes in our model was DHCR7 (z-score R = 2.22), a terminal enzyme in cholesterol biosynthesis. Using western blots, we confirmed DHCR7 protein levels were significantly lower in diseased cells (P < 0.05; Supplementary Fig. 7). Thus, PIUMet identified DHCR7 as one of the key regulatory molecules in this pathway, which can be further investigated for therapeutic purposes.
Furthermore, we examined the subnetwork associated with fatty acid metabolism (Fig. 4). Fatty acids are major components of neuronal membranes and the myelin sheath, and their balanced levels are essential in the brain22. Fatty acid dysregulation has been associated with HD23. The targeted metabolomic experiments verified significant changes in two fatty acids that are directly connected to disease features: eicosapentaenoic acid (EPA, P = 7.8 × 10‒6; Supplementary Fig. 8a) and dihomo-gamma-linolenic acid (DHGLA, P = 0.01; Supplementary Fig. 8b). Both EPA and DHGLA are essential fatty acids, and to our knowledge, there are no prior reports about their levels in HD neuronal tissues. Notably, because EPA has been reported to have neuroprotective effects in many systems, it has been tested as a therapeutic for HD24,25 despite the absence of any previous molecular data about its levels in disease cells. Our unbiased analysis of untargeted metabolomic data suggested a molecular mechanism behind EPA’s therapeutic effects, and revealed an unknown aspect of altered fatty acid metabolism in HD associated with DHGLA.
We experimentally tested the capability of our network approach to correctly infer connections between metabolites and proteins. One of the highest scoring proteins in the network was FASN (z-score R = 3.08), which is involved in de novo fatty acid biosynthesis26. In the resulting network, FASN was connected to intermediate metabolites in fatty acid synthesis pathways. Western blot experiments revealed a significant increase in the FASN enzyme in diseased cells (P = 0.007; Supplementary Fig. 9). Collectively, our results provided insight about dysregulation of fatty acid metabolism in HD.
Integrating metabolomics with other omics
To test the ability of PIUMet to integrate untargeted lipidomics and global phosphoproteomic data, we measured global levels of phosphoproteins in STHdh Q7 and STHdh Q111 cells by affinity purification followed by mass spectrometry. Thirty-one proteins showed significant changes in phosphorylation levels between the lines (P ≤ 0.01, Supplementary Table 3). Our integrative analysis of disease features with phosphoproteomics provided a more comprehensive picture of disease-associated pathways and components. The networks obtained by analyzing lipidomics and proteomics separately had little overlap. By contrast, multi-omic analysis not only inferred the majority of hidden components obtained from the analysis of lipidomics and phosphoproteomics individually, but it also revealed new disease-associated molecules (Fig. 5). These results emphasize that each type of molecular data resulted in identification of limited and distinct biological processes.
Figure 5.
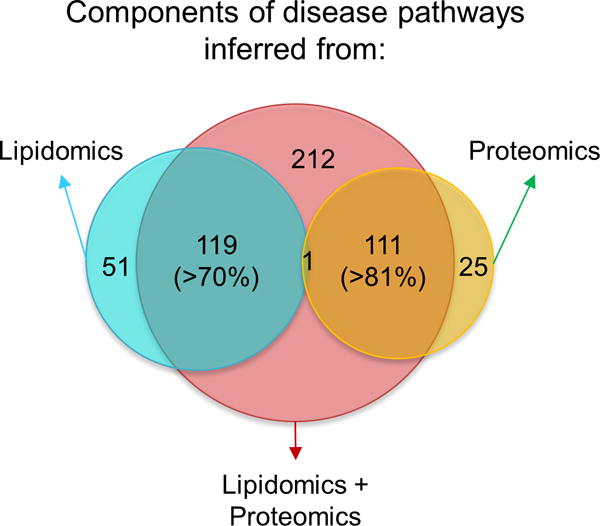
Comparison of disease-associated components identified in separate analyses of lipidomics and phosphoproteomics, and in an integrative analysis of those data.
We then analyzed high-scoring nodes from the networks. Including phosphoproteomic data increased the confidence (robustness score R) of many nodes, and the top-ranked ones were DHCR7 (z-score R = 3.96; Fig. 6a) and FASN (z-score R = 4.7; Fig. 6c), which we described above and verified experimentally. Similarly, the protein encoded by RASA1 (RasGAP) had the highest robustness score among the proteins that were originally found with only phosphoproteomic data, and their score increased with joint analysis with lipidomics (z-score of R = 4.70; Fig. 6b). We experimentally determined a significant increase in the level of RasGAP (P = 0.008; Supplementary Fig. 10a), which interacts with the mutated huntingtin protein27, but has an unknown role in HD progression.
Figure 6.
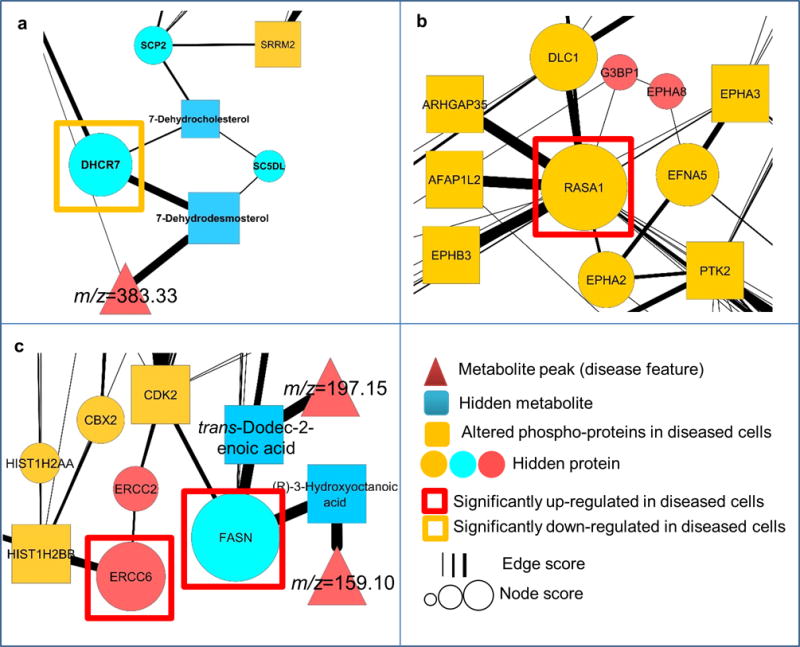
Regions of the resulting network obtained from integrative analysis of lipidomics and phosphoproteomics, displaying the dysregulation of high-scoring, hidden components. (a–c) High-scoring proteins belonging to three subsets of nodes. The first subset contains nodes that increase in robustness when lipidomics and phosphoproteomics are considered together compared to lipidomics alone. DHCR7 (a) and FASN (c) are high-scoring members of this subset, which are significantly (two-tailed Student’s t-test) altered in diseased cells. The second subset includes nodes that increase in robustness when lipidomics and phosphoproteomics are considered together compared to phosphoproteomics alone. RASA1 (b) is the highest-scoring node in this network, whose encoded protein is significantly upregulated in diseased cells. Finally, the third subset contains proteins that are only identified by multi-omic analysis of lipidomics and phosphoproteomics. We confirmed that ERCC6-encoded protein (c), a high-scoring node in this subset, is significantly upregulated in diseased cells.
Of the proteins that only appeared in the integrative analysis, we examined ERCC6-encoded protein CSB (z-score of R = 2.74), and found a significant increase in its levels (P < 0.05; Fig. 6c and Supplementary Fig. 10b). CSB is a DNA-excision repair protein involved in neurogenesis and neuronal development28, which may have a role in HD because defects in DNA-repair mechanisms has been associated with the disease29.
DISCUSSION
PIUMet is a network-based algorithm for integrative analysis of untargeted metabolomic data. Even with high-mass-accuracy instruments, most features detected in untargeted metabolomic experiments cannot be identified uniquely, and typically only a few are characterized unambiguously via additional time-consuming and costly MS/MS experiments. PIUMet leverages known metabolic reactions and protein-protein interactions to analyze the ambiguous assignment of metabolomics features. It can be used to identify dysregulated metabolic networks containing metabolites that are possible matches to the metabolomic features, and determine the robustness and disease-specificity of the results.
PIUMet is a general approach. Although we demonstrated its utility by analyzing data for HD, we did not tailor the algorithm to HD in any way. In fact, metabolomics changes in HD are studied relatively poorly compared to many other diseases such as diabetes, and even cancer. Therefore, we expect that PIUMet would be equally effective in identifying altered pathways using untargeted metabolomics from any disease, or any comparison of two biological states.
Untargeted metabolomics has an important role in understanding disease, because targeted metabolomic profiling captures few of the relevant metabolites. Only eight of 296 metabolites assigned to disease features in our data were detected with a targeted metabolomic platform, reflecting the bias of targeted platforms toward well-studied metabolites. As a result, methods such as PIUMet for analyzing untargeted metabolomics have great potential to systematically discover new molecular mechanisms.
PIUMet does not replace MS/MS for identifying metabolite peaks, but it prioritizes metabolite features for experimental validation, and provides the protein and small molecule context of these features. Considering the costs and time associated with performing these experiments6, PIUMet could make a considerable impact in current metabolomic studies.
We believe that PIUMet fills an important need by translating untargeted metabolomic data into relevant biological knowledge and contextualizing metabolomics with other system-level molecular data. We demonstrated that this integrative approach is crucial for understanding a complete picture of disease-associated processes. Although we established the integrative analysis of untargeted metabolomics with proteomics data, PIUMet can be also applied to analyze metabolomics in conjunction with genomic data, and further extended to include transcriptional data. Therefore, our multi-omic, integrative approach is likely to be of even greater use as more data are generated.
ONLINE METHODS
Rationale for PIUMet
PIUMet uses a graph-based approach to resolve the ambiguous identity of peaks identified by untargeted metabolomic experiments. These peaks (or features) are characterized by a unique m/z and a retention time (RT). PIUMet represents these peaks as nodes and connects each peak to the metabolites in the PPMI network with a mass matching the m/z value of the feature, after considering the weight of ionic adducts. PIUMet then searches for subnetworks that are enriched in these metabolites by solving the prize-collecting Steiner forest problem. The networks are evaluated using randomization strategies to identify results that are robust to choices of parameters.
The PPMI network
The PPMI interactome is a network of protein-protein and protein-metabolite interactions, which we constructed by the integration of three different databases. Protein-protein interactions were obtained from iRefIndex version 13 (ref. 30), and metabolite information and biochemical reactions were from HMDB version 3 (ref. 31) and Recon 2 (ref. 32) databases.
iRefIndex is a unified database of nine known protein-protein interaction (PPI) databases, in which the redundant PPIs among underlying databases are removed30. The iRefIndex database was downloaded in the provided PSI-MITAB format. We did not filter any interactions based on the source of their inference; instead, we used MIscore algorithm33 to calculate a confidence score for each interaction. To calculate these scores, we used PSISCORE Java API33. MIscore algorithm considers the number of publications, the type of interaction, and the used experiments to calculate confidence scores for molecular interactions.
HMDB or the human metabolome database is the most comprehensive resource of human metabolites31. HMDB contains various information for over 40,000 detected and expected metabolites. It additionally includes the association of transporters and enzymes with metabolites, obtained from KEGG and SMPDB34 pathways. We downloaded the HMDB database in XML format. We then parsed information about metabolites such as super class, molecular weight, chemical composition, and associated proteins from these XML files using ElementTree Library of Python. Finally, we built a network representing the links between metabolites and their associated enzymes and/or transporters using Python NetwrokX library.
Recon2 is a comprehensive database of metabolic reactions. First, the database was downloaded in SBML (Systems Biology Markup Language) format35, which is a standard XML-based format for representing metabolic reactions. We then parsed the information about the database entities such as enzymes, metabolites and metabolic reactions using LibSBML python library36. Finally, we generated a network representation of metabolic reactions, in which the reaction substrates are connected via edges to the reaction enzymes. The edges then link these enzymes to the reaction products (Fig. 2a). For each reaction, Recon2 provides a confidence score of zero to four; a score of zero indicates no supporting data about the confidence, and four indicates the evidence of biochemical data32. The edges are then weighted based on the reaction confidence score.
Next, we built a network of protein-protein and protein-metabolite interactions (PPMI) by combining the interactions obtained from iRefIndex, HMDB and Recon databases. PPMI is composed of two sets of metabolite (M) and protein (P) nodes. The set of interactome edges (E) shows interaction among proteins, as well as enzymes and transporters associated with metabolites. The PPMI interactome is defined as (P, M, E).
The associated confidence scores with the PPMI edges are obtained from the source databases. However, as the scale of scorings differs among databases, we scaled the edge weights to the PPI confidence score distribution. For this purpose, Recon 2 interactions with a confidence score of four were scaled to the maximum PPI score, and scores of 3, 2 and 1 were scaled to the PPI third quartile, median and second quartile scores, respectively. As there was no confidence score associated with interactions obtained from HMDB, we arbitrary assigned the edge weight to be the median of the PPI confidence score. Below we will address the inference of robust results in spite of uncertainties in the PPMI edge weights.
The prize-collecting Steiner forest (PCSF) optimization
PIUMet adapts the PCSF optimization that has been established to infer networks linking the dysregulation of proteins to changes in transcription37. The PCSF optimization identifies an optimum forest (a set of trees) representing simultaneously dysregulated pathways in a disease13. This forest is a subnetwork of the PPMI interactome in which experimentally detected dysregulated molecules (terminal set) are linked via undetected molecules (Steiner set); these nodes are connected by known molecular interactions. The optimum subnetwork is inferred by first assigning a prize to each node in the terminal set and costs to the PPMI edges. In addition, to obtain independent simultaneous pathways, an artificial node is connected to the terminal nodes via edges with weight ω. The algorithm then infers a forest solution, F, with NF nodes and EF edges, by minimizing the following objective function using an established message passing approach38:
| (1) |
where p(n) shows the associated prize to each node n ∈ NF, c(e) shows the cost of each edge e ∈ EF in the resultant forest F, and k shows the number of trees in the forest F. Here the terminal nodes’ prizes, p(n), are equal to –log (P value) of the significance of their alteration in the disease, calculated by two-tailed student’s t-test. The costs associated to each edge, c(e), are one minus the PPMI edges weights. Additionally, β is a tuning parameter that controls the size of the resultant forest13, which here is considered equals to 4. ω is further a tuning parameter regulating the size of k or the number of trees in the forest solution13. We considered different values of ω in the range of 10 to 25, based on the input terminal sets. To choose a value of ω, we considered the size of the resulting networks and the number of connected terminals. We started by examining smaller values. Increasing ω results in a larger network that connects more terminals, and we chose the value of ω that maximized the number of connected terminals. In addition to these parameters, the variable w is the equal and arbitrary weight assigned to edges between disease features and potentially matching metabolites. Here, we considered w equals to 0.99, which is the same as the maximum PPMI edge weight, while sensitivity analysis on the value of w resulted in more than 88% robust results (data is not shown).
Eliminating bias toward highly connected nodes
PIUMet algorithm infers resulting networks that are unbiased toward highly connected nodes. The presence of several nodes in the PPMI interactome with a high degree of connectivity leads to a network in which terminal nodes are always linked via a high degree node (Fig. 3c). To obtain results that are not biased toward highly connected nodes, PIUMet penalizes the results that have a high-degree node by introducing a negative prize to nonterminal nodes, which is a multiple of their degree. Since nodes’ degree distribution differ significantly between the metabolite and protein sets (t-test P = 8.16 × 10‒124), with an average of 35.18 in the metabolite set compared to 21.08 in the protein set, the negative prizes for protein and metabolite sets are defined as:
where p(n) is a prize of a none-terminal node n. P is the set of protein nodes in the PPMI interactome, and M represents the metabolite set. μ is a tuning parameter that controls the effect negative prizes; here we used μ = 0.015.
Obtaining robust results that capture complexity of metabolic networks
PIUMet infers robust, disease-associated pathways that capture the complexity of metabolic networks. Since the solution to equation (1) is a tree, it cannot capture the complex topology of metabolic reactions including interconnection of substrates, enzymes and products (Fig. 2d). To address this issue, we generated additional networks by adding small random noises to the PPMI edge weights. For this purpose, a random value in the range of [0, ε] is added to each PPMI edge weight. Here, ε is considered equal to 0.046, which is one half of the s.d. of the PPMI edge weight distribution. Therefore, the ε value will be the same for any other disease as long as the underlying interactome is unchanged. This process leads to inferring a family of networks including multiple possible paths that link terminals nodes. The union of these networks thus shows the complex interconnection of metabolic pathways. Furthermore, generating networks by adding random noise to the PPMI edge weights allows the distinction of the results that are robust in spite of the uncertainty in the interactome edge weights. Consequently, we calculated a robustness score for each node in the family of networks as below:
where for a family of R networks with N nodes, shows the robustness score of node ni ε N, and Fj(n) shows nodes in network j.
Calculating disease-specific score for resulting nodes and networks
To measure the specificity of the PIUMet results to the disease of interest, we generated networks by randomly selecting metabolite features with the same characteristics as disease features. We defined a detectable metabolite feature (DMF) set as a set of metabolite features mimicking the experimental data, as defined below: first, the DMF set included m/z values that are detectable with the mass spectrometer used in the experiments. Second, the DMF metabolite features must be matched to metabolites that belong to a superclass of metabolites that can be separated via the liquid chromatography step. To distinguish these metabolites, we obtained chemical taxonomy information, including superclass, from the HDMD database. Finally, these matched metabolites must belong to the PPMI network. The definition of the DMF set is:
The degree of each feature in the DMF set indicates the number of potential matched metabolites. Supplementary Figure 11 shows the degree distribution of the DMF set features compared with the terminal set features.
In the next step, for a terminal set with the size T, we randomly selected T features from the DMF set, in which the degree distribution of the selected features is similar to the terminal set. We repeated this process R times. Each of these R randomly chosen features is given as input to PIUMet, resulting in R networks. We compared these networks to the ones obtained from the experimental data to calculate disease-specific scores. This score indicates the frequency of a node in the networks obtained from randomly selected disease features and is defined as:
where ni is a node in the family of the networks obtained from disease features. For R random feature sets, RFj(n) is a network with n nodes that connects random features.
Additionally, we observed in the resulting networks from randomly selected disease features, the majority of the features remained unconnected (singletons), and a few were linked via a long path of protein-protein and protein-metabolite interactions (Fig. 3e). We quantified these properties by calculating the disease-specific score for each resulting network as the number of the connected metabolite features divided by the number of Steiner nodes. The disease-specific score was then calculated as:
Using the student t-test, we then compared the disease-specific scores of resulting networks from disease features and those obtained from randomly selected features.
Identifying background nodes
PIUMet distinguishes a relevant set of background nodes essential for downstream significance analysis of resulting networks39 such as gene ontology enrichment. Here the background nodes are a subset of the PPMI nodes that can connect metabolite features mimicking the experimental data (the DMF set features). To identify background nodes, we calculated the weighted shortest path length between the PPMI nodes and metabolites corresponding to DMF set features. A background set was defined as:
where B shows background nodes, DMF(M) is a set of metabolites corresponding to DMF set features, and PPMI(N) is the set of the PPMI nodes. Here we considered ∊ equals to 0.4; sensitivity analysis on the value of ∊ resulted in the similar outcomes (data not shown).
Experimental data collection on the STHdh cell line model of HD
To test our developed methodology, we collected various omic data of STHdh cell line15 model of HD. STHdh cells are the conditionally immortalized homozygote wild-type (STHdh Q7, Coriell CH00097) and mutant (STHdh Q111, Coriell CH00095) striatal neuronal progenitor cell lines, which were cultured as described15. Cells were maintained in a humid incubator at 33 °C and 5% CO2. Culture medium was changed every 2 d, and cells were subcultured when they reached 85% confluence. The passage number was kept below 14; to exclude mycoplasma contamination, cells were routinely tested with the PCR Mycoplasma Detection Kit (Applied Biological Materials Inc). The cell lines were genotyped by PCR as described15. Before each experiment, the temperature was raised at 39 °C for 48 h to halt proliferation and reduce cell-cycle differences between the two lines. Cells were subsequently washed twice with ice-cold phosphate-buffered saline, scraped on ice and pelleted by centrifugation at 450g for 5 min at 4 °C. Cell pellet were flash-frozen with liquid nitrogen and stored at −80 °C until use.
Global lipid profiling of STHdh cell lines
Lipids were extracted as described40. Cells were scraped from a 10-cm plate in 1 mL cold PBS and transferred to a glass vial which was vortexed with a cold mixture of 1 mL MeOH and 2 mL chloroform. The resulting mixture was centrifuged and the organic phase containing lipids was dried under a stream of N2 and stored at −80 °C before injection for LC-MS analysis.
Global lipidomics was performed with an Agilent 1200 Series HPLC online with an Agilent 6220 ESI-TOF (Agilent Technologies). Data were acquired in positive and negative ionization modes. For the negative mode, a Gemini (Phenomenex) or Inspire (Dikma Technologies) C18 column (5 μm, 4.6 mm × 50 mm) was used with a guard column (C18, 2 μm frit, 2 mm × 20 mm). Solvent A was 95:5 water:methanol with 0.1% ammonium hydroxide, and solvent B was 60:35:5 isopropanol:methanol:water with 0.1% ammonium hydroxide. For the positive mode, a Luna (Phenomenex) C5 or Bio-Bond (Dikma Technologies) C4 column (5 μm, 4.6 mm × 50 mm) was used with a guard column (C4, 2 μm frit, 2 mm × 20 mm). Solvent A was 95:5 water:methanol with 0.1% formic acid and 5 mM ammonium formate, and solvent B was 60:35:5 isopropanol:methanol:water with 0.1% formic acid and 5 mM ammonium formate. The identical gradient was used for both modes. The gradient was held at 0% B between 0 and 5 min, changed to 20% B at 5.1 min, increased linearly from 20% B to 100% B between 5.1 min and 45 min, held at 100% B between 45.1 min and 53 min, and returned to 0% B at 53.1 min and held at 0% B between 53.1 min and 60 min to allow column re-equilibration. The flow rate was maintained at 0.1 mL/min between 0 and 5 min to counter the increase in pressure due to chloroform injection. The flow rate was 0.4 mL/min between 5.1 min and 45 min, and 0.5 mL/min between 45.1 min and 60 min. Injection volume was 10–30 μL. The capillary, fragmentor and skimmer voltages were 3.5 kV, 100 V and 60 V, respectively. The drying gas temperature was 350 °C, drying gas flow rate was 10 l min−1 and nebulizer pressure was 45 p.s.i.. Data were collected in both profile and centroid modes using a mass range of 100–1500 Da. For untargeted analysis, raw data were converted to .mzXML format and analyzed by XCMS, which considers nonlinear alignments of features from different samples41. XCMS output files were filtered by statistical significance (P ≤ 0.05), fold change (≥3) and reproducibility across four independent data sets, and the remaining ions further verified by manual integration in Qualitative Analysis software (Agilent Technologies). Statistical significance was determined by two-tailed student’s t-test.
Experimental verification of altered metabolites inferred by PIUMet
Confirmation of altered metabolites inferred by PIUMet was done using two reverse-phase LC methods and MS data acquired in positive and negative ionization modes using two LC-MS systems comprised of Nexera X2 U-HPLC systems (Shimadzu Scientific Instruments) and either a Q Exactive hybrid quadrupole orbitrap mass spectrometers (Thermo Fisher Scientific) or a Exactive Plus orbitrap MS (Thermo Fisher Scientific).
For the measurement of lipids, LC-MS samples were extracted from cell pellets (3 × 107 cells) in isopropanol containing 1-dodecanoyl-2-tridecanoyl-sn-glycerol-3-phosphocholine as an internal standard (Avanti Polar Lipids). Extracted metabolites were injected into a Waters Aquity UPLC BEH C8 column (1.7 μm; Waters), eluted isocratically for 1 min at 80% mobile phase A (95:5:0.1 vol/vol/vol 10 mM ammonium acetate/methanol/acetic acid), followed by a 2-min linear gradient to 80% mobile phase B (99.9:0.1 vol/vol methanol/acetic acid) and a linear gradient to 100% mobile phase B over 7 min. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis with an ion spray voltage of 3.0 kV, capillary and probe heater temperature of 300 °C.
For the measurement of sphingosine-1-phosphate, metabolites were extracted from cell pellets (3 × 107 cells) in 80% methanol containing Prostaglandin E2-d4 (PGE2-d4) as an internal standard (Cayman Chemical Co.). Extracts were injected onto a 150 × 2 mm ACQUITY T3 column (Waters). The column was eluted isocratically at a flow rate of 450 μL/min with 25% mobile phase A (0.1% formic acid in water) for 1 min followed by a linear gradient to 100% mobile phase B (acetonitrile with 0.1% formic acid) over 11 min. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis is with an ion spray voltage of −3.5 kV, capillary temperature of 320 °C and probe heater temperature of 300 °C.
Raw data for both methods were processed using Progenesis CoMet and QI software (NonLinear Dynamics) for feature alignment, nontargeted signal detection, and signal integration. Targeted processing and manual inspection of features was conducted using TraceFinder software (Thermo Fisher Scientific).
Global phosphoprotein profiling of STHdh cell line model of HD
Mass spectrometry sample preparation
Samples were lysed with 8 M urea + 1 mM sodium orthovanadate (phosphatase inhibitor) and protein yield was quantified by BCA assay (Pierce). Samples were reduced with 10 μl of 10 mM DTT in 100 mM ammonium acetate pH 8.9 (1 h at 56 °C). Samples were alkylated with 75 μl of 55 mM iodoacetamide in 100 mM ammonium acetate pH 8.9 (1 h at room temperature). 1 mL of 100 mM ammonium acetate and 10 μg of sequencing grade trypsin (Promega PN:V5111) and digestion proceeded for 16 h at room temperature. Samples were acidified with 125 μl of trifluoroacetic acid (TFA) and desalted with C18 spin columns (ProteaBio, SP-150). Samples were lyophilized and subsequently labeled with iTRAQ 8plex (AbSciex) per manufacturer’s directions.
Immunoprecipitation
70 μl protein-G agarose beads (calbiochem IP08) were rinsed in 400 μl IP buffer (100 mM Tris, 0.3% NP-40, pH 7.4) and charged for 8 h with three phosphotyrosine-specific antibodies (12 μg 4G10 (Millipore), 12 μg PT66 (Sigma), and 12 μg PY100 (CST)) in 200 μl IP buffer. Beads were rinsed with 400 μl of IP buffer. Labeled samples were resuspended in 150 μl iTRAQ IP buffer (100 mM Tris, 1% NP-40, pH 7.4) + 300 μl milliQ water and pH was adjusted to 7.4 (with 0.5M Tris HCl pH 8.5). Sample was added to charged beads for overnight incubation. Supernatant was removed and beads were rinsed three times with 400 μl rinse buffer (100 mM Tris HCl, pH 7.4). Peptides were eluted in 70 μl of elution buffer (100 mM glycine, pH 2) for 30 min at room temperature.
Immobilized metal affinity chromatography (IMAC) purification
A fused silica capillary (FSC) column (200 μm inner diameter × 10 cm length) was packed with POROS 20MC beads (Applied Biosystems 1-5429-06). IMAC column was prepared by rinsing with solutions in the following order: 100 mM EDTA pH 8.9 (10 min), (10 min), 100 mM FeCl3 (20 min), 0.1% acetic acid (10 min). IP elution was loaded for 30 at a flow rate of 2 μl/min. The column was rinse with 25% MeCN, 1% HOAc, and 100 mM NaCl (10 min) and 0.1% acetic acid (10 min). Peptides were eluted with 50 μl 250 mM NaH2PO4 at 2 μl/min and collected on a 10 cm hand-made precolumn (fused silica: Polymicro Technologies cat.no. TSP100375, beads: YMC gel, ODS-A, 12 nm, S-10 μm, AA12S11). Precolumn was rinsed with 0.1% acetic acid before analysis.
Liquid chromatography mass spectrometry
Peptides were analyzed on a 240-min gradient (Agilent 1100 HPLC) from 100% A (0.1% formic acid) to 100% B (0.1% formic acid, 80% acetonitrile) spraying through a hand-made 10 cm analytical column (fused silica: Polymicro Technologies cat.no. TSP050375, beads: YMC gel, ODS-AQ, 12 nm, S-5 μm, AQ12S05, bead plug: YMC gel, ODS-A, 12 nm, S-10 μm, AA12S11) with integrated electrospray ionization tip, connected inline with the precolumn.
Data analysis
Thermo .RAW files were searched with MASCOT v2.4 using Proteome Discoverer (v1.2). Peptides that appeared in all replicates were included if their MASCOT scores exceeded 15 and they were designated as medium or high confidence by Proteome Discoverer. The P value obtained using a two-tailed t-test is reported between STHdh Q7 and STHdh Q111 biological replicates. We identified 35 peptides that were matched to 31 corresponding phosphoproteins using BLAST program blastp (Supplementary Table 3). If multiple peptides matched to one protein, the assigned P value to the protein was considered as the minimum P value of the corresponding peptides. Additionally, as the PPMI network was constructed from human data, we identified the human homologs of these phosphoproteins using NCBI HomoloGene42 database.
Inferring a network underlying changes in phosphoproteins
When proteomic data were used, the input to PIUMet consisted of phosphoproteins with significantly different levels of phosphorylation between STHdh Q7 and STHdh Q111 cells (P ≤ 0.01; Supplementary Table 3). PIUMet identified a subnetwork of the PPMI that connects these phosphoproteins, while calculating a robustness score for each resulting nodes. We also generated a family of random networks by randomly selecting phosphoproteins that mimic experimental data. For this purpose, we first identified a list of phosphoproteins from the Phosida database43. Of these phosphoproteins, 3,858 (>82%) were present in the PPMI network. Then, for an input size T, we randomly selected T of these phosphoproteins, in which the degree distribution of the selected phosphoproteins was similar to the real data. We repeated this process 100 times, and obtained the resulting networks. We then calculated disease-specific scores for the resulting nodes and networks. These scores showed that the resulting nodes from real data were specific to the disease (disease-specific score ≥ 93%), and the resulting networks from real data had significantly higher disease-specific scores compared to those from randomly selected disease features (P = 1.58 × 10‒78).
Inferring a network connecting untargeted lipidomic to phosphoproteomic data
We also ran PIUMet with both metabolomics disease features and significantly altered phosphoproteins (identified using a two-tailed student’s t-test) as inputs. PIUMet then identified a subnetwork of the PPMI linking changes in the global level of lipids to changes in phosphoproteins, and calculated robustness scores for the results. In addition, we calculated disease-specific scores for resulting nodes and networks, and found that the nodes were specific to the disease (disease-specific score ≥ 82%), and networks had significantly higher disease-specific scores compared to those obtained from randomly selected disease features and phosphoproteins (P = 1.30 × 10‒116).
Multiparameter high-content imaging for the analysis of cell apoptosis
STHdh Q7 and STHdh Q111 striatal cells were trypsinized, harvested in prewarmed growth medium and quantified using an automated cell counter (Countess II, Life Technologies). 6,000 cells per well were seeded in sterile, black, 96-well microplates with flat, clear bottoms. After 24 h, the complete medium was removed and replaced with phenol-red-free and serum-free medium containing either FTY720 phosphate (Sigma-Aldrich) or vehicle (DMSO, Sigma-Aldrich) and cells were incubated for 24 h at 33 °C. A multiple staining solution containing 1 μg/ml calcein-AM, 2 μg/ml propidium iodide and 1.5 μg/ml Hoechst 333442 (all from Life Technologies) was added to detect and quantify live, dead and total cells, respectively. After 20 min incubation, the Cellomics Arrayscan Platform (Thermo Scientific) was used for imaging acquisition. Seven fields per well were imaged at 10× magnification. Analysis was carried out using the Cellomics algorithm for cell viability. Cell loss was expressed as the percentage of propidium-iodide-positive cells. Two independent experiments were performed with 20 replicates each.
Protein extraction and western blot analysis
For total protein extraction, cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 12 mM sodium deoxycholate) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific). Nuclear extracts were prepared as described44. Total and nuclear protein extracts were quantified using the Bradford assay with bovine serum albumin as standard. Western blot experiments were carried out using the Odyssey infrared imaging system (Li-Cor Biosciences) as described45. The following primary antibodies were used: anti-CSB (Santa Cruz Biotechnology, sc-25370; dilution 1:200), anti-DHCR7 (Abcam, ab103296; dilution 1:500), anti-RASA1 (Abcam, ab40807; dilution 1:1,000), anti-FASN (Santa Cruz Biotechnology, sc-55580; dilution 1:500). An antibody against actin protein (Millipore, MAB1501; dilution 1:10,000) was used for normalization.
Code availability
The beta-version of PIUMet software is available for nonprofit academic use only at http://fraenkel-nsf.csbi.mit.edu/PIUMet/
Supplementary Material
Acknowledgments
We thank A. Soltis and S. Dalin. This work was supported by grants from US National Institute of Health R01-GM089903, U54-NS091046 and U01-CA184898 (E.F.), and National Cancer Institute U54 CA112967 (E.F. and F.M.W.) and P30 CA014051 (F.M.W.) as well as Searle Scholars Program (A.S.).
Footnotes
AUTHOR CONTRIBUTIONS
L.P. and E.F. designed the approach. L.P. implemented the algorithm and performed the computational analyses. P.M. prepared cell cultures and performed western blot and cell viability experiments. M.L and A.S. performed the untargeted lipidomic experiments. T.C. and F.M.W. measured global levels of phosphoproteins. J.A.-P. and C.B.C. performed targeted lipidomic experiments. L.P. and E.F. wrote the manuscript, and all the authors approved the final version.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the online version of the paper.
Editorial Summary
A network-based method and computational tool, PIUMet, reveals disease-associated molecular pathways from untargeted metabolomics data without requiring mass spectral feature identification.
COMPETING FINANCIAL INTERESTS Declaration
L.P. and E.F. are co-founders of ReviveMed, Inc., and have filed a provisional patent based on the work described here (Application number: 62/203,292).
References
- 1.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patti GJ, Yanes O, Siuzdak G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker M. Metabolomics: from small molecules to big ideas. Nat Methods. 2011;8:117–121. [Google Scholar]
- 4.Dunn WB, et al. Mass appeal: metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics. 2013;9:44–66. [Google Scholar]
- 5.Johnson CH, Ivanisevic J, Benton HP, Siuzdak G. Bioinformatics: the next frontier of metabolomics. Anal Chem. 2015;87:147–156. doi: 10.1021/ac5040693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho K, Mahieu NG, Johnson SL, Patti GJ. After the feature presentation: technologies bridging untargeted metabolomics and biology. Curr Opin Biotechnol. 2014;28:143–148. doi: 10.1016/j.copbio.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grapov D, Wanichthanarak K, Fiehn O. MetaMapR: pathway independent metabolomic network analysis incorporating unknowns. Bioinformatics. 2015;31:2757–2760. doi: 10.1093/bioinformatics/btv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo TC, Tian TF, Tseng YJ. 3Omics: a web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst Biol. 2013;7:64. doi: 10.1186/1752-0509-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karnovsky A, et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics. 2012;28:373–380. doi: 10.1093/bioinformatics/btr661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krumsiek J, et al. Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012;8:e1003005. doi: 10.1371/journal.pgen.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S, et al. Predicting network activity from high throughput metabolomics. PLOS Comput Biol. 2013;9:e1003123. doi: 10.1371/journal.pcbi.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeger-Lotem E, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet. 2009;41:316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuncbag N, et al. Simultaneous reconstruction of multiple signaling pathways via the prize-collecting steiner forest problem. J Comput Biol. 2013;20:124–136. doi: 10.1089/cmb.2012.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang SSC, Fraenkel E. Integrating proteomic, transcriptional, and interactome data reveals hidden components of signaling and regulatory networks. Sci Signal. 2009;2:ra40. doi: 10.1126/scisignal.2000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trettel F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 16.Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60. doi: 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Pardo A, et al. FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum Mol Genet. 2014;23:2251–2265. doi: 10.1093/hmg/ddt615. [DOI] [PubMed] [Google Scholar]
- 18.Di Menna L, et al. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol Res. 2013;67:1–9. doi: 10.1016/j.phrs.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Deogracias R, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valenza M, Cattaneo E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011;34:474–486. doi: 10.1016/j.tins.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Kreilaus F, Spiro AS, Hannan AJ, Garner B, Jenner AM. Brain cholesterol synthesis and metabolism is progressively disturbed in the R6/1 mouse model of Huntington’s disease: a targeted GC-MS/MS sterol analysis. J Huntingtons Dis. 2015;4:305–318. doi: 10.3233/JHD-150170. [DOI] [PubMed] [Google Scholar]
- 22.Yehuda S, Rabinovitz S, Mostofsky DI. Essential fatty acids and the brain: from infancy to aging. Neurobiol Aging. 2005;26(Suppl 1):98–102. doi: 10.1016/j.neurobiolaging.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 23.Block RC, Dorsey ER, Beck CA, Brenna JT, Shoulson I. Altered cholesterol and fatty acid metabolism in Huntington disease. J Clin Lipidol. 2010;4:17–23. doi: 10.1016/j.jacl.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puri BK, et al. Ethyl-EPA in Huntington disease: a double-blind, randomized, placebo-controlled trial. Neurology. 2005;65:286–292. doi: 10.1212/01.wnl.0000169025.09670.6d. [DOI] [PubMed] [Google Scholar]
- 25.Puri BK, et al. Reduction in cerebral atrophy associated with ethyl-eicosapentaenoic acid treatment in patients with Huntington’s disease. J Int Med Res. 2008;36:896–905. doi: 10.1177/147323000803600505. [DOI] [PubMed] [Google Scholar]
- 26.López M, Vidal-Puig A. Brain lipogenesis and regulation of energy metabolism. Curr Opin Clin Nutr Metab Care. 2008;11:483–490. doi: 10.1097/MCO.0b013e328302f3d8. [DOI] [PubMed] [Google Scholar]
- 27.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech Ageing Dev. 2008;129:441–448. doi: 10.1016/j.mad.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subba Rao K. Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol. 2007;3:162–172. doi: 10.1038/ncpneuro0448. [DOI] [PubMed] [Google Scholar]
- 30.Razick S, Magklaras G, Donaldson IM. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics. 2008;9:405. doi: 10.1186/1471-2105-9-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wishart DS, et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiele I, et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013;31:419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aranda B, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat Methods. 2011;8:528–529. doi: 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frolkis A, et al. SMPDB: The Small Molecule Pathway Database. Nucleic Acids Res. 2010;38:D480–D487. doi: 10.1093/nar/gkp1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hucka M, et al. SBML Forum The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 36.Bornstein BJ, Keating SM, Jouraku A, Hucka M. LibSBML: an API library for SBML. Bioinformatics. 2008;24:880–881. doi: 10.1093/bioinformatics/btn051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang SS, et al. Linking proteomic and transcriptional data through the interactome and epigenome reveals a map of oncogene-induced signaling. PLOS Comput Biol. 2013;9:e1002887. doi: 10.1371/journal.pcbi.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailly-Bechet M, Braunstein A, Pagnani A, Weigt M, Zecchina R. Inference of sparse combinatorial-control networks from gene-expression data: a message passing approach. BMC Bioinformatics. 2010;11:355. doi: 10.1186/1471-2105-11-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 40.Saghatelian A, et al. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry. 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 41.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41:D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–D260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ng CW, et al. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci USA. 2013;110:2354–2359. doi: 10.1073/pnas.1221292110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.