

Abstract
Immunotherapy is the new trend in cancer treatment due to the selectivity, long lasting effects, and demonstrated improved overall survival and tolerance, when compared to patients treated with conventional chemotherapy. Despite these positive results, immunotherapy is still far from becoming the perfect magic bullet to fight cancer, largely due to the facts that immunotherapy is not effective in all patients nor in all cancer types. How and when will immunotherapy overcome these hurdles? In this review we take a step back to walk side by side with the pioneers of immunotherapy in order to understand what steps need to be taken today to make immunotherapy effective across all cancers. While early scientists, such as Coley, elicited an unselective but effective response against cancer, the search for selectivity pushed immunotherapy to the side in favor of drugs focused on targeting cancer cells. Fortunately, the modern era would revive the importance of the immune system in battling cancer by releasing the brakes or checkpoints (anti-CTLA-4 and anti-PD-1/PD-L1) that have been holding the immune system at bay. However, there are still many hurdles to overcome before immunotherapy becomes a universal cancer therapy. For example, we discuss how the redundant and complex nature of the immune system can impede tumor elimination by teeter tottering between different polarization states: one eliciting anti-cancer effects while the other promoting cancer growth and invasion. In addition, we highlight the incapacity of the immune system to choose between a fight or repair action with respect to tumor growth. Finally we combine these concepts to present a new way to think about the immune system and immune tolerance, by introducing two new metaphors, the “push the accelerator” and “repair the car” metaphors, to explain the current limitations associated with cancer immunotherapy.
Keywords: CTLA-4, PD-1, PD-L1, Immunotherapy, Coley’s toxin
Introduction
Since war was declared on cancer in 1971, our arsenal of drugs against this enemy has steadily increased. The first class of drugs developed, conventional chemotherapy [1], provided significant benefits for the treatment and management of different cancers; however, conventional chemotherapy has two intrinsic defects: lack of selectivity [2] and long-term resistance [3]. The subsequent development of targeted therapies, including monoclonal antibody-based therapies, such as rituximab, overcame the lack of selectivity associated with conventional chemotherapy by targeting specific proteins involved in cancer cell stimulation, proliferation or apoptosis evasion [4]. While monoclonal antibody-based strategies have significantly increased in clinical practice over the past decade, mechanisms of acquired resistance remains a hurtle, likely due to genetic and epigenetic instability of cancer cells [5]. Thus, despite significant advances made in cancer treatment over the past 45 years, we are still far from developing therapies capable of effectively ablating cancer while avoiding adverse effects on healthy cells and loss of efficacy over time.
Starting with Coley’s toxin, followed by Paul Ehrlich’s hypothesis of tumor surveillance and contrasted by Burnet’s immunological tolerance theory [6], the idea that the immune system can play a pro- and/or anti-tumor role has been recognized and debated for years. As the immune system is armed to protect against pathogens, it has been long postulated that immune cells should recognize tumor cells as foreign, and effectively eliminate them before spreading to distant organs. This concept, today referred to as cancer immunotherapy, has the potential to be the magic bullet that investigators have been desperately searching for since the early 1900s [7].
The last century was a pendulum, swinging between big hope and deep disappointment in the immunotherapy field. Why has our view of immunotherapy shifted from promising to disappointing? Can immunotherapy be made more effective? In this review we look to the past to understand the current problems associated with immunotherapy. We present a new way to think about the immune system and immune tolerance. In addition to the standard “fuel the engine, release the brake” rules of immunotherapy, we introduce the “push the accelerator” and “repair the car” metaphors to explain part of the current limitations associated with cancer immunotherapy.
Immunotherapy: a revolutionary view of cancer treatment
Immunotherapy was born in 1890. Its father, William Coley [8], observed that a patient with an inoperable sarcoma that suffered a Streptococcus pyogenes infection twice obtained complete remission. Based on this observation, Coley treated approximately 1000 patients with inoperable cancers (specially sarcomas) with a mixture consisting of killed S. pyogenes and Serratia marcescens, achieving a complete remission in 10% of treated patients. Compared with actual 5-year survival rates for metastatic sarcoma (20%) [9], the effects of Coley’s toxin were promising and demonstrated that our immune system could effectively eradicate tumors with a low rate of adverse effects in a subset of patients. While the mechanism of action of Coley’s toxin was unknown at the time, Coley’s toxin essentially activated tumor-infiltrating leukocytes [heterogeneous populations of cells, including varying proportions of neutrophils, macrophages, T and B cells, and natural killer cells (NK)]. Following administration of Coley’s toxin, dendritic cells (DCs), professional antigen presenting cells (APCs), initiate an immune response by presenting the captured bacterial antigen to naïve CD4+ or CD8+ T cells [10] in lymphoid tissues, which results in T cell priming [11] and inflammatory interleukin production [IL-1, IL-2, tumor necrosis factor alpha (TNFα,) IL-12] [12, 13]. Clonal T cell expansion triggers a humoral immune response by activating B cells, or cellular immunity by activating Th1 effector cells or NK cells [14]. Thus, as tumors often display a high degree of leukocyte infiltration, it is reasonable that stimulation of tumor-infiltrating leukocytes can result in tumor cell targeting and elimination.
The first gauntlet: release the brake
In 1949, some years after Coley’s first experiments, Macfarlane Burnet stated, “if in embryonic life expendable cells from a genetically distinct race are implanted and established, no antibody response should develop against the foreign cell antigen when the animal takes on independent existence” [6, 15]. Peter Medawar would go one step further and propose that the immune system becomes tolerant to cancer cells due to the similarities that exist with normal healthy cells. In his early experiments, Medawar injected embryonic mouse donor cells into mice of a different strain, rendering the recipient mice tolerant to future grafts from the donor and not third-party strains [16]. These experiments set the initial groundwork for what would become the concept of acquired immunological tolerance. Even if the molecular mechanisms explaining Medawar and Burnet’s observations would not be discovered until 25 years later, Medawar and Burnet had already thrown down the first gauntlet to modern immunotherapy [16].
Immunological tolerance is a fundamental process, the lack of which would result in numerous pathologies including autoimmune illnesses. Since Burnet and Medawar won the Nobel Prize for their pioneering work in this field more than 50 years ago, an enormous amount of progress has been made to better understand immune tolerance and the numerous redundant mechanisms involved in this biological process. In 1959, Joshua Lederberg published nine propositions on immunity and tolerance [17]. The sixth proposition stated that “the immature antibody-forming cell is hypersensitive to an antigen–antibody combination: it will be suppressed if it encounters the homologous antigen at this time”, highlighting that each antibody-producing cell has a single specificity [17]. In 1978, Nossal and Pike experimentally demonstrated that bone marrow-derived cells cultured with an antigen became tolerant to the antigen in a time-dependent manner, with maximal tolerance being achieved only when the antigen was present continuously as the cultured bone marrow cells matured [18]. In the late 1980’s, Kappler demonstrated central tolerance in mice and indicated that tolerance induction may occur in the thymus [19]. Goodnow later showed that B cells that reactive to “self” antigens are eliminated or silenced in order to avoid autoimmunity [20]. And finally, Le Douarin demonstrated that the thymus is not only important for central tolerance but it produces cells [i.e. regulatory T (Treg) cells] that strongly regulate effector cells, discovering a third dominant form of immune tolerance [21].
Taken together these results clearly indicated that self-immune tolerance is maintained and regulated by multiple mechanisms, including similarity with self-antigens, regulatory immune cells, a suppressive versus activating cytokine balance and immune checkpoints. Thus, if this process is to be effectively manipulated in order to release the brake, we have to stimulate APCs with an antigen significantly different from “self” to recruit effector cells, balance cytokines to obtain a pro-inflammatory milieu, and finally, inhibit immune checkpoints to avoid tolerance.
Hope dies last
The idea of immune tolerance put forth by Medawar and Burnet dampened the hope generated by Coley’s experiments that activating the immune system could treat cancer. However, in 1953, Foley demonstrated that methylcholanthrene-induced tumor cells could produce immunogenic antigens in mice, although strain-dependent differences were observed [22]. Nathrath would add additional fuel to the fire by showing that the host immune system is capable of recognizing new molecular properties (antigens) displayed by tumor cell as a result of the changes accumulating during the transition from normal to malignant cells [23]. Interestingly, however, the greatest hope for immunotherapy would come from another illness described in the 1950s, autoimmune diseases such as lupus, which clearly confirmed that auto-reactive cells can elude self tolerance [24]. Finally, towards the later end of the 20th century, immunogenic tumor associated antigens were discovered in mice [25] and humans [26], resulting from cancer cell genomic instability. Unsettling still was the fact that while cancer cells could express a multitude of new and unknown antigens as a result of malignant transformation, the immune system was not stimulated to target these antigens/cells. The problem therefore was no longer the absence of immunogenic antigens on cancer cells, but rather understanding why our immune system “ignores/tolerates” cells harboring these antigens.
In the absence of inflammation, naïve T-cells circulate preferentially to secondary lymphoid tissue [27]. During an infection, APCs respond to inflammatory cytokines (IL-1, TNF-α) [28] and migrate via afferent lymph vessels to lymph nodes where they can interact with naïve T-cells. In a similar manner, macrophage or B cells can take up and process free antigen in the blood or spleen [29]. The interaction of T cell CD28 and DC CD80 (B7-1) or CD86 (B7-2) allows for specific T cells to proliferate in the paracortex [30] and become competent to receive further activation signals from antigen-bearing macrophages and B cells. This process results in the production of cytokines, loss of L-selectin (which is involved in lymph node entry) and increased expression of adhesion molecules like VLA 4 (which facilitates extravasation into non-lymphoid tissue) [31]. Macrophages and parenchymal cells produce inflammatory cytokines (IL-1, TNF-α) that increase expression of selectins and integrin ligands. Ultimately, activated T-cells express adhesion molecules that allow them to selectively enter inflamed tissues expressing counterpart adhesion molecules. In summary, immune cells can present antigens (dendritic cells, macrophages), produce cytokines (macrophage) and interact with cells of the adaptive immune system (B-cells, T-cells) in the lymph nodes, resulting in their subsequent activation via receptor/ligand interactions. Once activated, adaptive immune cells proliferate, alter their receptors and adhesion molecules, and finally migrate to initiate the destruction of foreign pathogens.
To avoid indiscriminate activation and self-destruction, the immune system has developed redundant mechanisms to tolerate self or non-dangerous antigens. Since central tolerance is not the underlying mechanisms by which cancer cells escape immune targeting, we refer the reader to several published reviews detailing the biology of central tolerance (Ref. [32, 33]). Peripheral tolerance, on the other hand, is the primary mechanism utilized by cancer cells to avoid the immune system. In the early 1990s, Jenkins and Schwartz demonstrated that T-cells need a co-stimulatory signal to fully activate, and if T-cells receive only TCR signals they become anergic [34]. The co-stimulatory signals must be received from APCs [35]: B7-1 (CD80) or B7-2 (CD86) on APCs binding to CD28 on T-cells is necessary to fully stimulate T-cells. While an important step forward in our understanding of T-cell regulation, the big discovery was not B7-1 or B7-2, but rather the receptor that inhibits the second co-stimulatory signal. James Allison, director of the UC Berkeley Cancer Research Laboratory, was intrigued with a molecule called cytotoxic T-lymphocyte antigen-4 (CTLA-4) [36], originally discovered in a cDNA library derived from activated T-cells. In the late 1990s, Allison and his group began to study how CTLA-4 inhibits T-cells and if this inhibition could explain why T-cells do not attack cancer cells. They demonstrated that CTLA-4, a homologue of CD28, bound with higher affinity (at least 10-fold) to both B7-1 and B7-2 [37] and inhibited CD4+ T-cell activation. Under certain conditions, T-cells up-regulate CTLA-4, which binds to B7-1 and B7-2 with a higher affinity than CD28, effectively hijacking the second co-stimulatory signal that T-cells require for full T-cell activation, proliferation, and effector function [38]. As a result, T-cells cannot be fully activated and thus undergo anergy. Since its discovery, several groups have worked diligently towards dissecting the role of CTLA-4. Studies with CTLA-4−/− mice confirmed its inhibitory function in vivo. Waterhouse et al. showed that mice lacking CTLA-4 died early on of fatal lymphoproliferative disorders [39], demonstrating that CTLA-4 acts as a negative regulator of T cell activation and is vital for the control of lymphocyte homeostasis. Based on ever increasing data demonstrating a role for CTLA-4 as a negative regulator of T-cell activation, Allison and colleagues went on to show that in vivo administration of antibodies to CTLA-4 promoted the rejection of tumors, including pre-established tumors, confirming that CTLA-4 blockage can allow for, and potentiate, an effective immune responses against tumors [40].
The first victory in the new era
These studies and others [41–43] suggested that monoclonal antibody-mediated CTLA-4 blockage could represent an effective anti-cancer therapy. To translate these findings to the clinical setting, the Medarex Corporation generated a series of monoclonal antibodies using a unique transgenic mouse (HuMAb), in which the endogenous murine immunoglobulin genes had been knocked out and replaced with the human loci [44]. Ipilimumab showed safety in a phase I study and efficacy in a phase III study (ClinicalTrials.gov Identifier: NCT00094653) with primary overall survival endpoints. Patients allocated to receive ipilimumab had a median overall survival of 10.1 versus 6.4 months for the control group [hazard ratio (HR), 0.68; P ≤ 0.003]. Based on the promising results, Bristol-Meyer’s Yervoy® (ipilimumab) was approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2011 for the treatment of metastatic melanoma [38].
While CTLA-4 was the first “checkpoint” inhibitor identified, the list of checkpoint immunomodulators continues to grow, with inhibitors of the programmed cell death protein 1/ligand pathway leading the way. Programmed cell death protein 1, also known as PD-1 and CD279 (cluster of differentiation 279) is a member of the CD28 superfamily expressed on activated CD4+ and CD8+ T-cells as well as NK and B-cells while programmed death-ligand 1 (PD-L1), also known as cluster of differentiation 274 (CD274) or B7 homolog 1 (B7-H1), is expressed predominantly on APCs. The main role of PD-1 is to act like a stopwatch to limit the activity of T-cells in the “battle field” during the effector phase of T-cell activation in peripheral tissues and the tumor microenvironment via the delivery of negative signals upon interaction with its two ligands (PD-L1 or PD-L2). PD-L1 and PD-L2 compete for PD-1 [45] and upon binding both inhibit T cell proliferation, cytokine production and cell adhesion [46], although some contradictory data have suggested a costimulatory function [47]. T-cells begin to express PD-1 when activated and its expression increase over time [48, 49], while PD-L1 is expressed on APCs present in the inflamed tissue. PD-1 is also highly expressed on Treg cells, where it may enhance their proliferation in the presence of ligand [50]. The expression on Treg cells also highlights the role PD-1 plays in regulating the induction and maintenance of peripheral tolerance and protection from autoimmune attack [51]. Our current understanding of the PD-1/PD-L1 pathway shows that engagement of PD-1 and PD-L1 leads to inactivation of effector T-cell molecules such as Zap70 to inhibit T-cell proliferation, thus limiting the inflammatory damage in inflamed tissues [52]. For example, during chronic infections the PD-1/PD-L1 pathway leads to anergy [53]. It appears as though several cancers, including lung, ovarian and colon carcinomas as well as melanomas, have evolved to over express PD-L1 [54]. Thus, chronic tumor associated antigen exposure in cancer can lead to high levels of PD-1 expression on T-cells, which can interact with PD-L1 expressed on cancer cells, inhibiting T-cell activation and perhaps inducing a state of anergy in immune cells present in the tumor. Blocking the PD-1/PD-L1 pathway can revert this condition, promoting cancer cell elimination by activated T-cells. In the past 5 years, Bristol-Myers Squibb has produced a fully humanized antibody against PD-1 named Opdivo (Nivolumab), which obtained FDA accelerated approval in 2014 [55] based on the “Study of Nivolumab (BMS-936558) Compared With Dacarbazine in Untreated, Unresectable, or Metastatic Melanoma”.
Today many check-point inhibitors are used by oncologist to achieve significant increases in survival rates (e.g. 1- and 2-year survival rates of 62 and 43%, respectively for melanoma or 1- and 2-year survival rates of 42 and 23%, respectively for lung cancer) and/or to achieve a durable partial or complete response in cancer patients (e.g. 31% for melanoma patients) [56, 57]. Table 1 summarizes the current FDA- and EMA-approved immune checkpoint inhibitors. Clinical studies have also investigated the efficacy of combination therapies using anti-PD-1/PD-L1 therapies together with other checkpoint inhibitors, such as the anti-CTLA4 treatment ipilimumab. The combination of nivolumab and ipilimumab increased overall survival in patients with untreated melanoma. The median progression-free survival was 11.5 months (95% confidence interval [CI], 8.9–16.7) with nivolumab plus ipilimumab, as compared with 2.9 months (95% CI, 2.8–3.4) with ipilimumab (hazard ratio for death or disease progression, 0.42; 99.5% CI, 0.31–0.57; P < 0.001), and 6.9 months (95% CI, 4.3–9.5) with nivolumab (hazard ratio for the comparison with ipilimumab, 0.57; 99.5% CI, 0.43–0.76; P < 0.001) [58]. It is important to stress that even if combination therapy allows us to obtain better results and improved medium overall survival, it is still far from becoming the perfect therapy. The current underlying problem with immune checkpoint inhibitors is that if there are no T-cells in the tumor border then there are no effector cells capable of eliminating the tumor cells. Therefore, even if we release the brake we cannot obtain clinically relevant results. Thus, the next strategy lies in “fueling the engine”, that is allowing T-cells to reach the tumor border.
Table 1.
Summary of immune therapies in clinical use
| Immune therapy | Target | Stage | Cancer type | Ref. |
|---|---|---|---|---|
| Ipilimumab | CTLA-4 | Clinical use | Advanced melanoma | 107 |
| Nivolumab | PD-1 | Clinical use | Melanoma Renal cancer NSLC |
108 109 110 |
| Pembrolizumab | PD-1 | Clinical use | Melanoma | 111 |
| Atezolizumab | PD-L1 | Clinical use | NSLC Clear renal cancer bladder cancer |
112 113 |
| Sipileucel | Peripheral blood mononuclear cells | Clinical use | Prostate cancer | 114 |
Fueling the engine
As stated above, immune checkpoints inhibitors are only effective if tumors are infiltrated with T-cells. Therefore, if a tumor lacks infiltrated T-cells, immune checkpoint inhibitors are essentially ineffective. Cancer vaccines have been extensively investigated as a strategy to induce T-cell infiltration in the tumor—“fuel the engine”. Currently, however, Sipileucel is the only “cellular immunotherapy” (i.e. vaccine therapy) approved by the FDA. Sipileucel consists of autologous peripheral blood mononuclear cells (PBMCs), obtained by leukapheresis and cultured with a prostatic acid phosphatase linked to granulocyte–macrophage colony-stimulating factor (GM-CSF) [59]. Results from the 9902B study in patients with prostate cancer demonstrated an overall survival of 25.8 months for patients receiving Sipileucel compared to 21.7 months for patients who received the control treatment [59].
While Sipileucel demonstrates the clear benefit of “fueling the engine”, the use of cancer vaccines in other tumor types is still in the experimental stages. For example, Kleponis et al. developed a GM-CSF-secreting pancreatic cancer vaccine (GVAX) that provided maturation signals to APCs at the local vaccine site. Stimulated APCs processed tumor antigens and presented them to T-effector cells, allowing T-cells to infiltrate the tumor [60]. The authors went on to show that PD-L1 expression was induced in the infiltrating cells (T-cells). This study demonstrated that vaccine-based therapies may have adequately primed pancreatic cancer for anti-PD-1/PD-L1 treatments, highlighting that this typically T-cell-effector poor/Treg-rich tumor [61] could be potentially treated with PD-1/PD-L1 inhibitors. Thus, cancer vaccine-based immunotherapy may overcome the resistance of certain cancers to immune checkpoint inhibitors, while immune checkpoint inhibitors may enhance the efficacy of the cancer-vaccine therapies. The goal of a combination strategy is to combine the strength of each immunotherapy approach, with cancer vaccines functioning to “fuel the engine” and immune checkpoint inhibitors working to “release the brake”.
Perplexing is the fact that PD-1 and CTLA-4 checkpoint inhibitors, even when helped by cancer vaccines, are not effective against all cancer types, nor do they work in every patient with the same cancer. Perhaps other immune cell types are negatively affecting cancer immunotherapy? The explanation we put forward to explain this dilemma is that in some cancers we have the machinery (the car) on a downhill slope, so if we release the brake (immune checkpoint inhibitors) the car can move. In contrast, when we are on an uphill slope or on a plain field, releasing the brake simply does not move the car. For such scenarios, we have to release the brake and push the accelerator.
Looking deep inside the immune system we can find a dynamic and complex environment of cells that are different in type, size, complexity, markers and function. Even the same cell can exist in two (or more) different polarized states. For example, macrophages can switch between a pro-inflammatory (classically activated) and a reparative (alternatively activated) state [62]. T-cells can be stimulated into T effector cells or Tregs cells, each of which can have a very distinct role within a tumor [63]. Thus, we need to take a step back and understand why, when and how a cell (i.e. macrophage, T-cell, etc.) can switch from a classically activated (inflammatory macrophage or T effector cell) to an alternatively activated (pro-tumorigenic macrophage or Treg) state and what mediates this change. Answering these questions may provide the key to push the accelerator.
Immune cells: Dr. Jekyll and Mr. Hide
The immune system is comprised of many cells, including but not limited to dendritic cells, mast cells, macrophages, neutrophils and lymphocytes. In addition, and to complicate the matter even more, immune cells are extremely plastic and each cell type can differentiate into at least two forms depending on their environment and the paracrine signals they receive (reviewed in [64–66]). Lymphocytes can differentiate into many subsets. Apart from B and T cells, T lymphocytes can differentiate into CD8+ or CD4+ T cells, the latter of which can in turn differentiate into Treg and Thelper cells [67]. DCs that present captured antigens to naïve T-cells, have two major subsets: myeloid (i.e. conventional DC or immunogenic DC) and plasmacytoid form (or tolerogenic DC) [68]. As described above, macrophages can exist in at least two forms [69], and in the context of a tumor, macrophages and neutrophils can differentiate into tumor-associated macrophages (TAMs) or neutrophils (TANs) [70], respectively.
The interest in the different states of immune cells, particularly within the tumor, stems from the different biological affects these cells produce depending on their state, polarization or differentiation (Fig. 1). For example, human DCs can exist as immunogenic or tolerogenic DCs [71], with immunogenic DCs functioning primarily to stimulate a Thelper [72] response while tolerogenic DCs function primarily to stimulate a Treg response [73]. Moreover, the ratio of different DCs depends on the cytokine milieu. In vitro studies clearly demonstrate that GM-CSF, interferon alpha (IFNα), or IL-15 can induce the differentiation of inflammatory DCs while IL-10, vitamin A or D3, or immunosuppressive drugs such as cyclosporine A induce tolerogenic DCs through E-cadherin mediated signaling [74, 75]. Thus, depending on the cytokine milieu, DCs can elicit a strong immune response or a tolerogenic state.
Fig. 1.
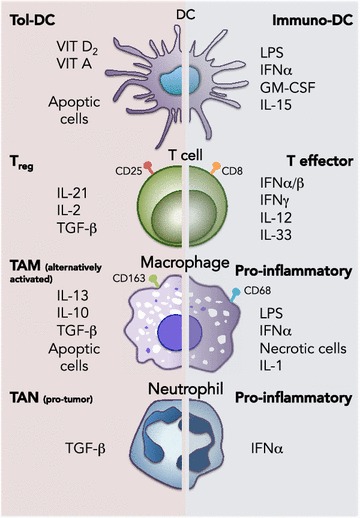
The two faces of immune cells: The Dr. Jekyll and Mr. Hide concept. Immune cells, including dendritic cells (DCs), T-cells, macrophages and neutrophils, are extremely plastic and can assume different roles/functions depending on factors encountered at the site of infection or within the tumor microenvironment. When stimulated by factors such as TGF-ß or when in contact with apoptotic cells, immune cells become pro-tumorigenic (left side) and differentiate/polarize towards tolerogenic DCs (Tol-DC), Treg cells, tumor-associated macrophages (TAMs) or tumor-associated neutrophils (TANs). In contrast, when stimulated by pro-inflammatory cytokines, such as IFNs, IL-1 or IL-12, immune cells become anti-tumorigenic (right side) and differentiate/polarize towards immunogenic DCs (immuno-DC), T effector cells, activated pro-inflammatory macrophages or neutrophils
Macrophages represent another important cell type that play a pivotal role in activating and shaping the immune response, and similar to DCs, a dichotomy has been proposed for macrophage activation: classically or alternatively activated [76, 77]. IFNα, LPS or inflammatory cytokines can induce classical activation while IL-4, IL-13, TGF-β and reparatory signals can induce alternative activation of macrophages. In the last decade, if macrophages have gone from a negligible player in tumor progression to a pivotal modulator of tumor growth and metastasis, it is due to the identification of TAMs. There is now solid and continuously growing evidence to show that TAMs actively promote all aspects of tumor growth and development including promotion of angiogenesis, matrix remodeling and suppression of adaptive immunity [78]. Recent studies also show that TAMs share many characteristics with alternatively activated macrophages such as (1) activation of the arginase pathway, implicated in arginine metabolism; (2) promotion of cell repair and proliferation in strong opposition with the NOS pathway that promote cells killing [79]; (3) production of IL-10 and vascular endothelial growth factor (VEGF) over other factors promoting cell survival [80]; (4) production of matrix metalloproteinases (MMPs), implicated in cancer initiation and metastasis [81, 82]; (5) activation of NFκB and STAT3 signaling, enhancing tumor progression by directly communicating with cancer stem cells (CSCs) [62], and (6) activation of STAT6, which possess potent inhibitory activity of T-cell activity [83].
T-cells, the soldiers of the immune system, have a fundamental role in immune surveillance, and even for these cells a dichotomy exists: a T-cell can defend the host from cancer while aiding tumor growth. Zhang et al. showed that ovarian carcinoma patients with high tumor infiltrating lymphocytes had improved 5-year survival rates compared to patients with low tumor infiltrating lymphocytes [84]; however, in other cancers, such as renal cancer, high tumor infiltrating lymphocytes translated into worse prognosis [85]. These clear opposing observations are now explained by differences in the type of infiltrating T-cells: Treg versus T effectors cells (TCD8+). Treg cells, a subgroup of T-cells expressing CD4 and CD25, regulate activation of other T-cells and are necessary to maintain peripheral tolerance to self-antigens. We have now come to understand that increased numbers of Treg cells present in the tumor can have a negative prognostic impact. For example, Sato et al. showed that a high TCD8+/Treg cell ratio translated into better overall survival while the opposite was seen for a high Treg/TCD8+ ratio [86]. Likewise, Li et al. demonstrated that the efficiency and percent depletion of Treg cells from a tumor can improve cancer outcome [67, 87]. Specifically, they show that in Foxp3.LuciDTR-4 mice, which show 90–95% Treg depletion, large established tumors completely regressed, unlike anti-CD25 antibody-mediated Treg elimination, which is less efficient (approximately 70%). Thus, high-level depletion of Treg cells is necessary for tumor regression.
Like a coin, immune cells have two faces, one with a strong potential to fight cancer and the other (the opposite one) strongly promoting cancer development and immune system escape. This Dr. Jekyll and Mr. Hide concept has been re-coined the “corrupted policemen” concept by Bonavita et al. in order to stress the fact that those cells born to protect the host can become corrupt and turn against it to favor cancer growth [88]. This switch is complex and partly mediated by cytokines: inflammatory cytokines promoting immune system cells to show their anti-cancer face while anti-inflammatory cytokines promote the pro-cancer side to dominate. But cytokines are only mediators. More important are the stimuli that induce immune host cells to convert into either cancer allies (pro-tumor macrophages, Treg cells, or tolerogenic DC) or cancer enemies (inflammatory macrophage, CD8+ T-cells, Thelper cells or immunogenic DC) (Fig. 1). If we discover which mechanism(s) induce the inflammatory anti-cancer response and which ones induce the pro-cancer response, in theory we could pharmacologically change the face of the coin, favoring an anti-cancer response.
To fight or to repair? That is the question
The first problem that oncoimmunotherapists faced was immune tolerance. Currently, a more challenging dilemma lies in the ability of the immune system to balance itself between two opposing actions: “fight” the enemy or “repair the damage”. A successful immune response can be accompanied by extensive tissue damage [89]. Fortunately, the immune system has the capacity to repair the resulting damage via Treg cells, macrophages and anti-inflammatory cytokines that send a repair message to the site of damage. In the context of cancer, however, the wrong choice can have detrimental effects on tumor eradication, as alluded to above. Numerous studies have shown that tumor growth and even post treatment tumor cell death can be perceived by the immune system as a repair signal, initiating a wound healing response that can favor sustained tumor growth and even tumor chemoresistance via immune cell secreted factors (reviewed in [90, 91]). Dissecting how the immune system senses and responds to damage will improve our efforts of inhibiting the immune system from favoring tumor growth over tumor destruction.
After immune system activation, the battlefield is replete with damaged cells, the majority being apoptotic cells (e.g. bacteria, neutrophils, epithelial cells). It is therefore logical to think that these cells could regulate the immune system or represent the signal that promotes a repair response. Although conventional and targeted therapies often aim to induce apoptosis, these strategies may themselves be carcinogenic [92].
The relation between cancer, immune cells and dead/apoptotic cells has gained increased attention over the past decade. Apoptosis obtained through activation of caspases or mitochondrial chain dysfunction was historically declared as non-immunogenic, while necrotic death accompanied by the release of proteins, lipids and other cellular debris from cells has been widely considered as strongly immunogenic [93, 94]. Chemotherapy-mediated cell death was accepted as an apoptosis-mediated process, inducing an immunosuppressive milieu of cytokines [95], but studies show that depending on the agent used and the degree of cell death induced, chemotherapy can also induce necrotic cells [96]. Moreover, other studies have shown that classically-induced apoptotic cells can profoundly affect the immune system [97], and the idea that chemotherapy-induced apoptosis is non-immunogenic may be overstated [98–100].
In cancer, there appears to be a ying-yang scenario with respect to the presence of dead cells and method of cell death induction. The presence of apoptotic cells can be sensed and translated into a “need to repair” action, allowing Treg/Th2 stimulation through cytokine expression (IL-10, IL-13) [100, 101]. On the other hand, necrotic cells or highly variable tumor associated antigens can be sensed and translated into a “need to fight” action, allowing T-CD4+ stimulation thought inflammatory cytokine expression (TNF-α, IL-1, etc.) [93]. Williams et al. showed that DCs exposed to apoptotic Jurkat cells or apoptotic primary T-cells failed to maturate and were unable to support CD4+ allogeneic T-cell proliferation, as compared to DCs exposed to lipopolysaccharide (LPS) or necrotic cells [102]. Conversely, phosphatidylserine exposed on apoptotic epithelial cells suppressed IFN-β production by dendritic cells via inhibitory signalling mediated by the cell-surface glycoprotein CD300a and thus suppressed Treg cell proliferation [103]. Moreover, Kleinclauss et al. in 2006 demonstrated that apoptotic cells induce CD4+ T-cells to express CD25+ (a marker of Treg cells) inducing a state of tolerance [104]. Taken together, these studies demonstrate that the immune system can sense the type of danger and activate a specific action. In our laboratory we have discovered that apoptotic pancreatic tumor cells, as opposed to necrotic or live cells, can strongly induce immune system suppression or a pro-Th2 state, promoting repair signals that favor pancreatic tumor growth and chemoresistance (unpublished data). Our observations are in line with data published by Wu et al. where they show that intravenous administration of donor apoptotic splenocytes promotes the generation of tolerogenic DCs and the expansion of Treg cells in the pancreas; in vivo clearance of either DCs or Treg cells abrogated immune tolerance induction [105]. Thus, in pancreatic cancer where chemotherapies such as gemcitabine induce tumor cell apoptosis, an immune response that favors tumor growth may be activated.
Conclusion
The field of cancer immunotherapy has been driven by scientists such as William Coley whose important results set the foundation for modern day immunotherapy, as well as by scientists who while opposed the concept of immunotherapy made pinnacle discoveries that favored the evolution of immunotherapy into one of the most promising techniques in place today to battle cancer. The scientific community is convinced that immunotherapy is only a step away from becoming the magic bullet to defeat cancer. While the future looks promising, there are still many hurdles that need to be overcome.
We now have the capacity to “fuel the engine” by using vaccine strategies to accumulate effector T-cells at the tumor border, and “release the brake” to allow T-cells to fight the cancer by inhibiting immune system checkpoints. This strategy allows us to fight cancer when the battlefield is downhill or when the tumor entity itself is “immunogenic” due to (1) profound differences in tumor associated antigens between normal cells and tumor cells or (2) the presence of immunogenic necrotic cells. This strategy, however, is ineffective when the battlefield is plain or uphill, or when the tumor itself promotes a “need to repair” response due to chemotherapy-induced apoptosis or other unknown factors. Thus, when faced with this scenario, an approach to ensure that the immune system senses the cancer as danger in order to promote a “need to fight” over a “need to repair” response is essential. Consequently, the “push the accelerator” component of the universal magic bullet is still lacking. This action may possibly be achieved by educating APCs (DCs and macrophages) to sense every cancer as “dangerous”, ensuring an anti-tumor immune response in all cases. Moreover, understanding that certain immune cells (e.g. macrophages) can shift the immune response in one direction over another, combination therapies may be necessary to transiently eliminate other immune cells. Ultimately, the goal is to unmask those factors that promote a “need to repair” response while at the same time enhancing those factors that are sensed as a danger signal by the immune system. Only then will immunotherapy truly become the magic bullet for cancer treatment.
Authors’ contributions
GD prepared, designed and wrote the manuscript. HLM and BSJ assisted in the manuscript design and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank their respective laboratories and clinical collaborators for helpful discussions.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by NIH R00 CA154605 and Louisiana Board of Regents LEQSF(2016-17)-RD-C-14 (H.L.M.), a Rámon y Cajal Merit Award from the Ministerio de Economía y Competitividad, Spain (B.S.Jr) and a Clinic and Laboratory Integration Program (CLIP) grant from the Cancer Research Institute, NY (B.S.Jr).
Abbreviations
- DC
dendritic cells
- APC
antigen presenting cells
- IL
interleukin
- NK
natural killer
- Treg
T-regulatory cell
- CTLA-4
cytotoxic T-Lymphocyte antigen-4
- PD-1
programmed cell death protein 1
- PD-L1
programmed death-ligand 1
- PBMC
peripheral blood mononuclear cells
- IFN
interferon
- TNF
tumor necrosis factor
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- TAM
tumor-associated macrophage
- TAN
tumor-associated neutrophil
Contributor Information
Gabriele D’Errico, Email: derricogab@hotmail.com.
Heather L. Machado, Phone: 504-988-1753, Email: hmachado@tulane.edu
Bruno Sainz, Jr., Phone: +34 91-497-3385, Email: bruno.sainz@uam.es
References
- 1.DeVita VT, Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 2.Liang XJ, Chen C, Zhao Y, et al. Circumventing tumor resistance to chemotherapy by nanotechnology. Methods Mol Biol. 2010;596:467–488. doi: 10.1007/978-1-60761-416-6_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2005;14(Suppl 1):35–48. doi: 10.1159/000086183. [DOI] [PubMed] [Google Scholar]
- 4.Attarwala H. Role of antibodies in cancer targeting. J Nat Sci Biol Med. 2010;1:53–56. doi: 10.4103/0976-9668.71675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reslan L, Dalle S, Dumontet C. Understanding and circumventing resistance to anticancer monoclonal antibodies. MAbs. 2009;1:222–229. doi: 10.4161/mabs.1.3.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnet F, Fenner F. The production of antibodies. 2. Melbourne: Macmillan; 1949. [Google Scholar]
- 7.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–158. [PMC free article] [PubMed] [Google Scholar]
- 9.Steen S, Stephenson G. Current treatment of soft tissue sarcoma. Proc (Bayl Univ Med Cent) 2008;21:392–396. doi: 10.1080/08998280.2008.11928435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alloatti A, Kotsias F, Magalhaes JG, et al. Dendritic cell maturation and cross-presentation: timing matters! Immunol Rev. 2016;272:97–108. doi: 10.1111/imr.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morelli AE, O’Connell PJ, Khanna A, et al. Preferential induction of Th1 responses by functionally mature hepatic (CD8alpha− and CD8alpha+) dendritic cells: association with conversion from liver transplant tolerance to acute rejection. Transplantation. 2000;69:2647–2657. doi: 10.1097/00007890-200006270-00027. [DOI] [PubMed] [Google Scholar]
- 12.Agrawal S, Agrawal A, Said HM. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am J Physiol Cell Physiol. 2016;00141:02016. doi: 10.1152/ajpcell.00141.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romagnani S. T-cell subsets (Th1 versus Th2) Ann Allergy Asthma Immunol. 2000;85:9–18. doi: 10.1016/S1081-1206(10)62426-X. [DOI] [PubMed] [Google Scholar]
- 14.Chen J, Zurawski G, Zurawski S, et al. A novel vaccine for mantle cell lymphoma based on targeting cyclin D1 to dendritic cells via CD40. J Hematol Oncol. 2015;8:35. doi: 10.1186/s13045-015-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liston A. Immunological tolerance 50 years after the Burnet Nobel Prize. Immunol Cell Biol. 2011;89:14–15. doi: 10.1038/icb.2010.138. [DOI] [PubMed] [Google Scholar]
- 16.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 17.Lederberg J. Genes and antibodies. Science. 1959;129:1649–1653. doi: 10.1126/science.129.3364.1649. [DOI] [PubMed] [Google Scholar]
- 18.Nossal GJ, Pike BL. Mechanisms of clonal abortion tolerogenesis. I. Response of immature hapten-specific B lymphocytes. J Exp Med. 1978;148:1161–1170. doi: 10.1084/jem.148.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marrack P, Kappler J. T cell tolerance. Semin Immunol. 1990;2:45–49. [PubMed] [Google Scholar]
- 20.Goodnow CC, Adelstein S, Basten A. The need for central and peripheral tolerance in the B cell repertoire. Science. 1990;248:1373–1379. doi: 10.1126/science.2356469. [DOI] [PubMed] [Google Scholar]
- 21.Le Douarin N, Corbel C, Bandeira A, et al. Evidence for a thymus-dependent form of tolerance that is not based on elimination or anergy of reactive T cells. Immunol Rev. 1996;149:35–53. doi: 10.1111/j.1600-065X.1996.tb00898.x. [DOI] [PubMed] [Google Scholar]
- 22.Foley EJ. Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res. 1953;13:835–837. [PubMed] [Google Scholar]
- 23.Nathrath WB. Organ and tumour antigens in malignant disease: a review. J R Soc Med. 1978;71:755–761. doi: 10.1177/014107687807101010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hargraves MM. Discovery of the LE cell and its morphology. Mayo Clin Proc. 1969;44:579–599. [PubMed] [Google Scholar]
- 25.DeLeo AB, Jay G, Appella E, et al. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci USA. 1979;76:2420–2424. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boon T, Cerottini JC, Van den Eynde B, et al. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 27.Salmi M, Hellman J, Jalkanen S. The role of two distinct endothelial molecules, vascular adhesion protein-1 and peripheral lymph node addressin, in the binding of lymphocyte subsets to human lymph nodes. J Immunol. 1998;160:5629–5636. [PubMed] [Google Scholar]
- 28.Steinman RM. Lasker basic medical research award. Dendritic cells: versatile controllers of the immune system. Nat Med. 2007;13:1155–1159. doi: 10.1038/nm1643. [DOI] [PubMed] [Google Scholar]
- 29.Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39:806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasilevko V, Ghochikyan A, Holterman MJ, et al. CD80 (B7-1) and CD86 (B7-2) are functionally equivalent in the initiation and maintenance of CD4+ T-cell proliferation after activation with suboptimal doses of PHA. DNA Cell Biol. 2002;21:137–149. doi: 10.1089/10445490252925404. [DOI] [PubMed] [Google Scholar]
- 31.Sprent J. T and B memory cells. Cell. 1994;76:315–322. doi: 10.1016/0092-8674(94)90338-7. [DOI] [PubMed] [Google Scholar]
- 32.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sprent J, Kishimoto H. The thymus and central tolerance. Philos Trans R Soc Lond B Biol Sci. 2001;356:609–616. doi: 10.1098/rstb.2001.0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 35.Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- 36.Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328:267–270. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- 37.van der Merwe PA, Bodian DL, Daenke S, et al. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185:393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res. 2011;17:6958–6962. doi: 10.1158/1078-0432.CCR-11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 40.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 41.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Elsas A, Sutmuller RP, Hurwitz AA, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med. 2001;194:481–489. doi: 10.1084/jem.194.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lute KD, May KF, Jr, Lu P, et al. Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti-CTLA-4 antibodies. Blood. 2005;106:3127–3133. doi: 10.1182/blood-2005-06-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keler T, Halk E, Vitale L, et al. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques. J Immunol. 2003;171:6251–6259. doi: 10.4049/jimmunol.171.11.6251. [DOI] [PubMed] [Google Scholar]
- 45.Ghiotto M, Gauthier L, Serriari N, et al. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int Immunol. 2010;22:651–660. doi: 10.1093/intimm/dxq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 47.Dong H, Zhu G, Tamada K, et al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 48.Ishida Y, Agata Y, Shibahara K, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agata Y, Kawasaki A, Nishimura H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 50.Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol. 2011;350:17–37. doi: 10.1007/82_2010_116. [DOI] [PubMed] [Google Scholar]
- 52.Sheppard KA, Fitz LJ, Lee JM, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 53.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 54.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm0902-1039c. [DOI] [PubMed] [Google Scholar]
- 55.Niezgoda A, Niezgoda P, Czajkowski R. Novel approaches to treatment of advanced melanoma: a review on targeted therapy and immunotherapy. Biomed Res Int. 2015;2015:851387. doi: 10.1155/2015/851387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keating GM. Nivolumab: a review in advanced nonsquamous non-small cell lung cancer. Drugs. 2016;76:969–978. doi: 10.1007/s40265-016-0589-9. [DOI] [PubMed] [Google Scholar]
- 58.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 60.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–631. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nummer D, Suri-Payer E, Schmitz-Winnenthal H, et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst. 2007;99:1188–1199. doi: 10.1093/jnci/djm064. [DOI] [PubMed] [Google Scholar]
- 62.Sainz B, Jr, Carron E, Vallespinos M, et al. Cancer stem cells and macrophages: implications in tumor biology and therapeutic strategies. Mediat Inflamm. 2016;2016:9012369. doi: 10.1155/2016/9012369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bui JD, Uppaluri R, Hsieh CS, et al. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res. 2006;66:7301–7309. doi: 10.1158/0008-5472.CAN-06-0556. [DOI] [PubMed] [Google Scholar]
- 64.Diller ML, Kudchadkar RR, Delman KA, et al. Balancing inflammation: the link between Th17 and regulatory T cells. Mediat Inflamm. 2016;2016:6309219. doi: 10.1155/2016/6309219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaw TJ, Martin P. Wound repair: a showcase for cell plasticity and migration. Curr Opin Cell Biol. 2016;42:29–37. doi: 10.1016/j.ceb.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 66.DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. 2016;16:149–163. doi: 10.1038/nri.2015.18. [DOI] [PubMed] [Google Scholar]
- 67.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mantovani A, Cassatella MA, Costantini C, et al. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 71.Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol. 2007;37(Suppl 1):S53–S60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 72.Fujii S, Liu K, Smith C, et al. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199:1607–1618. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol. 2010;108:111–165. doi: 10.1016/B978-0-12-380995-7.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee JI, Ganster RW, Geller DA, et al. Cyclosporine A inhibits the expression of costimulatory molecules on in vitro-generated dendritic cells: association with reduced nuclear translocation of nuclear factor kappa B. Transplantation. 1999;68:1255–1263. doi: 10.1097/00007890-199911150-00007. [DOI] [PubMed] [Google Scholar]
- 75.Penna G, Adorini L. 1 alpha, 25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000;164:2405–2411. doi: 10.4049/jimmunol.164.5.2405. [DOI] [PubMed] [Google Scholar]
- 76.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 77.Kidd JF, Pilkington MF, Schell MJ, et al. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem. 2002;277:6504–6510. doi: 10.1074/jbc.M106802200. [DOI] [PubMed] [Google Scholar]
- 78.Sica A, Schioppa T, Mantovani A, et al. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 79.Rath M, Muller I, Kropf P, et al. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Golpon HA, Fadok VA, Taraseviciene-Stewart L, et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. 2004;18:1716–1718. doi: 10.1096/fj.04-1853fje. [DOI] [PubMed] [Google Scholar]
- 81.Zhao Y, Zhou FL, Li WP, et al. Slit2Robo1 signaling promotes the adhesion, invasion and migration of tongue carcinoma cells via upregulating matrix metalloproteinases 2 and 9, and downregulating Ecadherin. Mol Med Rep. 2016;14:1901–1906. doi: 10.3892/mmr.2016.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jakubowska K, Pryczynicz A, Januszewska J, et al. Expressions of matrix metalloproteinases 2, 7, and 9 in carcinogenesis of pancreatic ductal adenocarcinoma. Dis Markers. 2016;2016:9895721. doi: 10.1155/2016/9895721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huber S, Hoffmann R, Muskens F, et al. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood. 2010;116:3311–3320. doi: 10.1182/blood-2010-02-271981. [DOI] [PubMed] [Google Scholar]
- 84.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 85.Geissler K, Fornara P, Lautenschlager C, et al. Immune signature of tumor infiltrating immune cells in renal cancer. Oncoimmunology. 2015;4:e985082. doi: 10.4161/2162402X.2014.985082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li X, Kostareli E, Suffner J, et al. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur J Immunol. 2010;40:3325–3335. doi: 10.1002/eji.201041093. [DOI] [PubMed] [Google Scholar]
- 88.Bonavita E, Galdiero MR, Jaillon S, et al. Phagocytes as corrupted policemen in cancer-related inflammation. Adv Cancer Res. 2015;128:141–171. doi: 10.1016/bs.acr.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 89.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 90.Beaman KD, Jaiswal MK, Katara GK, et al. Pregnancy is a model for tumors, not transplantation. Am J Reprod Immunol. 2016;76:3–7. doi: 10.1111/aji.12524. [DOI] [PubMed] [Google Scholar]
- 91.Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641–648. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 92.Harris CC. The carcinogenicity of anticancer drugs: a hazard in man. Cancer. 1976;37:1014–1023. doi: 10.1002/1097-0142(197602)37:2+<1014::AID-CNCR2820370805>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 93.Iyer SS, Pulskens WP, Sadler JJ, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 95.Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Okada M, Adachi S, Imai T, et al. A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood. 2004;103:2299–2307. doi: 10.1182/blood-2003-05-1605. [DOI] [PubMed] [Google Scholar]
- 97.Campisi L, Cummings RJ, Blander JM. Death-defining immune responses after apoptosis. Am J Transplant. 2014;14:1488–1498. doi: 10.1111/ajt.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferguson TA, Herndon J, Elzey B, et al. Uptake of apoptotic antigen-coupled cells by lymphoid dendritic cells and cross-priming of CD8(+) T cells produce active immune unresponsiveness. J Immunol. 2002;168:5589–5595. doi: 10.4049/jimmunol.168.11.5589. [DOI] [PubMed] [Google Scholar]
- 99.Griffith TS, Kazama H, VanOosten RL, et al. Apoptotic cells induce tolerance by generating helpless CD8+ T cells that produce TRAIL. J Immunol. 2007;178:2679–2687. doi: 10.4049/jimmunol.178.5.2679. [DOI] [PubMed] [Google Scholar]
- 100.Tomimori Y, Ikawa Y, Oyaizu N. Ultraviolet-irradiated apoptotic lymphocytes produce interleukin-10 by themselves. Immunol Lett. 2000;71:49–54. doi: 10.1016/S0165-2478(99)00163-7. [DOI] [PubMed] [Google Scholar]
- 101.Weigert A, Tzieply N, von Knethen A, et al. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell. 2007;18:3810–3819. doi: 10.1091/mbc.E06-12-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Williams CA, Harry RA, McLeod JD. Apoptotic cells induce dendritic cell-mediated suppression via interferon-gamma-induced IDO. Immunology. 2008;124:89–101. doi: 10.1111/j.1365-2567.2007.02743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakahashi-Oda C, Udayanga KG, Nakamura Y, et al. Apoptotic epithelial cells control the abundance of Treg cells at barrier surfaces. Nat Immunol. 2016;17:441–450. doi: 10.1038/ni.3345. [DOI] [PubMed] [Google Scholar]
- 104.Kleinclauss F, Perruche S, Masson E, et al. Intravenous apoptotic spleen cell infusion induces a TGF-beta-dependent regulatory T-cell expansion. Cell Death Differ. 2006;13:41–52. doi: 10.1038/sj.cdd.4401699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu C, Zhang Y, Jiang Y, et al. Apoptotic cell administration enhances pancreatic islet engraftment by induction of regulatory T cells and tolerogenic dendritic cells. Cell Mol Immunol. 2013;10:393–402. doi: 10.1038/cmi.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]