

Abstract
Recent safety issues involving non-active implantable medical devices (NAIMDs) have highlighted the need for better pre-market and post-market evaluation. Some stakeholders have argued that certain features of medicine safety evaluation should also be applied to medical devices. Our objectives were to compare the current processes and methodologies for the assessment of NAIMD safety profiles with those for medicines, identify potential gaps, and make recommendations for the adoption of new methodologies for the ongoing benefit–risk monitoring of these devices throughout their entire life cycle. A literature review served to examine the current tools for the safety evaluation of NAIMDs and those for medicines. We searched MEDLINE using these two categories. We supplemented this search with Google searches using the same key terms used in the MEDLINE search. Using a comparative approach, we summarized the new product design, development cycle (preclinical and clinical phases), and post-market phases for NAIMDs and drugs. We also evaluated and compared the respective processes to integrate and assess safety data during the life cycle of the products, including signal detection, signal management, and subsequent potential regulatory actions. The search identified a gap in NAIMD safety signal generation: no global program exists that collects and analyzes adverse events and product quality issues. Data sources in real-world settings, such as electronic health records, need to be effectively identified and explored as additional sources of safety information, particularly in some areas such as the EU and USA where there are plans to implement the unique device identifier (UDI). The UDI and other initiatives will enable more robust follow-up and assessment of long-term patient outcomes. The safety evaluation system for NAIMDs differs in many ways from those for drugs, but both systems face analogous challenges with respect to monitoring real-world usage. Certain features of the drug safety evaluation process could, if adopted and adapted for NAIMDs, lead to better and more systematic evaluations of the latter.
Key Points
| The collection of safety information and its integration into the risk management process for medical devices is not consistent. |
| Collaboration between all stakeholders is needed to develop a more proactive safety evaluation process. |
| This new process should incorporate real-world data to develop a risk assessment model that is suitable for all medical devices. |
Introduction
Medical devices play an increasingly important role in healthcare worldwide. A medical device is defined as “any instrument, apparatus, appliance, material or other article, whether used alone or in combination, including the software necessary for its proper application intended by the manufacturer to be used for human beings for the purpose of diagnosis, prevention, monitoring, treatment or alleviation of disease, replacement or modification of the anatomy or of a physiological process, and control of conception”. Unlike a drug, a medical device does not achieve its principal intended action in the human body by pharmacological, immunological, or metabolic means, but it may be assisted in its function by such means [1]. Medical devices are classified into different categories depending on the risk of harm that comes with their use. The dimension, complexity, and function of medical devices vary significantly, ranging from bandages, disposable gloves, and wheelchairs to more complicated forms such as active implants (i.e., pacemakers) and computerized systems used in cataract surgery.
This paper focuses on non-active implantable medical devices (NAIMDs) and uses these as a proxy to extrapolate the conclusions of the research, where possible, to other types of medical devices. An implantable medical device is one that is partly or totally inserted into the human body or a natural orifice and expected to stay there for 30 days or more or that is used to replace an epithelial surface or the surface of the eye and is expected to stay in use for 30 days or more. Examples of implantable medical devices include dental implants, breast implants, hip implants, or intraocular lenses. Both insertion or application and removal of implantable medical devices requires surgical or medical procedures. To be classified as an NAIMD, the medical device must not have an integral power source [2]. All NAIMDs fall into the European medical device risk class IIb and III.
Recent concerns involving NAIMDs, such as the Poly Implant Prothèse (PIP) breast implant [3] and the metal-on-metal hip implant, have shown many questions remain about the safety and effectiveness of NAIMDs after market approval, thus highlighting the need for better post-market monitoring. When compared with medicines, medical devices pose unique challenges in terms of ensuring their safe and effective use. Such challenges include user variability and user learning curves and the technological complexity or permanent nature of some implants. This latter challenge is mainly a potential problem with regard to safety and less with regard to effectiveness. In fact, the effectiveness of implantable devices is usually higher than that of drugs because implantable devices, in contrast to drugs, do not suffer from patient non-adherence.
To address this need for improvement, some stakeholders have argued that certain features of drug regulation should be applied to medical devices. This entails the recommendation or opinion that NAIMDs should undergo an assessment of their benefit–risk profile prior to being placed in the market—as well as continuous safety surveillance monitoring throughout the product life cycle. However, adoption of the medicinal product benefit–risk evaluation framework in its entirety may be difficult because of the significant differences between medicines and NAIMDs.
For all the reasons mentioned above, worldwide medical device regulations are undergoing ongoing changes geared towards improving pre-market and post-market evaluations of device safety.
In this paper, we compare the processes and methodologies used in the assessment of the safety profile of medical devices with those for medicines to identify potential gaps and make recommendations for the adoption of new approaches and methodologies in the medical device context. To make this comparison more practical, we used a specific group—NAIMDs, rather than the entire spectrum of medical devices—as an example.
Literature Review Methodology
A literature review served to examine the current tools for the safety evaluation of NAIMDs and medicines. We searched MEDLINE using these two categories. We supplemented this search with Google searches using the same key terms used in the MEDLINE search.
Safety Evaluation: A Life Cycle Approach
For both medicines and NAIMDs, the goal is to evaluate safety throughout the entire life cycle of the product. The way this is done differs substantially between the two. To understand the differences between the safety evaluations, it is important to focus on the different types of adverse events and to evaluate the factors contributing to these adverse events. The differences and similarities between medicines and NAIMDs are shown in Fig. 1.
Fig. 1.

Factors contributing to adverse events: medicines and non-active implantable medical devices (NAIMDs)
The design of a NAIMD plays a key role when evaluating possible errors that may arise while using the product. A study involving healthcare employees from three hospital systems indicated that a lack of training is associated with most errors. To minimize such errors, user training should focus on more effective error-prevention strategies such as retraining of the user during the NAIMD label review and double checks during critical steps of NAIMD implantation [4]. The three main causes of adverse events have been found to be user challenges, design problems, and lack of effective training; many of these could be minimized with adequate training and more user-friendly medical devices.
For medicines, the factors contributing to adverse events are mostly pharmacological effect, medication errors, drug interactions, and incorrect dosing.
These differences in factors contributing to adverse events influence the safety assessment performed during development and the post-market phase of a new product. Some metabolic conditions could contribute to adverse events for special populations for medicines. For NAIMDs, other characteristics such as anatomical differences—particularly in size—need to be considered.
New Product Development
The pre-market safety assessment for both NAIMDs and medicines is a process that comprises in-depth planning, evaluation, and reporting throughout the development of the product: from discovery and development to preclinical and clinical testing.
Discovery
The new product development pathway starts with the discovery phase. For both medicines and NAIMDs, the time dedicated to new concept and ideation depends on the level of breakthrough technology of the product, which can be a very lengthy and complex process.
As an example of breakthrough technology for NAIMDs, Sir Harold Ridley, inspired by one of his interns, developed the idea of implanting an intraocular lens (IOL) and, on 29 November 1949, was the first to successfully do so [5]. During World War II, Sir Ridley had treated pilots who had fragments of shattered cockpit in their eyes. He learned that the acrylic plastic material of the cockpit did not lead to long-term damage of the eye. Therefore, he chose the acrylic plastic material for his new invention, the first IOL. Soon after Sir Ridley’s discovery, several manufacturers rapidly used his new idea to produce similar IOLs with improved characteristics.
In 1964, Sir James W. Black developed the first clinically important beta blocker, propranolol, revolutionizing the medical treatment of angina pectoris [6]. Beta blockers have been a key contribution to clinical medicine and pharmacology in the twentieth century. Following Sir Black’s breakthrough, other beta blockers were developed as medicines.
Another type of product innovation, also through breakthrough technology, is the modification of the structure of existing products. This innovating process is more rapid for medical devices than for medicines. In general, modifying the molecular structure of an existing medicinal product to obtain a new medicinal product involves a long and protracted process, whereas the incremental changes made to medical devices can be released to the market much faster. A new medicinal product will have to undergo mandatory preclinical and clinical trials prior to approval and market authorization, whereas this is not always required to place a device in the market.
Development
The NAIMD pathway starts with the creation of a new product. Once the NAIMD has been ideated, the new prototype enters the iterative development cycle where continuous amendments and incremental design improvements will be made based on feedback from physicians/users, technology developments, preclinical testing, manufacturing improvements, and clinical studies. After such feedback, new ideas are transformed into prototypes, which are again tested, re-done, optimized, and then finalized.
In contrast, during the discovery of new medicinal products, many compounds are generated with the objective of detecting the best candidates for further development. The candidate drugs are frequently selected using in vitro testing models and enter formulation development in a continuous and unidirectional process.
Preclinical
After the ideation and development phases, the preclinical testing starts. For medicines, candidates not excluded in the initial steps are tested for efficacy and safety in animals. These animal studies are planned to ascertain a safe dose with which to start studies in humans, to learn which organs may be more affected by potential toxic effects, and to understand pharmacokinetic and dynamic parameters.
Manufacturers of NAIMDs and medicines are required to test the safety of the new products via ex vivo and in vivo studies. The role of animal testing for NAIMDs differs significantly from that of medicines. Contrary to the process with medicines, where all new products require organ-specific animal models, the majority of new NAIMDs do not require animal testing because they often use materials that are biocompatible with human tissue, such as stainless steel or ceramic. However, some devices with novel materials (i.e., materials that have not previously been used in a marketed medical device with the same type and duration of contact) might require biocompatibility testing in animals [7].
Clinical
Although a large amount of information is obtained from animal testing, this is not sufficient to rule out human trials. No animal or in vitro testing is sufficiently comparable to that in humans; human trials are inevitably required for medicines. For NAIMDs, clinical trials are used only in certain circumstances, for example when the biocompatibility and safety of NAIMDs cannot be assured during preclinical trials (both ex vivo and in vivo) (Fig. 2). This is considered on a product-by-product basis and depends on the NAIMD materials, components, clinical procedures, characteristics of the anatomical site for implantation of the NAIMD, or target populations.
Fig. 2.

Overview of the main differences during new product development between non-active implantable medical devices (NAIMDs) and medicines
Therefore, for some NAIMDs, unlike for medicines, extrapolating clinical data from published clinical investigations or other studies of similar devices in the scientific literature, or from clinical experience of a similar device may be sufficient to obtain approval to market at least in most countries. For NAIMDs that require clinical studies to obtain regulatory approval, the studies are usually smaller (average number of patients: <500) than pharmaceutical clinical trials, which are ruled by the size required to show efficacy [8]. The technical aspects of medical devices make it difficult to decide how much clinical data are required for a new NAIMD: substantially equivalent NAIMDs, and those with a completely new design or indication will all require different ways of evaluating NAIMD safety. For NAIMDs, the amount of clinical data required to obtain the market approval is not clearly defined.
As can be seen in Fig. 3 [9, 10], safety assessment for NAIMDs is an iterative process of detecting, assessing, managing, and communicating the benefits and potential risks while the product is not yet approved. Although the pre-market safety assessment for medicines could be iterative, it is often unidirectional rather than cyclical.
Fig. 3.
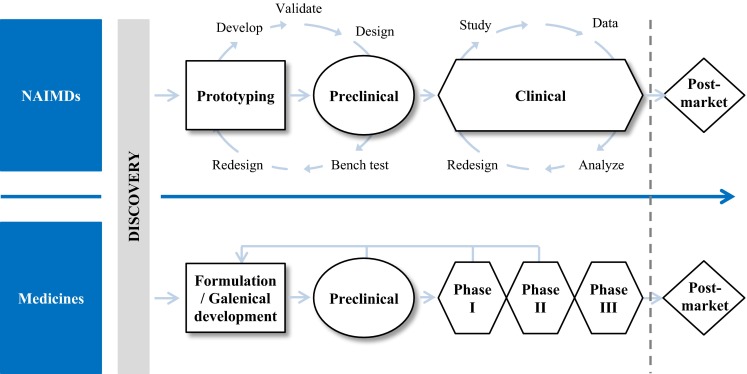
The medicinal product and the non-active implantable medical devices (NAIMDs) development pathway
Post-Market Surveillance
After the discovery, development, and preclinical and clinical testing phases are successful, the product is licensed for marketing and enters the post-market phase. Different types of data related to the actual use of the product are collected, and these post-market data are integrated into the risk-management plan (RMP) of the product. The data-collection processes and the integration of such data are similar but differ in some aspects between NAIMDs and medicines.
Importance of Data Sources
Data sources are a key aspect in the safety evaluation process. It is important to consider the different post-market surveillance data sources and their limitations. Table 1 provides examples of types of post-market data sources for both NAIMDs and medicines.
Table 1.
Examples of post-market data sources for non-active implantable medical devices and medicines
| NAIMDs | Medicines | |
|---|---|---|
| Spontaneous Reports | MAUDE (FDA), MEDSUN (FDA), MHRA (UK) | AERS (FDA), EudraVigilance (EEA), VigiBase (WHO) |
| Patient Registries | SCAAR, EUREQUO, AOANJRR | ESID, Atassia Teleangiectasia (Italy) |
| Prescription Databases | Underdeveloped | The Intensive Medicines Monitoring Programme (New Zealand), NorPD |
| Claims Data Sources | Medicare & Medicaid (USA) | FDA Sentinel, Medicare & Medicaid (USA) |
| EMR Databases | Underdeveloped | EUADR, FDA Minisentinel, General Practice Research Database (UK) |
| Public Information on Safety Issues | Medical Device Safety (FDA), Catalan Agency for Health Technology Assessment and Research | CDER (FDA), PRAC (EMA) |
| Post-Authorization Studies | Post-market clinical follow-up studies (EU), post-approval studies (USA), 522 Studies (USA) | Interventional study (efficacy study, PASS, PAS) and non-interventional study (efficacy study, PASS, PAS) |
AERS Adverse Event Reporting System, AOANJRR Australian Orthopaedic Association National Joint Replacement Registry, CDER Center for Drug Evaluation and Research, EEA European Economic Area, EMA European Medicines Agency, EMR electronic medical records, ESID European Society of Immunodeficiencies, EUADR Exploring and Understanding Adverse Drug Reactions, EUREQUO European Registry of Quality Outcomes for Cataract and Refractive Surgery, FDA US Food and Drug Administration, MAUDE Manufacturer and User Facility Device Experience, MedSun Medical Product Safety Network, MHRA Medicines and Healthcare products Regulatory Agency, NAIMD non-active implantable medical devices, NorPD Norwegian Prescription Database, PAS post-authorization study, PASS post-authorization safety study, PRAC Pharmacovigilance Risk Assessment Committee, SCAAR Swedish Coronary Angiography and Angioplasty Registry, WHO World Health Organization
Many data sources are common to both products, but some have been explored more in one than in the other. For both NAIMDs and medicines, passive post-market data sources are easily accessible and well established. On the other hand, active data sources are further advanced for medicines than for NAIMDs.
The main differences between NAIMDs and medicines is that prescription or pharmacy dispensing and electronic medical records (EMRs) or claims databases for NAIMDs are underdeveloped. This is primarily because of the lack of a unique device identifier (UDI). UDIs will enhance post-market surveillance activities by providing a standard and unambiguous way to document device use in EMRs or healthcare utilization databases.
The integration of the UDI into such databases could potentially support public health-related activities such as reducing use errors and the reporting and assessing of adverse events and other problems related to the NAIMD. It would also enable tracking of product withdrawals, assessment of patient outcomes and risk–benefit profiles of NAIMDs across different populations, as well as provide a viable source of device identification information to the various stakeholders.
Integrating UDI information into such databases will increase the use of ‘real-world’ data in the decision-making process. The US FDA has indicated that establishing a medical device safety evaluation system to gain real-world evidence is one of its strategic priorities for 2016–2017. In Europe, on 25 May 2016, an agreement was reached with the European parliament representatives, and the UDI will become reality in the near future [11]. This evidence will then aid in the regulatory decision-making process. The new system aims to lead to a better and faster identification of safety signals by collecting post-market data in a timely manner. Today’s vast amount of electronic clinical data will be used to determine safety signals and support risk–benefit analysis when the quality of data can be guaranteed and advanced analytics can be applied [12].
Risk Management
In the life cycle approach, new safety data need to be included in the RMPs. Again, the general processes are very similar for medicines and NAIMDs, but some differences exist that should be considered (Table 2).
Table 2.
Risk management processes for non-active implantable medical devices and medicines
| NAIMDs | Medicines | |
|---|---|---|
| AE Coding | Product problem: FDA and ISO Patient Outcome: SNOMED, MedDRA®, ICD, FDA |
MedDRA® |
| Signal management process | Signal detection, signal validation, signal prioritization, signal escalation, regulatory actions and connections with other processes not as clearly regulated | Signal detection, signal validation, signal prioritization, signal escalation, regulatory actions clearly regulated |
| Benefit–risk analysis | Underdeveloped, RMF | PSURs/PBRERs, RMPs |
| Regulatory actions | Withdrawal, Recall, Restriction, Ban, DFU update, Dear Doctor Letter (USA), Field Safety Notice (EU) | Withdrawal, SmPC update, black box warning, Dear Doctor Letter |
AE adverse event, DFU directions for use, FDA US Food and Drug Administration, ICD International Classification of Diseases, ISO International Organization for Standardization, MedDRA Medical Dictionary for Regulatory Activities, PBRER periodic benefit–risk evaluation reports, PSUR periodic safety update reports, RMF risk management file, RMP risk management plan, SmPC Summary of Product Characteristics, SNOMED Systematized Nomenclature of Medicine
Adverse Event Coding
Worldwide, the accepted adverse event coding for medicines is that of the Medical Dictionary for Regulatory Activities (MedDRA®). This is the result of exhaustive work by many stakeholders and a comprehensive maintenance system by a private company (Maintenance and Support Services Organization [MSSO]) in charge of ensuring codes reflect changes and innovation (biologicals and other new products require constant additions to and refining of the dictionary).
The coding system for adverse events and product problems for NAIMDs is more heterogeneous than that for drugs. Different standardized nomenclatures exist for product problems (FDA codes and International Organization for Standardization [ISO] codes) and for patient outcomes (Systematized Nomenclature of Medicine-Clinical Terms [SNOMED CT], MedDRA®, International Classification of Diseases [ICD], and FDA Patient Problem Codes). As seen in Table 3 [13–16], the standardized nomenclature systems vary significantly with regards to number of terms, granularity, hierarchy, and availability in different languages.
Table 3.
Standardized nomenclature for describing patient outcomes
| MedDRA® | SNOMED CT | ICD | US FDA patient problem codes | |
|---|---|---|---|---|
| Number of terms | 70,000 | 311,000 | 70,000 | 700 |
| Hierarchy | High | Medium | Medium | Low |
| Number of languages available | 11 | 5 | 42 | 1 |
| Owner | International Conference on Harmonization | International Health Terminology Standards | World Health Organization | US Food and Drug Administration |
ICD International Classification of Diseases, MedDRA Medical Dictionary for Regulatory Activities, SNOMED CT Systematized Nomenclature of Medicine Clinical Terms
Signal Management Process
The aim of signal detection for both medicines and NAIMDs is to promptly identify risks associated with the use of a product [17]. Decisions as to whether a finding represents a ‘safety signal’ and whether it warrants further investigation can be challenging.
Quantitative signal detection is followed by a signal validation process that confirms whether or not the signal is real (Fig. 4). This is often verified through qualitative analysis of case evaluation. Thereafter, the signal is prioritized on the basis of the strength of the signal, whether or not the signal represents a new finding, the clinical importance and potential public health implications of the issue, and the potential for preventive measures to mitigate the adverse public health impact.
Fig. 4.

Signal management process for both non-active implantable medical devices (NAIMDs) and medicines
After signal prioritization, the manufacturer or marketing authorization holder decides whether or not the signal must be escalated and whether or not any regulatory actions should be taken as risk minimization measures to address the safety issue [17].
Although the signal management process is the same, the legislation requirements are better described in pharmaceutical regulations [18–20]. This is not the case for NAIMDs; guidelines giving practical advice on signal management are yet to be developed. Drug regulations were developed earlier than medical device regulations, which explains and results in the poor description of legislation requirements for medical devices.
For medicines, new pharmacovigilance regulations in the EU have highlighted the relevance of signal management, and the European Medicines Agency’s recently established Pharmacovigilance Risk Assessment Committee (PRAC) has been instructed to supervise all aspects of the use of medicines, including signal management and prioritization [21]. Numerous worldwide initiatives are investigating new methods to facilitate earlier signal detection, mainly through mining of routinely collected data from electronic healthcare records (EHRs) [22].
Post-Market Benefit–Risk Analysis
Post-market benefit–risk analysis can be defined as a comparative assessment of benefits (positive effects) and risks (potential harms) of a particular product (medicinal product or medical device) after it has been introduced to the market. This is an iterative and dynamic process comprising four phases (Fig. 5). In the first stage, the benefits and risks should be defined. Thereafter, activities aimed at benefit optimization and risk mitigation or minimization should be outlined. During the third stage, the product should be assessed on the basis of its effectiveness and safety throughout its entire life cycle. In the fourth stage, the RMP should be revised if the benefit–risk profile of the product has changed.
Fig. 5.

Post-market benefit-risk analysis for both non-active implantable medical devices (NAIMDs) and medicines. RMP risk management plan
The process is the same for both NAIMDs and medicines. However, the requirements for a benefit–risk analysis framework are more defined for medicines because of the more developed legislation. For medicines, there is a regulatory requirement to submit periodic safety update reports (PSURs)/periodic benefit–risk evaluation reports (PBRERs). PSURs/PBRERs are pharmacovigilance documents intended to provide an evaluation of the benefit–risk balance of a medicinal product. These reports are submitted by marketing authorization holders at defined time points during the post-authorization phase [23]. PSURs/PBRERS are not currently required for NAIMDs.
In terms of risk-management document submission, there is a regulatory requirement for both medicines and NAIMDs: updated RMPs for medicines [24] and updated risk-management files (RMFs) for NAIMDs [25].
Regulatory Actions
Regulatory actions are well defined for both types of products. However, what constitutes a regulatory action differs, often by country or region, and there is no harmonization across jurisdictions. Moreover, the regulatory approval process for medical devices also differs widely across jurisdictions. For example, in the USA, the FDA approves the marketing of a new medical device and has tools to restrict the use of or ban a device and remove unsafe products from the market [17]. Conversely, in Europe, the pre-market evaluation of a device is performed by the notified body, which includes the assessment and verification of the clinical evaluation. Once medical devices bear the CE marking, they can circulate freely within the EU. In the post-market environment, it is sometimes difficult for the EU Member States to stop production, CE labelling, or distribution of medical devices [26]. Medical devices marketed first in the EU have a higher risk of post-marketing safety issues than medical devices first marketed in the USA [27].
Discussion
Role of the Patient
The patient needs to be aware of potential risks and able to easily communicate their personal experience relating to the safety and effectiveness of the device. Patient associations should be involved in defining the new regulations and guidelines for safety evaluation systems for medical devices.
Some initiatives have already been undertaken to try to develop a systematic methodology to calculate and include patient information into the medical device safety evaluation system [28] and encourage patient engagement [29]. The goal should be to obtain a more patient-centric system. The patient should be a key stakeholder in public health.
Recommendations
The basic systems for safety evaluation of medical devices and medicines are not very different from a conceptual perspective; however, gaps currently exist in the safety evaluation of medical devices. This paper has identified these gaps, and some recommendations on how to fill these gaps follow.
As seen in Fig. 6, the recommendations are ordered in three categories: harmonization and centralization, safety evaluation tools, and user training and customer service.
Fig. 6.
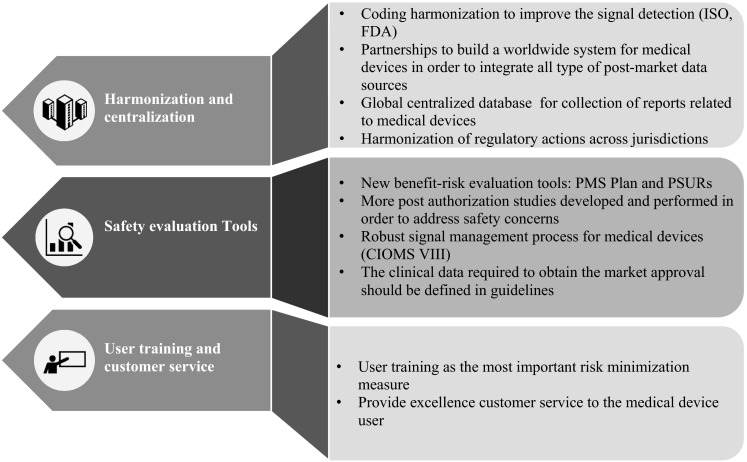
Recommendations to cover gaps in the safety evaluation of medical devices. CIOMS Council for International Organizations of Medical Sciences, FDA US Food and Drug Administration, ISO International Organization for Standardization, PMS post-market surveillance, PSUR periodic safety update reports
Harmonization and Centralization
Adverse event coding should be harmonized to improve the signal detection process. It is recommended that a global and centralized database, such as the World Health Organization (WHO) Vigibase, be established for the assembly of all medical device reports.
Moreover, a worldwide evaluation system should be developed for medical devices and should include representatives from the different stakeholders [12]. This system also does not yet exist for medicines and could use real-world evidence to support regulatory decision making. To ensure successful implementation of this system, three steps need to be incorporated: (1) the UDIs need to be consistently assembled within electronic health information, (2) all stakeholders need to ensure a continual use of the EHRs, including UDIs, and lastly (3) to link patient data, all data sources need to have interoperable linking capabilities [30]. This is a long-term goal because it involves policy change. Therefore, these three steps could take years or even decades.
For these harmonization and centralization recommendations to succeed, there must be active collaboration and support from all stakeholders.
Further to the recommendations listed above, there must be regulatory methodology harmonization: the regulatory approval process and the definitions of regulatory actions need to be aligned across jurisdictions to enable a more robust signal management process.
Safety Evaluation Tools
Post-market surveillance data are very important for medical devices because they provide valuable information regarding user variability. Relevant authorities could make more safety evaluation tools available to the different stakeholders to improve safety assessment:
Regulatory documents providing further guidance on the different steps in the signal management process. For instance, the following signal detection guidance has already been established for medicines: the report of the Council for International Organizations of Medical Sciences (CIOMS) Working Group VIII and the guidelines on good pharmacovigilance practices (GVP) module IX—signal management [19, 20].
A post-market surveillance (PMS) plan should be submitted for every medical device or group of medical devices to serve as a summary of all collected post-market information and as a guide to utilizing such information [2].
A post-market clinical follow-up study plan should also be part of the PMS plan [2].
PSURs for every medical device or group of medical devices to reinforce the benefit–risk analysis process.
The clinical data required (from both a quantitative and a qualitative perspective) to obtain the market approval should be defined in guidelines and should be consistent with the risk associated with the product and/or how innovative the device is. Medical devices with a high level of innovation (new material, new product, new surgical procedure) and/or a high level of risk should require more clinical data. For these types of products, more evidence should be generated during the pre-market phase to better define expected risks. Single-blind randomized controlled trials (RCTs) with these types of implantable medical devices should be conducted when required and if possible. Previous experience with similar devices should also be considered as evidence when deciding the clinical data required.
User Training and Customer Service
As previously stated (Fig. 1), user error and the user’s learning curve are two vital factors contributing to adverse events with medical devices. Manufacturers should primarily focus on user training as the most important risk minimization tool. Proper training should be provided to reduce user variability. Providing excellent customer service to the medical device user will supply the manufacturer with substantial knowledge about the medical device safety profile. To guarantee outstanding customer service, training standards need to be implemented to ensure consistent quality. Moreover, all involved staff members need to be trained on these standards and briefed about the company’s vision on customer service. To measure the efforts of staff and to evaluate the success of the training, a framework should be designed to measure quality and its consistency. Lastly, the company can only learn and grow from opinions and feedback from their customers. This should be received through customer surveys, which should be shared with the team and made public to all stakeholders [12].
Conclusions
Traditionally, the collection of safety information and its integration into the risk management process of medical devices has been neither consistent nor performed for all products.
To address this weakness, health authorities have started to work on new regulatory documents. Patients must be the key pillars and public health the cornerstone of this new system. Now is the time for collaboration between all stakeholders to develop a more proactive safety evaluation process. This new process should incorporate real-world data to develop a risk assessment model that is suitable for all medical devices.
Compliance with Ethical Standards
Funding
No sources of funding were used to assist in the preparation of this study.
Conflict of interest
Josep Pane and Irene Rebollo are employees of Alcon, Novartis which manufactures NAIMDs. Preciosa Coloma, Katia Verhamme and Miriam Sturkenboom have no conflicts of interest that are directly relevant to the content of this study.
Footnotes
Disclaimer: The views expressed in this article are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of Alcon (Novartis) or Erasmus University Medical Center or one of its committees or working parties.
References
- 1.ISO 13485: Medical Devices. Quality Management Systems. Requirements for Regulatory Purposes. 2016.
- 2.Proposal for a Regulation of the European Parliament and of the Council on medical devices, and amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 (2016).
- 3.New York Daily News. EU to tighten medical controls after breast implant scandal. 2012. http://www.nydailynews.com/life-style/health/eu-tighten-medical-controls-pip-breast-implant-scandal-article-1.1169444. Accessed 7 Feb 2016.
- 4.Johnson TR, Tang X, Graham MJ, Brixey J, Turley JP, Zhang J, et al. Attitudes toward medical device use errors and the preventions of adverse events. Jt Comm J Qual Patient Saf. 2007;33(11):689–694. doi: 10.1016/S1553-7250(07)33079-1. [DOI] [PubMed] [Google Scholar]
- 5.Sarwar H, Modi N. Sir Harold Ridley: innovator of cataract surgery. J Perioper Pract. 2014;24(9):210–212. doi: 10.1177/175045891402400905. [DOI] [PubMed] [Google Scholar]
- 6.Stapleton MP. Sir James Black and Propranolol. The role of the basic sciences in the history of cardiovascular pharmacology. Tex Heart Inst J. 1997;24((4)):336–42. [PMC free article] [PubMed]
- 7.US Food and Drug Administration. Why are animals used for testing medical products? 2016. http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194932.htm.
- 8.Ferguson M. Medical devices are different to medicinal products in the Health Technology Assessment process. J Compar Effect Res. 2014;3(3):217–219. doi: 10.2217/cer.14.17. [DOI] [PubMed] [Google Scholar]
- 9.Coloma PM. Phase 0 clinical trials: theoretical and practical implications in oncologic drug development. Open Access J Clin Trials. 2013;5:119–126. doi: 10.2147/OAJCT.S32978. [DOI] [Google Scholar]
- 10.U.S. Food and Drug Administration. CDRH Innovation Initiative. 2011. http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHInnovation/UCM242528.pdf. Accessed 7 Feb 2016.
- 11.The European Council and Council of the European Union. Medical devices: deal reached on new EU rules. 2016 http://www.consilium.europa.eu/en/press/press-releases/2016/05/25-medical-devices/. Accessed 5 Sep 2016.
- 12.U.S. Food and Drug Administration. 2016-2017 Strategic Priorities Center for Devices and Radiological Health. 2016. http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHVisionandMission/UCM481588.pdf. Accessed 5 Sep 2016.
- 13.MedDRA. Understanding MedDRA—The Medical Dictionary for Regulatory Activities. 2013. http://www.meddra.org/sites/default/files/page/documents/meddra2013.pdf. Accessed 7 Feb 2016.
- 14.U.S. Food and Drug Administration. Coding Tools / Resource Files. 2014. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/ReportingAdverseEvents/EventProblemCodes/ucm134751.htm. Accessed 18 Sep 2016.
- 15.International Health Terminology Standards Development Organisation. SNOMED CT. The Global Language of Healthcare. 2016. http://ihtsdo.org/snomed-ct/. Accessed 7 Feb 2016.
- 16.World Health Organization. Classification of Diseases (ICD). 2016. http://www.who.int/classifications/icd/en/. Accessed 5 Sep 2016.
- 17.U.S. Food and Drug Administration. Strengthening our national system for medical device postmarket surveillance. 2012. http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM301924.pdf. Accessed 7 Feb 2016.
- 18.Commission Implementing Regulation (EU) No 520/2012 (2012).
- 19.European Medicines Agency . Guideline on good pharmacovigilance practices, Module IX: signal management. London: EMA; 2012. [Google Scholar]
- 20.CIOMS Working Group. Practical aspects of signal detection in pharmacovigilance. Geneva; 2010.
- 21.Pacurariu AC, Coloma PM, van Haren A, Genov G, Sturkenboom MC, Straus SM. A description of signals during the first 18 months of the EMA pharmacovigilance risk assessment committee. Drug Saf. 2014;37(12):1059–1066. doi: 10.1007/s40264-014-0240-1. [DOI] [PubMed] [Google Scholar]
- 22.Coloma PM, Trifirò G, Patadia V, Sturkenboom MC. Postmarketing safety surveillance: where does signal detection using electronic healthcare records fit into the big picture? Drug Saf. 2013;36(3):183–197. doi: 10.1007/s40264-013-0018-x. [DOI] [PubMed] [Google Scholar]
- 23.European Medicines Agency . Guideline on good pharmacovigilance practices, Module VII, Periodic Safety Update Report. London: EMA; 2012. [Google Scholar]
- 24.European Medicines Agency . Guideline on good pharmacovigilance practices (GVP), Module V, Risk management systems. London: EMA; 2012. [Google Scholar]
- 25.ISO 14971: Medical devices. Application of risk management to medical devices. 2012.
- 26.Belgian Health Care Knowledge Centre. Towards a guided and phased introduction of high-risk medical devices in Belgium. 2015. https://kce.fgov.be/publication/report/towards-a-guided-and-phased-introduction-of-high-risk-medical-devices-in-belgium. Accessed 5 Sep 2016.
- 27.Hwang TJ, Sokolov E, Franklin JM, Kesselheim AS. Comparison of rates of safety issues and reporting of trial outcomes for medical devices approved in the European Union and United States: cohort study. BMJ. 2016;353:I3323. doi: 10.1136/bmj.i3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.U.S. Food and Drug Administration. Patient Preference Initiative. 2016. http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHPatientEngagement/ucm462830.htm. Accessed 5 Sep 2016.
- 29.Medical Device and Diagnostic Industry. How Medical Device Makers Can Encourage Patient Engagement. 2016. http://www.mddionline.com/blog/devicetalk/how-medical-device-makers-can-encourage-patient-engagement-11-09-15. Accessed 5 Sep 2016.
- 30.Engelberg Center for Health Care Reform at Brookings. Strengthening Patient Care: Building an Effective National Medical Device Surveillance System. 2015. https://www.brookings.edu/wp-content/uploads/2015/02/mds_finalreport_2016-09-12.pdf. Accessed 7 Feb 2016.