

Abstract
Angiotesin II (Ang II) plays an important role in cardiac remodeling. Fibroblast growth factor inducible-14 (Fn14) is the smallest member of the tumor necrosis factor superfamily of receptors. Currently, little is known about the functional role of Fn14 in the heart. Chiefly, we observe the up-regulation of extracellular matrix in in vivo model. We therefore assess the expression and regulation of Fn14 in cardiomyocytes and in vivo models induced by Ang II. In order to study the regulation of Fn14, cardiac remodeling was established in rats and neonatal cardiomyocytes were used in in vitro model. As well, Ang II is able to strongly induce Fn14 expression in in vivo and in vitro models. Fn14 is mediated via RhoA pathways, since siRNA against RhoA prevented the expression of Fn14 in cardiomyocytes. Pretreatment of cardiomyoctes with siRNA against NF-κB and IκBα also decreased Fn14 expression induced by Ang II. We here describe for the first time Ang II regulation of Fn14 in in vivo and in vitro models via RhoA, NF-κB and NF-κB driven gene signaling pathway. In conclusion, Fn14 may be important in regulating the process of cardiac remodeling induced by Ang II.
Keywords: Fn14, RhoA, NF-κB, Ang II
Introduction
Cardiac remodeling is characterized by excessive deposition of extracellular matrix (ECM) proteins [1-3]. It is one of the most important pathophysiological processes involved in cardiac remodeling and can cause dysfunction of systole and diastole, resulting in heart failure and arrhythmias [4,5]. However, the underlying mechanism involved in myocardial remodeling is incompletely understood. Researches demonstrate that the inflammatory reaction plays an essential part in myocardial remodeling [3]. Angiotesin II (Ang II), one of the cytokines involved in the cardiac remodeling process, has been confirmed closely related to cardiac remodeling [6,7]. Excessively elevated Ang II results in left ventricular dysfunction and cardiomyopathy [8-10], and Ang II level has been shown to be elevated in experimental models of diabetic cardiomyopathy [8,11,12].
Fibroblast growth factor inducible-14 (Fn14) is a type I transmembrane protein of 102 amino acids in length after removal of signal peptide, making it the smallest member of the tumor necrosis factor superfamily of receptors [13-15]. First described by Winkles and co-workers in 1999, Fn14 is highly expressed in many tissues including heart, brain, kidney and liver [14,16-18]. The tumor necrosis factor receptor-associated factor site links Fn14 to the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways to regulate cardiac remodeling [19-22]. We find that Fn14 is up-regulated in cardiac remodeling models and is activated by addition of Ang II stimulation. Additionally, Fn14 expression is very low in normal heart tissue but elevated in cardiac remodeling models. Several studies have revealed that Fn14 affected the pathogenesis of cardiac remodeling, heart failure and dilated cardiomyopathoy [23,24]. However, little is known about the potential role of Fn14 and its mechanism of action in cardiac remodeling.
Cardiac remodeling is dependent on continual reorganization of the actin cytoskeleton [25,26]. Members of the Rho family of small GTPase, such as RhoA, Rac1 and Cdc42, are key mediators of cardiac remodeling [27,28]. RhoA mediates the process of cardiac remodeling. RhoA switches between an inactive guanosine diphosphate (GDP)-bound form and an active guanosine triphosphate (GTP)-bound form [29,30]. RhoA is localized to membrane, followed by its interaction with effector molecules such as ROCK to trigger downstream cellular functions [31].
Cardiomyocyte is a major cell type in the heart [32] that plays a vital role in the development of cardiac remodeling through the synthesis of the extracellular matrix (ECM) [33-35], a process that requires factors such as collagen I, collagen III, and connective tissue growth factor (CTGF), which are the marker genes of cardiac remodeling [3,31]. Cardiac remodeling can be induced by various stimuli [31,36,37], including Ang II [31,38]. Ang II acts through several signaling pathways in cells, some of which involve small GTPase, including RhoA.
So far, little was known about the potential role of Fn14 in cardiac physiology and pathology. Therefore, we hypothesized that Fn14 promotes the formation of cardiac remodeling via the RhoA/NF-κB/IκBα pathway. To test the hypothesis, we investigated the effect of Fn14 on ECM production and the mechanism induced by Ang II. In this study, we aimed to evaluate a role of RhoA in Fn14-mediated cardiac remodeling and show that Ang II can induce Fn14 expression via RhoA/NF-κB/IκBα signaling pathway. Our findings demonstrated that Fn14 promotes ECM production by activating the RhoA/NF-κB/IκBα signaling pathway. Hence, Fn14 may be important in regulating the process of cardiac remodeling.
Materials and methods
Animals
Eight-week-old Sprague-Dawley (SD) rats were purchased from Zhejiang University experimental animal center and housed in a pathogen-free laboratory at Sir Run Run Shaw Hospital, medicine of college, Zhejiang University. The study was conducted in accordance with the guidelines and requirements from the Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011) and the Institutional Animal Care and Use Committee of Zhejiang University. All experimental protocols were approved by the Ethics Committee of Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University.
Cell culture
Neonatal cardiomyocytes were prepared from the ventricles of 1-3-day-old SD rats that were obtained from Zhejiang University experimental animal center as previously described [39]. Each heart was cut into small segments and digested by 0.1% collagenase type II and 0.12% trypsin at 37°C. The digestion was performed eight times for five minutes each. The neonatal cardiomyocytes were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% (v/v) new bovine serum (NBS) (HyClone, Thermo) in an environment containing 5% CO2 for two hour. The suspended cells were collected and plated in dishes. Cardiomyocytes were treated with reagents 72 hours after seeding.
Establishment of in vitro and in vivo models
SD rats that were infused with Ang II (Sigma Chemical, St. Louis, MO) were used as in vivo model. Ang II was administered at a rate of 65 ng/min for 14 days via a subcutaneously implanted osmotic mini-pump (Alzet, model 2002; Durect Corp., Cupertino, CA) [31]. In vitro model was established that cardiomyocytes were cultured for 4 hours in DMEM-10%NBS with 1 μM Ang II.
RNA extraction and quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) from cardiomyocytes or from the left ventricles of SD rats using a standard protocol [31]. cDNA synthesis was performed with 1 μg of total RNA using the miScript II RT Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. qRT-PCR and data analysis were performed with the ABI 7500 cycler (Applied Biosystems, CA, USA). β-actin was used as the endogenous control for mRNA expression. The primers that we designed were as follows: collagen I forward, 5’-GAGCCTAACCATCTGGCATCT-3’, reverse, 5’-AGAACGAGGTAGTCTTTCAGCAAC-3’; collagen III forward, 5’-GAGCGGAGAATACTGGGTTGAT-3’, reverse, 5’-GGTATGTAATGTTCTGGGAGGC-3’; CTGF, forward, :5’-CAGGGAGTAAGGGACACGA-3’, reverse, 5’-ACAGCAGTTAGGAACCCAGAT-3’; Fn14 forward, 5’-GTGTTGGGATTCGGCTTG-3’, reverse, 5’-GCAGAAGTCGCTGTGTGGT-3’; NF-κB forward, 5’-GCGGGGCATGCGTTTCCGTT3’, reverse, 5’-GGTATCTGTGCTTCTCTCCCCAGGA-3’; β-actin, forward, 5’-TCATCACTATTGGCAACGAGC-3, reverse, 5’-AACAGTCCGCCTAGAAGCAC-3’.
Western blots
Total protein from cardiomyocytes that were cultured in 6-well plates and SD heart tissue were extracted in a RIPA lysis buffer (Beyotime, Shanghai, China), which was supplemented with 1 mM PMSF [31]. Protein concentrations were determined using a BCA assay kit (Beyotime, Shanghai, China). Equal amounts of protein (20 μg) were separated on 10% or 12% (for RhoA and Rac1 analysis) sodium dodecyl sulphate polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). The membranes were blocked with 5% non-fat milk-TBST and incubated overnight with primary antibodies at 4°C, followed by 1 hour of incubation with horseradish peroxidase-conjugated secondary antibodies at room temperature. The bands were visualized with an enhanced chemiluminescence reagent (Amersham, Haemek, Israel) on a LAS-4000 image reader system (Fujifilm, Tokyo, Japan). To ensure equal protein loading, the β-actin protein was used as the endogenous control.
The anti-collagen I, anti-collagen III, anti-CTGF, anti-NF-κB and anti-Fn14 antibodies were purchased from Abcam Public Limited Company (Abcam, Cambridge, UK). The anti-β-actin antibody was purchased from Sigma Company (Chemical, St. Louis, MO).
Small interfering RNA (siRNA) against RhoA and Fn14
The siRNA against Fn14 and RhoA was designed and synthesised by GenePharma Co. (Shanghai, China), and a negative control was designed with a randomly chosen nonsense sequence. The effective siRhoA sequence was as followed: sense, 5’-AUCCUAGUUGGGAACAAGATT-3’; antisense, 5’-UCUUGUCCCAACUAGGAUTT-3’. The effective siFn14 sequence was as followed: sense, 5’-CGCCGGAGAGAAAAGUUUATT-3’; antisense, 5’-UAAACUUUUCUCUCCGGCGGC-3’. Cardiomyocytes were detached and cultured at 60~80×104 cells/well into six-well plates. After being cultured overnight, the cells were transfected with 50 nM siRNA. The cells were then cultured for another 24 hours and then treated with Ang II.
Statistics
All experiments were performed at least three times. The data were presented as the mean ± standard error of the mean (SEM). Statistical analysis was conducted with SPSS 20.0 software, using one-way ANOVA for multiple group comparisons or Student’s t test for two-group comparisons. P < 0.05 was considered to be statistically significant.
Results
Expression of ECM and histological observation in in vivo model rats
The in vivo model was established in SD rats that were infused with Ang II at a rate of 65 ng/min for 14 days via an osmotic mini-pump, and age-matched controls were included. When compared with control group, the protein and mRNA levels of collagen I were enriched to approximately 3.4/2.5-fold (Figure 1A) in A group; the protein and mRNA levels of collagen III were up-regulated to approximately 3.0/4.0-fold (Figure 1A) in A group; the protein and mRNA level of CTGF were enriched to approximately 1.8/2.3-fold (Figure 1A) in A group.
Figure 1.

Expression of ECM and Histological observation in in vivo model rats. A. Western blot and mRNA of collagen I, collagen III and CTGF expression in in vivo model rats. Relative expression levels are normalised to β-actin and expressed as the mean ± SEM, n = 5-7. **p < 0.01 vs. C group. B. Western blot and mRNA of collagen I, collagen III and CTGF expression in in vivo model rats. Relative expression levels are normalised to β-actin and expressed as the mean ± SEM, n = 5-7. **p < 0.01 vs. C group. C. Western blot and mRNA of NF-κB and Fn14 expression in in vivo model rats. Relative expression levels are normalised to β-actin and expressed as the mean ± SEM, n = 5-7. **p < 0.01 vs. C group. D. Masson trichrome staining showed cardiac fibrosis at magnification x 40. Scale bar: 50 μm. n = 5-7. *p < 0.05 vs. C group.
At the same time, the protein and mRNA levels of NF-κB and Fn14 were in rich of in vivo models. When compared with control group, the protein and mRNA levels of NF-κB were enriched to approximately 9.1/9.9-fold (Figure 1B) in A group; the protein and mRNA levels of Fn14 were up-regulated to approximately 4.8/7.1-fold (Figure 1B) in A group.
Morphological changes were highlighted by Masson trichrome staining (Figure 1C). When compared with C group, the histological score was at approximately 2.3-fold in A group.
Expression of NF-κB and Fn14 in in vitro models induced by Ang II
The in vitro models were established by Ang II in cardiomyocytes. NF-κB and Fn14 expression were both up-regulated in a time-dependent manner. Notably, both mRNA and protein levels of NF-κB and Fn14 peaked after 4 hours of Ang II stimulation (Figure 2). Taken together, the above results strongly suggested that NF-κB and Fn14 up-regulation played an important role in Ang II-induced in vitro models.
Figure 2.
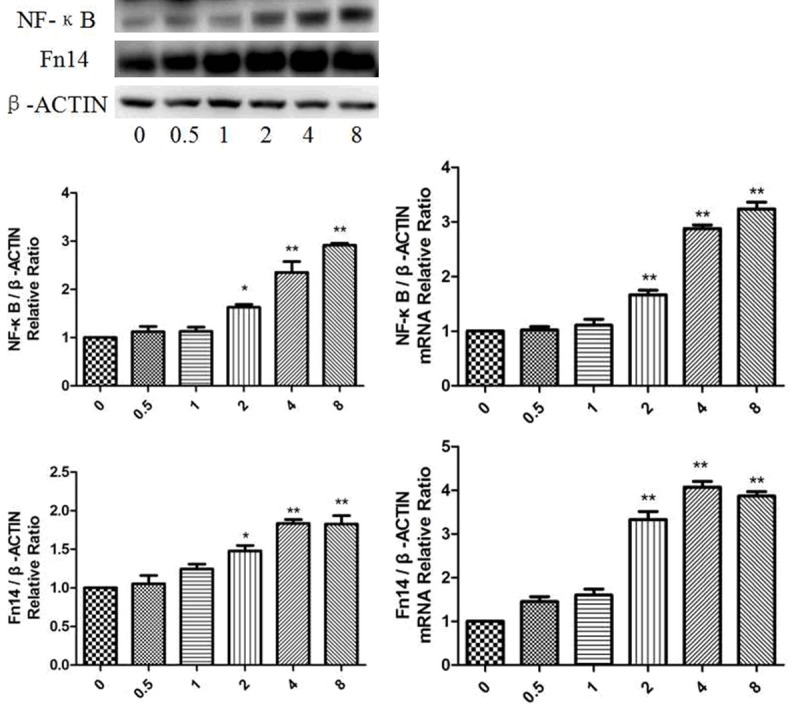
Expression of NF-κB and Fn14 in in vitro models of Ang II treatment. NF-κB and Fn14 proteins and mRNA expression were up-regulated by Ang II in a concentration-dependent manner. Relative expression levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. *p < 0.05 and **p < 0.01 vs. C group.
Effect of Fn14 knock-down on RhoA and Rac1 activity induced by Ang II in cardiomyocytes
The active form of RhoA was elevated to approximately 2.8-fold of the control group after a 15-minute treatment with Ang II (Figure 3A). siFn14 markedly reduced RhoA activation to approximately 1.1-fold of the control group. At the same time, the level of active Rac1 could not be elevated by Ang II when the cells were pretreated with siFn14 (Figure 3B). These results suggested that RhoA activation induced by Ang II was partially attenuated by siFn14.
Figure 3.
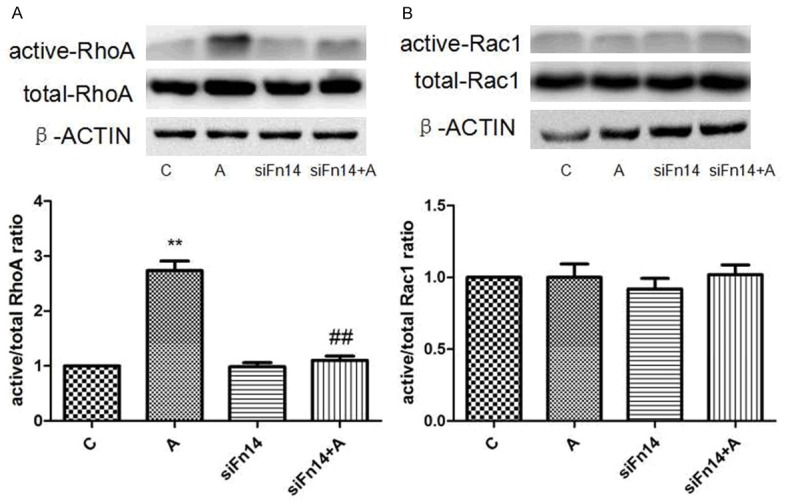
Effect of Fn14 knock-down on RhoA and Rac1 activity induced by Ang II in cardiomyocytes. A. Relative protein expression levels of active and total RhoA are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group. B. Relative protein expression levels of active and total Rac1 are normalised to β-actin and expressed as the mean ± SEM, n = 3.
Effect of Fn14 and RhoA knock-down on phosphorylation of IκBα and IKKβ induced by Ang II in cardiomyocytes
Given Fn14 could ameliorate RhoA activation, we sought to investigate the role of Fn14 and RhoA in Ang II induced in vitro models. We validated that A groups could effectively enhance the activity of phosphorylation of IκBα but not IKKβ (Figure 4A, 4B). The expression of IκBα phosphorylation was enriched to approximately 1.8-fold in A groups compared with control group. Then we meaningfully found siFn14 could down-regulate approximately 1.1-fold expression of IκBα phosphorylation in A+siFn14 group (Figure 4A), and siRhoA could down-regulate approximately 0.9-fold expression of IκBα phosphorylation in A+siRhoA group (Figure 4A).
Figure 4.
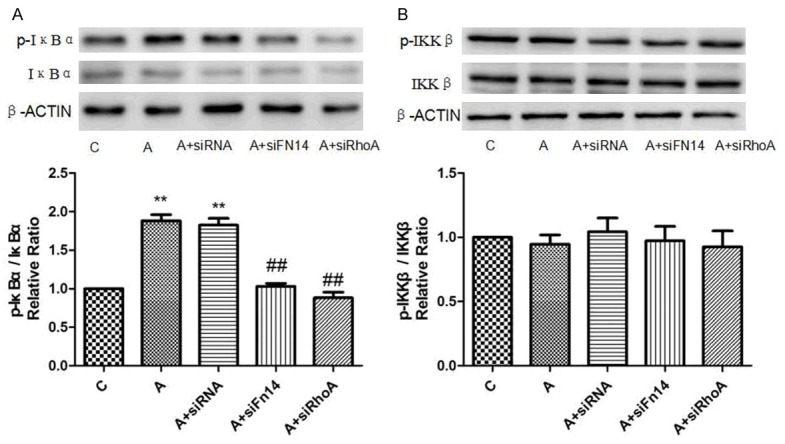
Effect of Fn14 and RhoA knock-down on phosphorylation of IκBα and IKKβ induced by Ang II in cardiomyocytes. A. Relative protein expression levels of active and total IκBα are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group. B. Relative protein expression levels of active and total IKKβ are normalised to β-actin and expressed as the mean ± SEM, n = 3.
Effect of RhoA and IκBα knock-down by siRNA on Ang II-induced response in cardiomyocytes
Effect of RhoA and Fn14 siRNA on the protein levels of RhoA and Fn14 in cardiomyocytes were shown in Figure 5A, 5B. siRhoA down-regulated the protein and mRNA expression of Fn14 induced by Ang II (Figure 5C, 5E). The Fn14 protein and mRNA were elevated to approximately 3.2/4.1-fold of the control group induced by Ang II while siRhoA markedly reduced Fn14 protein and mRNA to approximately 2.0/2.2-fold of the control group (Figure 5C, 5E). At the same time, the Fn14 protein and mRNA were elevated to approximately 3.2/3.8-fold of the control group induced by Ang II while siIκBα markedly reduced Fn14 protein and mRNA to approximately 1.6/2.7-fold of the control group (Figure 5D, 5F). Taken together, siRhoA and siIκBα could down-regulated Fn14 expression induced by Ang II in cardiomyocytes.
Figure 5.

Effect of RhoA and IκBα silencing by siRNA on Ang II-induced response in cardiomyocytes. A. Effect of siRhoA on the protein level of RhoA in cardiomyocytes. Protein expression levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. NC siRNA group. B. Effect of siFn14 on the protein level of Fn14 in cardiomyocytes. Protein expression levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. NC siRNA group. C. RhoA knock-down down-regulates Fn14 protein expression induced by Ang II. Realtive protein levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group. D. RhoA and IκBα knock-down down-regulates Fn14 protein expression induced by Ang II. Realtive protein levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, #p < 0.05 and ##p < 0.01 vs. Ang II group. E. RhoA knock-down down-regulates Fn14 mRNA expression induced by Ang II. Realtive mRNA levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group. F. RhoA and IκBα knock-down down-regulates Fn14 mRNA expression induced by Ang II. Realtive mRNA levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group.
Effect of NF-κB knock-down and inhibition on Ang II-induced Fn14 expression cardiomyocytes
The present study showed that NF-κB expression was related with Ang II-induced models. The Fn14 protein and mRNA levels were elevated to approximately 3.1/4.0-fold of the control group induced by Ang II while siNF-κB markedly reduced Fn14 protein and mRNA levels to approximately 1.9/1.8-fold of the control group (Figure 6A-C). At the same time, the Fn14 protein and mRNA were elevated to approximately 3.0/4.1-fold of the control group induced by Ang II while NF-κB inhibitor BAY11-7082 markedly reduced Fn14 protein and mRNA to approximately 1.8/2.1-fold of the control group (Figure 6B-D).
Figure 6.

Effect of NF-κB knock-down and inhibition on Ang II-induced Fn14 expression cardiomyocytes. (A and C) NF-κB knock-down down-regulates Fn14 protein and mRNA expression induced by Ang II. Realtive protein and mRNA levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group. (B and D) NF-κB inhibition down-regulates Fn14 protein and mRNA expression induced by Ang II. Realtive protein and mRNA levels are normalised to β-actin and expressed as the mean ± SEM, n = 3. **p < 0.01 vs. C group, ##p < 0.01 vs. Ang II group.
Discussion
Ang II, the main effector peptide of the RAS, is a known activator of in vivo [40] and in vitro [41] models of cardiac remodeling. Using in vivo models, we demonstrate that the expression of ECM, Fn14 and NF-κB is significantly increased in in vivo models that have been stimulated by Ang II. Under the given experimental conditions, Ang II successfully induces a cardiac remodeling response [31,38] and also evokes the time-dependent elevation of Fn14 and NF-κB expression via the RhoA/NF-κB/IκBα pathway. This result suggests that Fn14 might play an important role in the Ang II-mediated in vitro and in vivo cardiac remodeling.
Inflammatory cytokines play a crucial role in cardiac remodeling [42]. They have not only been related to typical inflammatory cardiac diseases like cardiomyopathy, but also a variety of evidence supports their importance in the process of cardiac remodeling and heart failure. Among the major inflammatory cytokines investigated so far in this context is Ang II. Here, we report for the first time the dynamic regulation of Fn14 in cardiomyocytes by Ang II stimulation. It has been proposed that injured tissues express Fn14 in response to different cytokines. In our research, we found that a similar mechanism exists in the heart. Ang II is a member of cytokines that is phosphorylatedly expressed and has been related to inhibition of cardiac hypertrophy, cardiac fibrosis, heart failure and cardiac remodeling. These effects critically depend on the expression of one of Ang II receptors Fn14.
Ang II is a main mediator of hormonal activation in cardiac remodeling. Blocking the myocardial hormones has been proven valuable in many experimental and clinical studies [43]. Interestingly, Ang II strongly up-regulates Fn14 expression in cardiomyocytes. Since we performed our experiments in neonatal cardiomyocytes and rats models, difference of the effects of Ang II might be observed. Nevertheless, very recent experimental data support our findings in the setting of dilated cardiomyopathy. More and more researches may be needed in the near future.
Adrenergic agents, Ang II as well as several other mediators of cardial hypertrophy exert their effects via G-Protein coupled receptors and several downstream pathways [42], including RhoA kinase activation and related NF-κB/IκBα pathways. Overexpression of RhoA in the heart leads to the development of heart failure with bradycardia [44], impaired contractile function and induction of interstitial fibrosis in in vivo models [45]. In our study, RhoA silencing by siRhoA inhibited Ang II-induced expression of Fn14 in cardiomyocytes. RhoA is activated in the hearts of Ang II-stimulated cardiac remodeling in vivo shown by Yang J et al. [46]. We confirm this finding by showing that a 15-minute treatment with Ang II is able to promote RhoA activity, which is reversible by siFn14. Our study shows here that up-regulation of Fn14 induced by Ang II is mediated, at least partly, via RhoA pathway. We cannot exclude that other pathways are also implicated in the observed regulation of Fn14 by Ang II activation; however, we also observe a powerful reduction of Fn14 in neonatal cardiomyocytes by using siRNA against RhoA instead of less specific inhibitors for these pathways.
Ang II mediates its varieties of biological effects both dependently and independently on Fn14 [24,47]. It has been described that Ang II induces NF-κB activation in other cells types [48]. NF-κB activation in cardiomyocytes has been linked to cardiac hypertrophy, fibrosis, inflammation, heart failure and cardiac remodeling. Fn14 mediates strong NF-κB activation in cardiomyocytes. At the same time, we observed expression of ECM via NF-κB activation. All molecules have been implicated in the pathogenesis of cardiac remodeling, e.g. via attraction of mononuclear cells to cardiac interstitium, where they contribution to cardiac remodeling. In line with these reports is our finding of increased expression of Fn14 and NF-κB after Ang II stimulation concomitantly to the expression of the Ang II-Fn14 axis.
Finally, the fact that we found an up-regulation of Fn14 in in vivo and in vitro cardiac remodeling implies a potential role of Ang II-Fn14 axis in this setting. The heart of rats induced by Ang II is subjected to a remodeling process with cardiac hypertrophy, fibrosis and dilated cardiomyopathy. We observed up-regulation of Fn14 both in cardiomyocytes as well as in vivo model induced by Ang II, supporting the notion that Fn14 is a novel myocardial cytokines receptor. Interestingly, a recent experiment report has implicated a role for Ang II and Fn14 in the setting of cardiac remodeling.
In summary, we describe here for the first time Ang II regulation of Fn14 pathway in in vivo and in vitro models. Moreover, Ang II strongly induced RhoA, NF-κB and NF-κB driven gene expression via Fn14. These findings, together with the induction of Ang II as well as in the setting of cardiac remodeling, suggest a role of Fn14, which promotes ECM production by activating the RhoA/NF-κB/IκBα pathway in cardiac remodeling induced by Ang II.
A limitation of this study is that we could not evaluate the Ang II-Fn14 signaling pathways because of our experimental conditions. At present, it is still unclear whether the activation of the Ang II-Fn14 axis in cardiac remodeling has positive or negative effects on cardiac remodeling, since Ang II is a multifunctional cytokine with not only detrimental but also potential beneficial properties. Furthermore, a similar ambivalent role in the heart has already been described for other cytokines. It will therefore be interesting to further evaluate the contribution of the Ang II-Fn14 activation in the progression of cardiac remodeling.
Acknowledgements
We thank the Biomedical Research Center in Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University for the use of equipment. This study was supported by Zhejiang Provincial Medical and Health Science and Technology Plan (No. 2015KYB206), Zhejiang Provincial Traditional Chinese Medical Technology Plan (No. 2016ZB072), Natural Science Funds of Zhejiang Province, China (Project No. LQ16H020001) and Department of Health of Zhejiang Province (Project No. WST14SB02). We also thank the Biomedical Research Center in Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University for the use of equipment.
Disclosure of conflict of interest
None.
Authors’ contribution
Fu Guosheng designed the study. Li Zhengwei, Shen Zhida, He Jialin and Chen Shengyu carried out experiments. Zhang Jiefang, Luan Yi and Du Lailing analyzed the data. Li Zhengwei wrote the manuscript. All authors reviewed the manuscript.
References
- 1.Zhang J, Chang L, Chen C, Zhang M, Luo Y, Hamblin M, Villacorta L, Xiong JW, Chen YE, Zhu X. Rad GTPase inhibits cardiac fibrosis through connective tissue growth factor. Cardiovasc Res. 2011;91:90–98. doi: 10.1093/cvr/cvr068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosin NL, Falkenham A, Sopel MJ, Lee TD, Legare JF. Regulation and role of connective tissue growth factor in AngII-induced myocardial fibrosis. Am J Pathol. 2013;182:714–726. doi: 10.1016/j.ajpath.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 4.Xu X, Pang J, Yin H, Li M, Hao W, Chen C, Cao JM. Hexarelin suppresses cardiac fibroblast proliferation and collagen synthesis in rat. Am J Physiol Heart Circ Physiol. 2007;293:H2952–2958. doi: 10.1152/ajpheart.00004.2007. [DOI] [PubMed] [Google Scholar]
- 5.Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–530. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 6.Yagi S, Aihara K, Ikeda Y, Sumitomo Y, Yoshida S, Ise T, Iwase T, Ishikawa K, Azuma H, Akaike M, Matsumoto T. Pitavastatin, an HMG-CoA reductase inhibitor, exerts eNOS-independent protective actions against angiotensin II induced cardiovascular remodeling and renal insufficiency. Circ Res. 2008;102:68–76. doi: 10.1161/CIRCRESAHA.107.163493. [DOI] [PubMed] [Google Scholar]
- 7.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120:3350–3364. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong B, Yu QT, Dai HY, Gao YY, Zhou ZL, Zhang L, Jiang H, Gao F, Li SY, Zhang YH, Bian HJ, Liu CX, Wang N, Xu H, Pan CM, Song HD, Zhang C, Zhang Y. Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol. 2012;59:739–747. doi: 10.1016/j.jacc.2011.09.071. [DOI] [PubMed] [Google Scholar]
- 9.Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122:717–728. doi: 10.1161/CIRCULATIONAHA.110.955369. 18 p following 728. [DOI] [PubMed] [Google Scholar]
- 10.Cheng CP, Cheng HJ, Cunningham C, Shihabi ZK, Sane DC, Wannenburg T, Little WC. Angiotensin II type 1 receptor blockade prevents alcoholic cardiomyopathy. Circulation. 2006;114:226–236. doi: 10.1161/CIRCULATIONAHA.105.596494. [DOI] [PubMed] [Google Scholar]
- 11.Masuda T, Muto S, Fujisawa G, Iwazu Y, Kimura M, Kobayashi T, Nonaka-Sarukawa M, Sasaki N, Watanabe Y, Shinohara M, Murakami T, Shimada K, Kobayashi E, Kusano E. Heart angiotensin II-induced cardiomyocyte hypertrophy suppresses coronary angiogenesis and progresses diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2012;302:H1871–1883. doi: 10.1152/ajpheart.00663.2011. [DOI] [PubMed] [Google Scholar]
- 12.Hao PP, Yang JM, Zhang MX, Zhang K, Chen YG, Zhang C, Zhang Y. Angiotensin-(1-7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2015;308:H1007–1019. doi: 10.1152/ajpheart.00563.2014. [DOI] [PubMed] [Google Scholar]
- 13.Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, Alberts GF, Copeland NG, Gilbert DJ, Jenkins NA, Richards CM, Winkles JA. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274:33166–33176. doi: 10.1074/jbc.274.46.33166. [DOI] [PubMed] [Google Scholar]
- 14.Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, Peifley KA, Winkles JA. The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol. 2000;156:1253–1261. doi: 10.1016/S0002-9440(10)64996-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA, Fanslow WC. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- 16.Campbell S, Burkly LC, Gao HX, Berman JW, Su L, Browning B, Zheng T, Schiffer L, Michaelson JS, Putterman C. Proinflammatory effects of TWEAK/Fn14 interactions in glomerular mesangial cells. J Immunol. 2006;176:1889–1898. doi: 10.4049/jimmunol.176.3.1889. [DOI] [PubMed] [Google Scholar]
- 17.Polavarapu R, Gongora MC, Winkles JA, Yepes M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappa B pathway activation. J Neurosci. 2005;25:10094–10100. doi: 10.1523/JNEUROSCI.3382-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yepes M, Brown SA, Moore EG, Smith EP, Lawrence DA, Winkles JA. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am J Pathol. 2005;166:511–520. doi: 10.1016/S0002-9440(10)62273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salzmann S, Lang I, Rosenthal A, Schafer V, Weisenberger D, Carmona Arana JA, Trebing J, Siegmund D, Neumann M, Wajant H. TWEAK inhibits TRAF2-mediated CD40 signaling by destabilization of CD40 signaling complexes. J Immunol. 2013;191:2308–2318. doi: 10.4049/jimmunol.1202899. [DOI] [PubMed] [Google Scholar]
- 20.Salzmann S, Seher A, Trebing J, Weisenberger D, Rosenthal A, Siegmund D, Wajant H. Fibroblast growth factor inducible (Fn14)-specific antibodies concomitantly display signaling pathway-specific agonistic and antagonistic activity. J Biol Chem. 2013;288:13455–13466. doi: 10.1074/jbc.M112.435917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar M, Makonchuk DY, Li H, Mittal A, Kumar A. TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-beta-activated kinase 1. J Immunol. 2009;182:2439–2448. doi: 10.4049/jimmunol.0803357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doerner JL, Wen J, Xia Y, Paz KB, Schairer D, Wu L, Chalmers SA, Izmirly P, Michaelson JS, Burkly LC, Friedman AJ, Putterman C. TWEAK/Fn14 Signaling Involvement in the Pathogenesis of Cutaneous Disease in the MRL/lpr Model of Spontaneous Lupus. J Invest Dermatol. 2015;135:1986–1995. doi: 10.1038/jid.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, Guan J, Lim CC, Naito Y, Thompson JS, Sam F, Ambrose C, Parr M, Crowell T, Lincecum JM, Wang MZ, Hsu YM, Zheng TS, Michaelson JS, Liao R, Burkly LC. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation. 2009;119:2058–2068. doi: 10.1161/CIRCULATIONAHA.108.837286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mustonen E, Sakkinen H, Tokola H, Isopoussu E, Aro J, Leskinen H, Ruskoaho H, Rysa J. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 during cardiac remodelling in rats. Acta Physiol (Oxf) 2010;199:11–22. doi: 10.1111/j.1748-1716.2010.02080.x. [DOI] [PubMed] [Google Scholar]
- 25.Wojciak-Stothard B, Zhao L, Oliver E, Dubois O, Wu Y, Kardassis D, Vasilaki E, Huang M, Mitchell JA, Harrington LS, Prendergast GC, Wilkins MR. Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ Res. 2012;110:1423–1434. doi: 10.1161/CIRCRESAHA.112.264473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartmann S, Ridley AJ, Lutz S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front Pharmacol. 2015;6:276. doi: 10.3389/fphar.2015.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 29.Gasmi-Seabrook GM, Marshall CB, Cheung M, Kim B, Wang F, Jang YJ, Mak TW, Stambolic V, Ikura M. Real-time NMR study of guanine nucleotide exchange and activation of RhoA by PDZ-RhoGEF. J Biol Chem. 2010;285:5137–5145. doi: 10.1074/jbc.M109.064691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi K, Sasaki T, Mammoto A, Hotta I, Takaishi K, Imamura H, Nakano K, Kodama A, Takai Y. Interaction of radixin with Rho small G protein GDP/GTP exchange protein Dbl. Oncogene. 1998;16:3279–3284. doi: 10.1038/sj.onc.1201874. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Bi X, Wang M, Zhang J, Song J, Shen X, Han J, Fu G, Ye Y. Inhibition of farnesyl pyrophosphate synthase prevents angiotensin II-induced cardiac fibrosis in vitro. Clin Exp Immunol. 2014;176:429–437. doi: 10.1111/cei.12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genet G, Guilbeau-Frugier C, Honton B, Dague E, Schneider MD, Coatrieux C, Calise D, Cardin C, Nieto C, Payre B, Dubroca C, Marck P, Heymes C, Dubrac A, Arvanitis D, Despas F, Altie MF, Seguelas MH, Delisle MB, Davy A, Senard JM, Pathak A, Gales C. Ephrin-B1 is a novel specific component of the lateral membrane of the cardiomyocyte and is essential for the stability of cardiac tissue architecture cohesion. Circ Res. 2012;110:688–700. doi: 10.1161/CIRCRESAHA.111.262451. [DOI] [PubMed] [Google Scholar]
- 33.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frederick JR, Fitzpatrick JR 3rd, McCormick RC, Harris DA, Kim AY, Muenzer JR, Marotta N, Smith MJ, Cohen JE, Hiesinger W, Atluri P, Woo YJ. Stromal cell-derived factor-1alpha activation of tissue-engineered endothelial progenitor cell matrix enhances ventricular function after myocardial infarction by inducing neovasculogenesis. Circulation. 2010;122:S107–117. doi: 10.1161/CIRCULATIONAHA.109.930404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan A, Moe GW, Nili N, Rezaei E, Eskandarian M, Butany J, Strauss BH. The cardiac atria are chambers of active remodeling and dynamic collagen turnover during evolving heart failure. J Am Coll Cardiol. 2004;43:68–76. doi: 10.1016/j.jacc.2003.07.030. [DOI] [PubMed] [Google Scholar]
- 36.Liu C, Cao F, Tang QZ, Yan L, Dong YG, Zhu LH, Wang L, Bian ZY, Li H. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J Nutr Biochem. 2010;21:1238–1250. doi: 10.1016/j.jnutbio.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Bian Z, Cai J, Shen DF, Chen L, Yan L, Tang Q, Li H. Cellular repressor of E1A-stimulated genes attenuates cardiac hypertrophy and fibrosis. J Cell Mol Med. 2009;13:1302–1313. doi: 10.1111/j.1582-4934.2008.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, Wang Y. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol. 2011;301:H538–547. doi: 10.1152/ajpheart.01114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schorb W, Booz GW, Dostal DE, Conrad KM, Chang KC, Baker KM. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ Res. 1993;72:1245–1254. doi: 10.1161/01.res.72.6.1245. [DOI] [PubMed] [Google Scholar]
- 40.Silvestre JS, Heymes C, Oubenaissa A, Robert V, Aupetit-Faisant B, Carayon A, Swynghedauw B, Delcayre C. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation. 1999;99:2694–2701. doi: 10.1161/01.cir.99.20.2694. [DOI] [PubMed] [Google Scholar]
- 41.Samuel CS, Unemori EN, Mookerjee I, Bathgate RA, Layfield SL, Mak J, Tregear GW, Du XJ. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. doi: 10.1210/en.2004-0209. [DOI] [PubMed] [Google Scholar]
- 42.Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, Frey N. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol. 2010;105:301–313. doi: 10.1007/s00395-009-0046-y. [DOI] [PubMed] [Google Scholar]
- 43.Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, Liu FF, Zhang K, Zhang C. Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat Rev Cardiol. 2014;11:413–426. doi: 10.1038/nrcardio.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW 2nd, Ross J Jr, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627–1634. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, Kitakaze M, Liao JK. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–1437. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Mou Y, Wu T, Ye Y, Jiang JC, Zhao CZ, Zhu HH, Du CQ, Zhou L, Hu SJ. Cardiac-specific overexpression of farnesyl pyrophosphate synthase induces cardiac hypertrophy and dysfunction in mice. Cardiovasc Res. 2013;97:490–499. doi: 10.1093/cvr/cvs347. [DOI] [PubMed] [Google Scholar]
- 47.Nasimi A, Kafami M. Vasopressin and sympathetic system mediate the cardiovascular effects of the angiotensin II in the bed nucleus of the stria terminalis in rat. Neurosci Res. 2016;108:34–9. doi: 10.1016/j.neures.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Lv P, Miao SB, Shu YN, Dong LH, Liu G, Xie XL, Gao M, Wang YC, Yin YJ, Wang XJ, Han M. Phosphorylation of smooth muscle 22alpha facilitates angiotensin II-induced ROS production via activation of the PKCdelta-P47phox axis through release of PKCdelta and actin dynamics and is associated with hypertrophy and hyperplasia of vascular smooth muscle cells in vitro and in vivo. Circ Res. 2012;111:697–707. doi: 10.1161/CIRCRESAHA.112.272013. [DOI] [PubMed] [Google Scholar]