

Abstract
Hematological malignancy originated from B-cell line, multiple myeloma (MM), is a kind of plasma cells in bone marrow hyperplasia and cause of osteoclast-mediated skeletal destruction disease. MiR-34a plays an important epigenetic regulating role in malignant tumors and presents a therapeutic potential. In this study, we investigated the effects of overexpression of miR-34a in MM cancer stem cells (CSCs) on tumor growth and bone lesions. Here we showed that miR-34a overexpression inhibited cell proliferation, colony formation, and increased CSC apoptosis in vitro. The apparent epigenetic modulation induced by miR-34a overexpression was found no only in MM RPMI8226 cells but also in CSC xenograft MM. Both bioinformatics prediction and dual-luciferase reporter assay showed that transforming growth interaction factor 2 (TGIF2) was sufficient to confer miR-34a regulation. The results of qRT-PCR and Western blot assays demonstrated that the expression of TGIF2 was significant decreased in tumor tissues from NOD/SCID mice injected with miR-34a-MM CSCs. We conclude that miR-34a overexpression in MM CSCs significantly suppressed the tumorigenicity and lytic bone lesions in mouse model by inducing apoptosis and inhibiting TGIF2 expression.
Keywords: Multiple myeloma, miR-34a, cancer stem cells, transforming growth interaction factor 2
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy characterized by the presence of a monoclonal immunoglobulin, clonal plasma cell proliferation, bone lesions, hypercalcemia, anemia, renal insufficiency, and an increase of infection risk [1-3]. MM treatment has improved in the last 15 years with the introduction of immunomodulatory drugs, proteasome inhibitors, monoclonal antibodies, cell cycle-specific drugs, and deacetylase inhibitors etc., which has significantly ameliorated patient’s response rates and prolonged survival time. Nevertheless, most MM patients eventually succumb to the disease that is still incurable. As such, more effective treatments are urgently needed [3-6]. Emerging evidence suggests microRNAs (miRNA) regulation has been considered to be significantly related to the control of cancer progress. One of the miRNA-34 family, miR-34a is thought to be a potential tumor growth inhibitor that results in tumor cell’s G1 phase arrest and apoptosis. Research reports have showed that there are abnormal miR-34a expression in prostate cancer, kidney carcinoma, glioblastoma tumor, hepatocellular carcinoma, and MM etc. [7-10]. Since miR-34a can block MM cells secreting the osteoclast-activating factor by inhibiting expression of its target gene TGIF2 (Transforming growth interaction factor 2, also known as TG-interaction factor 2), it leads to preventing the formation of osteoclasts, reducing bone resorption and osteoporosis symptoms as well as and bone metastases in animal [11]. However, much less information is involved in the effect of miR-34a on MM cancer stem cells (CSCs) that are responsible for initiating tumors and maintaining the pull of residual disease, and resulting in eventual recurrence in MM patients. Therefore, how does miR-34a regulate the biological characteristics of MM CSCs remains unknown. Accordingly, it needs to be explored. To this regard, our purpose in this study was to investigate the epigenetically regulation function of miR-34a in the human MM RPMI8226 cells as well as MM CSCs. To reach this purpose, we constructed the recombinant containing miR-34a and transfected it into the MM cells and further isolated the MM CD138-CD34-CSCs from the stably transfected cells. Here we showed a direct functional relationship between the miR-34a overexpression and the inhibition of the capability of MM cell proliferation and colony forming. In particular, up-regulating miRNA-34a expression in MM CD138-CD34-CSCs was correlated with inhibition of the CSC tumori-genicity and lytic bone lesions in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice. This study provides primary research that the miRNA-34a acts as a tumor suppressor in inhibiting the MM CD138-CD34-CSC xenograft growth and the lytic bone lesions not only by inducing apoptosis but by inhibiting the expression of transcriptional inhibitor TGIF2 in MM bearing mouse model.
Materials and methods
Cell line
Human MM RPMI 8226 cell line was purchased from the Institute of Basic Medical Sciences Chinese Academy, People’s Republic of China. Cells were cultured in complete medium consisting of RPMI 1640, 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum at 37°C in a humidified incubator containing 5% CO2.
NOD/SCID mice
NOD/SCID mice at 5-6 weeks of age with 16-18 grams in weight were purchased from Slac laboratory animal center, Shanghai, China. All the mice were maintained in a pathogen-free facility that has a 12-hour light/dark cycle and relative humidity ranged from 40% to 50% at 24°C. All the animal experiments were performed in compliance with the Guidelines of the Animal Research Ethics Board of Southeast University. Full details of approval of the study can be found in the approval ID: 20080925.
Isolation of MM CD138-CD34-CSCs
CD138-cells were isolated from human MM RPMI 8226 cells using mouse anti-human CD138 monoclonal antibody coupled to magnetic microbeads (Miltenyi Biotec, Germany) followed by magnetic column selection, using immune magnetic activated cell sorting (Miltenyi Biotec, Germany). Resulting cells were additionally depleted of normal hematopoietic progenitors by using mouse antihuman CD34 antibody (Miltenyi Biotec, Germany). We named CD138-CD34-cells in human MM RPMI 8226 cell line for the MM stem-like cells (MM CSCs) as described in our previous reports [12-14].
Generation of recombinant miR-34a and stable expression colony
To generate the miR-34a expression recombinant, we amplified an insert (full-length human miR-34a) by PCR from MM RPMI 8226 cells. The cDNA fragment was cloned into the Not I and Xba I sites of pIRES mammalian expression vector (Promega, USA). The target gene in recombinant was confirmed by DNA sequence.
MM RPMI 8226 cells grown to 70% confluent in complete medium were transferred to a six-well plate and cells were transfected with mixtures including the Pullulan-spermine (Ps) nanomaterials, serum-free medium with recombinant miR-34a, and the plates were incubated at 37°C in 5% CO2 atmosphere. Growth medium may be replaced after incubation for 4 h, followed by selection with 800 µg/ml of G418 (Clontech, CA) 24 h later. MM RPMI 8226 cells was transfected with blank plasmid as vector control. At same time, cells were transfected with mixtures including the Lipofectamin 2000 reagent, serum-free medium with recombinant miR-34a as positive control. After about 48 h, total cellular RNA was isolated from MM RPMI 8226 cells for detecting expression of miR-34a by qRT-PCR. The remaining cells were used for screening stable expression colony by limiting the dilution assay. The stable expression of miR-34a colony was named for miR-34a-MM cells, and the vector control colony was named for vector-MM cells [15,16].
RNA isolation and quantitative RT-PCR
Total cellular RNA was isolated from the different cells or tumor tissues by using a Qiagen RNeasy Kit (Qiagen, CA). The sequences of the primers are as follows: miR-34a-primer forward, 5’-TCTATTTGCCATCGTCTA-3; reverse, CAGGCAGCTCATTTGG AC; TGIF2 forward, 5’-AGAGTGCACTGCATGATAGCGTTCT-3’; reverse, 5’-CCAAT GATAAAACAACCATTTGG-3’; β-actin forward, 5’-GGACTTCGAGCAAGAGATGG-3’; reverse, 5’-AGCACTGTGTTGGCGTACAG-3’. The qRT-PCR analysis was performed on an ABI step one plus real-time system (Applied Biosystems, USA). The mRNA-level of the genes of interest were expressed as the ratio of a gene of interest to β-actin mRNA for each sample. The comparative Ct (ΔΔCt) method was used to determine the expression fold change [17].
Cell proliferation
5×105 MM RPMI 8226 cells transfected with recombinant miR-34a or recombinant-vector suspension were respectively seeded into 96-well plates (100 μl/well) at 37°C, 5% CO2 for 24 h and then 10 μl of the cell-counting kit-8 solution (CCK-8, Dojindo, Japan) was added to each well for 1.5 h incubation. The absorbance at 450 nm was recorded using a microplate reader (Model 550, Bio-Rad, USA) [18].
Colony formation in soft agar media
The assay protocol of colony formation was described in our previous reports [12,19]. Briefly, one hundred single-cell suspension miR-34a-MM cells or vector-MM cells were resuspended in 0.8 mL growth media containing 0.3% low melting temperature agarose (Promega, Madison, WI, USA) and were plated in triplicate on 24-well plate over a base layer of 0.8 mL growth media containing 0.6% low melting temperature agarose. The plates were incubated for 14-15 days until colonies were formed. Colony diameters larger than 75 µm or colony cells more than 50 cells were counted as 1 positive colony.
Cell apoptosis assay
1×106 miR-34a-MM cells and vector-MM cells were incubated at 37°C in 5% CO2 for 24 hours, and cells were resuspended in binding buffer. 100 mL cell suspension was incubated with Annexin V-PE and 7-AAD for 15 mins at room temperature in the dark. The rate of apoptosis was analyzed by flow cytometer (FCM, FACS Caliber, BD, USA) [20].
Animal experiments
5×106 miR-34a-MM cells and vector-MM cells or 1×106 miR-34a-MM CSCs and vector-MM CSCs were mixed with matrigel (100 μL in volume), and injected respectively subcutaneously into the dorsal side in NOD/SCID mice. The palpable tumors at the injection sites were examined. Three mice/group were used in the study, and the experiment was repeated twice. The tumour growth in NOD/SCID mice was monitored once three days for tumor volume, the general health indicators, such as overall behavior, feeding, body weight and appearance of fur after treatment. The endpoint for this study was the diameter of tumor ≥20 mm, and then mice were anesthetized with phenobarbital sodium and executed. Mouse tumor tissues and bones were collected for further analysis [13,21].
Detection of bone lesions
MM-bearing mice were anesthetized with phenobarbital sodium and executed by cervical dislocation. The bone mineral densities (BMD) of femurs, humerus, and vertebra in body were respectively detected by in vivo Micro-CT imaging (MCT-1108, China), and the parameters of voltage 45 mV with electric current 90 mA was used in the assay [22,23].
Detecting activity of the luciferase gene linked to the 3’UTR of TGIF2
The psiCHECK2 firefly luciferase reporter plasmids with the wild-type or mutated 3’UTR sequences of TGIF2 were transiently transfected into human MM RPMI 8226 cells along with 25 nM miR-34a mimics or negative control. Luciferase activity was performed using a Double-Luciferase Assay system (Promega) per the manufacturer’s instructions after 72 h [24,25].
Western blot
Total cell or tumor tissue lysates were prepared and analyzed by using the Western blot. Briefly, 1×106 different cells were collected and lyzed in a protein extraction buffer according to the manufacturer’s protocol. The PVDF membrane was blocked with 4% dry milk in the Tris-buffered saline with Tween-20 for 1 h at 20°C, and was incubated with the rabbit antibody specific to mouse/human TGIF2 or β-actin (Santa cruz Biotechnology, CA, USA) for overnight at 4°C. The membrane was then incubated with the goat anti-rabbit fluorescence secondary antibody for 1 h at 20°C, and the subsequent steps were performed according to the Western Blot Kit’s protocol (Pierce Company). Immunoreactive bands were detected by the Odyssey scanning instrument (LICOR Odyssey, USA) [26,27].
Statistical analysis
The SPSS 19.0 software package was used for data analysis. Data are expressed in the mean and standard deviations. The Student-Newman-Keuls test and one-way ANOVA were used to compare the variables among different groups. Differences were considered statistically significant if P value <0.05.
Results
MiR-34a expression in recombinant transfected MM cells and MM CSCs
As was described in the method section, the recombinant pIRES miR-34a was transfected into MM cells by using Ps [28], and the transient expression of miR-34a detected by qRT-PCR was significant increased in the cells transfected with the Ps-pIRES miR-34a compared with the cells transfected with the Ps-pIRES (P<0.005), and the transfection efficacy of Ps was even higher than that of lipofectamine2000 (Figure 1A). The stably transfected cells were screened by a single clone selection assay (Figure 1B), and this coincides with changed expression of miR-34a in the stably transfected cells as is shown in Figure 1C. In addition, the MM CD138-CD34-CSCs, isolated from the human MM RPMI 8226 cells by using magnetic activated cell sorting system, were also lower expression of miR-34a than that of MM non--CSCs or that of RPMI 8226 cells, which were statistically significant (P<0.05 and P<0.005) in Figure 1D.
Figure 1.
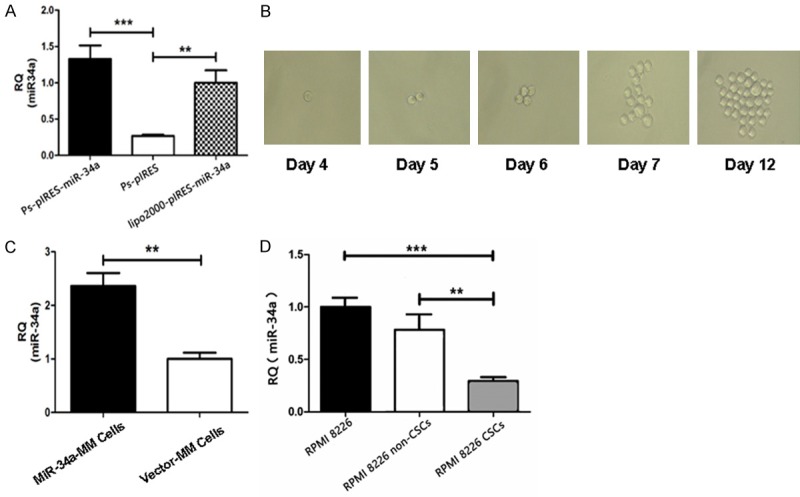
Detection of the miR-34a expression in MM cells and MM CSCs by qRT-PCR. A. Transient miR-34a expression in MM RPMI8226 cells 48 hours after the cells transfected with the pIRES miR-34a and the pIRES by Pullulan-spermine (Ps) or with the pIRES miR-34a by lipofectamine2000. B. Selection of miR-34a expressive clone. C. Detection of miR-34a expression in the stably transfected clone. D. Detection of the miR-34a expression in MM RPMI8226 cells, MM RPMI8226 CSCs, and MM RPMI8226 non-CSCs. **P<0.01 and ***P<0.005.
MiR-34a overexpression inhibits cell proliferation and colony formation but promotes apoptosis
To evaluate the modulation action of miR-34a, we first tested the effects of miR-34a overexpression on the RPMI 8226 cells based on cellular proliferation, colony forming, and cellular apoptosis. Figure 2A shows that the inhibitive rates of cell proliferation in both of miR-34a-RPMI 8226 cells and vector-RPMI 8226 cells detected by CCK8 assay. Higher inhibitive rate was found in miR-34a-MM cells than in vector-MM cells, in particular on 4 days, which was statistically significant (P<0.01).
Figure 2.
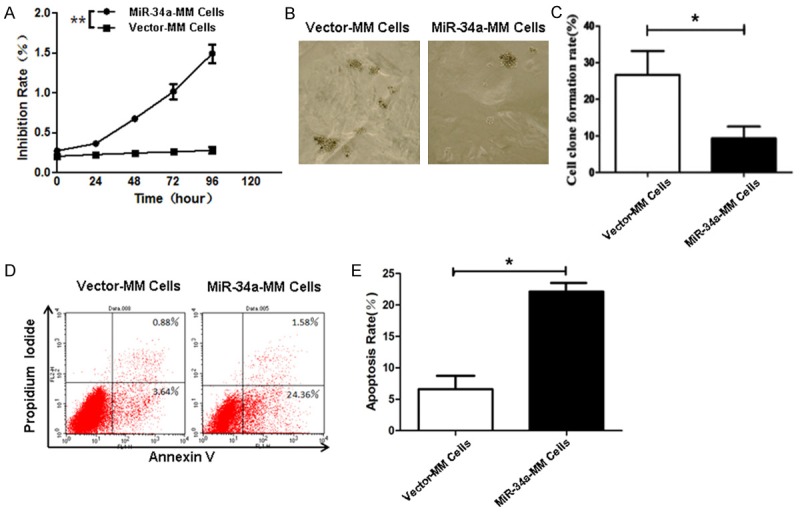
Effects of miR-34a overexpression on cell proliferation, colony formation, and apoptosis. A. Proliferation of miR-34a-MM and Vector-MM cells detected by CCK8 assay. B. Colony forming rates of miR-34a-MM and Vector-MM cells 14 days after incubation in the soft agar medium. C. Quantification of colony forming rate. D. Apoptosis of miR-34a-MM and Vector-MM cells was analyzed by FCM as described in the Method section. E. Quantification of apoptotic rate. *P<0.05 and **P<0.01.
The colony forming rate, analyzed by the soft agar colony forming assay as shown in Figure 2B, was around 26% for vector-MM cells and 9% for miR-34a-MM cells. It was significant inhibited in miR-34a-MM cells compared with vector-MM cells (P<0.05). To further assess the influence of miR-34a overexpression on the cellular apoptosis, we performed FCM to analyze the apoptosis of MM cells stably transfected with recombinant pIRES miR-34a or blank vector. Figure 2D displays the representative flow cytometric dot plots, and the overexpression of miR-34a clearly induced the cell apoptosis (24.36% ± 1.58) compared to vector-MM cells (3.64% ± 0.88), which was statistically significant (P<0.05) as is shown in Figure 2E. These positive data indicated that the miR-34a overexpression markedly decreased the cell proliferation and colony forming as well as increased the apoptosis of RPMI 8226 cells in vitro.
Overexpression of miR-34a in MM cells and MM CSCs decreases the tumorigenicity and lytic bone lesions in NOD/SCID mice
Since the overexpression of miR-34a exhibited significant effects on the cellular proliferation, colony forming and apoptosis of RPMI 8226 cells in vitro, we attempted to know whether the effects could impact the tumorigenicity, lytic bone lesions, and MM progression in vivo NOD/SCID mice. The representative tumor pictures in Figure 3A were taken from the mice injected with the miR-34a-MM cells or the vector-MM cells when the tumor bearing mice were photographed on Day 52. It was found that the mice injected with 5×106 miR-34a-MM cells or vector-MM cells generated tumors in around 21 days, and the vector-MM cells formed bigger tumor sizes than that of miR-34a-MM cells; whereas no tumor was found in one mouse injected with miR-34a-MM cells throughout the 52-day observation. Figure 3B shows the tumor growth dynamic curves drawn from the tumors of mice injected with the miR-34a-MM and vector-MM cells. There was a significant difference in tumor volumes between the two group cells (P<0.01). Consequently, the MM-bearing mice’s survival rate was significant longer in mice injected with miR-34a-MM cells than that of mice injected with vector-MM cells (Figure 3C, P<0.05).
Figure 3.
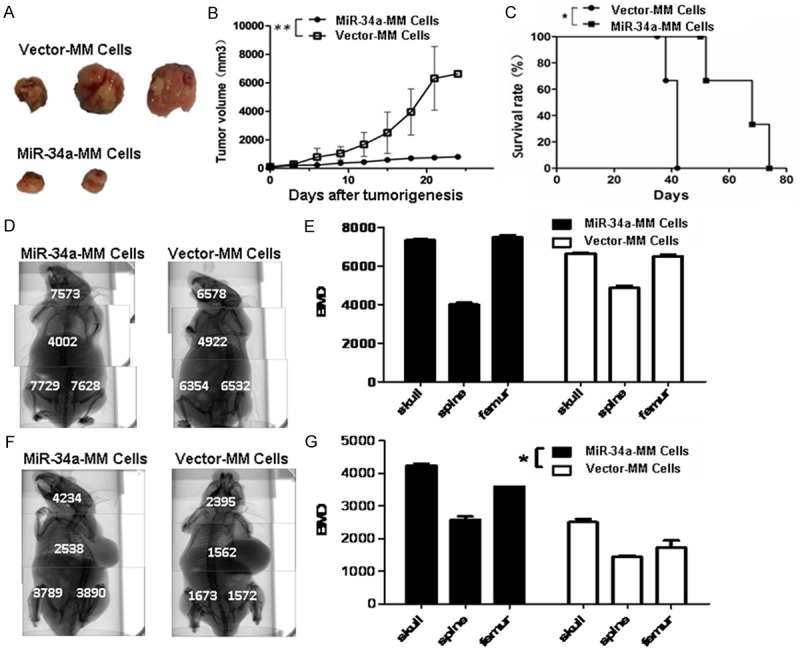
Effects of miR-34a overexpression in MM cells on the tumorigenicity and bone lesions in mice. A. Images of tumor sizes from the mice injected with the miR-34a-MM cells or the vector-MM cells on Day 52. B. Quantification analysis of tumor volumes in the tumor growth different times. C. MM bearing mice’s survival. D. Images of whole mouse BMD, as measured by Micro-CT, before tumor formation. E. Quantification analysis of BMD before tumor formation. F. Images of whole mouse BMD after tumor formation. G. Quantification analysis of BMD after tumor formation. *P<0.05 and **P<0.01.
Next, we wanted to know whether this inhibitive tumor growth effect would have an action on the decrease of skeleton lesions in MM-bearing mice. From top images in Figure 3D, we found the BMD of the skulls, vertebrates and femurs was no significant diffrences between the mice injected with miR-34a-MM and vector-MM cells before tumor formation (Figure 3E, P>0.05); whereas the BMD of those exbibited marked difference between the mice injected with miR-34a-MM cells and the mice injected with vector-MM cells after tumor formation (Figure 3F), which was statistically significant (P<0.005) in Figure 3G.
Further, we observed the regulation action of miR-34a in MM CSCs. Figure 4A indicates the images taken from the mice injected with 1×106 miR-34a-MM CD138-CD34-CSCs or vector-MM CD138-CD34-CSCs when the MM bearing mice were photographed on Day 39. Smaller tumors were found in miR-34a-CD138-CD34-CSCs group (1109 mm3 ± 76 mm3) than that in vector-CD138-CD34-CSCs group (3469 mm3 ± 146 mm3), and there was a significant difference between the two groups (P<0.01) in Figure 4B. Moreover, this targeted therapeutic effect was also reflected in the amelioration of lytic bone lesions in mice. The representative clinical images of BMD of the vertebrates and femurs were photographed on Day 39 in Figure 4C, which shows the the BMD of mice injected with miR-34a-MM CSCs was significantly increased in top panel compared with the mice injected with vector-MM CSCs in bottom panel. The difference is statistically significant (P<0.01 or P<0.005) as is shown in Figure 4D. These data demonstrated that the stable miR-34a overexpression in the MM cells and MM CSCs significantly inhibited their tumorigenicity, skeleton lesions, and MM progression in NOD/SCID mice.
Figure 4.
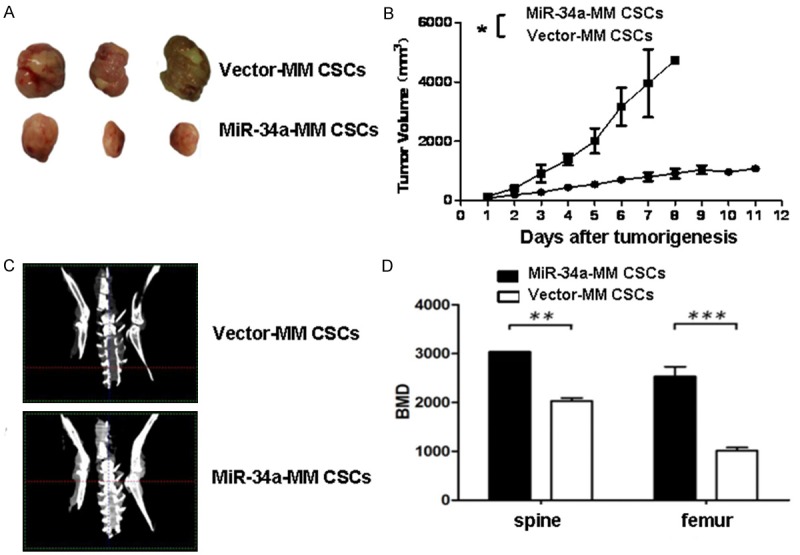
MiR-34a overexpression in MM CSCs reduces the tumorigenicity and bone lesions in mice. A. Images of tumor sizes from the mice injected with the miR-34a-MM CDCs or the vector-MM CSCs on Day 39. B. Quantification analysis of tumor volumes in the tumor growth different times. C. Images of mouse BMD scaned by in vivo Micro-CT on Day 39. D. Quantification analysis of BMD in spine and femur of mice. *P<0.05, **P<0.01, and***P<0.005.
MiR-34a inhibits MM growth and decreases bone lesions by targeting TGIF2
To elucidate the mechanisms underlying the suppressive effects of miR-34a on the MM cells and MM CD138-CD34-CSCs in tumorigenic potential and skeleton lesions, we used the open-target prediction programs of TargetScan, picTar, and miRnada to predict the targets of miR-34a. The data showed miR-34a may target the TGIF2, which is a transcriptional inhibitor that inhibit the formation of osteoclasts [11,26]. The gene sequence of TGIF2 contained 7 basic continuous base complementary sites for the seed region of mature miR-34a (Figure 5A). The dual-luciferase reporter assay result exhibited that the luciferase activity of the reporter containing the wild-type of TGIF2 gene was significantly decreased following treatment with miR-34a mimics (29% vs 100%, *P<0.005), which suggested that the direct down-regulation of TGIF2 was miR-34a-dependent as is shown in Figure 5B.
Figure 5.
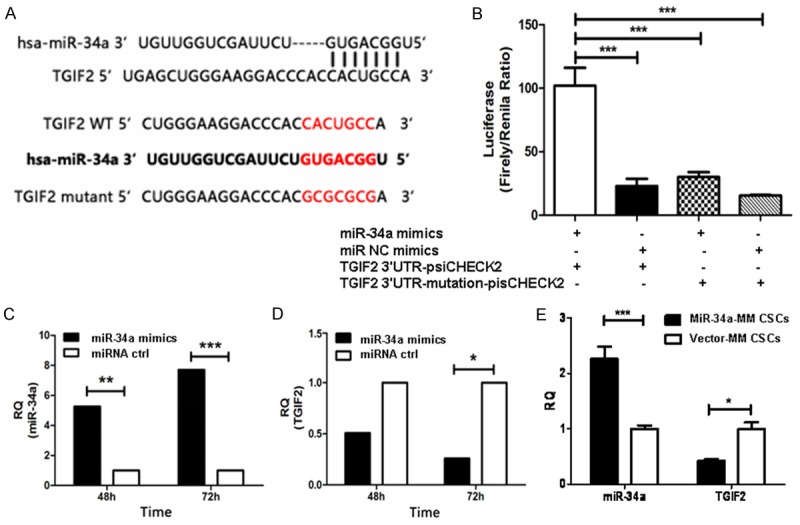
MiR-34a targets TGIF2 resulting in suppressing MM progression and decreasing bone lesions in mice. A. Top panel: predicted duplex formation between human TGIF2 3’UTR and miR-34a. Bottom panel: sequences of the wild-type or mutant TGIF2 3’UTR of human (hsa) and mice, which are the miR-34a binding sites. B. Luciferase activity of the wild-type or mutant TGIF2 3’UTR reporter gene in MM RPMI8226 cells transfected with the miR-34a mimics or miRNA negative control (miRNA ctrl). C. Quantification analysis of miR-34a expression in MM RPMI8226 cells transfected with the miR-34a mimics or miRNA ctrl. D. Quantification analysis of TGIF2 expression in MM RPMI8226 cells transfected with the miR-34a mimics or miRNA ctrl. E. Quantification analysis of miR-34a and TGIF2 expression in tumor tissues from the mice injected with miR-34a-MM CSCs or vector-MM CSCs.
To further confirm whether TGIF2 is the direct target gene for miR-34a, we used qRT-PCR to test the changes of TGIF2 expression in response to forced miR-34a overexpression in MM RPMI8226 cells. Figure 5C gives miR-34a expression was significant increased in miR-34a mimics transducted MM RPMI8226 cells compared with in the miR-ctrl mimics transducted RPMI8226 cells in 48 h (P<0.01) or in 72 h (P<0.005); whereas the TGIF2 expression in Figure 5D was significant decreased in miR-34a mimics transducted MM RPMI8226 cells compared with miR-ctrl transducted RPMI8226 cells, in particular 72 h after transduction (P<0.05). Consistent with the miR-34a mimics transducted cells, the expression of miR-34a and TGIF2 were also true in tumor tissues from the MM bearing NOD/SCID mice injected with miR-34a-MM CSCs or vector-MM CSCs, which was statistically significant (P<0.05) as is shown in Figure 5E. These data were further confirmed by Western blot assay in Figure 6, in where TGIF2 expression was significantly reduced in miR-34a-MM cells (Figure 6A) and in tumor tissues from the mice injected with miR-34a-MM CSCs (Figure 6C) compared with their corresponding controls. The statistical significant differences (P<0.05 or P<0.01) were shown in Figure 6B and 6D. These results demonstrated the miR-34a did directly suppress the TGIF2 gene expression, suggesting the overexpression of miR-34a was association with inhibition of MM cell tumorigenicity and bone lesions via down regulation of TGIF2 expression.
Figure 6.
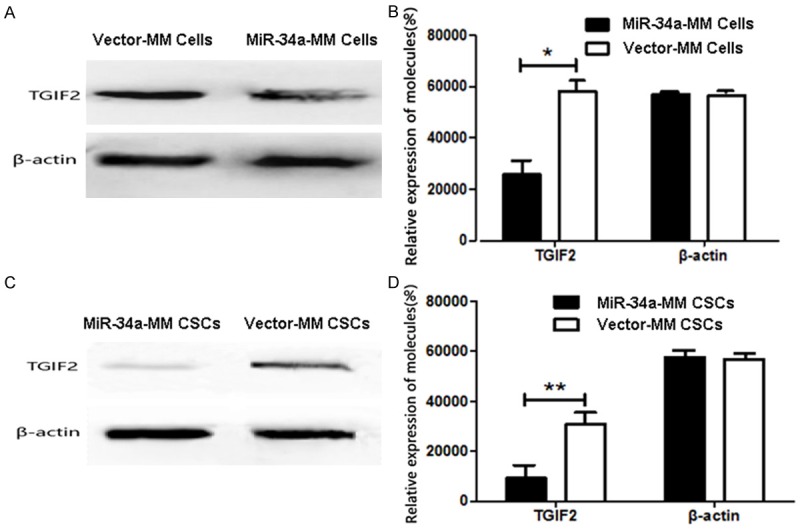
TGIF2 expression in MM cells and MM tissues. A. TGIF2 expression analyzed by Western blot assay in miR-34a MM and Vector-MM cells. B. Semi-quantification analysis of miR-34a expression in MM cells. C. TGIF2 expression in MM tissues from the mice injected with miR-34a-MM CSCs or vector-MM CSCs. D. Semi-quantification analysis of miR-34a expression in MM tissues. *P<0.05 and **P<0.01.
Discussion
There is growing evidence that CSCs exist in several cancers including MM, but there is still no evidence that they can be targeted therapeutically [29]. Latest study approaches based on miR-34 family has received much attention to potentially regulating tumor progression. However, it is unknown whether the MM CSCs, the “seed cells” in MM, could be closely associated with the epigenetic regulating role of miR-34a. To this end, we identified the miR-34a expression in human MM RPMI 8226 cells and MM CSCs. The results showed the miR-34a expression in MM CSCs was lower than in MM cells, and in order to up regulate miR-34a expression in MM CSCs, we first forced miR-34a overexpression in MM cells with transfection of recombinant containing miR-34a by pullulan-spermine, then we isolated the MM CSCs, marked by CD138-CD34-cell phenotypes, from recombinant stable transfected miR-34a-MM cells, and investigated the properties of the miR-34a-overexpressed cells in vitro and in vivo. The conspicuous epigenetic regulation action induced by miR-34a overexpression was found not only in MM RPMI 8226 cells but also in MM CSC xenograft tumors. The data from our current investigation showed that miR-34a overexpression were highly efficient in inhibiting the MM cell growth, clone formation, and inducing cell apoptosis when compared with control cells in vitro. Moreover, the miR-34a-overexpression in MM CSCs exhibited in vivo against MM CSC xenografts in NOD-SCID mice. We observed significant MM growth inhibition, which was of benefit to mouse survival and lytic bone lesion amelioration. The efficacy was reflected in incerase of BMD of the spine and femur in MM bearing mice.
To understand the efficient mechanisms, we analyzed the miR-34a and TGIF2 expression in MM RPMI 8226 cells and tumor tissues from mice, which manifested efficient epigenetic modulation induced by miR-34a. This is because miR-34a mimics transducted cells caused low TGIF2 expression, and miR-34a-MM CSC xenograft tumor tissue indicated down-regulation of TGIF2 expression. It is known that TGIF2 is transcriptional repressor of TGFβ1 signaling via the Smad pathway that is associated with inhibition of osteoblastic growth and differentiation [11,30]. We guess that in this study, our developed miR-34a-MM cells and miR-34a-MM CSCs suppressed the osteoclasts growth and differentiation, resulting in promoting the amelioration of lytic bone lesions in MM bearing mice by inhibiting TGIF2 expression. Further investigation of the mechanisms of miR-34a overexpression in suppressing the tumorigenicity of MM CSCs still is a necessary for the miR-34a-therapeutics in MM patients [31].
In summary, the data of the present study demonstrated that the miR-34a overexpression significantly reduced the tumorigenicity and the lytic bone lesions of MM CD138-CD34-CSCs in NOD/SCID mice via down-regulation of TGIF2 expression and induction of apoptosis. The findings suggests that the miR-34a overexpression may represent a novel tool to target MM CSCs and to effectively treat the refractory and relapsed MM in clinic.
Acknowledgements
We thank Prof. Jian Liu and Prof. Haiyan Wu (Institute of Basic Medical Sciences, Chinese Academy of Medical Science, Beijing, 100730, China) kindly provided the Pullulan-spermine (Ps) nanomaterials for this study. The study has been supported by the National Natural Science Foundation of China (No. 81572887), and partly supported by the Graduate Research and Innovation Projects in Jiangsu Province of China (KYZZ16_0126) as well as partly supported by the Collaborative Innovation Center of Suzhou NanoScience and Technology.
Disclosure of conflict of interest
None.
References
- 1.Saini N, Mahindra A. Therapeutic strategies for the treatment of multiple myeloma. Discov Med. 2013;15:251–258. [PubMed] [Google Scholar]
- 2.Yang C, Xiong F, Dou J, Xue J, Zhan X, Shi F, Li M, Wu S, Luo S, Zhang T, Zhang Y, Ming J, Gu N. Target therapy of multiple myeloma by PTX-NPs and ABCG2 antibody in a mouse xenograft model. Oncotarget. 2015;6:27714–27724. doi: 10.18632/oncotarget.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.San Miguel JF, Paiva B, Lasarte JJ. Engineering anti-myeloma responses using affinity-enhanced TCR-engineered T cells. Cancer Cell. 2015;28:281–283. doi: 10.1016/j.ccell.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Borrello I. Can we change the disease biology of multiple myeloma? Leuk Res. 2012;36(Suppl 1):S3–12. doi: 10.1016/S0145-2126(12)70003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atanackovic D, Hildebrandt Y, Templin J, Cao Y, Keller C, Panse J, Meyer S, Reinhard H, Bartels K, Lajmi N, Sezer O, Zander AR, Marx AH, Uhlig R, Zustin J, Bokemeyer C, Kröger N. Role of interleukin 16 in multiple myeloma. J Natl Cancer Inst. 2012;104:1005–1020. doi: 10.1093/jnci/djs257. [DOI] [PubMed] [Google Scholar]
- 6.Abe M, Harada T, Matsumoto T. Concise review: defining and targeting myeloma stem cell-like cells. Stem Cells. 2014;32:1067–1073. doi: 10.1002/stem.1643. [DOI] [PubMed] [Google Scholar]
- 7.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–5022. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 9.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Wang CM, Jiang ZZ, Yu XJ, Fan CG, Xu FF, Zhang Q, Li LI, Li RF, Sun WS, Zhang ZH, Liu YG. MicroRNA-34c targets TGFB-induced factor homeobox 2, represses cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Oncol Lett. 2015;10:3095–3102. doi: 10.3892/ol.2015.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC, Xie XJ, He L, Mangala LS, Lopez-Berestein G, Sood AK, Mendell JT, Wan Y. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature. 2014;512:431–435. doi: 10.1038/nature13375. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Dou J, Pan M, Wen P, Li Y, Tang Q, Chu L, Zhao F, Jiang C, Hu W, Hu K, Gu N. Isolation and identification of cancer stem-like cells from murine melanoma cell lines. Cell Mol Immunol. 2007;4:467–472. [PubMed] [Google Scholar]
- 13.Yang C, Wang J, Chen D, Chen J, Xiong F, Zhang H, Zhang Y, Gu N, Dou J. Paclitaxel-Fe3O4 nanoparticles inhibit growth of CD138(-) CD34(-) tumor stem-like cells in multiple myeloma-bearing mice. Int J Nanomedicine. 2013;8:1439–1449. doi: 10.2147/IJN.S38447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi F, Yang F, He X, Zhang Y, Wu S, Li M, Zhang Y, Di W, Dou J, Gu N. Inhibitory effect of epirubicin-loaded lipid microbubbles with conjugated anti-ABCG2 antibody combined with therapeutic ultrasound on multiple myeloma cancer stem cells. J Drug Target. 2016;24:34–46. doi: 10.3109/1061186X.2015.1052075. [DOI] [PubMed] [Google Scholar]
- 15.Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, Schadendorf D, Paschen A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012;72:460–471. doi: 10.1158/0008-5472.CAN-11-1977. [DOI] [PubMed] [Google Scholar]
- 16.Chen D, Zhang Y, Wang J, Chen J, Yang C, Cai K, Wang X, Shi F, Dou J. MicroRNA-200c overexpression inhibits tumorigenicity and metastasis of CD117+CD44+ovarian cancer stem cells by regulating epithelial-mesenchymal transition. J Ovarian Res. 2013;6:50. doi: 10.1186/1757-2215-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He X, Wang J, Zhao F, Yu F, Chen D, Cai K, Yang C, Chen J, Dou J. Antitumor efficacy of viable tumor vaccine modified by heterogenetic ESAT-6 antigen and cytokine IL-21 in melanomatous mouse. Immunol Res. 2012;52:240–249. doi: 10.1007/s12026-012-8332-4. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Zhou D, He X, Wang Y, Hu W, Jiang L, Dou J. Effect of downregulated beta-catenin on cell proliferative activity, the sensitivity to chemotherapy drug and tumorigenicity of ovarian cancer cells. Cell Mol Biol (Noisy-le-grand) 2011;57(Suppl):OL1606–1613. [PubMed] [Google Scholar]
- 19.Ouyang L, Shen LY, Li T, Liu J. Inhibition effect of Oncostatin M on metastatic human lung cancer cells 95D in vitro and on murine melanoma cells B16BL6 in vivo. Biomed Res. 2006;27:197–202. doi: 10.2220/biomedres.27.197. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Hu H, Song L, Cai L, Wei R, Jin W. Epirubicin-mediated expression of miR-302b is involved in osteosarcoma apoptosis and cell cycle regulation. Toxicol Lett. 2013;222:1–9. doi: 10.1016/j.toxlet.2013.06.242. [DOI] [PubMed] [Google Scholar]
- 21.Delude C. Tumorigenesis: Testing ground for cancer stem cells. Nature. 2011;480:S43–5. doi: 10.1038/480S43a. [DOI] [PubMed] [Google Scholar]
- 22.Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, Ikeda H, Perrone G, Gorgun G, Patel K, Desai N, Sportelli P, Kapoor S, Vali S, Mukherjee S, Munshi NC, Anderson KC, Raje N. Dual inhibition of Akt/Mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther. 2010;9:963. doi: 10.1158/1535-7163.MCT-09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C, Xiong F, Wang J, Dou J, Chen J, Chen D, Zhang Y, Luo S, Gu N. Anti-ABCG2 monoclonal antibody in combination with paclitaxel nanoparticles against cancer stem-like cellactivity in multiple myeloma. Nanomedicine (Lond) 2014;9:45–60. doi: 10.2217/nnm.12.216. [DOI] [PubMed] [Google Scholar]
- 24.Chiyomaru T, Fukuhara S, Saini S, Majid S, Deng G, Shahryari V, Chang I, Tanaka Y, Enokida H, Nakagawa M, Dahiya R, Yamamura S. Long non-coding RNA HOTAIR is targeted and regulated by miR-141 in human cancer cells. J Biol Chem. 2014;289:12550–12565. doi: 10.1074/jbc.M113.488593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Cai K, Wang J, Wang X, Cheng K, Shi F, Jiang L, Zhang Y, Dou J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells. 2014;32:2858–2868. doi: 10.1002/stem.1795. [DOI] [PubMed] [Google Scholar]
- 26.Melhuish TA, Taniguchi K, Wotton D. Tgif1 and Tgif2 regulate axial patterning in mouse. PLoS One. 2016;11:e0155837. doi: 10.1371/journal.pone.0155837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Pu Q, Cui B, Lin J. MicroRNA-34a inhibits tumor invasion and metastasis in gastric cancer by targeting Tgif2. Int J Clin Exp Pathol. 2015;8:8921–8928. [PMC free article] [PubMed] [Google Scholar]
- 28.Ebner OA, Selbach M. Quantitative proteomic analysis of gene regulation by miR-34a and miR-34c. PLoS One. 2014;9:e92166. doi: 10.1371/journal.pone.0092166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delude C. Tumorigenesis: testing ground for cancer stem cells. Nature. 2011;480:S43–5. doi: 10.1038/480S43a. [DOI] [PubMed] [Google Scholar]
- 30.Sharma A, Sinha NR, Siddiqui S, Mohan RR. Role of 5’TG3’-interacting factors (TGIFs) in Vorinostat (HDAC inhibitor)-mediated Corneal Fibrosis Inhibition. Mol Vis. 2015;21:974–84. [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Fei X, Shi F, Dou J, Wu S, Wu D, Zhang Y, Pan M, Luo S, Gu N. Homoharringtonine delivered by high proportion PEG of long- circulating liposomes inhibits RPMI8226 multiple myeloma cells in vitro and in vivo . Am J Transl Res. 2016;8:1355–1368. [PMC free article] [PubMed] [Google Scholar]