

Abstract
Diabetes mellitus is frequently accompanied by chronic complications like delayed wound healing, which is consider to be attributed to the accumulation of advanced glycosylation end product (AGE). However, the impacts of AGE on epidermal stem cells (ESCs) are largely unknown. This study aims to address the influence and mechanism of AGE on ESCs. ESCs isolated from rats were cultured in AGE-modified bovine serum albumin and transfected with small interfering RNA to knock down AGE-specific receptor (AGER). Expression of stem cell markers integrin β1 (ITGB1) and keratin 19 (KRT19), cell viability, apoptosis and reactive oxygen species (ROS) were examined. Wnt pathway-related factors Wnt family member 1 (WNT1), WNT3A, β-catenin, v-myc avian myelocytomatosis viral oncogene homolog (MYC), cyclin D1 (CCND1) and matrix metallopeptidase 7 (MMP7) were quantified. The interaction between forkhead box O1 (FOXO1) and β-catenin was assessed by co-immunoprecipitation. Results indicated that AGE down-regulated ITGB1 and KRT19 expression, suppressed ESC viability and promoted apoptosis, and ROS level (P < 0.01), implying decreased capacities of ESCs. AGE also promoted AGER and FOXO1, while AGER knockdown had the opposite effects. Moreover, AGER knockdown elevated the level of WNT1, WNT3A, MYC, CCND1 and MMP7 that were suppressed by AGE (P < 0.01). Immunoprecipitation analysis showed that FOXO1 could compete with lymphoid enhancer binding factor 1 to interact with β-catenin, which might help to elucidate the mechanism of AGE repressing ESCs. This study helps to understand the mechanism of accumulated AGE in affecting ESC capacities, and provides potential therapeutic targets to meliorate diabetic wound healing.
Keywords: Diabetic wound healing, epidermal stem cell, advanced glycosylation end product, forkhead box O1, Wnt
Introduction
Diabetes mellitus is a kind of metabolic disease severely threatening the health of human beings. Statistically, there are nearly 3 hundred million diabetes mellitus patients worldwide, and the number is predicted to soar to 4.39 hundred million in 2030 [1]. Diabetes mellitus has acquired deadly features partly due to its various complications, among which diabetic wound healing is quite typical. Diabetic wound healing can be delayed and impaired by numerous physiological factors that are already discovered, such as reduced production of growth factors [2], elevation of reactive oxygen species (ROS) [3] and dysregulation of matrix metallopeptidases (MMPs) [4]. Effective improvement of diabetic wound healing can obviously lower amputation rate, and intensive research has focused on the potential of auxiliary methods and pivotal factors [5,6].
Advanced glycosylation end product (AGE) is produced by the Maillard reaction between proteins and various saccharide derivatives. AGE plays causative roles in many degenerative diseases including diabetic mellitus and related complications. Studies have suggested that AGE is accumulated in some cell types of diabetic mellitus patients and participates in the pathogenesis of delayed diabetic wound healing by interfering with a number of components [7]. As the membrane receptor for AGE, AGE-specific receptor (AGER) is also involved in the development and progression of many diseases, whose depressor is a research hotspot for therapeutics [8,9]. In diabetic patients, AGER is higher expressed in inflammatory cells of wound tissue, together with the increased ROS level in neutrophils [10]. These findings might provide ample evidences for the pivotal role of AGE and AGER in the mechanism of delayed diabetic wound healing.
Wnt pathway, including the canonical and non-canonical ones, has been addressed in cell proliferation and survival. Evidences have also been found the involvement of Wnt pathway, the canonical Wnt/β-catenin including downstream transcription factors T-cell factors (TCFs) and lymphoid enhancer binding factor 1 (LEF1) particularly, in improving wound healing [11,12]. For example, Wnt pathway provides signals for the epithelium to facilitate epithelial tissue patterning during wound repair [13]. Furthermore, it is the effector for other regulators such as pigment epithelium-derived factor to influence wound healing in diabetic patients [14]. Thus Wnt pathway holds great potential to decipher the mechanism of diabetic wound healing.
Epidermal stem cells (ESCs) are the stem cells specifically existed in the skin tissue, which participate in skin development, wound repair and reconstruction. Studies have found impaired capacities of ESCs during diabetic wound healing [15,16]. However, the causal relationship between AGE and the suppression in ESC capacities is unclear. Therefore, we performed this study to uncover the effects and mechanisms of AGE-AGER in ESCs in order to elucidate how these factors influence diabetic wound healing. Rat ESCs were isolated and cultured for AGE treatment and AGER knockdown, and then cell viability, apoptosis, expression of ESC markers and ROS level were compared. Expression of Wnt pathway factors and interaction between proteins were also analyzed. These investigations will help to understand the mechanism of impaired diabetic wound healing and provided promising therapeutic targets to improve wound healing in diabetic patients.
Materials and methods
Animals and cells
This study was approved by a local ethics committee and conducted based on the instructions of our institute. Neonatal Sprague-Dawley rats of 1-3 d old (10 individuals, 6-8 g, HFK Bio-Technology, Beijing, China) were sacrificed by neck dislocation. The skin of the back was sampled and digested in 0.25% Dispase II (Roche Diagnosis, Mannheim, Germany) overnight at 4°C. Then the dermis was peeled off and the epidermis was digested in 0.05% Trypsin (Gibco, Carlsbad, CA, USA) for 5 min at 37°C. Dulbecco’s modified eagle medium supplemented with 20% fetal bovine serum (FBS) was added to terminate the digestion. Single-cell suspension was seeded in culture plates pre-coated with 100 mg/L collagen type IV (Equl, Shanghai, China) in phosphate-buffered saline (PBS) and incubated at 37°C for 20 min, after which the non-adhensive cells were discarded. The medium was replaced with keratinocyte serum-free medium (KSFM, Gibco). Cells were cultured in humid air with 5% CO2 at 37°C. The medium was changed every other day. The identification of ESCs was performed by antibodies against integrin β1 (ab179471, Abcam, Cambridge, UK), keratin 19 (KRT19, ab191208) and KRT10 (ab76318) [17,18] using flow cytometry Attune Nxt (Invitrogen, Carlsbad, CA, USA). The percent of integrin β1- and KRT19-positive cells was over 85%, and that of KRT10-positive cells was less than 10%.
Cell treatment and transfection
According to previous research, 200 μg/mL AGE-modified bovine serum albumin (AGE-BSA, BioVision, Milpitas, CA, USA) was added to the cultured ESCs [19]. As a control, the same concentration of BSA (BioVision) was used. The cultured ESCs were treated for 48 h before the following experiments. For cell viability assay, the cells were treated with AGE-BSA or BSA for 72 h, during which they were sampled at preset time points.
The cells were transfected with small interfering RNA (siRNA) for Ager (si-AGER). The siRNAs and siRNA control (si-control) were synthesized by Genechem (Shanghai, China). Transfection was performed in 24-well plates when the confluence reached about 80%. In each well, 5 pmol si-AGER or si-control was added together with the Lipofactamine® RNAiMAX (Invitrogen). The plates were incubated at 37°C for 72 h, during which cells were sampled for the following detection.
MTT assay
MTT assay was performed to detect viability of ESCs at 0, 1, 2 and 3 d post treatment or transfection using MTT Cell Proliferation Assay Kit (ATCC, Manassas, VA, USA). MTT Reagent (10 μL) was added to each well of 24-well plates and the plates were incubated for 4 h. Then 100 μL Detergent Reagent was added and the plates were incubated in the dark until the purple precipitates were dissolved. The absorbance at 570 nm was detected by a microplate reader HBS-1096B (DeTie, Nanjing, China).
Cell apoptosis assay
Apoptotic cells were detected by flow cytometry after ESCs were stained by fluorescein isothiocyanate (FITC) and propidium iodide (PI) using Annexin V-FITC Apoptosis Detection Kit (Vazyme, Nanjing, China). ESCs were collected after Trypsin treatment and centrifugation, and then washed twice in cold PBS. Cells were suspended in 100 μL Binding Buffer, after which 5 μL Annexin V-FITC and 5 μL PI Staining Solution were added. Then cells were incubated in the dark at room temperature for 10 min. Finally, 400 μL Binding Buffer was added and the detection was immediately performed by flow cytometry. The percent of FITC-positive and PI-negative cells were calculated to assess apoptotic cells.
ROS assay
Cellular ROS level in epidermal stem cells were detected by Reactive Oxygen Species Assay Kit (Solarbio, Beijing, China) according to the manufacturer’s instructions. In the 24-well plates, 2,7-dichlorofluorescin diacetate (DCFH-DA) was added to a final concentration of 1 μM. The cells were incubated at 37°C for 30 min and washed twice in PBS. Fluorescence signals were detected by a fluorescence microscope DM500 (Leica Microsystems, Wetzlar, Germany) and quantified by software Qwin (Leica Microsystems).
Immunoprecipitation
Immunoprecipitation was performed using Classic Magnetic IP/Co-IP Kit (Pierce, Carlsbad, CA, USA) to analyze the interaction between β-catenin and forkhead box O1 (FOXO1). ESCs were washed twice in PBS and lysed in IP Lysis Buffer on ice for 5 min. The protein lysate was collected by centrifugation. Protein A/G Magnetic Beads (25 μL) were incubated with anti-β-catenin antibody (ab22656) for 1 h at room temperature, and then added to the protein lysate and incubated overnight at 4°C. The beads were then collected and washed in IP Wash Buffer for 5 times. Proteins were dissolved in Elution Buffer and detected by Western blot.
qRT-PCR
Total RNA samples of the epidermal stem cells were extracted using Trizol (Invitrogen) and purified by RNA Cleanup Kit (Qiagen, Dusseldorf, Germany) based on the instructions of the manufacturers. The complementary DNA (cDNA) was synthesized using PrimeScript 1st Strand cDNA Synthesis Kit (Takara, Dalian, China) with 1 μg RNA from for each sample. qRT-PCR was performed on LightCycler 480 (Roche, Basel, Switzerland) catalyzed by SYBR Green I Master (Roche), each sample containing 20 ng cDNA and the specific primers for targeted genes (Table 1). Data were calculated by the 2-ΔΔCt method normalized with glyceraldehyde-3-phosphate dehydrogenase (Gapdh).
Table 1.
Primers used in qRT-PCR
| Primer | Targeted gene (GenBank Accession) | Sequence (5’ to 3’) | Produce size (bp) |
|---|---|---|---|
| Itgb1-Fw | Itgb1 (NM_017022) | GGGACACGGGTGAAAATCCT | 152 |
| Itgb1-Rv | AGAGCCCCAAAGCTACCCTA | ||
| Krt19-Fw | Krt19 (BC126075) | CATGGCAGAGAAGAACCGGA | 125 |
| Krt19-Rv | GGAGTTCCGTGACCTCAGTC | ||
| Ager-Fw | Ager (NM_053336) | GTCTCCTTCAGCTTCCGACC | 117 |
| Ager-Rv | AGCATGGATCATGTGGGCTC | ||
| Foxo1-Fw | Foxo1 (NM_001191846) | CAGCCAGGCACCTCATAACA | 143 |
| Foxo1-Rv | TCAAGCGGTTCATGGCAGAT | ||
| Wnt1-Fw | Wnt1 (NM_001105714) | CGTTGCTGTCCCTGTGGTAT | 105 |
| Wnt1-Rv | CAGGTGTGGTGGTTAGGGAC | ||
| Wnt3a-Fw | Wnt3a (NM_001107005) | GACCTTGAGGCCACGTTACA | 90 |
| Wnt3a-Rv | TTGGGCTCGCAGAAGTTAGG | ||
| Ctnnb1-Fw | Ctnnb1 (NM_053357) | ACTCCAGGAATGAAGGCGTG | 109 |
| Ctnnb1-Rv | GAACTGGTCAGCTCAACCGA | ||
| Myc-Fw | Myc (NM_012603) | TGAAAAGAGCTCCTCGCGTT | 139 |
| Myc-Rv | AAATAGGGCTGCACCGAGTC | ||
| Ccnd1-Fw | Ccnd1 (NM_171992) | TCAAGTGTGACCCGGACTG | 138 |
| Ccnd1-Rv | GACCAGCTTCTTCCTCCACTT | ||
| Mmp7-Fw | Mmp7 (NM_012864) | CAAAGGACGACATTGCAGGC | 94 |
| Mmp7-Rv | GAAGGGCGTTTGCTCATTCC | ||
| Gapdh-Fw | Gapdh (NM_017008) | TCCTGCACCACCAACTGCTTAG | 102 |
| Gapdh-Rv | AGTGGCAGTGATGGCATGGACT |
Western blot
Protein samples of ESCs were extracted by ProteoPrep Total Extraction Sample Kit (Sigma-Aldrich, Shanghai, China) according to the manufacturer’s instructions. The proteins (20 μg) of each sample were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were first blocked in 5% skim milk (in PBS) for 2 h at room temperature and then incubated overnight at 4°C in the specific primary antibodies against GAPDH (ab8245, Abcam), which was used as an internal reference, AGER (ab3611), FOXO1 (ab52857), Wnt family member 1 (WNT1, ab85060), WNT3a (Sangon Biotech, Shanghai, China), β-catenin (ab32572), v-myc avian myelocytomatosis viral oncogene homolog (MYC, ab32072), cyclin D1 (ab134175) and MMP7 (ab189277). After being washed in PBS for 3 times (5 min for each time), the membranes were incubated in secondary antibodies conjugated with horseradish peroxidase for 2 h at room temperature. Then the membranes were washed in PBS again and EasyBlot ECL Kit (Sangon Biotech) was used to develop signals, which were then analyzed by ImageJ 1.49 (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All the experiments were performed in triplicate. Results were presented as mean ± standard error of mean. Data were analyzed by one-way analysis of variance and Student’s t test in SPSS 20 (SPSS, Chicago, IL, USA). Differences between groups were considered statistically significant if P < 0.05.
Results
AGE inhibits capacities of ESCs and promotes FOXO1
We first detected the effects of AGE on ESCs from four indexes: ESC markers, cell viability, cell apoptosis and ROS level. qRT-PCR indicates that the mRNA levels of two ESC markers integrin β1 (Itgb1) and Krt19 were obviously suppressed after 48 h of AGE-BSA treatment (P < 0.01, Figure 1A), which might imply the weakened characteristics of ESCs. MTT assay showed that the cell viability was significantly suppressed after 1, 2 or 3 d of AGE-BSA treatment (P < 0.01 or P < 0.001, Figure 1B). Meanwhile, the percent of apoptotic cells was markedly elevated by AGE-BSA (P < 0.01, Figure 1C). The ROS level of ESCs was also elevated by AGE-BSA treatment (P < 0.01, Figure 1D). These results implied that accumulation of AGE might affect the capacities of ESCs.
Figure 1.
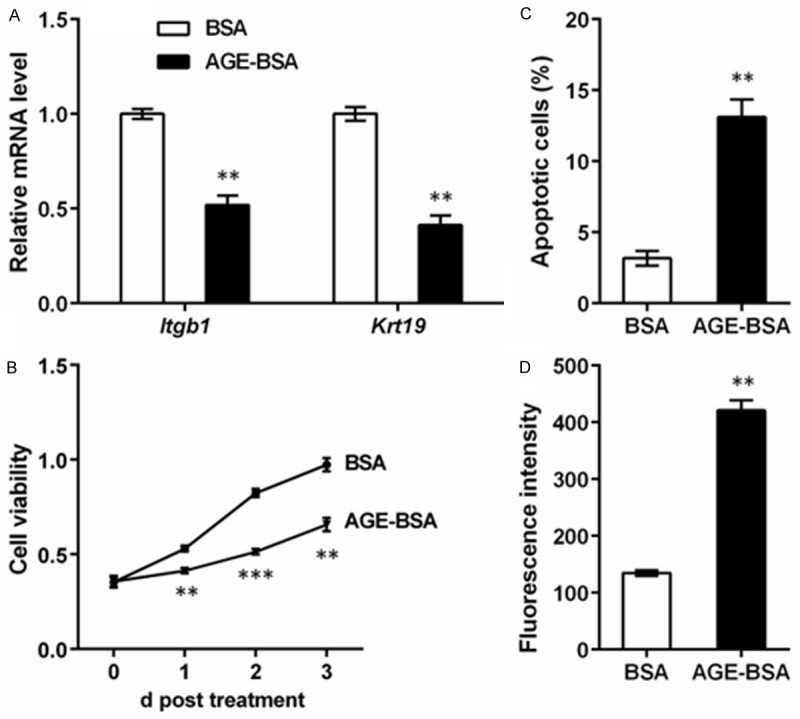
Advanced glycosylation end product (AGE) inhibits capacities of epidermal stem cells (ESCs). Rat ESCs were isolated and cultured with AGE-modified bovine serum albumin (AGE-BSA) or BSA (as a control) treatment. Cells were detected by qRT-PCR, flow cytometry and reactive oxygen species (ROS) assay after 48 h of treatment. A. qRT-PCR shows that mRNA levels of ESC markers integrin β1 (Itgb1) and keratin 19 (Krt19) are suppressed by AGE-BSA treatment. B. Flow cytometry shows that the percent of apoptotic cells is increased by AGE-BSA treatment. C. MTT assay shows that the viability of ESCs is inhibited by AGE-BSA treatment when detected at 1, 2 and 3 d post treatment. D. ROS level is elevated by AGE-BSA treatment. **P < 0.01 and ***P < 0.001 compared to the BSA group.
Then we detected the expression change of AGER. qRT-PCR showed significant elevation in Ager mRNA after AGE-BSA treatment (P < 0.001, Figure 2A), and western blot found similar expression patterns in its proteins (Figure 2B). Intriguingly, FOXO1, a factor that has been reported up-regulated in diabetic mellitus [20], was also significantly promoted in both mRNA (P < 0.05) and protein levels. It might be conjectured from these phenomena that AGER and FOXO1 might be involved in the functions of AGE in ESCs.
Figure 2.
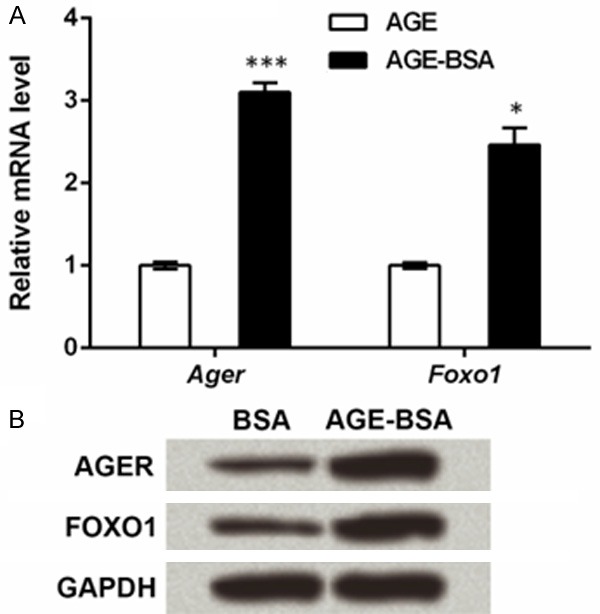
Advanced glycosylation end product (AGE) elevates expression of AGE-specific receptor (AGER) and forkhead box O1 (FOXO1) in epidermal stem cells (ESCs). Rat ESCs were isolated and cultured with AGE-modified bovine serum albumin (AGE-BSA) or BSA (as a control) treatment. Cells were detected by qRT-PCR and Western blot after 48 h of treatment. A. qRT-PCR shows that mRNA levels of Ager and Foxo1 are elevated by AGE-BSA treatment. *P < 0.05 and ***P < 0.001 compared to the BSA group. B. Western blot shows the expression of AGER and FOXO1 proteins are increased by AGE-BSA treatment. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an internal reference.
AGER knockdown elevates capacities of ESCs
In order to investigate the possible mechanism of AGE on ESCs and elucidate the function of AGER in the mechanism, we knocked down AGER in the cultured ESCs with AGE-BSA treatment. After cell transfection, the Ager mRNA level was significantly suppressed (P < 0.01, Figure 3A), and its protein expression was also reduced (Figure 3B), suggesting the successful knockdown of AGER. Meanwhile, we discovered that AGER knockdown was accompanied by mRNA and protein down-regulation of FOXO1 (P < 0.01), implying that AGER knockdown affected the expression of FOXO1.
Figure 3.
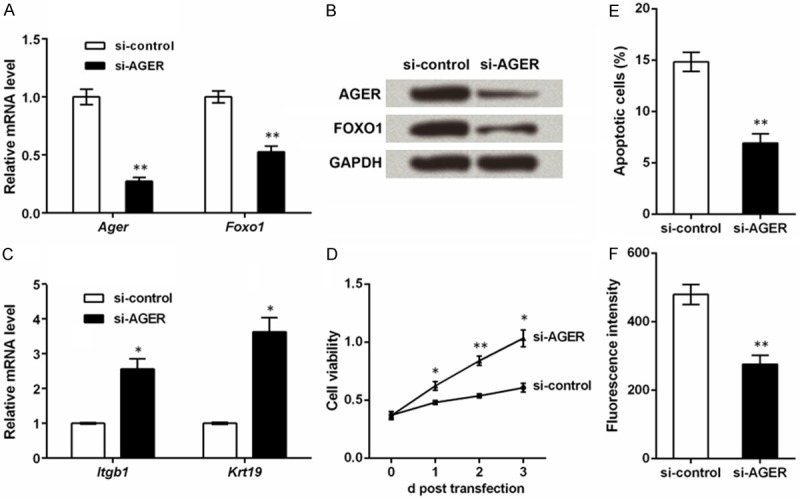
Knockdown of advanced glycosylation end product-specific receptor (AGER) improves capacities of epidermal stem cells (ESCs). Rat ESCs were isolated and cultured with AGE-modified bovine serum albumin (AGE-BSA) for 48 h, after which they were transfected with the specific small interfering RNA for Ager (si-AGER) or the control (si-control). Detection of AGER levels by qRT-PCR (A) and Western blot (B) at 48 h post transfection suggests the successful knockdown of AGER. Meanwhile, forkhead box O1 (FOXO1) is also suppressed. (C) qRT-PCR shows that mRNA levels of ESC markers integrin β1 (Itgb1) and keratin 19 (Krt19) are suppressed by AGER knockdown at 48 h post transfection. (D) MTT assay shows that cell viability is elevated by AGER knockdown when detected at 1, 2 and 3 d post transfection. (E) Flow cytometry indicates that the percent of apoptotic cells is reduced by AGER knockdown at 48 h post transfection. (F) ROS level is suppressed by AGER knockdown at 48 h post transfection. *P < 0.05 and **P < 0.01 compared to the si-control group.
ESC markers, cell viability, cell apoptosis and ROS level were also assessed. Itgb1 and Krt19 mRNA levels were elevated along with the knockdown of AGER (P < 0.05, Figure 3C). Cell viability detected at 1, 2 and 3 d post transfection were all significantly promoted by AGER knockdown (P < 0.05 or P < 0.01, Figure 3D), while cell apoptosis was suppressed by AGER knockdown (P < 0.01, Figure 3E). Moreover, si-AGER could also reduce ROS level in ESCs (P < 0.01, Figure 3F). Taken together, AGER knockdown might generate opposite effects against AGE-BSA treatment, improving capacities of ESCs.
AGE affects Wnt pathway
Factors in Wnt pathway were also detected in the ESCs transfected with si-AGER. qRT-PCR showed that AGE-BSA treatment could significantly down-regulate the mRNA level of Wnt1, Wnt3a, Myc, cyclin D1 (Ccnd1) and Mmp7 compared to BSA treatment (P < 0.05 or P < 0.01, Figure 4A). However, transfection with si-AGER further elevated levels of these factors and β-catenin (Ctnnb1) compared to the cells only treated with AGE-BSA (P < 0.01). Western blot showed similar protein expression patterns of these factors (Figure 4B). Thus AGE might have suppressive effects on Wnt pathway, while knockdown of AGER could reverse the effect of AGE.
Figure 4.
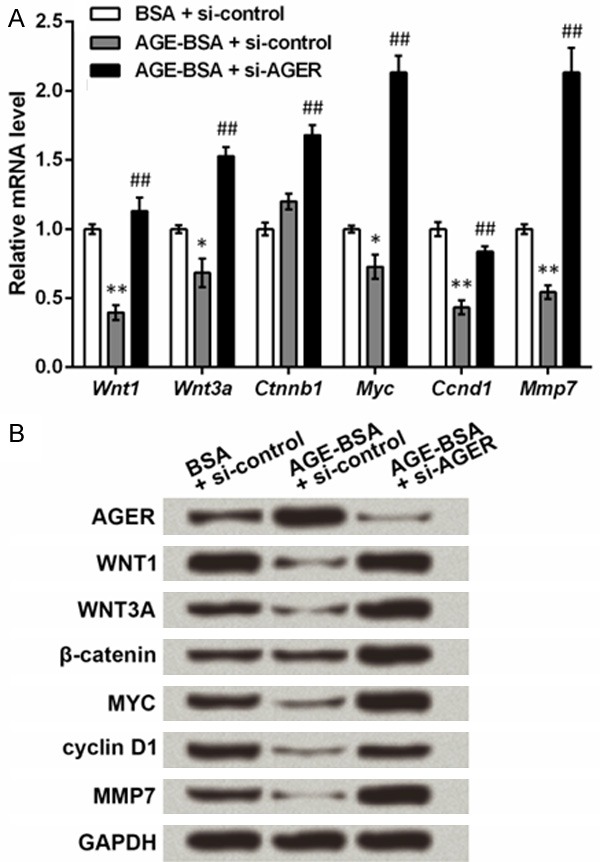
Advanced glycosylation end product (AGE) treatment and knockdown of AGE-specific receptor (AGER) can regulate Wnt pathway. Rat epidermal stem cells were isolated and cultured with AGE-modified bovine serum albumin (AGE-BSA) or BSA (as a control) treatment for 48 h, after which they were transfected with the specific small interfering RNA for Ager (si-AGER) or the control (si-control). qRT-PCR and Western blot were performed at 48 h post transfection. A. qRT-PCR shows that AGE and AGER regulate the mRNA level of Wnt pathway factors including Wnt family member 1 (Wnt1), Wnt family member 3a (Wnt3a), β-catenin (Ctnnb1), v-myc avian myelocytomatosis viral oncogene homolog (Myc), cyclin D1 (Ccnd1) and matrix metallopeptidase 7 (Mmp7). *P < 0.05 and **P < 0.01 compared to the BSA + si-control group. ##P < 0.01 compared to the AGE-BSA + si-AGER group. B. Western blot shows that AGE and AGER regulate the protein level of Wnt pathway factors. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an internal reference.
In Figures 2 and 3, results showed that FOXO1 was elevated or suppressed along with the AGE-induced AGER promotion or AGER knockdown. Thus we speculated that FOXO1 was also involved in the regulation of Wnt pathway. Since β-catenin is a crucial junction which can be bind by lymphoid enhancer binding factor 1 (LEF1) to activate Wnt pathway, we performed immunoprecipitation using antibodies against β-catenin to test whether FOXO1 could interact with β-catenin. Results showed that FOXO1 protein could be detected in the immunoprecipitates of β-catenin (Figure 5), but its level was increased by AGE-BSA treatment, and then reduced by si-AGER. Simultaneously, it was the other way around that LEF1 protein level in the immunoprecipitates was reduced by AGE-BSA and then increased by si-AGER, implying that the interaction between β-catenin and LEF1 might be taken away by FOXO1. Thus FOXO1 competed with LEF1 to interact with β-catenin, by which it might participate in the regulation of Wnt pathway.
Figure 5.
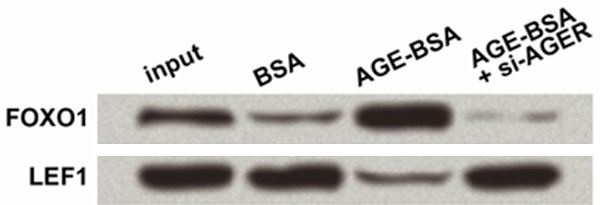
Forkhead box O1 (FOXO1) competes with lymphoid enhancer binding factor 1 (LEF1) to interact with β-catenin protein. Rat epidermal stem cells were isolated and cultured with AGE-modified bovine serum albumin (AGE-BSA) or BSA (as a control) treatment for 48 h, after which they were transfected with the specific small interfering RNA for Ager (si-AGER) or the control (si-control). Immunoprecipitation was performed at 48 h post transfection with the antibodies against β-catenin. Immunoprecipitation products were detected by Western blot with primary antibodies against FOXO1 and LEF1.
Discussion
Delayed diabetic wound healing has long been a thorny problem for diabetic patients. In this study, we detected the relationship between the accumulated AGE and the repressed ESCs by treating rat ESCs with AGE-BSA to mimic AGE accumulation during diabetic mellitus. The treatment was found to suppress expression of ESC markers, inhibit viability, and promote apoptosis and ROS level, and meanwhile, the expression of AGER and FOXO1 was elevated. Further, knockdown of AGER by the siRNA had opposite effects to AGE. We also found that AGE treatment and AGER knockdown could regulate expression of Wnt pathway factors adversely, and that FOXO1 competed with LEF1 to interact with β-catenin.
In the cultured rat ESCs, AGE-BSA treatment diminished expression of two ESC markers integrin β1 and KRT19, suggesting the debilitated ESC features. The two markers have also been reported to regulate proliferation and apoptosis of skin cells. For example, knockdown of ITGB1 in human keratinocyte stem cells could inhibit cell proliferation and induce differentiation [21], and overexpression of KRT19 is observed in skin cancer [22]. Consistently in this study, the suppressed integrin β1 and KRT19 levels by AGE-BSA were accompanied by reduced ESC viability and increased cell apoptosis and ROS levels. These findings were in accordance with existed reports in mesenchymal stem cells, endothelial progenitor cells, where AGE elevates cell apoptosis and induces production of ROS [23-25]. Thus these results implied that AGE accumulation represses capacities of ESCs during diabetic mellitus, which may delay wound healing.
When detecting the mRNA and protein level of AGER and FOXO1, we found that both factors were induced by the exposure to AGE. Previous studies in osteoblastic cell lines also observed induced AGER expression by AGE treatment [26]. As to FOXO1, it can be elevated by AGE and is associated with aggravated apoptosis of fibroblasts and retinal pericytes, possibly via p38 and JNK/MAPK [27,28]. Further AGER knockdown in rat ESCs caused elevation in integrin β1, KRT19 and cell viability, as well as the decrease in apoptotic cell percent and ROS level, which were opposite to AGE-BSA treatment and similar results have been reported [29,30]. Meanwhile, knockdown of AGER led to the inhibition of FOXO1, which is a factor suppressing proliferation and inducing apoptosis upon oxidative stress in some cell types [31,32]. From this point of view, the AGE-AGER-FOXO1 axis may be active for the repression of ESC capacities during diabetic wound healing.
The canonical Wnt pathway is crucial for sustaining cell survival and growth, thus providing promising targets for many diseases [33,34]. Moreover, it can be modulated by ROS and constitute a crosstalk during important biological processes [35]. In this study, we explored changes in this pathway, hoping to find possible mechanisms of AGE in ESCs. As expected, WNT1, WNT3A and β-catenin in Wnt pathway and the downstream factors MYC, cyclin D1 and MMP7 were all regulated by AGE-BSA treatment or AGER knockdown, implying that the regulation of Wnt pathway by AGE-AGER may play significant roles in modulating capacities of ESCs. Similar results show that overexpression of AGER inhibits Wnt pathway and reduces expression of Wnt targets MYC and cyclin D1 to suppress osteoblast proliferation [36], raising the possibility that Wnt pathway is a vital responder of AGE-AGER to modulate ESCs.
In the regulation of Wnt pathway factors, significant mRNA level changes were detected in Myc, Ccnd1 and Mmp7, all of which are targets for the transcription factor β-catenin [37-39], thus we suspected that the transcriptional activity of β-catenin was regulated. We analyzed the immunoprecipitation of FOXO1, which is also a transcription factor, by β-catenin, and found that when FOXO1 was up-regulated by AGE-BSA treatment, β-catenin immunoprecipitated more FOXO1 and less LEF1 than the BSA control, while the inverse phenomenon was observed when AGER was knocked down. Since LEF1 is an important regulator of β-catenin activity by directly interacting with β-catenin [40,41], it is tempting to deduce that FOXO1 may compete with LEF1 to interact with β-catenin, thus reducing the LEF1-mediated transportation of β-catenin to the nucleus. The blocked β-catenin activity may further lead to the suppressed transcription of its targets Myc, Ccnd1 and Mmp7. Hence the AGE-AGER-FOXO1 axis may suppress Wnt pathway and targets of β-catenin, which is a potential mechanism to explain the suppressed ESC capacities during diabetic wound healing.
It was intriguing that the mRNA and protein changes of β-catenin by AGE-BSA treatment were not significant, while changes in WNT1 and WNT3A were remarkable. Furthermore, si-AGER was able to induce significant changes in β-catenin levels. One possible reason is that AGE may cause multiple alterations other than AGER elevation and impose a comprehensive effect on β-catenin, which needs to be verified by further research. With more details about AGE mechanism revealed, the AGE-AGER-FOXO1-Wnt axis will be of great importance to improve the capacities of ESCs and meliorate diabetic wound healing.
To sum up, accumulation of AGE and its induced AGER-FOXO1 suppress Wnt pathway in ESCs, which may contribute to the repression in ESC capacities and delayed diabetic wound healing. This potential mechanism will offer promising therapeutic targets for promoting diabetic wound healing.
Acknowledgements
This work received the supports by Medical Foundation of Wu Jieping (320.6750.14139); National Natural Science Foundation of China (81401996) and Natural Science Foundation of Guangdong Province (2015A030310099).
Disclosure of conflict of interest
None.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Lin S, Xiao Y, Huang Y, Tan Y, Cai L, Li X. Acceleration of diabetic wound healing with chitosan-crosslinked collagen sponge containing recombinant human acidic fibroblast growth factor in healing-impaired STZ diabetic rats. Life Sci. 2008;82:190–204. doi: 10.1016/j.lfs.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Knobel D, Crawford JL, Butala P, Davidson EH, Wetterau M, Sultan SM, Szpalski C, Warren SM, Saadeh PB. Local reactive oxygen species scavenging improves diabetic wound healing. Journal of the American College of Surgeons. 2010;211:S80. [Google Scholar]
- 4.Botusan IR, Sunkari VG, Savu O, Catrina AI, Grunler J, Lindberg S, Pereira T, Yla-Herttuala S, Poellinger L, Brismar K, Catrina SB. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci U S A. 2008;105:19426–19431. doi: 10.1073/pnas.0805230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tellechea A, Silva EA, Min J, Leal EC, Auster ME, Pradhan-Nabzdyk L, Shih W, Mooney DJ, Veves A. Alginate and DNA Gels Are Suitable Delivery Systems for Diabetic Wound Healing. Int J Low Extrem Wounds. 2015;14:146–153. doi: 10.1177/1534734615580018. [DOI] [PubMed] [Google Scholar]
- 6.Tan Q, Chen B, Yan X, Lin Y, Xiao Z, Hou X, Dai J. Promotion of diabetic wound healing by collagen scaffold with collagen-binding vascular endothelial growth factor in a diabetic rat model. J Tissue Eng Regen Med. 2014;8:195–201. doi: 10.1002/term.1513. [DOI] [PubMed] [Google Scholar]
- 7.Peppa M, Stavroulakis P, Raptis SA. Advanced glycoxidation products and impaired diabetic wound healing. Wound Repair Regen. 2009;17:461–472. doi: 10.1111/j.1524-475X.2009.00518.x. [DOI] [PubMed] [Google Scholar]
- 8.Farmer DG, Ewart MA, Mair KM, Kennedy S. Soluble receptor for advanced glycation end products (sRAGE) attenuates haemodynamic changes to chronic hypoxia in the mouse. Pulm Pharmacol Ther. 2014;29:7–14. doi: 10.1016/j.pupt.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Wear-Maggitti K, Lee J, Conejero A, Schmidt AM, Grant R, Breitbart A. Use of topical sRAGE in diabetic wounds increases neovascularization and granulation tissue formation. Ann Plast Surg. 2004;52:519–521. doi: 10.1097/01.sap.0000122857.49274.8c. [DOI] [PubMed] [Google Scholar]
- 10.Niu YW, Miao MY, Dong W, Dong JY, Cao XZ, Lu SL. Effects of advanced glycation end products and its receptor on oxidative stress in diabetic wounds. Zhonghua Shao Shang Za Zhi. 2012;28:32–35. [PubMed] [Google Scholar]
- 11.Zhang DL, Gu LJ, Liu L, Wang CY, Sun BS, Li Z, Sung CK. Effect of Wnt signaling pathway on wound healing. Biochem Biophys Res Commun. 2009;378:149–151. doi: 10.1016/j.bbrc.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Targeting Wnt signaling to improve wound healing after myocardial infarction. Methods Mol Biol. 2013;1037:355–380. doi: 10.1007/978-1-62703-505-7_21. [DOI] [PubMed] [Google Scholar]
- 13.Fathke C, Wilson L, Shah K, Kim B, Hocking A, Moon R, Isik F. Wnt signaling induces epithelial differentiation during cutaneous wound healing. BMC Cell Biol. 2006;7:4. doi: 10.1186/1471-2121-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi W, Yang C, Dai Z, Che D, Feng J, Mao Y, Cheng R, Wang Z, He X, Zhou T, Gu X, Yan L, Yang X, Ma JX, Gao G. High levels of pigment epithelium-derived factor in diabetes impair wound healing through suppression of Wnt signaling. Diabetes. 2015;64:1407–1419. doi: 10.2337/db14-1111. [DOI] [PubMed] [Google Scholar]
- 15.Morasso MI, Tomic-Canic M. Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell. 2005;97:173–183. doi: 10.1042/BC20040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian X, Bai S, Tian W. Dynamic change of epidermal stem cells in the wound healing course of diabetic rats. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2007;21:693–697. [PubMed] [Google Scholar]
- 17.Zhang L, Ge Y, Fuchs E. miR-125b can enhance skin tumor initiation and promote malignant progression by repressing differentiation and prolonging cell survival. Genes Dev. 2014;28:2532–2546. doi: 10.1101/gad.248377.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu HW, Cheng B, Li JF, Wu HJ, Li KY, Sun TZ, Fu XB. Characterization of angiotensin-converting enzyme expression during epidermis morphogenesis in humans: a potential marker for epidermal stem cells. Br J Dermatol. 2009;160:250–258. doi: 10.1111/j.1365-2133.2008.08970.x. [DOI] [PubMed] [Google Scholar]
- 19.Li DX, Deng TZ, Lv J, Ke J. Advanced glycation end products (AGEs) and their receptor (RAGE) induce apoptosis of periodontal ligament fibroblasts. Braz J Med Biol Res. 2014;47:1036–1043. doi: 10.1590/1414-431X20143996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagashima T, Shigematsu N, Maruki R, Urano Y, Tanaka H, Shimaya A, Shimokawa T, Shibasaki M. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol Pharmacol. 2010;78:961–970. doi: 10.1124/mol.110.065714. [DOI] [PubMed] [Google Scholar]
- 21.Tang HY, Wu SH, Chen J, Liang HP, Su YY, Sun RJ, Luo XD. Influence of integrin β1 on the proliferation and differentiation of human keratinocyte stem cells. Chin J Exp Surg. 2007;24:22–25. [Google Scholar]
- 22.Ming M, Qiang L, Zhao B, He YY. Mammalian SIRT2 inhibits keratin 19 expression and is a tumor suppressor in skin. Exp Dermatol. 2014;23:207–209. doi: 10.1111/exd.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Li H, Guo R, Wang Q, Zhang D. Antioxidants inhibit advanced glycosylation end-product-induced apoptosis by downregulation of miR-223 in human adipose tissue-derived stem cells. Sci Rep. 2016;6:23021. doi: 10.1038/srep23021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Song M, Yu S, Gao P, Yu Y, Wang H, Huang L. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol Cell Biochem. 2010;335:137–146. doi: 10.1007/s11010-009-0250-y. [DOI] [PubMed] [Google Scholar]
- 25.Shi L, Yu X, Yang H, Wu X. Advanced glycation end products induce human corneal epithelial cells apoptosis through generation of reactive oxygen species and activation of JNK and p38 MAPK pathways. PLoS One. 2013;8:e66781. doi: 10.1371/journal.pone.0066781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cortizo AM, Lettieri MG, Barrio DA, Mercer N, Etcheverry SB, McCarthy AD. Advanced glycation end-products (AGEs) induce concerted changes in the osteoblastic expression of their receptor RAGE and in the activation of extracellular signal-regulated kinases (ERK) Mol Cell Biochem. 2003;250:1–10. doi: 10.1023/a:1024934008982. [DOI] [PubMed] [Google Scholar]
- 27.Alikhani M, Maclellan CM, Raptis M, Vora S, Trackman PC, Graves DT. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am J Physiol Cell Physiol. 2007;292:C850–C856. doi: 10.1152/ajpcell.00356.2006. [DOI] [PubMed] [Google Scholar]
- 28.Alikhani M, Roy S, Graves DT. FOXO1 plays an essential role in apoptosis of retinal pericytes. Mol Vis. 2010;16:408–415. [PMC free article] [PubMed] [Google Scholar]
- 29.Jin Q, Chen H, Luo A, Ding F, Liu Z. S100A14 stimulates cell proliferation and induces cell apoptosis at different concentrations via receptor for advanced glycation end products (RAGE) PLoS One. 2011;6:e19375. doi: 10.1371/journal.pone.0019375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xanthis A, Hatzitolios A, Fidani S, Befani C, Giannakoulas G, Koliakos G. Receptor of advanced glycation end products (RAGE) positively regulates CD36 expression and reactive oxygen species production in human monocytes in diabetes. Angiology. 2009;60:772–779. doi: 10.1177/0003319708328569. [DOI] [PubMed] [Google Scholar]
- 31.Prasad SB, Yadav SS, Das M, Govardhan HB, Pandey LK, Singh S, Pradhan S, Narayan G. Down Regulation of FOXO1 Promotes Cell Proliferation in Cervical Cancer. J Cancer. 2014;5:655–662. doi: 10.7150/jca.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang JQ, Gao BW, Wang J, Ren QL, Chen JF, Ma Q, Zhang ZJ, Xing BS. Critical Role of FoxO1 in Granulosa Cell Apoptosis Caused by Oxidative Stress and Protective Effects of Grape Seed Procyanidin B2. Oxid Med Cell Longev. 2016;2016:6147345. doi: 10.1155/2016/6147345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertrand FE, Angus CW, Partis WJ, Sigounas G. Developmental pathways in colon cancer: crosstalk between WNT, BMP, Hedgehog and Notch. Cell Cycle. 2012;11:4344–4351. doi: 10.4161/cc.22134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramachandran I, Thavathiru E, Ramalingam S, Natarajan G, Mills WK, Benbrook DM, Zuna R, Lightfoot S, Reis A, Anant S, Queimado L. Wnt inhibitory factor 1 induces apoptosis and inhibits cervical cancer growth, invasion and angiogenesis in vivo. Oncogene. 2012;31:2725–2737. doi: 10.1038/onc.2011.455. [DOI] [PubMed] [Google Scholar]
- 35.Wen JW, Hwang JT, Kelly GM. Reactive oxygen species and Wnt signalling crosstalk patterns mouse extraembryonic endoderm. Cell Signal. 2012;24:2337–2348. doi: 10.1016/j.cellsig.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 36.Li G, Xu J, Li Z. Receptor for advanced glycation end products inhibits proliferation in osteoblast through suppression of Wnt, PI3K and ERK signaling. Biochem Biophys Res Commun. 2012;423:684–689. doi: 10.1016/j.bbrc.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 37.Yochum GS, Cleland R, Goodman RH. A Genome-Wide Screen for β-Catenin Binding Sites Identifies a Downstream Enhancer Element That Controls c-Myc Gene Expression. Mol Cell Biol. 2008;28:7368–7379. doi: 10.1128/MCB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang I, Choi YS, Jeon MY, Jeong S. NF-κB p65 represses β-catenin-activated transcription of cyclin D1. Biochem Biophys Res Commun. 2010;403:79–84. doi: 10.1016/j.bbrc.2010.10.118. [DOI] [PubMed] [Google Scholar]
- 39.Uno S, Endo K, Jeong Y, Kawana K, Miyachi H, Hashimoto Y, Makishima M. Suppression of β-catenin signaling by liver X receptor ligands. Biochem Pharmacol. 2009;77:186–195. doi: 10.1016/j.bcp.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Feigin ME, Malbon CC. OSTM1 regulates beta-catenin/Lef1 interaction and is required for Wnt/beta-catenin signaling. Cell Signal. 2008;20:949–957. doi: 10.1016/j.cellsig.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTcf-3 Transcription Factor Mediates β-Catenin-Induced Axis Formation in Xenopus Embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]